

Abstract
Recent strides in anti-cancer therapeutics have improved longevity and led to a growing population of cancer survivors, who are increasingly likely to die of other causes. Treatment-induced cardiotoxicity is a complication of several therapeutic agents with acute and long-term consequences for cancer patients. Vascular endothelial dysfunction is a precursor and hallmark of ischemic coronary disease and may play a role in anti-cancer therapy-induced cardiotoxicity. This review summarizes clinical evidence for endothelial dysfunction following anti-cancer therapy and extends the discussion to include the impact of therapeutic agents on conduit arteries and the microcirculation. We highlight the role of innate immune system activation and cross-talk between inflammation and oxidative stress as pathogenic mechanisms underlying anti-cancer therapy-induced vascular toxicity. Understanding the impact of anti-cancer agents on the vascular endothelium will inform therapeutic approaches to prevent or reverse treatment-induced cardiotoxicity and may serve as an important tool to predict, monitor, and prevent adverse cardiovascular outcomes in patients undergoing treatment.
Keywords: Cardio-oncology, endothelium, inflammation, cancer-therapy related cardiac dysfunction, microcirculation, chemotherapy
1. Introduction
Advances in the efficacy of anti-cancer therapies (CTx) together with progress in screening and diagnosis have improved patient outcomes and led to a marked increase in cancer survivorship in recent decades (Siegel et al., 2021). The growing population of cancer survivors has revealed the need for research focused on understanding and mitigating the detrimental, off-target effects of CTx. Cardiotoxicity is a well-established complication of CTx that has given rise to the field of cardio-oncology, which aims to prevent, predict, and manage the cardiovascular consequences of CTx (Kostakou et al., 2019). Recently, the impact of CTx on the vasculature has emerged as a putative contributor to cardiovascular morbidity in cancer patients and survivors. The aim of this review is to summarize evidence of CTx-induced macrovascular and microvascular toxicity with an emphasis on endothelial function. We highlight clinical studies investigating the impact of CTx on vascular health and discuss the role of inflammation in mediating CTx-induced vascular perturbations. We also explore the putative role of vascular toxicity in the overall cardiovascular response to CTx.
2. Cardiovascular disease in cancer survivors
Epidemiological studies suggest that cancer survivors develop premature cardiovascular disease and are at elevated risk of adverse cardiovascular events compared to peers without a prior cancer diagnosis (Meinardi et al., 2000; Mulrooney et al., 2009; Park et al., 2017; Patnaik et al., 2011; Weberpals et al., 2018). Cardiovascular disease is the leading non-cancer cause of death in cancer patients, and in several cancer types, mortality from cardiovascular disease surpasses that of cancer itself (Sturgeon et al., 2019; Zaorsky et al., 2017). The heightened cardiovascular risk is most prominent in patients diagnosed at younger ages or with cancer sites associated with better prognoses (Sturgeon et al., 2019). Among cancer survivors diagnosed prior to 55 years of age, mortality from cardiovascular causes is ten-fold higher than in the general population after controlling for age, race, and sex (Sturgeon et al., 2019). Continued progress in treatment options and early detection are likely to unveil further cardiovascular complications as cancer survival rates improve; indeed, cardiovascular-specific mortality increased 20-fold from the year 2000 to 2016 in a registry study of 2.7 million cancer patient deaths (Oh et al., 2020). The persistent elevated risk of cardiovascular morbidity and mortality in cancer survivors is multifactorial and likely involves common risk factors shared between cancer and cardiovascular disease, pathophysiological mechanisms linked to cancer itself, and adverse cardiovascular effects of CTx.
Cardiotoxicity has long been recognized as a complication of CTx which has acute and long-term implications for cancer patients (Becker et al., 1999; Singal & Iliskovic, 1998). The clinical manifestations of CTx-induced cardiotoxicity include heart failure, hypertension, myocardial ischemia, thromboembolism, and cardiac arrhythmias (Yeh & Bickford, 2009). Many CTx agents, including both classic, systemic chemotherapies and newer, targeted therapies, are associated with acute cardiovascular toxicity that may prompt interruption or discontinuation of treatment. Cardiotoxicity experienced during or shortly after treatment contributes to the exacerbated rate of cardiovascular mortality among cancer patients, which is greatest in the first year following diagnosis (Sturgeon et al., 2019). Ultimately, the life-saving anti-neoplastic effects of CTx must be balanced with the acute cardiovascular risk. Although cardiotoxicity that develops during the course of treatment is often reversible (depending on the treatment regimen), latent cardiovascular complications of CTx arise in many patients in the months and years following therapy. Thus, the risk of cardiotoxicity presents a barrier to treatment in some patients, and the deleterious effects of CTx on the cardiovascular system persist long after completion of treatment.
Cancer patients are treated with a broad range of therapies which differ in terms of cardiovascular effects (Yeh & Bickford, 2009). Several classic cytotoxic therapies, including many chemotherapies and radiotherapy, are associated with cardiotoxicity. Subclasses of cytotoxic chemotherapies known to increase the risk of cardiac dysfunction include anthracyclines (e.g., doxorubicin), alkylating agents (e.g., cyclophosphamide), and antimetabolites (e.g., clofarabine). Classes that increase the risk of ischemia include antimetabolites (e.g., 5-Fluorouracil) and antimicrotubule agents (e.g., paclitaxel), while alkylating agents (e.g., cisplatin) increase the risk of thromboembolic events. In recent decades, molecular targeted therapies revolutionized cancer treatment, and targeted therapies are now widely used both alone and in combination with chemotherapy and/or radiation. This class of CTx interferes with specific molecules to impede cancer progression and includes monoclonal antibodies and small-molecule inhibitors, such as tyrosine kinase inhibitors (TKIs). Despite the specificity of targeted therapies, cardiovascular complications have emerged as important clinical side effects (Raschi et al., 2010). By design, some targeted therapies directly impact the cardiovascular system, such as anti-vascular endothelial growth factor (VEGF) treatments (e.g., sunitinib), which are associated with hypertension and ischemia. Others have off-target effects that increase the risk of cardiac dysfunction (e.g., trastuzumab, dasatinib). Recent innovations gave rise to immunotherapies such as immune checkpoint inhibitors, T-cell therapies, and anti-cancer vaccines, which harness the immune system to destroy cancer cells. Although epidemiological data are scarce, cardiovascular toxicity linked to immunotherapies is increasingly being recognized as a serious complication which can present as myocarditis, vasculitis, acute coronary syndrome, or atherosclerosis, among other manifestations (Drobni et al., 2020; Stein-Merlob et al., 2021). Thus, it is clear that novel, specific treatment options do not circumvent cardiovascular complications, which can arise directly or indirectly from a broad range of CTx classes.
Although research investigating cardiotoxicity induced by CTx has predominantly focused on the direct impact of CTx on cardiomyocytes, vascular dysfunction following CTx could play a key role in the development of cardiovascular complications (Cameron et al., 2016; Herrmann et al., 2016; Mulrooney et al., 2012; Soultati et al., 2012). Systemic CTx agents encounter the vascular endothelium before diffusing to underlying tissue, and mounting evidence suggests that CTx diminishes endothelial function, reduces nitric oxide (NO) bioavailability, and promotes endothelial cell activation. This has important implications for both cardiac and peripheral tissues, as endothelial dysfunction leads to a range of issues including tissue ischemia, diminished vascular barrier function, atherosclerosis, and altered hemodynamics (Figure 1).
Figure 1: Causes and consequences of endothelial toxicity arising from anti-cancer therapy.
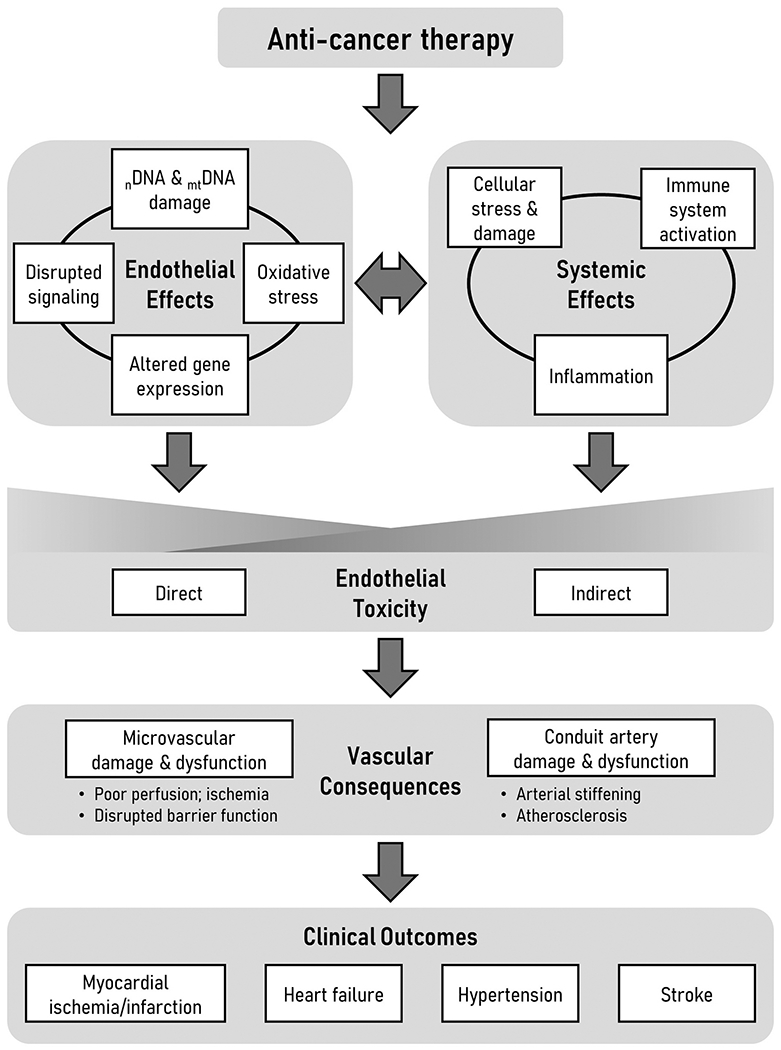
Many forms of anti-cancer therapy induce endothelial damage directly through primary effects on endothelial cells and indirectly through systemic effects such as innate immune system activation. There is interplay between endothelial and systemic effects wherein changes to the endothelium exacerbate systemic factors while systemic factors amplify endothelial effects. Endothelial toxicity causes damage and dysfunction at the level of the microcirculation and conduit arteries that precedes and potentially contributes to adverse clinical outcomes in patients. nDNA, nuclear DNA; mtDNA, mitochondrial DNA
3. Endothelial control of vascular homeostasis
The vascular endothelium is a critical regulator of vasomotor tone that communicates with underlying vascular smooth muscle cells to regulate tissue perfusion. The endothelium constantly monitors chemical and mechanical cues and facilitates metabolic dilator mechanisms to fine tune oxygen delivery to meet metabolic demand. Endothelial cells sense changes in shear stress exerted by blood moving through the vessel lumen. In response to an increase in blood velocity (i.e., shear stress), the endothelium initiates vasodilation. This important physiological function, referred to as flow-mediated dilation (FMD), is widely used in clinical research studies as an index of endothelial health (Celermajer et al., 1992; Thijssen et al., 2019). In healthy conduit arteries and resistance arterioles, FMD is attributed primarily to enzymatic production of the vasodilator NO (Joannides et al., 1995), which has anti-inflammatory, anti-proliferative, and anti-thrombotic characteristics in addition to its vasoactive properties. However, FMD is altered by many disease states that promote elevated production of reactive oxygen species (ROS) such as superoxide, which scavenges NO (Touyz & Schiffrin, 2004). In this situation, FMD is typically abolished in conduit vessels, while FMD persists in the microcirculation but is instead attributed to endothelial release of the pro-inflammatory mediator hydrogen peroxide (H2O2) (Miura et al., 2003; Phillips et al., 2007). Thus, a dysfunctional endothelium characterized by impaired vasodilation, reduced NO bioavailability, and elevated ROS levels sets the stage for tissue ischemia and promotes progression of atherosclerosis and thrombotic events among other inflammatory processes (Gokce et al., 2002; Libby et al., 2009).
The systemic nature of endothelial dysfunction has implications for all vital organ systems. Tissues with high metabolic demand, such as the heart, already extract a near-maximal amount of oxygen from the blood at rest. Thus, the heart must regulate local perfusion on a second-to-second basis to maintain adequate oxygenation. A dysfunctional endothelium blunts the coronary flow response to increased metabolic demand; therefore, the myocardium is particularly susceptible to the consequences of impaired endothelial function. Endothelial dysfunction is recognized as a key contributor to several cardiovascular ailments, and endothelial dysfunction precedes and independently predicts adverse cardiac events (Broxterman et al., 2019; Gokce et al., 2002).
In addition to controlling vasomotor tone, the endothelium regulates vascular integrity and controls angiogenesis (Murakami & Simons, 2009). The endothelium serves as an interface that regulates the exchange of solutes and fluid between tissues and the bloodstream. Inflammatory stimuli disrupt the continuity of endothelial cell junctions, which increases vascular permeability to permit leukocyte extravasation. Under pathological conditions, poor endothelial integrity may lead to uncontrolled vascular leakage, tissue edema, and rupture. Thus, endothelial barrier function is crucial for tissue homeostasis. Endothelial cell proliferation and migration are also central to the formation and recruitment of new blood vessels. Angiogenic processes are disrupted in the dysfunctional endothelium, which may lead to pathological vascular remodeling. Moreover, endothelium-derived signals regulate the phenotype of underlying vascular smooth muscle, and endothelial dysfunction promotes vascular smooth muscle proliferation and remodeling (Li et al., 2018).
The physiological ramifications of endothelial dysfunction differ depending on the extent to which large arteries and small arterioles are impacted. In the following sections, we discuss evidence of CTx-induced endothelial dysfunction at the level of conduit arteries and the microcirculation. For detailed discussion regarding the impact of radiotherapy on endothelial function, we direct readers to three recent reviews (Baselet et al., 2019; Korpela & Liu, 2014; Venkatesulu et al., 2018). We primarily focus on results from human studies, where available. A summary of studies that have investigated the impact of CTx on human endothelial function is presented in the Table.
Table.
Studies investigating the impact of CTx on human vascular endothelial function
| Treatment | Population | Design | Outcomes | Reference |
|---|---|---|---|---|
| Acute effects (within 48 h of treatment) | ||||
| Anthracyclines (Doxorubicin) | Breast cancer (n=10) | Prospective Measurements before and 30 min after initial doxorubicin infusion | Compared to baseline: ↓ baFMD ↔ baNMD ↓ plasma nitrate and nitrite levels ↔ cardiac echo parameters |
(Duquaine et al., 2003) |
| Anthracycline-based therapy (AC, HOP, or ABVD) | Lymphoma Non-Hodgkin’s (n=18) Hodgkin’s disease (n=4) |
Prospective Measurements before and 6, 12, 24, and 48 h after doxorubicin administration |
Compared to baseline: ↓ baFMD ↔ baNMD Correlation between acute ΔbaFMD and ΔLVEF after 1–2 y of follow-up (r=0.66) |
(Nagy et al., 2001) |
| Anthracycline-based therapy (AC) | Breast cancer (n=16) | Prospective Measurements before, during, and 15 min after AC infusion or saline infusion (at a separate experimental visit) |
Compared to saline: ↓ calf blood flow and vascular conductance during and 15 min after AC ↑ circulating endothelial microparticles 15 min after AC ↑ total peripheral resistance and blood pressure during and 15 min after AC |
(Sales et al., 2019) |
| Anthracyclines (Doxorubicin) | Human adipose or coronary arterioles Children (n=15) Adults (n=20) |
Measurements in paired vessels exposed to doxorubicin (100 nM) or vehicle control overnight | Compared to vehicle control: ↓ microvascular FMD |
(Hader et al., 2019) |
| Carboplatin and paclitaxel (TC) | Ovarian or endometrial cancer Surgery + TC (n=28) Surgery only (n=19) |
Prospective Cross-sectional Measurements before and 1 h after TC infusion; PWV 12 mo post-therapy |
Compared to baseline: ↓ baFMD 1 h after TC infusion ↑ PWV in TC group 12 mo after discontinuation of therapy |
(Sekijima et al., 2011) |
| Cisplatin-based therapy (BEP or EP) | Testicular cancer Low-intensity cisplatin (n=7) High-intensity cisplatin (n=10) Surveillance (n=10) |
Prospective Cross-sectional Measurements before and 24 h after cisplatin administration |
Compared to baseline: ↓ baFMD in high-intensity cisplatin group only ↑ urine albumin to creatinine ratio in low- and high-intensity cisplatin groups |
(Cameron et al., 2020) |
| Imatinib or nilotinib | Human adipose arterioles from non-diseased adults (n=30) | Measurements in paired vessels exposed to drug (10 μM imatinib or 100 nM nilotinib) or vehicle control overnight | Compared to vehicle control: ↓ microvascular NO production in response to flow ↑ microvascular H2O2 production in response to flow |
(Durand et al., 2020) |
| Short- and medium-term effects (weeks to months after treatment) | ||||
| Anthracycline-based therapy (AC) | Breast cancer AC (n=15) AC + HIIT exercise training (n=15) |
Prospective Interventional Measurements before and 9 w after starting therapy ± exercise intervention |
Compared to baseline: ↓ baFMD in AC group; ↑ baFMD in AC + HIIT group ↑ CIMT in AC group; ↔ CIMT in AC + HIIT group |
(Lee et al., 2019) |
| Anthracycline-based therapy (AC) | Breast cancer AC (n=10) AC + aerobic exercise training (n=10) |
Prospective Interventional Measurements before and 12 w after starting therapy ± exercise intervention |
Compared to baseline: ↔ baFMD in AC group; ↑ baFMD in AC + exercise group (P=0.07) ↑ IL-2 in AC group; ↓ IL-2 in AC + exercise group Tumor microarray analysis: transcripts converging on NF-κB downregulated in exercise group vs. AC only |
(Jones et al., 2013) |
| Anthracycline-based therapy (EC-T or EC-TH) | Breast cancer (n=33) | Prospective Measurements before treatment and 2 w and 1 y following completion of therapy |
Compared to baseline: ↓ forearm vasodilation to ACh infusion (relative to uninfused arm) at 1 y ↔ forearm vasodilation to SNP ↔ PWV ↑ CD163, CD206/MR at 2 w |
(Fredslund et al., 2021) |
| Bevacizumab (Monotherapy or combined therapy) | Colon or breast cancer Colon cancer (n=12) Breast cancer (n=2) |
Prospective Measurements before, during (6 w), and > 3 mo after bevacizumab treatment |
Compared to baseline: ↓ baFMD during and > 3 mo after treatment ↔ baNMD ↓ capillary density during treatment; reversed after discontinuation ↑ PWV > 3 mo after discontinuation |
(Steeghs et al., 2010) |
| Cisplatin-based therapy (BEP) | Testicular cancer (n=65) | Prospective Measurements before and ≤ 10 w after completion of therapy |
Compared to baseline: ↔ baFMD ↑ CIMT ↑ plasma vWF |
(Nuver et al., 2005) |
| Cisplatin-based therapy (BEP or EP) | Testicular cancer Low-intensity cisplatin (n=7) High-intensity cisplatin (n=10) Surveillance (n=10) |
Prospective Cross-sectional Measurements before and 6 w, 3 mo, 6 mo, and 9 mo after cisplatin administration |
Compared to baseline: ↔ baFMD ↑ urine albumin to creatinine ratio at 6 w; returned to baseline levels by 3 mo ↑ plasma vWF at 6 w, returned to baseline levels by 3 mo ↔ hs-CRP, ICAM-1, hs-TNI |
(Cameron et al., 2020) |
| Sunitinib | Renal cell carcinoma (n=20) | Prospective Measurements before and after 40 d treatment with sunitinib |
Compared to baseline: ↓ baFMD ↔ CIMT |
(Lai et al., 2020) |
| Telatinib | Advanced solid tumors (n=18) | Prospective Measurements before and after 5 w of telatinib treatment |
Compared to baseline: ↓ baFMD ↓ baNMD ↑ PWV ↓ capillary density ↓ skin blood flow |
(Steeghs et al., 2008) |
| Vandetanib | Breast cancer, stage IV (n=17) | Prospective Measurements before and after 6 w of vandetanib treatment |
Compared to baseline: ↓ brachial artery diameter ↓ plasma nitrites ↔ baFMD ↔ baNMD |
(Mayer et al., 2011) |
| Various anti-neoplastic agents | Breast, ovarian, or cervical cancer Cervical cancer, low-dose cisplatin (n=10) Breast cancer, paclitaxel (n=5) Breast cancer, epirubicin & paclitaxel (n=6) Ovarian cancer, paclitaxel & carboplatin (n=16) |
Prospective Cross-sectional Measurements before and 1 mo after completion of treatment |
Compared to baseline: ↓ baFMD: paclitaxel alone or combined with epirubicin or carboplatin ↔ baFMD: low-dose cisplatin ↔ baNMD: paclitaxel or low-dose cisplatin ↓ baNMD: paclitaxel combined with epirubicin or carboplatin (p=0.06) Inverse correlation between ΔbaFMD and ΔTNFα (r=−0.32) in full cohort |
(Vassilakopoulou et al., 2010) |
| Various anti-neoplastic agents | Breast cancer or non-Hodgkins lymphoma Breast cancer (n=5) Non-Hodgkin’s lymphoma (n=2) Cancer-free age- and sex-matched controls (n=7) |
Retrospective Cross-sectional Measurements 13±5 d following last treatment dose (mean±SE) |
Compared to controls: ↓ baFMD in cancer patients ↓ skin microvascular reactivity to ACh in cancer patients |
(Sutterfield et al., 2018) |
| Long-term effects (years after treatment) | ||||
| Anthracyclinc-based therapy | Childhood cancer survivors, several types (n=14) Cancer-free age- and sex-matched controls (n=14) |
Retrospective Cross-sectional Measurements 20±19 mo following last treatment dose (mean±SD) |
Compared to controls: ↓ baFMD in cancer survivors |
(Chow et al., 2006) |
| Anthracycline-based vs. non-anthracycline chemotherapy | Childhood cancer survivors, several types CTx with anthracyclines (n=67) CTx without anthracyclines (n=29) Cancer-free age- and sex-matched controls (n=72) |
Retrospective Cross-sectional Measurements at 11±6 y survival (mean±SD) |
Compared to controls: ↓ baFMD in cancer survivors; ↓↓ in anthracycline group ↔ baNMD ↑ aortic stiffness in cancer survivors; ↑↑ in anthracycline group |
(Jenei et al., 2013) |
| Anthracycline-based therapy (Nordic regimen; maxi-CHOP) | Childhood acute lymphoblastic leukemia survivors (n=21 Cancer-free age- and sex-matched controls (n=21) |
Retrospective cohort Cross-sectional Interventional Median time since diagnosis: 16 y (range: 11–21 y) Measurements before and after 16 w home-based resistance exercise program (cancer survivors only) |
Compared to controls: ↓ baFMD in cancer survivors ↔ CIMT Compared to baseline: ↑ baFMD after exercise training; driven by male subjects ↓ CIMT after exercise training |
(Järvelä et al., 2013) |
| Anthracycline-based therapy (AIEOP-BFM treatment protocols) | Childhood acute lymphoblastic leukemia survivors (n=52) Cancer-free age- and sex-matched controls (n=34) |
Retrospective Cross-sectional Mean survival: 2.3 y (range: 0.3–8.5 y) |
Compared to controls: ↓ baFMD in cancer survivors ↔ CIMT ↑ vWF in cancer survivors |
(Giordano et al., 2017) |
| Anthracycline and/or trastuzumab-based therapy (AC-T, AC-TH, or TCH) | HER2-positive breast cancer survivors (n=26) Cancer-free age- and sex-matched controls (n=10) |
Retrospective cohort Cross-sectional Treatment completed 20 ± 10 mo prior to study (mean±SD) |
Compared to controls: ↔ baFMD ↔ baNMD |
(Jones et al., 2007) |
| Cisplatin-based therapy (BEP, EP, PVB, CAP-5, other) | Ovarian cancer survivors (n=21) Age- and sex-matched controls from population study (n=2,926) |
Retrospective Cross-sectional Median survival: 14 y (range: 3–21 y) |
Compared to controls: ↑ microalbuminuria in cancer survivors Raynaud’s phenomenon in 38% of cancer survivors |
(De Vos et al., 2004) |
| Cisplatin-based therapy (Primarily BEP or EP) | Testicular cancer survivors Chemotherapy (n=90) Surgery only (n=44) Cancer-free age- and sex-matched controls (n=47) |
Retrospective cohort Cross-sectional Median survival: 7 y (range: 3–13 y) |
Compared to controls: ↑ microalbuminuria in chemotherapy group ↔ baFMD ↔ baNMD ↔ CIMT ↑ carotid artery stiffness in chemotherapy group |
(Nuver et al., 2004) |
| Cisplatin-based therapy (BEP or EP) | Testicular cancer survivors Cisplatin (n=24) Chemotherapy-naïve controls (n=15) |
Retrospective Cross-sectional Median time from diagnosis: 6 y (range: 3–27 y) |
Compared to controls: ↓ baFMD in chemotherapy group ↔ baNMD ↔ CIMT ↑ circulating endothelial cells and sICAM-1 in chemotherapy group |
(Vaughn et al., 2008) |
| Cisplatin-based therapy (BEP) | Testicular cancer survivors Cisplatin (n=12) Surveillance controls (n=14) |
Retrospective Cross-sectional Treatment 1–7 y prior to study |
Compared to controls: ↔ forearm vasodilation to ACh, bradykinin, or SNP |
(Cameron et al., 2020) |
| Various anti-neoplastic agents | Adult survivors of childhood acute lymphoblastic leukemia Chemotherapy only (n=25) Chemotherapy + radiation (n=50) Cancer-free age-matched controls (n=59) |
Retrospective cohort Cross-sectional Mean survival: 25±5 y (mean±SD) |
Compared to controls: ↓ baFMD in cancer survivors regardless of radiation status ↔ baNMD |
(Dengel et al., 2008) |
| Various anti-neoplastic agents | Cancer survivors; type unspecified (n=8) Cancer-free, age-matched controls (n=9) |
Retrospective Cross-sectional Mean time post-treatment: 5.8 y (range: 2.6–15.6 y) |
Compared to controls: ↔ baFMD ↓ skeletal muscle microvascular function during exercise in cancer survivors |
(Ederer et al., 2016) |
Abbreviations: ABVD, doxorubicin/bleomycin/vinblastine/dacarbazine; AC, doxorubicin/cyclophosphamide; ACh, acetylcholine; AC-T, doxorubicin/cyclophosphamide/docetaxel; AC-TH, doxorubicin/cyclophosphamide/docetaxel/trastuzumab; baFMD, brachial artery flow-mediated dilation; baNMD, brachial artery nitroglycerin-mediated dilation; BEP, bleomycin/etoposide/cisplatin; CAP-5, cyclophosphamide/doxorubicin/cisplatin; CD163, cluster of differentiation 163; CD206/MR, cluster of differentiation 206/mannose receptor; CHOP, cyclophosphamide/doxorubicin/vincristine/prednisone; CIMT, carotid artery intima-media thickness; CTx, anti-cancer therapy; EC-T, epirubicin/cyclophosphamide/docetaxel or paclitaxel; EC-TH, epirubicin/cyclophosphamide/docetaxel or paclitaxel/trastuzumab; EP, etoposide/cisplatin; FMD, flow-mediated dilation; HER2, human epidermal growth factor receptor 2; HIIT, high-intensity interval training; H2O2, hydrogen peroxide; HOP, doxorubicin/vincristine/prednisone; hs-CRP, high-sensitivity C-reactive protein; hs-TNI, high-sensitivity troponin I; ICAM-1, intercellular adhesion molecule-1; IL-2, interleukin 2; LVEF, left-ventricular ejection fraction; NF-κB, nuclear factor κ B; NO, nitric oxide; PWV, pulse-wave velocity; PVB, cisplatin/vinblastine/bleomycin; SNP, sodium nitroprusside; TC, paclitaxel/carboplatin; TCH, docetaxel/carboplatin/trastuzumab; TNFα, tumor necrosis factor α vWF, von Willebrand factor
4. Conduit artery endothelial dysfunction following anti-cancer therapy
4.1. Acute effects
Evidence that the widely-used chemotherapy agent doxorubicin interacts with endothelial NO synthase (eNOS) to promote formation of the free radical superoxide in place of NO (Vásquez-Vivar et al., 1997) prompted initial studies investigating the impact of CTx agents on vascular endothelial function. In lymphoma patients, brachial artery FMD (baFMD) is impaired 6 h following bolus administration of doxorubicin and does not return to baseline levels within 48 h (Nagy et al., 2001). Notably, the acute change in baFMD in this small cohort of patients correlated with the decline in left-ventricular ejection fraction (LVEF) observed over 1–2 y of follow-up. Indeed, ΔbaFMD (r = 0.66) was a stronger predictor of ΔLVEF than even the cumulative dose of doxorubicin (r = −0.47) (Nagy et al., 2001). Duquaine et al. observed impaired endothelium-dependent relaxation to acetylcholine in aortas isolated from rabbits 12 h following a single injection of doxorubicin and showed that ex vivo exposure of aortic segments to high concentrations (100–500 μM) of doxorubicin for 10 min induced endothelial superoxide production (Duquaine et al., 2003). Moreover, they demonstrated that conduit artery endothelial function is diminished in breast cancer patients immediately following infusion of doxorubicin (Duquaine et al., 2003). Thus, it appears that CTx regimens which include doxorubicin have a rapid impact on conduit artery endothelial function. Similarly, baFMD is reduced in patients with ovarian or endometrial cancer 1 h after an initial dose of platinum-based chemotherapy (Sekijima et al., 2011) and in patients with testicular cancer 24 h after an initial dose of cisplatin-based chemotherapy (Cameron et al., 2020). Arterial infusion of 5-fluorouracil (5-FU) in rabbits evokes systemic morphological changes in the endothelium within 15 min that persist for at least 30 days and manifest as patchy disruption of the endothelial layer and increased thrombus formation observed by electron microscopy (Cwikiel et al., 1995, 1996). In patients, 5-FU administration induces acute brachial artery vasoconstriction (Südhoff et al., 2004); however, a study of rabbit aortic rings exposed to 5-FU suggests this does not reflect an impairment in endothelium-dependent vasodilatory capacity (Mosseri et al., 1993). The immediate impacts of other classes of CTx on conduit artery endothelial function have not been as extensively studied.
4.2. Short- and medium-term effects
Data regarding the impact of CTx on endothelial function during the course of treatment or shortly after completion of therapy are limited. A cross-sectional study of cancer patients undergoing adjuvant therapy with a range of cardiotoxic CTx agents revealed impaired endothelial function in patients studied approximately two weeks following the most recent dose compared to well-matched healthy control participants (Sutterfield et al., 2018). Although cisplatin-based chemotherapy causes an acute reduction in FMD in patients with testicular cancer, conduit artery endothelial function returns to pre-therapy baseline levels during the course of treatment and there is no evidence of dysfunction in the months following therapy (Cameron et al., 2020). This is in line with the observation that FMD is maintained one month following low-dose cisplatin therapy in patients with cervical cancer (Vassilakopoulou et al., 2010). In contrast, treatment with paclitaxel was associated with a decline in conduit artery endothelial function one month following treatment in a small group of women undergoing treatment for breast or ovarian cancer, and a greater reduction was observed in those treated with a combination of paclitaxel and anthracyclines (Vassilakopoulou et al., 2010). In prospective studies of women undergoing breast cancer treatment with anthracyclines and cyclophosphamide, baFMD was reduced following 9 weeks of treatment in one cohort (Lee et al., 2019) but unchanged at 12 weeks of treatment in another cohort (Jones et al., 2013). Studies of male mice exposed to a single, intraperitoneal injection of doxorubicin demonstrate impaired NO-dependent vasodilation to acetylcholine in isolated carotid arteries four weeks after doxorubicin administration in conjunction with increased aortic mitochondrial ROS generation (Clayton et al., 2020). Endothelium-dependent dilation was preserved in mice that received concurrent treatment with a mitochondrial antioxidant, which suggests that conduit artery endothelial dysfunction observed several weeks following doxorubicin administration is mediated by excessive mitochondrial ROS production (Clayton et al., 2020). Small studies of patients undergoing anti- VEGF therapy with bevacizumab or telatinib reported a reduction in baFMD 5–6 weeks after starting treatment that persists > 3 months post-treatment; however, a reduction in vascular smooth muscle vasodilator capacity may have contributed to these results (Steeghs et al., 2010; Steeghs et al., 2008). In contrast, six weeks of treatment with vandetanib, a VEGF receptor 2 & 3 TKI, did not impact baFMD in breast cancer patients; however, vandetanib reduced brachial artery diameter and constitutive NO production (Mayer et al., 2011). A decline in baFMD has been reported after 40 days of treatment with the multi-target TKI sunitinib (Lai et al., 2020). It is important to note that endothelial dysfunction reported during or shortly after treatment with specific CTx agents is often the first manifestation of cardiovascular complications. Moreover, the acute decline in endothelial corresponds with subsequent cardiac dysfunction in some circumstances (Nagy et al., 2001).
4.3. Long-term effects
Several studies provide evidence of a prolonged impact of CTx on endothelial function in humans. Many of the data supporting this come from long-term follow-up studies of childhood cancer survivors. Brachial FMD was impaired in a small group of pediatric cancer patients studied 20 ± 19 months (mean ± SD) following anthracycline treatment compared to age-matched control patients (age at time of study: 4–21 y) (Chow et al., 2006). A subsequent study of 96 pediatric cancer survivors extended these findings and showed that baFMD remains lower in cancer survivors 11 ± 6 years (mean ± SD) after chemotherapy compared to healthy controls matched for age, sex, bodyweight, and blood pressure (age at time of study: 14 ± 5 y); moreover, the impairment is worse in those who received anthracyclines (Jenei et al., 2013). Young adult (age: 30 ± 7 y, mean ± SD) survivors of acute lymphoblastic leukemia treated with diverse chemotherapies exhibited impaired baFMD approximately 25 years following diagnosis compared to healthy peers, and there was no difference among those treated with chemotherapy only vs. chemotherapy combined with radiation (Dengel et al., 2008). Collectively, these cross-sectional studies demonstrate impaired conduit artery endothelial function in cancer survivors years after treatment compared to healthy cohorts, yet the findings do not discern the impact of CTx from the impact of cancer itself or associated comorbidities. Metabolic abnormalities and other cardiovascular risk factors were reported alongside impaired baFMD in survivors of childhood acute lymphoblastic leukemia studied an average of 2.3 years following CTx (age at time of study: 4–19 y), and several risk factors predicted endothelial dysfunction (Giordano et al., 2017). Thus, while these studies indicate endothelial dysfunction in long-term survivors of childhood cancer, further research is needed to establish the time course and causative mechanisms.
Few studies have examined the long-term impact of CTx on endothelial function in survivors who were diagnosed with cancer as adults. Brachial FMD was not significantly different in human epidermal growth factor receptor 2 (HER2)-positive breast cancer patients (n = 26) ~20 months after completion of adjuvant chemotherapy compared to age-matched healthy controls (n = 10) (Jones et al., 2007). However, exploratory analyses suggested that patients randomized to receive treatment with trastuzumab (docetaxel/carboplatin/trastuzumab; TCH or doxorubicin/cyclophosphamide/docetaxel/trastuzumab; AC-TH regimens) had worse endothelial function than those who did not receive trastuzumab (doxorubicin/cyclophosphamide/docetaxel; AC-T regimen) (Jones et al., 2007), which merits further research to investigate potential differences between treatment regimens. Future studies should also incorporate several time points to define the time course of endothelial dysfunction following therapy, which is particularly relevant for agents with reversible cardiotoxicity such as trastuzumab. A small study of eight long-term cancer survivors evaluated 3–16 years (median: 3.4 y) following treatment with a broad range of adjuvant chemotherapies found no difference in baFMD compared to nine healthy controls matched for age, sex, and cardiovascular risk factors (Ederer et al., 2016). In contrast, baFMD was impaired in a small cohort of testicular cancer survivors who were treated with cisplatin-based chemotherapy compared to chemotherapy-naïve survivors studied 3–27 years following diagnosis despite similar demographics and cardiovascular risk factors (Vaughn et al., 2008). This suggests divergent short- and long-term effects, as the acute impact of cisplatin appears to be transient during treatment (Cameron et al., 2020). Thus, it appears that CTx may have long-term implications for conduit artery endothelial function in some circumstances, although additional studies are needed to distinguish the effects of different classes of CTx and examine the association between CTx and endothelial function in patients treated as adults. Future studies should prospectively evaluate endothelial function in cancer patients before and after CTx and include a comparison group of patients managed without CTx (e.g., surgical resection or surveillance) to define the time course of events and discern the impact of CTx from that of cancer itself.
5. Role of the microcirculation
Although clinical assessment of endothelial function in humans is predominantly based on measurement of conduit arteries, crucial physiological processes such as regulation of tissue blood flow and solute exchange occur at the level of the microcirculation. Tissue perfusion is controlled primarily by resistance vessels; thus, microvascular function is paramount to ensuring adequate delivery of oxygen and nutrients to vital organs. The microcirculation is particularly important in cardiac tissue, where high levels of basal oxygen extraction necessitate rapid adjustments in blood flow to match oxygen supply to metabolic demand. Coronary microvascular dysfunction has emerged as a key cardiovascular risk factor that is a substantially better predictor of major adverse cardiovascular events than large coronary artery disease (Van De Hoef et al., 2014), and myocardial microvascular dysfunction predicts adverse cardiovascular events in breast cancer patients (Divakaran et al., 2021). In addition to controlling tissue oxygenation, the microcirculation regulates angiogenesis, coordinates local inflammatory responses, and serves as a barrier that regulates exchange of solutes and fluids. Disruption of this barrier function results in greater vascular permeability, which permits migration of leukocytes and may alter tissue exposure to toxic CTx agents. Thus, damage to the microcirculation may contribute to the detrimental impact of CTx on cardiovascular outcomes.
6. Microvascular dysfunction following anti-cancer therapy
6.1. Vasomotor function
Some CTx agents directly impact microvascular structure and function. Histological assessments of cardiac tissue reveal a reduction in microvessel density following anthracycline treatment in animal models (Galán-Arriola et al., 2021; Yin et al., 2016), and imaging studies demonstrate a reversible reduction in capillary density in humans during anti-VEGF therapy (Coschignano et al., 2021; Steeghs et al., 2010; Steeghs et al., 2008). Endothelium-dependent vasodilation is effectively abolished in human coronary or adipose arterioles following overnight exposure to a clinically-relevant dose of doxorubicin (Hader et al., 2019). Although flow-induced vasodilation is preserved in adipose arterioles exposed to the BCR-ABL TKIs imatinib or nilotinib overnight, vasodilation is facilitated by the pro-inflammatory mediator H2O2 rather than NO (Durand et al., 2020). This switch in the mediator of vasodilation caused by BCR-ABL TKIs is similar to the vasodilatory phenotype observed in patients with coronary artery disease (Beyer et al., 2017; Phillips et al., 2007) and may explain the growing number of reports of systemic and pulmonary hypertension in this patient population (Roa-Chamorro et al., 2021; Weatherald et al., 2020). To date, sparse data exist regarding long-term cardiac function in patients treated with BCR-ABL TKIs, which have only been used as a first-line therapy for approximately 10 years.
The microvascular insult induced by some CTx agents has immediate implications for tissue blood flow regulation. A rapid, profound (−33%) deterioration of gonadal tissue perfusion and hindlimb blood flow has been reported in mice within 3 min of doxorubicin injection (Bar-Joseph et al., 2011). In breast cancer patients, peripheral blood flow and vascular conductance decline while blood pressure rises during infusion of doxorubicin plus cyclophosphamide, and circulating endothelial microparticle levels are elevated 15 min post-infusion (Sales et al., 2019). In contrast, intravenous administration of paclitaxel does not impact femoral artery blood flow or gonadal perfusion in mice in the 20 min following injection (Bar-Joseph et al., 2011). However, it is important to note that these studies did not specifically probe endothelial vasomotor function, which appears to be especially sensitive to CTx and has not been assessed in the microcirculation in response to paclitaxel. Moreover, changes in basal blood flow do not provide a complete picture, as compensatory mechanisms may mask the effects of CTx. Future studies that stress the vasculature may expose more subtle decrements.
The time course of microcirculatory function during and after treatment regimens has not been studied extensively. In a porcine model, local administration of doxorubicin via direct intra-coronary infusion stimulates a progressive decline in myocardial perfusion and coronary microcirculatory function characterized by structural damage and impaired vasodilation (Galán-Arriola et al., 2021). Notably, microvascular defects occur at lower doses and earlier time points than contractile defects, which demonstrates that CTx-induced alterations in microvascular function precede cardiotoxicity. In humans, cutaneous microvascular endothelial function is impaired in patients undergoing adjuvant therapy with a wide range of CTx agents compared to healthy controls (Sutterfield et al., 2018). A recent, longitudinal study of breast cancer patients undergoing adjuvant therapy reported reduced vasodilation to acetylcholine in the forearm resistance vasculature one year following completion of treatment; however, the impairment in endothelial function was not yet evident two weeks after therapy (Fredslund et al., 2021). Preliminary results from a prospective study of breast cancer patients indicate that 21% of trastuzumab recipients and 43% of combined trastuzumab and doxorubicin recipients exhibit a deterioration of microvascular reactive hyperemia index 3–18 months following treatment compared to pre-therapy values (defined as a ≥ 20% reduction) (Hazim et al., 2020). Studies of long-term cancer survivors have reported a greater incidence of microalbuminuria in patients treated with cisplatin (De Vos et al., 2004; Meinardi et al., 2001; Nuver et al., 2004), which suggest the detrimental impact on microvascular function is sustained years after treatment. Long-term cancer survivors also exhibit accelerated skeletal muscle deoxygenation during exercise compared to healthy controls (Ederer et al., 2016). Interestingly, this observation occurred in the absence of conduit artery endothelial dysfunction, which suggests that changes in the microcirculation have lasting functional implications for tissue perfusion and may play a greater role in long-term cardiovascular consequences than conduit artery endothelial dysfunction.
6.2. Vascular barrier function, fibrosis, and microvascular remodeling
In addition to regulating tissue perfusion, the microvascular endothelium is a crucial regulator of vascular barrier function. In capillaries and post-capillary venules, the endothelium regulates the distribution of water and solutes between tissues and the bloodstream. When the endothelial barrier is compromised, proteins and water leak from the vasculature and cause tissue edema, which may lead to organ dysfunction. Cancer cells secrete VEGF to stimulate tumor vascularization, which also promotes greater vascular permeability. The cancer-induced increase in vascular permeability is exacerbated by some CTx agents (Wolf & Baynes, 2006), which may result in exposure of parenchymal tissue to higher doses of toxic CTx agents.
Tissues with tight endothelial junctions that carefully regulate solute transport under physiological conditions, such as the brain and heart, may be particularly susceptible to damage from changes in vascular permeability. Indeed, chimeric antigen receptor–modified T (CAR-T) cell infusion induces endothelial activation and capillary leak in patients with B-cell malignancies, and the blood-brain barrier is disrupted in patients with severe neurotoxicity (Gust et al., 2017; Hay et al., 2017). In cell culture models, doxorubicin and trastuzumab interrupt coronary microvascular endothelial barrier formation, which results in increased drug permeability, whereas doxorubicin and trastuzumab did not impact barrier function in brain or dermal endothelial cells (Wilkinson et al., 2016). Interestingly, liposome-encapsulated doxorubicin, which is unable to cross tight endothelial junctions and is therefore preferentially distributed to tissues with a fenestrated or inflamed endothelial barrier, is less damaging to endothelial cells and has a favorable cardiovascular risk profile compared to conventional doxorubicin (Kaushal et al., 2004; Rafiyath et al., 2012; Toldo et al., 2013). Disruption of microvascular barrier function and excessive endothelial permeability also appear to contribute to pulmonary effusion and pulmonary hypertension induced by the BCR-ABL TKI dasatinib (Dasgupta et al., 2017; Fazakas et al., 2018; Kreutzman et al., 2017; Phan et al., 2018), whereas the BCR-ABL TKIs imatinib and nilotinib do not alter barrier function in pulmonary endothelial cells (Fazakas et al., 2018). The physiological relevance of changes in endothelial barrier integrity has not yet been explored in intact human vessels.
Excessive vascular permeability coincides with structural remodeling of the microcirculation and surrounding tissues. In a tumor-bearing mouse model, treatment with doxorubicin and trastuzumab leads to perivascular cardiac fibrosis concomitant with increased coronary microvascular permeability, which suggests that extravasation of CTx agents may activate cardiac fibroblasts (Hoffman et al., 2021). It is important to note that this model lacks a normal immune system and may therefore underestimate the effects. In line with the findings of cardiac fibrosis in tumor-bearing mice, myofibroblast proliferation and collagen accumulation have been described in regions of coronary microvascular damage in a porcine model of anthracycline cardiotoxicity (Galán-Arriola et al., 2021). This was accompanied by irreversible histological changes indicative of structural damage to arterioles at early stages of doxorubicin administration, and higher cumulative doses led to capillary rarefaction. Collectively, these findings suggest that a second wave of CTx-induced vascular damage occurs by increasing vascular permeability, thus allowing for greater uptake of subsequently-administered CTx agents and leading to a feed-forward worsening of structural damage to the microvasculature and promotion of local inflammation and fibrosis. The cause and underlying mechanisms of this feed-forward cycle have not yet been identified.
7. Association between endothelial function and clinical outcomes
Altogether, these studies suggest that CTx frequently induces endothelial dysfunction within the macro and microcirculation. CTx-induced endothelial perturbations impact blood flow control, vascular leakage, and vascular remodeling, and also have implications for physiological processes such as thromboembolism and atherosclerosis, which have recently been reviewed (Grover et al., 2021; Mukai et al., 2018). Vascular complications vary between therapies with different mechanisms of action; however, endothelial dysfunction is observed in response to several classical chemotherapeutics as well as newer targeted therapies. Currently, there is sparse information regarding the impact of immunotherapies on endothelial function, though initial reports indicate that the endothelium is involved in cytokine release syndrome in patients treated with immunotherapies. The long-term impact of immunotherapies on endothelial function has not yet been established and represents an important area for investigation.
7.1. Role of endothelial dysfunction in long-term cardiovascular risk
It is currently unclear whether the decline in endothelial function in response to CTx predicts or contributes to acute cardiotoxicity or long-term cardiovascular risk in cancer patients. Notably, low baseline baFMD prior to treatment predicts subsequent reductions in left ventricular ejection fraction following anthracycline treatment in breast cancer patients (Anastasiou et al., 2017). This suggests that poor basal endothelial function predisposes patients to anthracycline cardiotoxicity and may be useful as a risk stratification tool. However, few studies have investigated whether endothelial dysfunction induced by CTx contributes to clinical outcomes. In a cohort of lymphoma patients followed for an average of 19 months, the acute (same-day) reduction in brachial FMD following administration of the first dose of doxorubicin was an independent predictor of longitudinal reductions in LVEF (Nagy et al., 2001). This suggests a link between the adverse effect of anthracyclines on endothelial function and subsequent development of cardiotoxicity, although it does not distinguish whether endothelial dysfunction per se or concomitant risk factors drive cardiotoxicity outcomes. Altogether, these studies suggest that conduit artery endothelial dysfunction precedes and may be associated with anthracycline-induced cardiotoxicity, though further research is needed to determine whether endothelial dysfunction directly contributes to clinical outcomes. Future studies should also investigate whether microvascular endothelial dysfunction is predictive of cardiotoxicity and evaluate its potential mechanistic role. Although we speculate that CTx agents that cause chronic or irreversible endothelial dysfunction are more likely to induce cardiac dysfunction compared to CTx agents with a lesser impact on the endothelium, it is difficult to discern whether this is the case due to variability in the definition and incidence of cardiotoxicity among published studies and heterogeneity in the populations studied, dosage, and combinations of multiple CTx agents.
7.2. Potential sex differences
Mounting evidence denotes sex differences in CTx-induced cardiotoxicity, although the impact of sex varies widely across different treatment types (D’Amario et al., 2020; Hohneck et al., 2021; Meiners et al., 2018; Özdemir et al., 2018). Outside of the context of cancer, sex and sex hormones have been shown to influence endothelial function (Stanhewicz et al., 2018); thus, it is likely that interactions between CTx, sex, and endothelial function combine to shape clinical outcomes. Currently, it is unclear whether sex modulates the impact of CTx on the endothelium. Further studies are warranted to investigate this and to evaluate the impact of therapies that modulate sex hormones, such as tamixofen, aromatase inhibitors, and androgen deprivation therapy. Future research should also establish whether there is a differential contribution of endothelial dysfunction to adverse cardiovascular outcomes based on sex.
8. Vascular stiffening following anti-cancer therapy
Arterial stiffening is an independent predictor of cardiovascular disease and all-cause mortality which precedes and contributes to many chronic disorders. The impact of CTx on arterial stiffness has recently been reviewed (Parr et al., 2020; Solomou et al., 2019). In their meta-analysis, Parr et al. (2020) note a substantial increase in arterial stiffness following CTx compared to pre-treatment baseline in longitudinal studies. They also report that cancer survivors with a history of CTx have greater arterial stiffness than cancer-free controls matched for age, sex, and cardiovascular risk factors. Several pathophysiological mechanisms contribute to augmented vascular stiffness, which are predominantly attributed to vascular smooth muscle perturbations. However, signaling molecules released from the endothelium (e.g., NO and endothelin-1) also regulate vascular stiffness; thus, endothelial dysfunction can influence arterial wall stiffness (Wilkinson & McEniery, 2004). Indices of endothelial function and vascular stiffness (e.g., pulse-wave velocity; PWV) are often collected jointly in clinical research studies, including several of the studies summarized in the Table. Prospective studies of patients undergoing CTx with carboplatin and paclitaxel, bevacizumab, or telatinib have reported impaired baFMD during treatment and increased large artery stiffness in the weeks and months after therapy in the same patients (Sekijima et al., 2011; Steeghs et al., 2010; Steeghs et al., 2008). However, a longitudinal study of breast cancer patients undergoing anthracycline-based therapy reported a decline in microvascular endothelial function (measured indirectly via forearm blood flow) one year after cessation of treatment that was not accompanied by changes in PWV (Fredslund et al., 2021). This is in contrast to a cross-sectional study of long-term childhood cancer survivors, which reported that impaired baFMD and elevated aortic stiffness in survivors were further exacerbated in those who received anthracyclines (Jenei et al., 2013). These findings suggest that CTx-induced endothelial dysfunction and vascular stiffening often coincide; however, they do not provide insight regarding a causal relationship. Further research is needed to determine whether changes in vascular stiffness predict CTx-induced cardiotoxicity.
9. Pathogenic mechanisms of anti-cancer therapy-induced vascular toxicity
The pathophysiology of CTx-induced vascular toxicity is multifactorial and dependent upon the types of CTx employed. Many therapies directly damage the endothelium through mechanisms such as damage to nuclear and/or mitochondrial DNA, altered gene expression, disrupted signaling mechanisms, and oxidative stress (Figures 1 & 2) (Tocchetti et al., 2019). The outcomes of direct endothelial toxicity range from aberrant cellular function (e.g., diminished production of NO; impaired angiogenesis) to endothelial cell apoptosis and necrosis (Soultati et al., 2012). CTx agents also impact the endothelium indirectly through secondary or systemic effects, such as activation of the immune system and inflammatory pathways (Klee et al., 2017).
Figure 2: Proposed mechanisms of endothelial toxicity arising from anti-cancer therapy (CTx).
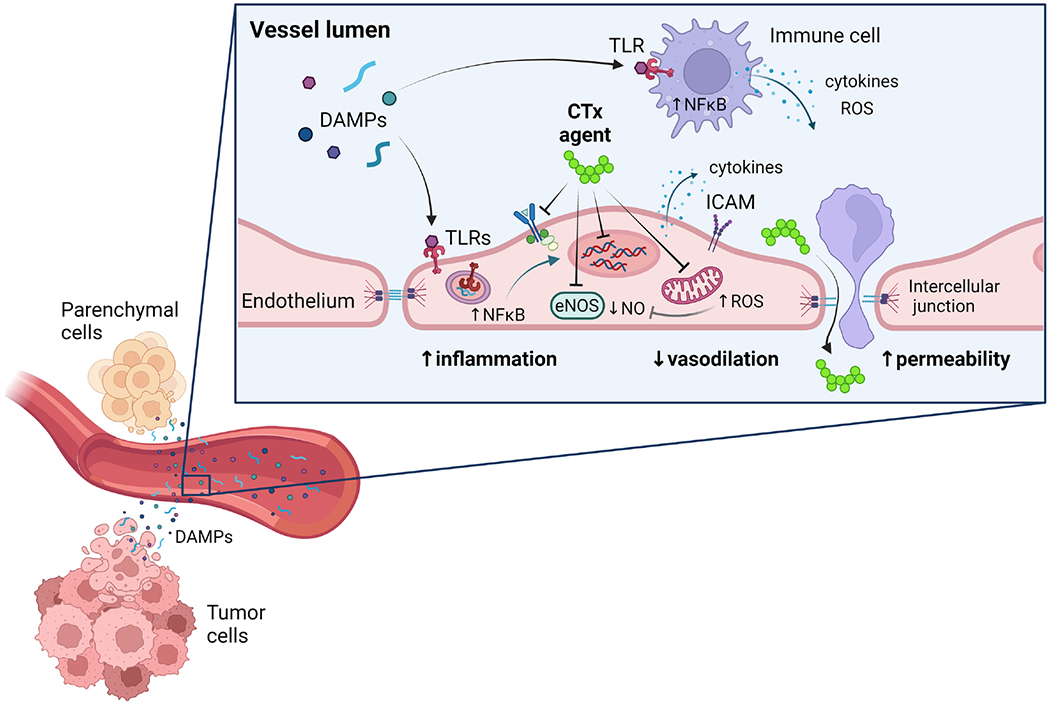
CTx agents directly impact endothelial cells through several mechanisms that interfere with signaling pathways (e.g., tyrosine kinase inhibitors), alter gene expression, reduce nitric oxide (NO) bioavailability, and promote generation of reactive oxygen species (e.g., mitochondria-derived hydrogen peroxide). CTx-induced damage to tumor and parenchymal cells leads to the release of damage-associated molecular patterns (DAMPs), which circulate in the bloodstream and activate toll-like receptors (TLRs) in endothelial cells and immune cells. This initiates an innate immune response via transcription factors such as nuclear factor kappa B (NFκB). Systemic and local inflammation induce cytokine release and surface expression of intercellular adhesion molecules (ICAM), which promote leukocyte transmigration. Augmented vascular permeability also increases exposure of underlying tissue to CTx agents. Ultimately, CTx promotes vascular inflammation, impairs vasodilation, and increases vascular permeability. eNOS, endothelial nitric oxide synthase
Although the specific contributing mechanisms differ among heterogeneous classes of therapies, several common themes have emerged, including oxidative stress and inflammation. Oxidative stress is an important factor in both cancer progression and cancer treatment, and disruptions to oxidative homeostasis have been implicated in vascular dysfunction associated with advancing age and chronic disease. Cancer and CTx are also associated with inflammation, which is exacerbated by pro-oxidant environments. The role of oxidative stress in CTx-mediated cardiovascular dysfunction has been recently reviewed elsewhere (Attanasio et al., 2021; Cadeddu Dessalvi et al., 2021; Carrasco et al., 2021; Tocchetti et al., 2019; Vincent et al., 2013); therefore, in the following sections, we focus on inflammation as a common underlying cause of vascular toxicity related to several classes of CTx.
10. Direct endothelial toxicity
The cytotoxic and molecular mechanisms of several CTx agents directly affect endothelial cells. Anti-angiogenic therapies, which interfere with VEGF signaling or its receptors, intentionally target the endothelium. In contrast, endothelial perturbations are off-target side effects of many treatment classes, which target cancer cells at the expense of the endothelium. The pathophysiological mechanisms underlying direct endothelial cytotoxicity vary based on each CTx agent’s mechanism of action, and the specific pathways involved with several CTx classes have been reviewed elsewhere (Morbidelli et al., 2016; Soultati et al., 2012; Tocchetti et al., 2019). Several CTx agents, most notably anthracyclines, generate free radicals and induce oxidative stress within endothelial cells (Clayton et al., 2020; He et al., 2019; Vásquez-Vivar et al., 1997; Wolf & Baynes, 2006). Agents that damage DNA or disrupt the cytoskeleton also interfere with aspects of endothelial function such as growth and migration (Mabeta & Pepper, 2009). A wide range of CTx agents induce endothelial apoptosis through mechanisms including caspase activation and Fas ligand upregulation (Mabeta & Pepper, 2009; Nuver et al., 2009; Soultati et al., 2012; Wu et al., 2002). In some cases, this can lead to disruption of the endothelial layer and exposure of the intimal surface; thus, thrombogenesis secondary to endothelial cell damage may also contribute to cardiotoxicity (Grover et al., 2021).
11. Role of inflammation
Chronic inflammation is a hallmark of both cancer and cardiovascular disease which accelerates pathophysiological processes (Hansson, 2005; Koene et al., 2016; Libby & Kobold, 2019; Ridker et al., 2017; van’t Klooster et al., 2019). In addition to the low-grade inflammation associated with cancer itself, CTx promotes immune system activation through both direct and indirect mechanisms. Although this prompts tissue repair and is cytoprotective in the short term, exaggerated or persistent immune system activation is maladaptive and has deleterious consequences for cardiovascular health (Libby et al., 2009; Ridker et al., 2017). Chronic inflammation leads to vascular dysfunction in a variety of circumstances (Pierce et al., 2009; Steyers & Miller, 2014); thus, inflammation arising from CTx may contribute vascular damage and cardiotoxicity.
Cancer itself is associated with elevated levels of inflammatory biomarkers, and CTx prompts an increase in circulating cytokines and immune mediators (Demissei et al., 2020; Todorova et al., 2020; Wang et al., 2016; Yu et al., 2018). In addition to evoking systemic inflammation, some cytotoxic chemotherapeutic agents, including doxorubicin, cisplatin, and bleomycin, directly induce inflammatory signaling cascades in endothelial cells (Abou El Hassan et al., 2003; Nuver et al., 2010) Novel therapies that manipulate the immune system to promote destruction of cancer cells, such as CAR-T therapy and immune checkpoint inhibitors, trigger a systemic inflammatory response that results in cytokine release syndrome in many patients (Ceschi et al., 2020; Shimabukuro-Vornhagen et al., 2018). Endothelial cell activation is a hallmark of cytokine release syndrome, which is characterized by hypotension and capillary leak syndrome (Hay et al., 2017). Although few intervention studies have directly assessed the role of inflammation in CTx-induced vascular dysfunction, a recent study demonstrated that tissue necrosis factor α (TNF-α)-mediated vascular inflammation drives aortic stiffening in doxorubicin-treated mice (Clayton et al., 2021). In addition to the cardiovascular consequences, vascular inflammation arising from CTx has potential to initiate a feed-forward cycle that promotes cancer growth. In a pre-clinical model, myocardial infarction accelerates breast cancer progression by promoting immune system-mediated inflammation (Koelwyn et al., 2020). Thus, inflammation induced by CTx may contribute to vascular damage, which can exacerbate inflammation and worsen cardiovascular and cancer outcomes alike.
11.1. Damage-associated molecular patterns and the innate immune system in anti-cancer therapy
Although some CTx agents directly impact the cardiovascular system (e.g., anti-VEGF therapies), the mechanisms underlying treatment-induced cardiotoxicity are less clear for other forms of CTx. CTx agents are designed to promote tumor cell death through either broad cytotoxic mechanisms or targeted molecular or immune mechanisms. The ensuing death of a vast number of cells within a short time period has been proposed as a potential mediator of inflammation and cardiovascular damage following CTx (Ghigo et al., 2016; Klee et al., 2017). Rupture of cell membranes allows intracellular contents to diffuse and enter the circulation, where they serve as damage signals that initiate tissue repair processes (Kono & Rock, 2008). In addition to passive release of contents from dying cells, cellular stress can also promote active secretion of intracellular components (Vénéreau et al., 2015). Pattern recognition receptors (PRRs) of the innate immune system recognize damage-associated molecular patterns (DAMPs) released from dying or injured cells and facilitate inflammation. Although this mechanism is initially protective, chronic activation promotes adverse changes in vascular structure and function that have been proposed to contribute to CTx-induced cardiotoxicity (Klee et al., 2017).
DAMPs encompass a wide range of molecules, including nucleic acids, proteins, and lipids, that are normally compartmentalized and serve functional purposes within cells. Classical examples of DAMPs include high mobility group box 1 (HMGB1), heat shock protein 70, calreticulin, and mitochondrial DNA (Apetoh et al., 2007b; Fiuza et al., 2003; Massé et al., 2004; Obeid et al., 2007; Zhang et al., 2010). Toll-like receptors (TLRs) and other PRRs recognize specific motifs to identify the presence of DAMPs and trigger an inflammatory response. Rapid death of tumor and parenchymal cells following CTx results in a massive release of DAMPs to the circulation, and there is evidence that CTx leads to innate immune system activation through PRRs (Apetoh et al., 2007a, 2007b; Hayashi et al., 2020; Inoue et al., 2021; Solari et al., 2020; Wang et al., 2016; Yu et al., 2018). DAMPs have pleiotropic effects and can both promote and inhibit inflammation. In some circumstances, this pathway is beneficial and can harness the immune system to attack tumor cells (i.e., immunogenic cell death), which has prompted interest in manipulating DAMPs/PRRs to improve cancer outcomes (González-Reyes et al., 2011; Hayashi et al., 2020; Hernandez et al., 2016; Krysko et al., 2012; Obeid et al., 2007). However, DAMPs have also been shown to promote cancer progression and induce treatment resistance (Hernandez et al., 2016; Rajput et al., 2013). These contradictory effects suggest hormesis whereby low-level DAMPs signaling is beneficial to a degree, yet excessive levels or chronic activation may prompt unrestrained inflammation that promotes tumor growth (Hernandez et al., 2016). The net result is likely dependent on many factors, including the type of cancer, biochemical processes targeted by specific classes of CTx, and modifications of DAMPs that alter their effects, such as redox alterations. New strategies to manipulate the balance between immunostimulatory and inhibitory DAMPs have emerged as an approach to promote immunogenic cell death following CTx (Hayashi et al., 2020), which may also hold promise for limiting off-target consequences of inflammation.
The considerable release of DAMPs following CTx may contribute to the adverse cardiovascular side effects of CTx. Chronic activation of PRRs has been observed in cardiovascular diseases, including atherosclerosis, hypertension, chronic heart failure, and cerebrovascular disease (McCarthy et al., 2014, 2015; Oka et al., 2012), and preclinical models suggest a causative role for DAMPs in the pathogenesis of cardiovascular disease (Ding et al., 2013; Hosseini et al., 2016; Ishikawa et al., 2021; Ito et al., 2007; Ueda et al., 2019; Yu & Feng, 2018). The endothelium serves as an interface that detects circulating DAMPs and actively coordinates the ensuing inflammatory cascade through processes such as cytokine secretion, adhesion molecule expression, and changes in vascular permeability that permit leukocyte migration (Bai et al., 2020; Shao et al., 2020). In addition to initiating an innate immune response, activation of PRRs modulates several aspects of vascular function (McCarthy et al., 2019; Wenceslau et al., 2014). For instance, genetic or pharmacological inhibition of TLR4 or TLR9 in rodents protects against conduit artery and resistance vessel endothelial dysfunction (Liang et al., 2013; McCarthy et al., 2015, 2017), while activation of TLR2 or TLR9 reduces NO bioavailability and increases endothelial permeability (Speer et al., 2013; Sun et al., 2013). In the context of CTx, doxorubicin increases levels of DAMPs, and pharmacological blockade or genetic deficiency of TLR2, TLR4 and/or TLR9 or their downstream signaling components protects mice from doxorubicin-induced cardiac inflammation, apoptosis, fibrosis, and cardiomyopathy (Krysko et al., 2011; Ma et al., 2012; Nozaki et al., 2004; Riad et al., 2008; Yao et al., 2012). Altogether, these observations suggest that activation of the innate immune system by DAMPs released from damaged tumor and/or parenchymal cells represents a potential mechanism by which CTx may indirectly induce vascular dysfunction and cardiotoxicity.
Intriguingly, similar pathways related to innate immunity may be involved in the phenomenon of “reverse cardio-oncology” whereby cardiovascular disease promotes cancer development and progression (Aboumsallem et al., 2020). Reprogramming of the innate immune system following myocardial infarction accelerates breast cancer progression in preclinical models, and patients who experience cardiovascular events after a breast cancer diagnosis have an increased risk of cancer recurrence and cancer-related mortality (Koelwyn et al., 2020). Thus, the innate immune system appears to be a crucial link between cancer and cardiovascular disease.
11.2. Link between inflammation and oxidative stress in anti-cancer therapy
ROS are implicated in many of the cytotoxic effects of CTx and represent one mechanism which may link inflammation with vascular dysfunction. In addition to scavenging NO, excessive ROS promote eNOS uncoupling, which results in enzymatic production of superoxide rather than NO. The resulting loss of NO bioavailability is a hallmark of endothelial dysfunction which results in impaired vasodilation. Moreover, loss of the anti-inflammatory effects of NO contributes to a pro-inflammatory state; thus, an oxidative stress-mediated reduction in NO bioavailability may contribute to or exacerbate vascular inflammation.
11.2.1. Myeloperoxidase
The association between inflammation and oxidative stress is bidirectional: inflammation promotes oxidative stress while oxidative stress promotes inflammation (Ding et al., 2013). One example of inflammation-induced oxidative stress is myeloperoxidase, an enzyme secreted by leukocytes to generate ROS as part of the innate immune response. Elevated myeloperoxidase provokes endothelial dysfunction and has been proposed as a contributing factor to the development of atherosclerotic cardiovascular disease (Nicholls & Hazen, 2005). Circulating levels of myeloperoxidase negatively correlate with baFMD (Vita et al., 2004) and independently predict the presence coronary artery disease (Zhang et al., 2001) and risk of major adverse cardiovascular events (Baldus et al., 2003; Brennan et al., 2003). Recent studies have prompted interest in myeloperoxidase as a biomarker of CTx-induced cardiotoxicity because treatment of breast cancer patients with anthracycline-based chemotherapy prompts an increase in plasma myeloperoxidase that predicts subsequent cardiac dysfunction (Demissei et al., 2020; Ky et al., 2014; Putt et al., 2015; Todorova et al., 2020). Elevated plasma myeloperoxidase prior to treatment is also associated with adverse cardiac outcomes, which suggests that basal levels of oxidative stress influence susceptibility to the cardiovascular complications of this treatment regimen (Demissei et al., 2020; Todorova et al., 2020). Notably, although other inflammatory markers including high-sensitivity C-reactive protein and growth-differentiation factor 15 increase following treatment, data regarding their prognostic utility are conflicting, possibly due to the timing of measurements and cardiovascular outcomes assessed (Demissei et al., 2020; Ky et al., 2014; Putt et al., 2015; Todorova et al., 2020).
11.2.2. Mitochondrial damage
Mitochondria play a prominent role in the crosstalk between oxidative stress and inflammation. Many forms of CTx induce mitochondrial damage through a variety of direct and indirect mechanisms. Some CTx agents directly induce mitochondrial DNA (mtDNA) damage or inhibit mitochondrial respiratory complexes, which provokes aberrant mitochondrial ROS production and directly induces endothelial toxicity. Damage to mitochondria can also prompt the release of mtDNA Circulating mtDNA fragments serve as DAMPs that activate TLR9 on endothelial cells to trigger an innate immune response. This mechanism has not been assessed in patients undergoing CTx; however, circulating mtDNA and TLR9 activation stimulate an increase in vascular ROS that quenches NO bioavailability and impairs endothelial vasoreactivity in spontaneously hypertensive rats (McCarthy et al., 2015). CTx agents that promote ROS production can also cause indirect, oxidative damage to mtDNA, which stimulates mitochondrial ROS generation that further exacerbates oxidative stress. Moreover, many CTx agents impact mitochondrial biogenesis and inhibit autophagy/mitophagy, which results in accumulation of damaged, dysfunctional mitochondria. Mitochondrial dysfunction is implicated in endothelial dysfunction, and administration of a mitochondria-targeted antioxidant ameliorates doxorubicin-induced mitochondrial ROS production and prevents conduit artery endothelial dysfunction in mice (Clayton et al., 2020).
12. Clinical Implications
Vascular toxicity has emerged as an important consequence of CTx that impacts the vasculature at the level of the macro and microcirculation. Endothelial dysfunction induced by CTx precedes development of cardiotoxicity and may contribute to the pathogenesis of cardiovascular disease in cancer patients and survivors; thus, endothelial dysfunction and related parameters present an opportunity to predict and monitor cardiovascular disturbances. Understanding the pathogenesis of CTx-induced vascular dysfunction will also provide therapeutic targets for intervention to protect patients from acute and long-term cardiovascular disease associated with their treatment regimens.
12.1. Predicting & monitoring risk
There is a need for validated biomarkers and diagnostic approaches to identify CTx patients at risk of cardiotoxicity and monitor the efficacy of interventions aimed at restoring cardiovascular health. Baseline endothelial dysfunction and circulating factors related to inflammation and oxidative stress have shown promise as screening tools that predict risk of CTx-induced cardiomyopathy prior to initiating treatment (Anastasiou et al., 2017; Demissei et al., 2020; Yu et al., 2018), which may prove useful for guiding treatment decisions and recognizing patients who may require more intensive monitoring. Similarly, larger changes are observed in parameters related to vascular function and inflammation in patients who subsequently develop cardiotoxicity (Demissei et al., 2020; Finkelman et al., 2017; Nagy et al., 2001; Yu et al., 2018), which underscores the potential clinical utility of these measurements. Further research and larger clinical studies are warranted to validate these findings.
12.2. Therapeutic interventions
In the general population, endothelial dysfunction is a key antecedent and driver of cardiovascular disease; thus, the endothelium may be an important therapeutic target to prevent cardiotoxicity in patients undergoing CTx. Although antioxidant supplements have shown promising results in preclinical models, several clinical trials of antioxidant interventions have failed to protect against CTx-induced cardiotoxicity in humans, which has prompted recent interest in targeted strategies such as mitochondria-specific antioxidants (Sabbatino et al., 2021; Šimůnek et al., 2009). Anti-inflammatory interventions have shown promise for ameliorating vascular toxicity and cardiomyocyte damage in preclinical studies (Bruynzeel et al., 2007; Clayton et al., 2021; Henidi et al., 2020; Krishnamurthy et al., 2015; Zhu et al., 2010); however, additional research is needed to verify this in clinical studies. The effects of CTx on the vasculature can arise from on-target or off-target mechanisms, as many CTx agents target signaling pathways that are involved in tumor progression but also play a role in cardiovascular health. Therefore, strategies to minimize vascular damage should focus on signaling pathways that reduce adverse side effects without undermining the anti-tumor effects (Henidi et al., 2020). The innate immune system and ROS are valuable tools for tumor suppression, yet unrestrained inflammation and oxidative stress promote cancer growth and damage the vasculature. Strategies to ameliorate cardiovascular dysfunction while preserving anti-cancer efficacy will likely rely on targeted interventions to restrict vascular inflammation and ROS outside the tumor region, without counteracting the anti-cancer effects of treatment.
In addition to pharmacological approaches, lifestyle interventions may be valuable in combatting the cardiovascular side effects of CTx (Campia & Barac, 2016). A small study of breast cancer patients receiving anthracycline plus cyclophosphamide therapy demonstrated improvements in baFMD, aerobic capacity, and inflammatory markers in a cohort that completed a 12 week aerobic exercise program during adjuvant therapy, and whole genome microarray analysis revealed downregulation of transcripts related to nuclear factor κ B (NF-κB) signaling in the exercise group compared to the non-exercise group (Jones et al., 2013). Importantly, the exercise intervention did not adversely affect tumor biology, which suggests that exercise may mitigate vascular toxicity without interfering with the anti-cancer actions of treatment. Eight weeks of supervised high-intensity interval training (HIIT) during the course of anthracycline-based treatment improved baFMD in breast cancer patients, whereas baFMD deteriorated by nearly 50% over the same time period in a cohort who did not complete HIIT training (Lee et al., 2019). A home-based exercise program has also been reported to improve brachial artery FMD in long-term survivors of childhood acute lymphoblastic leukemia (Järvelä et al., 2013), which highlights the potential of exercise training to reverse chronic endothelial dysfunction following CTx. Additional clinical trials are needed to investigate exercise interventions in a broader range of patients and to examine the underlying mechanisms.
12.3. Conclusion
In conclusion, the deleterious impact of CTx on vascular endothelial function has acute and long-term consequences for cardiovascular health in cancer survivors. Clinical studies have demonstrated vascular toxicity associated with cancer treatment that may be mediated in part by systemic inflammation. Vascular damage can contribute to impaired cardiac performance, which highlights the need for new strategies to preserve endothelial function in this population.
Acknowledgements
This work was supported by NIH grants T32HL134643 (J.D.T.), R01HL133029 (A.M.B.), and R01HL135901 (D.D.G.) and by the We Care Fund. Figure 2 was created with Biorender.com.
Abbreviations
- ABVD
doxorubicin/bleomycin/vinblastine/dacarbazine
- AC
doxorubicin/cyclophosphamide
- ACh
acetylcholine
- AC-T
doxorubicin/cyclophosphamide/docetaxel
- AC-TH
doxorubicin/cyclophosphamide/docetaxel/trastuzumab
- baFMD
brachial artery flow-mediated dilation
- baNMD
brachial artery nitroglycerin-mediated dilation
- BEP
bleomycin/etoposide/cisplatin
- CAP-5
cyclophosphamide/doxorubicin/cisplatin
- CAR-T
chimeric antigen receptor–modified T cell
- CD163
cluster of differentiation 163
- CD206/MR
cluster of differentiation 206/mannose receptor
- CHOP
cyclophosphamide/doxorubicin/vincristine/prednisone
- CIMT
carotid artery intima-media thickness
- CTx
anti-cancer therapy
- DAMPs
damage-associated molecular patterns
- EC-T
epirubicin/cyclophosphamide/docetaxel or paclitaxel
- EC-TH
epirubicin/cyclophosphamide/docetaxel or paclitaxel/trastuzumab
- eNOS
endothelial nitric oxide synthase
- EP
etoposide/cisplatin
- FMD
flow-mediated dilation
- HER2
human epidermal growth factor receptor 2
- HIIT
high-intensity interval training
- H2O2
hydrogen peroxide
- HOP
doxorubicin/vincristine/prednisone
- hs-CRP
high-sensitivity C-reactive protein
- hs-TNI
high-sensitivity troponin I
- ICAM-1
intercellular adhesion molecule-1
- IL-2
interleukin 2
- LVEF
left-ventricular ejection fraction
- mtDNA
mitochondrial DNA
- NF-κB
nuclear factor κ B
- NO
nitric oxide
- PRR
pattern-recognition receptor
- PWV
pulse-wave velocity
- PVB
cisplatin/vinblastine/bleomycin
- ROS
reactive oxygen species
- SNP
sodium nitroprusside
- TC
paclitaxel/carboplatin
- TCH
docetaxel/carboplatin/trastuzumab
- TKI
tyrosine kinase inhibitor
- TLR
toll-like receptor
- TNFα
tumor necrosis factor α
- VEGF
vascular endothelial growth factor
- vWF
von Willebrand factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Abou El Hassan MAI, Verheul HMW, Jorna AS, Schalkwijk C, Van Bezu J, Van Der Vijgh WJF, & Bast A (2003). The new cardioprotector Monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. British Journal of Cancer, 89(2), 357–362. doi: 10.1038/sj.bjc.6601022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboumsallem JP, Moslehi J, & de Boer RA (2020). Reverse cardio-oncology: cancer development in patients With cardiovascular disease. Journal of the American Heart Association, 9(2), 1–12. doi: 10.1161/JAHA.119.013754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou M, Oikonomou E, Zagouri F, Siasos G, Antonopoulos AS, Psaltopoulou T, Bamias A, Dimopoulos MA, & Tousoulis D (2017). Flow-mediated dilation of brachial artery as a screening tool for anthracycline-induced cardiotoxicity. Journal of the American College of Cardiology, 70(24), 3072. doi: 10.1016/j.jacc.2017.09.1140 [DOI] [PubMed] [Google Scholar]
- Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, André F, Tursz T, Kroemer G, & Zitvogel L (2007). The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunological Reviews, 220(1), 47–59. doi: 10.1111/j.1600-065X.2007.00573.x [DOI] [PubMed] [Google Scholar]
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, … Zitvogel L (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature Medicine, 13(9), 1050–1059. doi: 10.1038/nm1622 [DOI] [PubMed] [Google Scholar]
- Attanasio U, Pirozzi F, Poto R, Cuomo A, Carannante A, Russo M, Ghigo A, Hirsch E, Tocchetti CG, Varricchi G, & Mercurio V (2021). Oxidative stress in anticancer therapies-related cardiac dysfunction. Free Radical Biology and Medicine, 169, 410–415. doi: 10.1016/j.freeradbiomed.2021.04.021 [DOI] [PubMed] [Google Scholar]
- Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, Aung LHH, Li P. feng, Yu T, & Chu X. ming. (2020). NLRP3 inflammasome in endothelial dysfunction. Cell Death and Disease, 11, 776. doi: 10.1038/s41419-020-02985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Münzel T, Simoons ML, & Hamm CW (2003). Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation, 108(12), 1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51 [DOI] [PubMed] [Google Scholar]
- Bar-Joseph H, Ben-Aharon I, Tzabari M, Tsarfaty G, Stemmer SM, & Shalgi R (2011). In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS ONE, 6(9), e23492. doi: 10.1371/journal.pone.0023492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselet B, Sonveaux P, Baatout S, & Aerts A (2019). Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cellular and Molecular Life Sciences, 76(4), 699–728. doi: 10.1007/s00018-018-2956-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K, Erckenbrecht JF, Häussinger D, & Frieling T (1999). Cardiotoxicity of the antiproliferative compound fluorouracil. Drugs, 57(4), 475–484. doi: 10.2165/00003495-199957040-00003 [DOI] [PubMed] [Google Scholar]
- Beyer AM, Zinkevich N, Miller B, Liu Y, Wittenburg AL, Mitchell M, Galdieri R, Sorokin A, & Gutterman DD (2017). Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease. Basic Research in Cardiology, 112(1), 5. doi: 10.1007/s00395-016-0594-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan M, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, & Hazen SL (2003). Prognostic value of myeloperoxidase in patients with chest pain. New England Journal of Medicine, 349(17), 1595–1604. [DOI] [PubMed] [Google Scholar]
- Broxterman RM, Witman MA, Trinity JD, Groot HJ, Rossman MJ, Park SY, Malenfant S, Gifford JR, Kwon OS, Park SH, Jarrett CL, Shields KL, Hydren JR, Bisconti AV, Owan T, Abraham A, Tandar A, Lui CY, Smith BR, & Richardson RS (2019). Strong relationship between vascular function in the coronary and brachial arteries. Hypertension, 74, 208–215. doi: 10.1161/HYPERTENSIONAHA.119.12881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruynzeel A, Abou MA, Hassan E, Schalkwijk C, Berkhof J, Bast A, Niessen H, & Van Der Vijgh W (2007). Anti-inflammatory agents and monoHER protect against DOX-induced cardiotoxicity and accumulation of CML in mice. British Journal of Cancer, 96, 937–943. doi: 10.1038/sj.bjc.6603640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadeddu Dessalvi C, Deidda M, Noto A, Madeddu C, Cugusi L, Santoro C, López-Fernández T, Galderisi M, & Mercuro G (2021). Antioxidant approach as a cardioprotective strategy in chemotherapy-induced cardiotoxicity. Antioxidants and Redox Signaling, 34(7), 572–588. doi: 10.1089/ars.2020.8055 [DOI] [PubMed] [Google Scholar]
- Cameron AC, McMahon K, Hall M, Neves KB, Rios FJ, Montezano AC, Welsh P, Waterston A, White J, Mark PB, Touyz RM, & Lang NN (2020). Comprehensive characterization of the vascular effects of cisplatin-based chemotherapy in patients with testicular cancer. JACC: CardioOncology, 2(3), 443–455. doi: 10.1016/j.jaccao.2020.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AC, Touyz RM, & Lang NN (2016). Vascular complications of cancer chemotherapy. Canadian Journal of Cardiology, 32(7), 852–862. doi: 10.1016/j.cjca.2015.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campia U, & Barac A (2016). Exercise and aerobic fitness to reduce cancer-related cardiovascular toxicity. Current Treatment Options in Cardiovascular Medicine, 18, 44. doi: 10.1007/s11936-016-0465-7 [DOI] [PubMed] [Google Scholar]
- Carrasco R, Castillo RL, Gormaz JG, Carrillo M, & Thavendiranathan P (2021). Role of oxidative stress in the mechanisms of anthracycline-induced cardiotoxicity: effects of preventive strategies. Oxidative Medicine and Cellular Longevity, 2021, 8863789. doi: 10.1155/2021/8863789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celermajer DS, Sorensen KE, Gooch VM, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE, & Spiegelhalter DJ (1992). Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. The Lancet, 340(8828), 1111–1115. doi: 10.1016/0140-6736(92)93147-F [DOI] [PubMed] [Google Scholar]
- Ceschi A, Noseda R, Palin K, & Verhamme K (2020). Immune checkpoint inhibitor-related cytokine release syndrome: analysis of WHO global pharmacovigilance database. Frontiers in Pharmacology, 11, 557. doi: 10.3389/fphar.2020.00557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AY, Chin C, Dahl G, & Rosenthal DN (2006). Anthracyclines cause endothelial injury in pediatric cancer patients: a pilot study. Journal of Clinical Oncology, 24(6), 925–928. doi: 10.1200/JCO.2005.03.5956 [DOI] [PubMed] [Google Scholar]
- Clayton ZS, Brunt VE, Hutton DA, Casso AG, Ziemba BP, Melov S, Campisi J, & Seals DR (2021). Tumor necrosis factor alpha-mediated inflammation and remodeling of the extracellular matrix underlies aortic stiffening induced by the common chemotherapeutic agent doxorubicin. Hypertension, 77(5), 1581–1590. doi: 10.1161/hypertensionaha.120.16759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton ZS, Brunt VE, Hutton DA, VanDongen NS, D’Alessandro A, Reisz JA, Ziemba BP, & Seals DR (2020). Doxorubicin-induced oxidative stress and endothelial dysfunction in conduit arteries is prevented by mitochondrial-specific antioxidant treatment. JACC: Cardio Oncology, 2(3), 475–488. doi: 10.1016/j.jaccao.2020.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschignano MA, De Ciuceis C, Agabiti-Rosei C, Brami V, Rossini C, Chiarini G, Malerba P, Famà F, Cosentini D, Muiesan ML, Salvetti M, Petelca A, Capellini S, Arnoldi C, Nardin M, Grisanti S, Rizzoni D, Berruti A, & Paini A (2021). Microvascular structural alterations in patients treated with antiangiogenic drugs. Frontiers in Cardiovascular Medicine, 8, 161. doi: 10.3389/fcvm.2021.651594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cwikiel M, Eskilsson J, Wieslander JB, Stjernquist U, & Albertsson M (1996). The appearance of endothelium in small arteries after treatment with 5-fluorouracil. An electron microscopic study of late effects in rabbits. Scanning Microscopy, 10(3), 805–819. [PubMed] [Google Scholar]
- Cwikiel M, Zhang B, Eskilsson J, Wieslander JB, & Albertsson M (1995). The influence of 5-fluorouracil on the endothelium in small arteries. An electron microscopic study in rabbits. Scanning Microscopy, 9(2), 561–576. [PubMed] [Google Scholar]
- D’Amario D, Camilli M, Migliaro S, Canonico F, Galli M, Arcudi A, Montone RA, Borovac JA, Crea F, & Savarese G (2020). Sex-related differences in dilated cardiomyopathy with a focus on cardiac dysfunction in oncology. Current Cardiology Reports, 22(10), 1–11. doi: 10.1007/s11886-020-01377-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta SK, Le A, Vijayan KV, & Thiagarajan P (2017). Dasatinib inhibits actin fiber reorganization and promotes endothelial cell permeability through RhoA-ROCK pathway. Cancer Medicine, 6(4), 809–818. doi: 10.1002/CAM4.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos FYFL, Nuver J, Willemse PHB, Van Der Zee AGJ, Messerschmidt J, Burgerhof JGM, De Vries EGE, & Gietema JA (2004). Long-term survivors of ovarian malignancies after cisplatin-based chemotherapy: Cardiovascular risk factors and signs of vascular damage. European Journal of Cancer, 40(5), 696–700. doi: 10.1016/j.ejca.2003.11.026 [DOI] [PubMed] [Google Scholar]
- Demissei BG, Hubbard RA, Zhang L, Smith AM, Sheline K, McDonald C, Narayan V, Domchek SM, DeMichele A, Shah P, Clark AS, Fox K, Matro J, Bradbury AR, Knollman H, Getz KD, Armenian SH, Januzzi JL, Tang WHW, … Ky B (2020). Changes in cardiovascular biomarkers with breast cancer therapy and associations with cardiac dysfunction. Journal of the American Heart Association, 9(2). doi: 10.1161/JAHA.119.014708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengel DR, Ness KK, Glasser SP, Williamson EB, Baker KS, & Gurney JG (2008). Endothelial function in young adult survivors of childhood acute lymphoblastic leukemia. Journal of Pediatric Hematology/Oncology, 30(1), 20–25. doi: 10.1097/MPH.0b013e318159a593 [DOI] [PubMed] [Google Scholar]
- Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, & Mehta JL (2013). Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Scientific Reports 2013 3:1, 3(1), 1–6. doi: 10.1038/srep01077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaran S, Caron JP, Zhou W, Hainer J, Bibbo CF, Skali H, Taqueti VR, Dorbala S, Blankstein R, Groarke JD, Nohria A, & Di Carli MF (2021). Coronary vasomotor dysfunction portends worse outcomes in patients with breast cancer. Journal of Nuclear Cardiology. doi: 10.1007/s12350-021-02825-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, Mosarla RC, Lee C, Zlotoff DA, Raghu VK, Hartmann SE, Gilman HK, Gong J, Zubiri L, Sullivan RJ, Reynolds KL, Mayrhofer T, Zhang L, Hoffmann U, & Neilan TG (2020). Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation, 142, 2299–2311. doi: 10.1161/CIRCULATIONAHA.120.049981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquaine D, Hirsch GA, Chakrabarti A, Han Z, Kehrer C, Brook R, Joseph J, Schott A, Kalyanaraman B, Vasquez-Vivar J, & Rajagopalan S (2003). Rapid-onset endothelial dysfunction with adriamycin: Evidence for a dysfunctional nitric oxide synthase. Vascular Medicine, 8(2), 101–107. doi: 10.1191/1358863x03vm476oa [DOI] [PubMed] [Google Scholar]
- Durand MJ, Hader SN, Derayunan A, Zinkevich N, McIntosh JJ, & Beyer AM (2020). BCR-ABL tyrosine kinase inhibitors promote pathological changes in dilator phenotype in the human microvasculature. Microcirculation, 27(7), 1–5. doi: 10.1111/micc.12625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederer AK, Didier KD, Reiter LK, Brown M, Hardy R, Caldwell J, Black CD, Larson RD, & Ade CJ (2016). Influence of adjuvant therapy in cancer survivors on endothelial function and skeletal muscle deoxygenation. PLoS ONE, 11(1), 1–15. doi: 10.1371/journal.pone.0147691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazakas C, Nagaraj C, Zabini D, Végh AG, Marsh LM, Wilhelm I, Krizbai IA, Olschewski H, Olschewski A, & Bálint Z (2018). Rho-kinase inhibition ameliorates dasatinib-induced endothelial dysfunction and pulmonary hypertension. Frontiers in Physiology, 9, 537. doi: 10.3389/fphys.2018.00537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelman BS, Putt M, Wang T, Wang L, Narayan H, Domchek S, DeMichele A, Fox K, Matro J, Shah P, Clark A, Bradbury A, Narayan V, Carver JR, Tang WHW, & Ky B (2017). Arginine-nitric oxide metabolites and cardiac dysfunction in patients with breast cancer. Journal of the American College of Cardiology, 70(2), 152–162. doi: 10.1016/j.jacc.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, & Suffredini AF (2003). Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood, 101(7), 2652–2660. doi: 10.1182/blood-2002-05-1300 [DOI] [PubMed] [Google Scholar]
- Fredslund SO, Buus NH, Højgaard Skjold C, Laugesen E, Jensen AB, & Laursen BE (2021). Changes in vascular function during breast cancer treatment. British Journal of Clinical Pharmacology, 1–11. doi: 10.1111/bcp.14837 [DOI] [PubMed] [Google Scholar]
- Galán-Arriola C, Vílchez-Tschischke JP, Lobo M, López GJ, de Molina-Iracheta A, Pérez-Martínez C, Villena-Gutiérrez R, Macías Á, Díaz-Rengifo IA, Oliver E, Fuster V, Sánchez-González J, & Ibanez B (2021). Coronary microcirculation damage in anthracycline cardiotoxicity. Cardiovascular Research, 0, 1–11. doi: 10.1093/cvr/cvab053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigo A, Li M, & Hirsch E (2016). New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1863(7), 1916–1925. doi: 10.1016/J.BBAMCR.2016.01.021 [DOI] [PubMed] [Google Scholar]
- Giordano P, Muggeo P, Delvecchio M, Carbonara S, Romano A, Altomare M, Ricci G, Valente F, Zito A, Scicchitano P, Cavallo L, Ciccone MM, Santoro N, & Faienza MF (2017). Endothelial dysfunction and cardiovascular risk factors in childhood acute lymphoblastic leukemia survivors. International Journal of Cardiology, 228, 621–627. doi: 10.1016/j.ijcard.2016.11.025 [DOI] [PubMed] [Google Scholar]
- Gokce N, Keaney JF, Hunter LM, Watkins MT, Menzoian JO, & Vita JA (2002). Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: A prospective study. Circulation, 105(13), 1567–1572. doi: 10.1161/01.CIR.0000012543.55874.47 [DOI] [PubMed] [Google Scholar]
- González-Reyes S, Fernández JM, González LO, Aguirre A, Suárez A, González JM, Escaff S, & Vizoso FJ (2011). Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunology, Immunotherapy, 60(2), 217–226. doi: 10.1007/s00262-010-0931-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover SP, Hisada YM, Kasthuri RS, Reeves BN, & Mackman N (2021). Cancer therapy-associated thrombosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 41, 1291–1305. doi: 10.1161/ATVBAHA.120.314378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Baker SH, Ozpolat T, Fink KR, Riddell SR, Maloney DG, & Turtle CJ (2017). Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery, 7(12), 1404–1419. doi: 10.1158/2159-8290.CD-17-0698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hader SN, Zinkevich N, Toro LEN, Kriegel AJ, Kong A, Freed JK, Gutterman DD, & Beyer AM (2019). Detrimental effects of chemotherapy on human coronary microvascular function. American Journal of Physiology - Heart and Circulatory Physiology, 317(4), H705–H710. doi: 10.1152/ajpheart.00370.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK (2005). Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine, 352, 1685–1695. [DOI] [PubMed] [Google Scholar]
- Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, López JA, Chen J, Chung D, Harju-Baker S, Cherian S, Chen X, Riddell SR, Maloney DG, & Turtle CJ (2017). Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood, 130(21), 2295–2306. doi: 10.1182/blood-2017-06-793141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Nikolos F, Lee YC, Jain A, Tsouko E, Gao H, Kasabyan A, Leung HE, Osipov A, Jung SY, Kurtova AV, & Chan KS (2020). Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nature Communications, 11, 6299. doi: 10.1038/s41467-020-19970-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazim AZ, Nhola LF, Villarraga HR, Patel SR, Sandhu N, Lerman A, Loprinzi CL, Ruddy KJ, & Herrman J (2020). Peripheral endothelial function changes during HER2-directed therapy differ based on whether or not a patient receives anthracycline. J Clinical Oncology, 38(15 Suppl), 1044. [Google Scholar]
- He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, Yin D, Huang Q, & He M (2019). Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Frontiers in Pharmacology, 10, 1531. doi: 10.3389/fphar.2019.01531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henidi HA, Al-Abbasi FA, El-Moselhy MA, El-Bassossy HM, Al-Abd AM, & Gil G (2020). Despite blocking doxorubicin-induced vascular damage, quercetin ameliorates its antibreast cancer activity. Oxidative Medicine and Cellular Longevity, 2020, 8157640. doi: 10.1155/2020/8157640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C, Huebener P, & Schwabe RF (2016). Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene, 35(46), 5931–5941. doi: 10.1038/onc.2016.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A, Toutouzas K, Leesar MA, Grines CL, & Marmagkiolis K (2016). Vascular toxicities of cancer therapies: the old and the new - an evolving avenue. Circulation, 133(13), 1272–1289. doi: 10.1161/CIRCULATIONAHA.115.018347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman RK, Kim B-J, Shah PD, Carver J, Ky B, & Ryeom S (2021). Damage to cardiac vasculature may be associated with breast cancer treatment-induced cardiotoxicity. Cardio-Oncology, 7(1), 1–13. doi: 10.1186/s40959-021-00100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohneck A, Custodis F, Rosenkaimer S, Hofheinz R, Maier S, Akin I, Borggrefe M, & Gerhards S (2021). Gender aspects in cardiooncology. European Journal of Public Health, 31(6), 1170–1176. doi: 10.1093/eurpub/ckab146 [DOI] [PubMed] [Google Scholar]
- Hosseini H, Li Y, Kanellakis P, Tay C, Cao A, Liu E, Peter K, Tipping P, Toh BH, Bobik A, & Kyaw T (2016). Toll-like receptor (TLR)4 and MyD88 are essential for atheroprotection by peritoneal B1a B cells. Journal of the American Heart Association, 5(11). doi: 10.1161/JAHA.115.002947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Tsutsumi H, Tanaka K, Iwama E, Shiraishi Y, Hirayama A, Nakanishi T, Ando H, Nakajima M, Shinozaki S, Ogata H, Uryu K, Okamura K, Kimura S, Ogawa T, Ota K, Yoneshima Y, Hamada N, Nakanishi Y, & Okamoto I (2021). Increased plasma levels of damage-associated molecular patterns during systemic anticancer therapy in patients with advanced lung cancer. Translational Lung Cancer Research, 10(6), 2475–2486. doi: 10.21037/tlcr-21-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Abe K, Takana-Ishikawa M, Yoshida K, Watanabe T, Imakiire S, Hosokawa K, Hirano M, Hirano K, & Tsutsui H (2021). Chronic inhibition of toll-like receptor 9 ameliorates pulmonary hypertension in rats. Journal of the American Heart Association, 10(7), e019247. doi: 10.1161/JAHA.120.019247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, Hashiguchi T, & Maruyama I (2007). High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. Journal of Thrombosis and Haemostasis, 5(1), 109–116. doi: 10.1111/J.1538-7836.2006.02255.X [DOI] [PubMed] [Google Scholar]
- Järvelä LS, Niinikoski H, Heinonen OJ, Lähteenmäki PM, Arola M, & Kemppainen J (2013). Endothelial function in long-term survivors of childhood acute lymphoblastic leukemia: Effects of a home-based exercise program. Pediatric Blood and Cancer, 60(9), 1546–1551. doi: 10.1002/pbc.24565 [DOI] [PubMed] [Google Scholar]
- Jenei Z, Bárdi E, Magyar MT, Horváth Á, Paragh G, & Kiss C (2013). Anthracycline causes impaired vascular endothelial function and aortic stiffness in long term survivors of childhood cancer. Pathology and Oncology Research, 19(3), 375–383. doi: 10.1007/s12253-012-9589-6 [DOI] [PubMed] [Google Scholar]
- Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, & Lüscher TF (1995). Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation, 91(5), 1314–1319. [DOI] [PubMed] [Google Scholar]
- Jones LW, Fels DR, West M, Allen JD, Broadwater G, Barry WT, Wilke LG, Masko E, Douglas PS, Dash RC, Povsic TJ, Peppercorn J, Marcom PK, Blackwell KL, Kimmick G, Turkington TG, & Dewhirst MW (2013). Modulation of circulating angiogenic factors and tumor biology by aerobic training in breast cancer patients receiving neoadjuvant chemotherapy. Cancer Prevention Research, 6(9), 925–937. doi: 10.1158/1940-6207.CAPR-12-0416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LW, Haykowsky M, Peddle CJ, Joy AA, Pituskin EN, Tkachuk LM, Courneya KS, Slamon DJ, & Mackey JR (2007). Cardiovascular risk profile of patients with HER2/neu-positive breast cancer treated with anthracycline-taxane-containing adjuvant chemotherapy and/or trastuzumab. Cancer Epidemiology Biomarkers and Prevention, 16(5), 1026–1031. doi: 10.1158/1055-9965.EPI-06-0870 [DOI] [PubMed] [Google Scholar]
- Kaushal V, Kaushal GP, & Mehta P (2004). Differential toxicity of antracyclines on cultured endothelial cells. Endothelium: Journal of Endothelial Cell Research, 77(5–6), 253–258. doi: 10.1080/10623320490904124 [DOI] [PubMed] [Google Scholar]
- Klee NS, McCarthy CG, Martinez-Quinones P, & Webb RC (2017). Out of the frying pan and into the fire: damage-associated molecular patterns and cardiovascular toxicity following cancer therapy. Therapeutic Advances in Cardiovascular Disease, 11(11), 297–317. doi: 10.1177/1753944717729141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelwyn GJ, Newman AAC, Afonso MS, van Solingen C, Corr EM, Brown EJ, Albers KB, Yamaguchi N, Narke D, Schlegel M, Sharma M, Shanley LC, Barrett TJ, Rahman K, Mezzano V, Fisher EA, Park DS, Newman JD, Quail DF, … Moore KJ (2020). Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nature Medicine 2020 26:9, 26(9), 1452–1458. doi: 10.1038/s41591-020-0964-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene RJ, Prizment AE, Blaes A, & Konety SH (2016). Shared risk factors in cardiovascular disease and cancer. Circulation, 133(11), 1104–1114. doi: 10.1161/CIRCULATIONAHA.115.020406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, & Rock KL (2008). How dying cells alert the immune system to danger. Nature Reviews Immunology, 5(4), 279–289. doi: 10.1038/nri2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpela E, & Liu SK (2014). Endothelial perturbations and therapeutic strategies in normal tissue radiation damage. Radiation Oncology, 9(1). doi: 10.1186/s13014-014-0266-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakou PM, Kouris NT, Kostopoulos VS, Damaskos DS, & Olympios CD (2019). Cardio-oncology: a new and developing sector of research and therapy in the field of cardiology. Heart Failure Reviews, 24(1), 91–100. doi: 10.1007/s10741-018-9731-y [DOI] [PubMed] [Google Scholar]
- Kreutzman A, Colom-Fernandez B, Jimenez AM, Hander M, Cuesta-Mateos C, Perez-García Y, Arevalo CD, Brück O, Hakanen H, Saarela J, Ortega-Carrion A, De Rosendo A, Juanes-García A, Steegmann JL, Mustjoki S, Vicente-Manzanares M, & Muñoz-Calleja C (2017). Dasatinib reversibly disrupts endothelial vascular integrity by increasing non-muscle myosin II contractility in a ROCK-dependent manner. Clinical Cancer Research, 23(21), 6697–6707. doi: 10.1158/1078-0432.CCR-16-0667 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy B, Rani N, Bharti S, Golechha M, Bhatia J, Nag TC, Ray R, Arava S, & Arya DS (2015). Febuxostat ameliorates doxorubicin-induced cardiotoxicity in rats. Chemico-Biological Interactions, 237, 96–103. doi: 10.1016/j.cbi.2015.05.013 [DOI] [PubMed] [Google Scholar]
- Krysko Dmitri V., Garg AD, Kaczmarek A, Krysko O, Agostinis P, & Vandenabeele P (2012). Immunogenic cell death and DAMPs in cancer therapy. Nature Reviews Cancer, 12(12), 860–875. doi: 10.1038/nrc3380 [DOI] [PubMed] [Google Scholar]
- Krysko DV, Kaczmarek A, Krysko O, Heyndrickx L, Woznicki J, Bogaert P, Cauwels A, Takahashi N, Magez S, Bachert C, & Vandenabeele P (2011). TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death & Differentiation 2011 18:8, 18(8), 1316–1325. doi: 10.1038/cdd.2011.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ky B, Putt M, Sawaya H, French B, Januzzi JL, Sebag IA, Plana JC, Cohen V, Banchs J, Carver JR, Wiegers SE, Martin RP, Picard MH, Gerszten RE, Halpern EF, Passeri J, Kuter I, & Scherrer-Crosbie M (2014). Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. Journal of the American College of Cardiology, 63(8), 809–816. doi: 10.1016/j.jacc.2013.10.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai S, Amabile MI, Mazzaferro S, Mitterhofer AP, Mazzarella A, Galani A, Imbimbo G, Cianci R, Pasquali M, & Molfino A (2020). Effects of sunitinib on endothelial dysfunction, metabolic changes, and cardiovascular risk indices in renal cell carcinoma. Cancer Medicine, 9(11), 3752–3757. doi: 10.1002/cam4.2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Kang I, Mack WJ, Mortimer J, Sattler F, Salem G, Lu J, & Dieli-Conwright CM (2019). Effects of high-intensity interval training on vascular endothelial function and vascular wall thickness in breast cancer patients receiving anthracycline-based chemotherapy: a randomized pilot study. Breast Cancer Research and Treatment, 177, 477–485. doi: 10.1007/s10549-019-05332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Qian M, Kyler K, & Xu J (2018). Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Frontiers in Cardiovascidar Medicine, 5, 151. doi: 10.3389/FCVM.2018.00151/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CF, Liu JT, Wang Y, Xu A, & Vanhoutte PM (2013). Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arteriosclerosis, Thrombosis, and Vascular Biology, 33(4), 777–784. doi: 10.1161/ATVBAHA.112.301087 [DOI] [PubMed] [Google Scholar]
- Libby P, & Kobold S (2019). Inflammation: a common contributor to cancer, aging, and cardiovascular diseases - expanding the concept of cardio-oncology. Cardiovascular Research, 115(5), 824–829. doi: 10.1093/cvr/cvz058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, & Hansson GK (2009). Inflammation in atherosclerosis, from pathophysiology to practice. Journal of the American College of Cardiology, 54(23), 2129–2138. doi: 10.1016/j.jacc.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H, Wang Q, Wang Z, Yan J, Fan G, Lindsey ML, & Hu Z (2012). Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS ONE, 7(7), e40763. doi: 10.1371/journal.pone.0040763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabeta P, & Pepper MS (2009). A comparative study on the anti-angiogenic effects of DNA-damaging and cytoskeletal-disrupting agents. Angiogenesis, 12(1), 81–90. doi: 10.1007/s10456-009-9134-8 [DOI] [PubMed] [Google Scholar]
- Massé D, Ebstein F, Bougras G, Harb J, Meflah K, & Grégoire M (2004). Increased expression of inducible HSP70 in apoptotic cells is correlated with their efficacy for antitumor vaccine therapy. International Journal of Cancer, 111(4), 575–583. doi: 10.1002/IJC.20249 [DOI] [PubMed] [Google Scholar]
- Mayer EL, Dallabrida SM, Rupnick MA, Redline WM, Hannagan K, Ismail NS, Burstein HJ, & Beckman JA (2011). Contrary effects of the receptor tyrosine kinase inhibitor vandetanib on constitutive and flow-stimulated nitric oxide elaboration in humans. Hypertension, 58(1), 85–92. doi: 10.1161/HYPERTENSIONAHA.110.168120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Goulopoulou S, & Webb RC (2019). Paying the toll for inflammation: immunoreceptor-mediated vascular dysfunction in hypertension. Hypertension, 73(3), 514–521. doi: 10.1161/HYPERTENSIONAHA.118.11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, & Webb RC (2014). Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. American Journal of Physiology - Heart and Circulatory Physiology, 306(2), 184–196. doi: 10.1152/ajpheart.00328.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, & Webb RC (2017). Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. American Journal of Hypertension, 30(2), 173–181. doi: 10.1093/ajh/hpw113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, & Webb RC (2015). Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovascular Research, 107(1), 119–130. doi: 10.1093/cvr/cvv137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinardi M, Gietema J, van Graaf W, van Veldhuisen D, Runne M, Sluiter W, de Vries E, Willemse P, Mulder N, van den Berg M, Koops H, & Sleijfer D (2001). Cardiovascular morbidity in long-term survivors of metastatic testicular cancer. Rehabilitation Oncology, 19(2), 27. doi: 10.1097/01893697-200119020-00016 [DOI] [PubMed] [Google Scholar]
- Meinardi MT, Gietema JA, Van Veldhuisen DJ, Van der Graaf WTA, De Vries EGE, & Sleijfer DT (2000). Long-term chemotherapy-related cardiovascular morbidity. Cancer Treatment Reviews, 26(6), 429–447. doi: 10.1053/ctrv.2000.0175 [DOI] [PubMed] [Google Scholar]
- Meiners B, Shenoy C, & Zordoky BN (2018). Clinical and preclinical evidence of sex-related differences in anthracycline-induced cardiotoxicity. Biology of Sex Differences, 9(1). doi: 10.1186/S13293-018-0198-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, & Gutterman DD (2003). Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circuation Research, 92(2), e31–e40. doi: 10.1161/01.res.0000054200.44505.ab [DOI] [PubMed] [Google Scholar]
- Morbidelli L, Donnini S, & Ziche M (2016). Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardio-Oncology, 2(1), 1–12. doi: 10.1186/s40959-016-0010-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosseri M, Fingert HJ, Varticovski L, Chokshi S, & Isner JM (1993). In vitro evidence that myocardial ischemia resulting from 5-fluorouracil chemotherapy is due to protein kinase C-mediated vasoconstriction of vascular smooth muscle. Cancer Research, 53(13), 3028–3033. [PubMed] [Google Scholar]
- Mukai M, Komori K, & Oka T (2018). Mechanism and management of cancer chemotherapy-induced atherosclerosis. Journal of Atherosclerosis and Thrombosis, 25(10), 994–1002. doi: 10.5551/JAT.RV17027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulrooney DA, Blaes AH, & Duprez D (2012). Vascular injury in cancer survivors. Journal of Cardiovascular Translational Research, 5(3), 287–295. doi: 10.1007/s12265-012-9358-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulrooney DA, Yeazel MW, Kawashima T, Mertens AC, Mitby P, Stovall M, Donaldson SS, Green DM, Sklar CA, Robison LL, & Leisenring WM (2009). Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the childhood cancer survivor study cohort. British Medical Journal, 339, b4606. doi: 10.1136/bmj.b4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, & Simons M (2009). Regulation of vascular integrity. Journal of Molecular Medicine, 87(6), 571–582. doi: 10.1007/s00109-009-0463-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Szabó F, Iványi J, Németh L, Kovács GL, Palatka J, Tarján J, Tóth K, & Röth E (2001). A method for detection of doxorubicin-induced cardiotoxicity: Flow-mediated vasodilation of the brachial artery. Experimental and Clinical Cardiology, 6(2), 87–92. [PMC free article] [PubMed] [Google Scholar]
- Nicholls SJ, & Hazen SL (2005). Myeloperoxidase and cardiovascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(6), 1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d [DOI] [PubMed] [Google Scholar]
- Nozaki N, Shishido T, Takeishi Y, & Kubota I (2004). Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Gradation, 110(18), 2869–2874. doi: 10.1161/01.CIR.0000146889.46519.27 [DOI] [PubMed] [Google Scholar]
- Nuver J, De Haas EC, Van Zweeden M, Gietema JA, & Meijer C (2009). Vascular damage in testicular cancer patients: A study on endothelial activation by bleomycin and cisplatin in vitro. Oncology Reports, 23(1), 247–253. doi: 10.3892/or_00000630 [DOI] [PubMed] [Google Scholar]
- Nuver J, Smit AJ, Sleijfer DT, Van Gessel AI, Van Roon AM, Van Der Meer J, Van Den Berg MP, Burgerhof JGM, Hoekstra HJ, Sluiter WJ, & Gietema JA (2004). Microalbuminuria, decreased fibrinolysis, and inflammation as early signs of atherosclerosis in long-term survivors of disseminated testicular cancer. European Journal of Cancer, 40(5), 701–706. doi: 10.1016/j.ejca.2003.12.012 [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, & Kroemer G (2007). Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature Medicine, 13(1), 54–61. doi: 10.1038/nm1523 [DOI] [PubMed] [Google Scholar]
- Oh CM, Lee D, Kong HJ, Lee S, Won YJ, Jung KW, & Cho H (2020). Causes of death among cancer patients in the era of cancer survivorship in Korea: Attention to the suicide and cardiovascular mortality. Cancer Medicine, 9(5), 1741–1752. doi: 10.1002/cam4.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, & Otsu K (2012). Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012 485:7397, 485(7397), 251–255. doi: 10.1038/nature10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir BC, Csajka C, Dotto GP, & Wagner AD (2018). Sex differences in efficacy and toxicity of systemic treatments: An undervalued issue in the era of precision oncology. Journal of Clinical Oncology, 36(26), 2680–2683. doi: 10.1200/JCO.2018.78.3290 [DOI] [PubMed] [Google Scholar]
- Park NJ, Chang Y, Bender C, Conley Y, Chlebowski RT, Van Londen GJ, Foraker R, Wassertheil-Smoller S, Stefanick ML, & Kuller LH (2017). Cardiovascular disease and mortality after breast cancer in postmenopausal women: Results from the Women’s Health Initiative. PLoS ONE, 12(9), 1–20. doi: 10.1371/journal.pone.0184174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr SK, Liang J, Schadler KL, Gilchrist SC, Steele CC, & Ade CJ (2020). Anticancer therapy-related increases in arterial stiffness: a systematic review and meta-analysis. Journal of the American Heart Association, 9(14), e015598. doi: 10.1161/JAHA.119.015598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik JL, Byers T, DiGuiseppi C, Dabelea D, & Denberg TD (2011). Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: A retrospective cohort study. Breast Cancer Research, 13(3). doi: 10.1186/bcr2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan C, Jutant E-M, Tu L, Thuillet R, Seferian A, Montani D, Huertas A, Bezu J. van, Breijer F, Noordegraaf AV, Humbert M, Aman J, & Guignabert C (2018). Dasatinib increases endothelial permeability leading to pleural effusion. European Respiratory Journal, 51(1), 1701096. doi: 10.1183/13993003.01096-2017 [DOI] [PubMed] [Google Scholar]
- Phillips SA, Hatoum OA, & Gutterman DD (2007). The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. American Journal of Physiology - Heart and Circulatory Physiology, 292(1), 93–100. doi: 10.1152/ajpheart.00819.2006 [DOI] [PubMed] [Google Scholar]
- Pierce GL, Lesniewski LA, Lawson BR, Beske SD, & Seals DR (2009). Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation, 119(9), 1284–1292. doi: 10.1161/CIRCULATIONAHA.108.804294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putt M, Hahn VS, Januzzi JL, Sawaya H, Sebag IA, Plana JC, Picard MH, Carver JR, Halpern EF, Kuter F, Passeri J, Cohen V, Banchs J, Martin RP, Gerszten RE, Scherrer-Crosbie M, & Ky B (2015). Longitudinal changes in multiple biomarkers are associated with cardiotoxicity in breast cancer patients treated with doxorubicin, taxanes, and trastuzumab. Clinical Chemistry, 61(9), 1164–1172. doi: 10.1373/clinchem.2015.241232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, & Liu D (2012). Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Experimental Hematology & Oncology, 1(1), 10. doi: 10.1186/2162-3619-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput S, Volk-Draper LD, & Ran S (2013). TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Molecular Cancer Therapeutics, 12(8), 1676–1687. doi: 10.1158/1535-7163.MCT-12-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschi E, Vasina V, Ursino MG, Boriani G, Martoni A, & de Ponti F (2010). Anticancer drugs and cardiotoxicity: Insights and perspectives in the era of targeted therapy. Pharmacology and Therapeutics, 125(2), 196–218. doi: 10.1016/j.pharmthera.2009.10.002 [DOI] [PubMed] [Google Scholar]
- Riad A, Bien S, Gratz M, Escher F, Westermann D, Heimesaat MM, Bereswill S, Krieg T, Felix SB, Schultheiss HP, Kroemer HK, & Tschöpe C (2008). Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. European Journal of Heart Failure, 10(3), 233–243. doi: 10.1016/j.ejheart.2008.01.004 [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, … Glynn RJ (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine, 377(12), 1119–1131. doi: 10.1056/nejmoa1707914 [DOI] [PubMed] [Google Scholar]
- Roa-Chamorro R, Jaén-Águila F, Puerta-Puerta JM, Torres-Quintero L, González-Bustos P, & Mediavilla-García JD (2021). Arterial hypertension assessment in a population with chronic myeloid leukemia. Scientific Reports, 11(1), 14637. doi: 10.1038/S41598-021-94127-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatino F, Conti V, Liguori L, Polcaro G, Corbi G, Manzo V, Tortora V, Carlomagno C, Vecchione C, Filippelli A, & Pepe S (2021). Molecules and mechanisms to overcome oxidative stress inducing cardiovascular disease in cancer patients. Life, 11(2), 105. doi: 10.3390/LIFE11020105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales ARK, Negrão MV, Testa L, Ferreira-Santos L, Ramalho Groehs RV, Carvalho B, Toschi-Dias XE, Rocha NG, Martins Laurindo FR, Debbas V, Rondon MUPB, Mano MS, Hajjar LA, Gehm Hoff PM, Filho RK, & Negrão CE (2019). Chemotherapy acutely impairs neurovascular and hemodynamic responses in women with breast cancer. American Journal of Physiology - Heart and Circulatory Physiology, 317(7), H1–H12. doi: 10.1152/ajpheart.00756.2018 [DOI] [PubMed] [Google Scholar]
- Sekijima T, Tanabe A, Maruoka R, Fujishiro N, Yu S, Fujiwara S, Yuguchi H, Yamashita Y, Terai Y, & Ohmichi M (2011). Impact of platinum-based chemotherapy on the progression of atherosclerosis. Climacteric, 14(1), 31–40. doi: 10.3109/13697137.2010.522278 [DOI] [PubMed] [Google Scholar]
- Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, Drummer C, Johnson C, Xu K, Jiang X, Wang H, & Yang X (2020). Vascular endothelial cells and innate immunity. Arteriosclerosis, Thrombosis, and Vascular Biology, 40, E138–E152. doi: 10.1161/ATVBAHA.120.314330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmier HJ, Schlößer HA, Schlaak M, Kochanek M, Böll B, & von Bergwelt-Baildon MS (2018). Cytokine release syndrome. Journal for ImmunoTherapy of Cancer, 6, 56. doi: 10.1186/s40425-018-0343-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE, & Jemal A (2021). Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians, 71(1), 7–33. doi: 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- Šimůnek T, ؐtěrba M, Popelová O, Adamcová M, Hrdina R, & Gerši V (2009). Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacological Reports, 61(1), 154–171. doi: 10.1016/S1734-1140(09)70018-0 [DOI] [PubMed] [Google Scholar]
- Singal PK, & Iliskovic N (1998). Doxorubicin-induced cardiomyopathy. The New England Journal of Medicine, 339, 900–905. [DOI] [PubMed] [Google Scholar]
- Solari JIG, Filippi-Chiela E, Pilar ES, Nunes V, Gonzalez EA, Figueiró F, Andrade CF, & Klamt F (2020). Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer, 20(1), 1–14. doi: 10.1186/s12885-020-06964-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomou E, Aznaouridis K, Masoura C, Cutajar I, Toutouzas K, Vlachopoulos C, & Tousoulis D (2019). Aortic wall stiffness as a side-effect of anti-cancer medication. Expert Review of Cardiovascular Therapy, 17(11), 791–799. doi: 10.1080/14779072.2019.1691528 [DOI] [PubMed] [Google Scholar]
- Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos MA, & Papadimitriou C (2012). Endothelial vascular toxicity from chemotherapeutic agents: Preclinical evidence and clinical implications. Cancer Treatment Reviews, 38(5), 473–483. doi: 10.1016/j.ctrv.2011.09.002 [DOI] [PubMed] [Google Scholar]
- Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Kränkel N, Kania G, Zewinger S, Akhmedov A, Shi Y, Martin T, Perisa D, Winnik S, Müller MF, Sester U, Wernicke G, Jung A, Gutteck U, Eriksson U, … Landmesser U (2013). Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity, 38(4), 754–768. doi: 10.1016/j.immuni.2013.02.009 [DOI] [PubMed] [Google Scholar]
- Stanhewicz AE, Wenner MM, & Stachenfeld NS (2018). Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. American Journal of Physiology - Heart and Circulatory Physiology, 315(6), H1569–H1588. doi: 10.1152/ajpheart.00396.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeghs N, Gelderblom H, Roodt JOT, Christensen O, Rajagopalan P, Hovens M, Putter H, Rabelink TJ, & De Koning E (2008). Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clinical Cancer Research, 14(11), 3470–3476. doi: 10.1158/1078-0432.CCR-07-5050 [DOI] [PubMed] [Google Scholar]
- Steeghs N, Rabelink TJ, op ’t Roodt J, Batman E, Cluitmans FHM, Weijl NI, de Koning E, & Gelderblom H (2010). Reversibility of capillary density after discontinuation of bevacizumab treatment. Annals of Oncology, 21(5), 1100–1105. doi: 10.1093/annonc/mdp417 [DOI] [PubMed] [Google Scholar]
- Stein-Merlob AF, Rothberg MV, Ribas A, & Yang EH (2021). Cardiotoxicities of novel cancer immunotherapies. Heart, 107(21), 1694–1703. doi: 10.1136/heartjnl-2020-318083 [DOI] [PubMed] [Google Scholar]
- Steyers CM, & Miller FJ (2014). Endothelial dysfunction in chronic inflammatory diseases. International Journal of Molecular Sciences, 15(7), 11324–11349. doi: 10.3390/ijms150711324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon KM, Deng L, Bluethmann SM, Zhou S, Trifiletti DM, Jiang C, Kelly SP, & Zaorsky NG (2019). A population-based study of cardiovascular disease mortality risk in US cancer patients. European Heart Journal, 40(48), 3889–3897. doi: 10.1093/eurheartj/ehz766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhoff T, Enderle MD, Pahlke M, Petz C, Teschendorf C, Graeven U, & Schmiegel W (2004). 5-Fluorouracil induces arterial vasocontractions. Annals of Oncology, 15(4), 661–664. doi: 10.1093/ANNONC/MDH150 [DOI] [PubMed] [Google Scholar]
- Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, & Itagaki K (2013). Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE, 8(3), e59989. doi: 10.1371/journal.pone.0059989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterfield SL, Caldwell JT, Post HK, Lovoy GM, Banister HR, & Ade CJ (2018). Lower cutaneous microvascular reactivity in adult cancer patients receiving chemotherapy. Journal of Applied Physiology, 125(4), 1141–1149. doi: 10.1152/japplphysiol.00394.2018 [DOI] [PubMed] [Google Scholar]
- Thijssen DHJ, Bruno RM, Van Mil ACCM, Holder SM, Faita F, Greyling A, Zock PL, Taddei S, Deanfield JE, Luscher T, Green DJ, & Ghiadoni L (2019). Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. European Heart Journal, 40(30), 2534–2547. doi: 10.1093/eurheartj/ehz350 [DOI] [PubMed] [Google Scholar]
- Tocchetti CG, Cadeddu C, Di Lisi D, Femminò S, Madonna R, Mele D, Monte I, Novo G, Penna C, Pepe A, Spallarossa P, Varricchi G, Zito C, Pagliaro P, & Mercuro G (2019). From molecular mechanisms to clinical management of antineoplastic drug-induced cardiovascular toxicity: a translational overview. Antioxidants and Redox Signaling, 30(18), 2110–2153. doi: 10.1089/ars.2016.6930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova VK, Hsu P-C, Wei JY, Lopez-Candales A, Chen JZ, Su LJ, & Makhoul I (2020). Biomarkers of inflammation, hypercoagulability and endothelial injury predict early asymptomatic doxorubicin-induced cardiotoxicity in breast cancer patients. American Journal of Cancer Research, 10(9), 2933. /pmc/articles/PMC7539772/ [PMC free article] [PubMed] [Google Scholar]
- Toldo S, Goehe RW, Lotrionte M, Mezzaroma E, Sumner ET, Biondi-Zoccai GGL, Seropian IM, Van Tassell BW, Loperfido F, Palazzoni G, Voelkel NF, Abbate A, & Gewirtz DA (2013). Comparative cardiac toxicity of anthracyclines in vitro and in vivo in the mouse. PLoS ONE, 8(3), 4–11. doi: 10.1371/journal.pone.0058421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, & Schiffrin EL (2004). Reactive oxygen species in vascular biology: Implications in hypertension. Histochemistry and Cell Biology, 122(4), 339–352. doi: 10.1007/s00418-004-0696-7 [DOI] [PubMed] [Google Scholar]
- Ueda H, Yamaguchi O, Taneike M, Akazawa Y, Wada-Kobayashi H, Sugihara R, Yorifuji H, Nakayama H, Omiya S, Murakawa T, Sakata Y, & Otsu K (2019). Administration of a TLR9 inhibitor attenuates the development and progression of heart failure in mice. JACC: Basic to Translational Science, 4(3), 348–363. doi: 10.1016/j.jacbts.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van’t Klooster CC, Ridker PM, Hjortnaes J, Van Der Graaf Y, Asselbergs FW, Westerink J, Aerts JGJV, Visseren FLJ, Asselbergs FW, Nathoe HM, De Borst GJ, Bots ML, Geerlings MI, Emmelot MH, De Jong PA, Leiner T, Lely AT, Van Der Kaaij NP, Kappelle LJ, … Westerink J (2019). The relation between systemic inflammation and incident cancer in patients with stable cardiovascular disease: A cohort study. European Heart Journal, 40(48), 3901–3909. doi: 10.1093/eurheartj/ehz587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Hoef TP, Van Lavieren MA, Damman P, Delewi R, Piek MA, Chamuleau SAJ, Voskuil M, Henriques JPS, Koch KT, De Winter RJ, Spaan JAE, Siebes M, Tijssen JGP, Meuwissen M, & Piek JJ (2014). Physiological basis and long-term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circulation: Cardiovascular Interventions, 7(3), 301–311. doi: 10.1161/CIRCINTERVENTIONS.113.001049 [DOI] [PubMed] [Google Scholar]
- Vásquez-Vivar J, Martasek P, Hogg N, Masters BSS, Pritchard KA, & Kalyanaraman B (1997). Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry, 36(38), 11293–11297. doi: 10.1021/bi971475e [DOI] [PubMed] [Google Scholar]
- Vassilakopoulou M, Mountzios G, Papamechael C, Protogerou AD, Aznaouridis K, Katsichti P, Venetsanou K, Dimopoulos MA, Ikonomidis I, & Papadimitriou CA (2010). Paclitaxel chemotherapy and vascular toxicity as assessed by flow-mediated and nitrate-mediated vasodilatation. Vascular Pharmacology, 55(3–4), 115–121. doi: 10.1016/j.vph.2010.05.002 [DOI] [PubMed] [Google Scholar]
- Vaughn DJ, Palmer SC, Carver JR, Jacobs LA, & Mohler ER (2008). Cardiovascular risk in long-term survivors of testicular cancer. Cancer, 112(9), 1949–1953. doi: 10.1002/cncr.23389 [DOI] [PubMed] [Google Scholar]
- Vénéreau E, Ceriotti C, & Bianchi ME (2015). DAMPs from cell death to new life. Frontiers in Immunology, 6, 422. doi: 10.3389/fimmu.2015.00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesulu BP, Mahadevan LS, Aliru ML, Yang X, Bodd MEL, Singh PK, Yusuf SW, Abe J. ichi, & Krishnan S (2018). Radiation-induced endothelial vascular injury: a review of possible mechanisms. JACC: Basic to Translational Science, 3(4), 563–572. doi: 10.1016/j.jacbts.2018.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent DT, Ibrahim YF, Espey MG, & Suzuki YJ (2013). The role of antioxidants in the era of cardio-oncology. Cancer Chemotherapy and Pharmacology, 72(6), 1157–1168. doi: 10.1007/s00280-013-2260-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita JA, Brennan ML, Gokce N, Mann SA, Goormastic M, Shishehbor MH, Penn MS, Keaney JF, & Hazen SL (2004). Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation, 110(9), 1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chen Q, Qi H, Wang C, Wang C, Zhang J, & Dong L (2016). Doxorubicin-induced systemic inflammation is driven by upregulation of toll-like receptor TLR4 and endotoxin leakage. Cancer Research, 76(22), 6631–6642. doi: 10.1158/0008-5472.CAN-15-3034 [DOI] [PubMed] [Google Scholar]
- Weatherald J, Bondeelle L, Chaumais M-C, Guignabert C, Savale L, Jaïs X, Sitbon O, Rousselot P, Humbert M, Bergeron A, & Montani D (2020). Pulmonary complications of Bcr-Abl tyrosine kinase inhibitors. European Respiratory Journal, 56(4). doi: 10.1183/13993003.00279-2020 [DOI] [PubMed] [Google Scholar]
- Weberpals J, Jansen L, Muller OJ, & Brenner H (2018). Long-term heart-specific mortality among 347 476 breast cancer patients treated with radiotherapy or chemotherapy: a registry-based cohort study. European Heart Journal, 39(43), 3896–3903. doi: 10.1093/eurheartj/ehy167 [DOI] [PubMed] [Google Scholar]
- Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, & Webb RC (2014). Mitochondrial damage-associated molecular patterns and vascular function. European Heart Journal, 35(18), 1172–1177. doi: 10.1093/eurheartj/ehu047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson EL, Sidaway JE, & Cross MJ (2016). Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biology Open, 5(10), 1362–1370. doi: 10.1242/bio.020362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson IB, & McEniery CM (2004). Arterial stiffness, endothelial function and novel pharmacological approaches. Clinical and Experimental Pharmacology and Physiology, 31(11), 795–799. doi: 10.1111/j.1440-1681.2004.04074.x [DOI] [PubMed] [Google Scholar]
- Wolf MB, & Baynes JW (2006). The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochimica et Biophysica Acta - General Subjects, 1760(2), 267–271. doi: 10.1016/j.bbagen.2005.10.012 [DOI] [PubMed] [Google Scholar]
- Wu S, Ko YS, Teng MS, Ko YL, Hsu LA, Hsueh C, Chou YY, Liew CC, & Lee YS (2002). Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. Journal of Molecular and Cellular Cardiology, 34(12), 1595–1607. doi: 10.1006/jmcc.2002.2110 [DOI] [PubMed] [Google Scholar]
- Yao Y, Xu X, Zhang G, Zhang Y, Qian W, & Rui T (2012). Role of HMGB1 in doxorubicin-induced myocardial apoptosis and its regulation pathway. Basic Research in Cardiology, 107, 267. doi: 10.1007/s00395-012-0267-3 [DOI] [PubMed] [Google Scholar]
- Yeh ETH, & Bickford CL (2009). Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. Journal of the American College of Cardiology, 53(24), 2231–2247. doi: 10.1016/j.jacc.2009.02.050 [DOI] [PubMed] [Google Scholar]
- Yin Z, Zhao Y, Li H, Yan M, Zhou L, Chen C, & Wang DW (2016). miR-320a mediates doxorubicin-induced cardiotoxicity by targeting VEGF signal pathway. Aging, 8(1), 192–207. doi: 10.18632/AGING.100876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L-R, Cao Z, Makhoul I, Daniels JR, Klimberg S, Wei JY, Bai JP, Li J, Lathrop JT, Beger RD, & Todorova VK (2018). Immune response proteins as predictive biomarkers of doxorubicin-induced cardiotoxicity in breast cancer patients. Experimental Biology and Medicine, 243(3), 248–255. doi: 10.1177/1535370217746383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, & Feng Z (2018). The role of Toll-like receptor signaling in the progression of heart failure. Mediators of Inflammation, 2018, 9874109. doi: 10.1155/2018/9874109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM, & Meyer JE (2017). Causes of death among cancer patients. Annals of Oncology, 28(2), 400–407. doi: 10.1093/annonc/mdw604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, & Hauser CJ (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature, 464(7285), 104–107. doi: 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, & Hazen SL (2001). Association between myeloperoxidase levels and risk of coronary artery disease. Journal of the American Medical Association, 286(17), 2136–2142. doi: 10.1001/jama.286.17.2136 [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, Zhu S, Zhang R, & Han W (2010). Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. European Journal of Pharmacology, 643(2–3), 247–253. doi: 10.1016/J.EJPHAR.2010.06.024 [DOI] [PubMed] [Google Scholar]