

Abstract
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related death worldwide with limited therapeutic options. Comprehensive investigation of protein posttranslational modifications in HCC is still limited. Lysine acetylation is one of the most common types of posttranslational modification involved in many cellular processes and plays crucial roles in the regulation of cancer. In this study, we analyzed the proteome and K-acetylome in eight pairs of HCC tumors and normal adjacent tissues using a timsTOF Pro instrument. As a result, we identified 9219 K-acetylation sites in 2625 proteins, of which 1003 sites exhibited differential acetylation levels between tumors and normal adjacent tissues. Interestingly, many novel tumor-specific K-acetylation sites were characterized, for example, filamin A (K865), filamin B (K697), and cofilin (K19), suggesting altered activities of these cytoskeleton-modulating molecules, which may contribute to tumor metastasis. In addition, we observed an overall suppression of protein K-acetylation in HCC tumors, especially for enzymes from various metabolic pathways, for example, glycolysis, tricarboxylic acid cycle, and fatty acid metabolism. Moreover, the expression of deacetylase sirtuin 2 (SIRT2) was upregulated in HCC tumors, and its role of deacetylation in HCC cells was further explored by examining the impact of SIRT2 overexpression on the proteome and K-acetylome in Huh7 HCC cells. SIRT2 overexpression reduced K-acetylation of proteins involved in a wide range of cellular processes, including energy metabolism. Furthermore, cellular assays showed that overexpression of SIRT2 in HCC cells inhibited both glycolysis and oxidative phosphorylation. Taken together, our findings provide valuable information to better understand the roles of K-acetylation in HCC and to treat this disease by correcting the aberrant acetylation patterns.
Keywords: acetyl-proteomics, cellular metabolism, hepatocellular carcinoma, nonhistone, PASEF, proteomics, SIRT2, timsTOF Pro
Abbreviations: 2-DG, 2-deoxy-glucose; ACN, acetonitrile; CC, cellular component; ECAR, extracellular acidification rate; FA, formic acid; FASN, fatty acid synthase; FC, fold change; FCCP, carbonyl cyanide 4-trifluoromethoxyphenylhydrazone; GO, gene ontology; GPI, glucose-6-phosphate isomerase; HCC, hepatocellular carcinoma; KDAC, lysine deacetylase; MDH2, malate dehydrogenase; NAM, Nicotinamide; NAT, normal adjacent tissue; OCR, oxygen consumption rate; PASEF, parallel accumulation serial fragmentation; PCA, principal component analysis; PGK1, phosphoglycerate kinase; PTM, posttranslational modification; R/A, rotenone/antimycin A; TCA, trichloroacetic acid; TIMS, trapped ion mobility spectrometry; TSA, Trichostatin A
Graphical abstract
Highlights
-
•
K-acetylation was generally reduced in HCC, especially in metabolic enzymes.
-
•
Deacetylase SIRT2 was upregulated in HCC tumors.
-
•
SIRT2 overexpression induced broad alteration of protein K-acetylation.
-
•
SIRT2 overexpression inhibited glycolysis and oxidative phosphorylation.
In Brief
The proteome and K-acetylome in eight pairs of HCC tumors and normal adjacent tissues was investigated using a timsTOF Pro instrument. We observed suppression of K-acetylation in HCC especially in metabolic enzymes. The roles of deacetylase SIRT2 were explored by examining the effects of SIRT2 overexpression in HCC cells. SIRT2 overexpression reduced K-acetylation on proteins involved in diverse cellular processes and inhibited glycolysis and oxidative phosphorylation. Our findings provide valuable information to understand the roles of K-acetylation in HCC.
Liver cancer posts a global health challenge with increasing incidence around the world (1). Hepatocellular carcinoma (HCC) is the most common type of liver cancer, accounting for about 90% of all cases (2). HCC patients are often diagnosed at the advanced stage of the disease with limited therapeutic options available (3). Overall, the prognosis of HCC remains poor with a 5-year survival rate of just 18% (4, 5). Targeted therapy based on multi-target kinase inhibitors, for example, sorafenib, and immune checkpoint blockade, for example anti-programmed cell death 1 and anti-programmed cell death ligand 1, are gaining success for HCC treatment, however, only a subset of patients respond to these therapies, and the overall survival benefit is not satisfactory (6, 7). To further explore the molecular characteristics of HCC is crucial for developing novel and more efficient HCC treatment strategies.
Lysine acetylation is an important type of posttranslational modification (PTM), and it was first discovered on histones by Vincent Allfrey et al. in 1964 (8). Histone acetylation and deacetylation as a major element of epigenetic regulation has been intensively investigated, and the alteration of histone acetylation modulates chromatin structure and thereby affects gene transcription (9, 10). Meanwhile, substantial evidence has established that acetylation occurs on a variety of nonhistone proteins in evolutionarily diverse organisms (11). Acetylation could affect protein structure, protein–protein interaction, protein subcellular localization, and enzymatic activity and has been associated with diverse cellular processes, for example, signal transduction, protein folding, autophagy, and metabolism (12). In human liver tissues, more than 1000 acetylation sites in proteins have been identified, and enzymes involved in a wide range of metabolic pathways are found to be acetylated (13), suggesting a potential role of acetylation in the regulation of cellular metabolism. Accumulating evidences have implicated acetylation in the metabolic reprogramming of cancer cells (14). For example, acetylation at K305 of pyruvate kinase isoform M2 decreases its enzyme activity (15), while acetylation at K433 promotes its nuclear translocation (16), and both acetylation events contribute to the role of pyruvate kinase isoform 2 in promoting cancer progression.
Acetylation occurs by nonenzymatic reaction with acetyl-CoA that potentially targets all solvent accessible lysine residues in cellular proteins (17, 18). Acetylation can also be catalyzed by acyltransferases (KATs) that target specific proteins, including both histone and nonhistone substrates (12, 19). Deacetylation is catalyzed by various lysine deacetylases (KDACs) that can be divided into four families: class I, class II, class III, and class IV (20). Sirtuin deacetylases, also referred to as class III KDACs, are evolutionarily conserved NAD-dependent deacetylases, and mammalian sirtuins markedly vary in their subcellular localization, enzymatic activity, and targets (21, 22). Specifically, SIRT2 is predominately localized in the cytoplasm but can also shuttle to the nucleus and mitochondria (23). SIRT2 was initially reported as an α-tubulin deacetylase protein (24), and further studies have shown that SIRT2 interacts with many histone and nonhistone substrates involved in various cellular processes (25). SIRT2 has been reported to modulate the acetylation and activities of several enzymes involved in glycolysis, for example, phosphoglycerate mutase 1, and enzymes from the pentose phosphate pathway, for example, glucose-6-phosphate dehydrogenase (14). With the advance in modern mass spectrometry, more and more K-acetylation sites are being characterized (11, 13, 26), however, how majority of these sites are regulated and more importantly how they are involved in diseases remain largely unclear.
Currently, immunoenrichment of acetylpeptides followed by LC-MS/MS identification is the mostly widely employed strategy for large-scale profiling of protein acetylation. Prefractionation using techniques such as strong cation exchange or gel-based separation is commonly applied to improve the detection coverage of acetylation sites (27). The recently introduced timsTOF Pro instrument implements a trapped ion mobility spectrometry (TIMS) that provides an additional dimension of separation to the liquid chromatography. With the parallel accumulation serial fragmentation (PASEF) method enabled by the dual TIMS design, this instrumental platform delivers high scanning speed and enables detection of more than 6000 proteins from HeLa cell digest in a single run (28). Such high speed and sensitivity of timsTOF Pro makes it promising for proteome and PTM analysis of complex clinical samples without extensive prefractionation. However, thus far, its application in clinical proteomics studies is still limited.
In this study, we investigated the proteome and K-acetylome in eight paired tumor and nontumor tissues from treatment-naive HCC patients using a timsTOF Pro mass spectrometer. All samples were not fractionated and directly analyzed by single runs with 90 min or 60 min gradients. Over 6000 proteins and 9000 K-acetylation sites were detected, showing a decent detection coverage. Label-free quantification identified a variety of differentially expressed proteins and revealed the landscape of K-acetylation alteration in HCC. Interestingly, the deacetylase SIRT2 was upregulated in tumor tissues, and its potential role in regulating protein acetylation in HCC cells was further explored by examining the effects of SIRT2 overexpression on the proteome and K-acetylome in Huh7 liver cancer cells. SIRT2 overexpression reduced K-acetylation of proteins involved in a wide range of cellular processes. Furthermore, overexpression of SIRT2 in liver cancer cells inhibited both glycolysis and oxidative phosphorylation as revealed by seahorse analysis.
Experimental Procedures
Antibodies and Reagents
Antibody against GAPDH and SIRT2 was purchased from Abcam. Triton X-100 was bought in Sangon Biotech. Protease Inhibitor Cocktail tablets were purchased from Merck Millipore. Trichostatin A (TSA) was bought in MedChemExpress. Nicotinamide (NAM), trichloroacetic acid (TCA), triethylammonium bicarbonate (TEAB), DTT, iodoacetamide, EDTA, 2-deoxy-glucose (2-DG), TFA, and formic acid (FA) were bought from Sigma-Aldrich. BCA protein quantification kit was bought from Beyotime Biotechnology. Trypsin was purchased from Promega. Anti-acetyl-lysine antibody beads were bought from PTM BIO. Acetonitrile (ACN) was purchased from ThermoFisher Scientific. TRIzol reagent was bought in Invitrogen. FastQuant RT kit was bought from TianGen. Oligomycin was bought from Toronto Research Chemicals.
Sample Collection and Preparation
This study was approval by the Medical Ethics Committee of the fifth Medical Center of General Hospital of PLA (R2016244DA040), and consent was obtained from the patients. The studies in this work abide by the Declaration of Helsinki principles. Resected tumor samples from eight treatment-naïve HCC patients were collected in the fifth Medical Center of General Hospital of PLA. Normal adjacent tissues (NATs) were obtained from the resected specimens at ∼2 cm away from the tumor margin. The patient information was provided in supplemental Table S1. The specimens were washed with PBS and stored in liquid nitrogen before use.
Protein Extraction and Digestion
The collected tissue samples were grinded in liquid nitrogen and transferred to a 5 ml centrifuge tube. Then, samples were disrupted using four volumes of lysis buffer (1% Triton X-100, 1% protease inhibitor cocktail III, 3 μM TSA, and 50 mM NAM) followed by 5 min of sonication (3-s on and 5-s off, amplitude 25%) on ice using an ultrasonic processor. To avoid artificial deacetylation during sample preparation, pan KDAC inhibitor TSA that targets Class I, II, and IV was added in the lysis buffer, and NAM was used to inhibit the deacetylation activity of sirtuins. The remaining debris was removed by centrifugation at 12000g at 4 °C for 10 min. Finally, the supernatant was collected, and protein concentration was measured with a BCA kit. For each sample, 3 mg of protein was used in the following procedure. TCA was added to the samples to the final concentration of 20%, and proteins were precipitated at 4 °C for 2 h. The pellet was collected by centrifugation at 4500g for 5 min and washed by three times with precooled acetone. Next, the pellet was redissolved in 200 mM triethylammonium bicarbonate. The samples were reduced with 5 mM DTT for 60 min at 37 °C and alkylated with 11 mM iodoacetamide for 45 min at room temperature in darkness. Trypsin was added at 1:50 trypsin-to-protein mass ratio for digestion at 37 °C overnight. Tryptic peptides were desalted with Strata X SPE columns (Phenomenex) and dried by vacuum centrifugation. Peptide concentration was measured by a Pierce Quantitative Colorimetric Peptide Assay kit (ThermoFisher Scientific), and 5 μg of peptides was used for proteome analysis.
Acetylpeptide Enrichment
For acetylpeptide enrichment, 2 mg tryptic peptides were first dissolved in the NETN lysis buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris–HCl, 0.5% NP-40, pH 8.0) and incubated with prewashed anti-lysine-acetylation antibody-conjugated agarose beads at 4 °C overnight with gentle shaking. Then, the beads were washed four times with NETN buffer and twice with water. The bound peptides were eluted from the beads with 0.1% TFA. Next, the eluted fractions were vacuum dried. For LC-MS/MS analysis, the enriched acetylpeptides were desalted with C18 ZipTips (Millipore) according to the manufacturer’s instructions, and all purified K-acetylpeptides were loaded for each sample.
Liquid Chromatography Tandem Mass Spectrometry
Peptides were analyzed by nano-LC-MS/MS using a NanoElute UHPLC system coupled online to a hybrid TIMS- quadrupole time of flight mass spectrometer (timsTOF Pro) with a modified nano-electrospray ion source. Liquid chromatography was performed with a constant flow of 450 nl/min on a reversed-phase column (25 cm × 100 μm i.d.) with a pulled emitter tip, packed with 1.9 μm C18-coated porous silica beads. Mobile phase A was water with 0.1% FA (v/v) and 2% ACN, and mobile phase B was 0.1% FA (v/v) and 100% ACN. For proteome, in a 90-min experiment, peptides were separated with a linear gradient from 6 to 24% B within 70 min, followed by an increase to 35% B within 14 min and further to 80% within 3 min then holding at 80% for the last 3 min. For acetylome, in a 60-min experiment, peptides were separated with a linear gradient from 6 to 22% B within 43 min, followed by an increase to 30% B within 13 min and further to 80% within 2 min then holding at 80% for the last 2 min. The electrospray voltage applied was 1.7 kV. The timsTOF Pro mass spectrometer was set to the PASEF mode. The mass resolution of the TOF analyzer was 30,000. Precursor ions with charge state of 2 to 5 and unknown were selected for HCD fragmentation, and 10 PASEF MS/MS scans were acquired for each cycle. The target intensity was 2e4 and ramp time was set to 100 ms. The dynamic exclusion time was set to 30 s to avoid repeated scanning of the precursor ions. MS and MS/MS spectra were recorded from m/z 100 to 1700.
Protein Identification and Quantification
The tandem mass spectra were searched against the reviewed Homo sapiens SwissProt database (20,375 sequence entries, released in September 2020) using MaxQuant (version: 1.6.6.0) (29). Trypsin was selected as the proteolytic enzyme, and two missed cleavages sites were allowed. For proteome, carbamidomethylation of cysteine residues was set as a fixed modification, and methionine oxidation was set as a variable modification. For acetylome, carbamidomethylation of cysteine residues was set as a fixed modification, and methionine oxidation and lysine acetylation were set as variable modifications. The positions of acetylation sites were determined by site probabilities calculated by PTM score, and localization probability >0.75 was used as cutoff. Peptides with the minimum length of seven amino acid were considered, and the required false discovery rate was set to 1% at both the peptide and protein level. The maximum number of peptide modifications was set to 5. Match Between Run was applied with default parameters. The mass tolerance for precursor ions was set as 20 ppm in first search and 20 ppm in main search, and the mass tolerance for fragment ions was set as 0.02 Da. Unique peptides ≥ 2 was applied as the filter criteria for protein identification and quantification. Quantification was performed by MaxQuant using the label-free quantification algorithm that employed extracted ion currents of peptides for comparison between samples. Unique peptides were used for quantification, and the other parameters were kept as default values. Label-free quantification intensities were extracted from the MaxQuant result files and log2 transformed for statistical analysis. For the acetylpeptide quantification, the intensities of acetylpeptides were normalized with the abundance of the corresponding proteins in the same sample to avoid the impact of protein expression changes on the measured acetylation level.
Bioinformatics Analysis
Principal component analysis (PCA) was performed by the R package FactoMineR (version:2.3.0) (30), and heatmaps were generated with the package pheatmap (version:1.0.12) https://CRAN.R-project.org. The volcano plot was generated by the package ggplot2 (version:3.3.3) (31) using “fold change (FC) > 1.5” and “p < 0.05” as the cutoff. Correlation analysis was performed by the package corrplot (version: 0.84) https://github.com/taiyun/corrplot and PerformanceAnalytics (version:2.0.4) https://CRAN.R-project.org. For the proteome data, the functional enrichment analysis was performed using the R package clusterprofiler (version: 3.16.1) (32) for Kyoto Encyclopedia of Genes and Genomes pathway enrichment (33). Gene Ontology (GO) analysis was performed using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) (34). Functional enrichment analysis for K-acetylome was performed by AGOTOOL (https://agotool.org/) using the abundance-corrected proteome as the control (35). MoMo (version: 5.4.1) was employed to find the statistically significant amino acid sequence motifs associated with the observed acetylation sites, and the shuffled peptides with the central residue conserved were used as the background (36). Protein structures from the Protein Data Base were analyzed by pymol software (37).
Cell Lines and Culture
Huh7 HCC cell line was purchased from Japanese Collection of Research Bioresources. The MHCC-97H cell line was purchased from the Cell Bank of Type Culture Collection. Cells were maintained in Dulbecco’s modified Eagle’s high glucose medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (100 μg/ml) at 37 °C in a humidified incubator in the presence of 5% CO2.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated from the cells using TRIzol reagent and complementary DNA was synthesized from 2 μg of total RNA using the FastQuant RT kit according to the manufacturer’s instructions. Quantitative mRNA analysis was performed on a 7500 Fast Real-Time PCR System (ABI) using the SuperReal SYBR Green PreMix (TianGen) following the manufacturer’s instructions. The mean Ct for each sample was normalized using GAPDH as the reference gene (for primer sequences, see supplemental Table S2).
Western Blotting
Cellular proteins were extracted with 1% SDS cell lysis buffer, and 20 μg of lysate was separated by 10% SDS-PAGE and transferred to a Immobilon-P membrane. The membrane was incubated in a blocking solution consisting of 5% milk in 10 mM Tris–HCl (pH 8.0), 150 mM NaCl, and 0.1% Tween 20 at room temperature for 1 h. Then, primary antibodies were incubated with the membranes at 4 °C overnight and washed with tris-buffered saline and tween 20 buffer three times. Next, the blots were incubated with the horseradish peroxidase-conjugated secondary antibody and developed by enhanced chemiluminescence. GAPDH was used as the internal standard. The band intensities were measured using ImageJ (V 1.8.0) for quantitative comparisons.
Transfection and RNA Interference
SIRT2-Flag was amplified via PCR and cloned into vector pcDNA3.1. Transfection was performed in the OPTI-MEM medium (Invitrogen) using LipoGene 2000Plus Transfection Reagent (Yuheng Bio) according to the manufacturer's instructions. For siRNA-mediated SIRT2 knockdown, cells were transfected with siRNA targeting SIRT2 or a negative control siRNA (siNC) using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Three siRNAs were synthesized by GENEWIZ (supplemental Table S2). Transfection efficiency was evaluated by quantitative real-time PCR and Western blotting, and the siRNA with the highest knockdown efficiency was selected for the following experiments.
Cell Proliferation Assay
Cells were plated at the density of 2000 cells per well in 96-well plates and allowed to grow for 24 h, 48 h, or 72 h. Then, 10 μl of Cell Counting Kit-8 solution (Solarbio) was added to each well. The samples were incubated at 37 °C for 2 h before the absorbance was measured at 450 nm.
Extracellular Flux Analysis
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of Huh7 cells were determined using the Seahorse XF extracellular flux analyzer (Agilent). Cells were plated at a density of 3 × 104 cells per well on 24-well Seahorse plates and allowed to attach overnight in the growth medium. Then, the adherent cells were washed by PBS, and the fresh assay medium was added before analysis. For the glycolysis stress test, ECAR was measured under basal condition and in response to 10 mM glucose, 1 mM oligomycin and 50 mM 2-DG successively to calculate the basal glycolytic rate, glycolytic capacity (in response to oligomycin), and glycolytic reserve (calculated as glycolytic capacity-basal glycolytic rate). For the mitochondrial stress test, OCR was measured under basal condition and in response to sequential injections of 1 μM oligomycin, 1 μM carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μM rotenone/antimycin A(R/A) to calculate basal respiration rate (baseline OCR-R/A OCR), maximal respiration rate (FCCP OCR-R/A OCR), and oxidative reserve (maximal respiration-basal respiration).
Experimental Design and Statistical Rationale
The proteomics and acetylomics experiments were performed as illustrated in Figure 1. Eight paired tumor and NATs from HCC patients were investigated. Statistical and bioinformatics analyses were performed using the R framework (version: 3.6.1). The statistical significance was evaluated by two-tailed unpaired Student’s t test. Proteins with a fold change >1.5 and a p value < 0.05 were defined as significantly differentially expressed. Cell experiments were performed in triplicates and analyzed by two-sided Student′s t test for comparison between two groups. Data were presented as means ± sd, and p < 0.05 was considered as a significant difference.
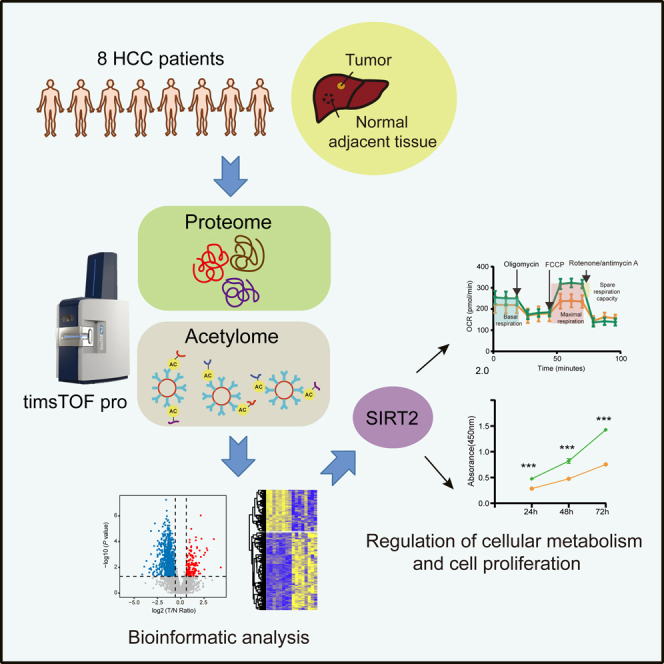
Fig. 1.

The schematic workflow of the proteome and K-acetylome analysis approach employed in this study. Acetylpeptides were isolated by immunoprecipitation with anti-K-acetyl-lysine antibody-conjugated beads. The timsTOF Pro instrument was operated at the PASEF mode and used for both proteome and acetylome analyses. PASEF, parallel accumulation serial fragmentation.
Results
Quantitative Proteomics Analysis of HCC
In this study, we characterized the proteome and K-acetylome in eight paired tumor and NATs from treatment-naïve HCC patients by a four-dimensional label-free quantitative proteomics strategy using a timsTOF Pro mass spectrometer (Fig. 1). Proteomics analysis identified a total of 6014 proteins, of which 5361 proteins were quantified across all samples. On average, 4678 proteins per tumor and 4475 proteins per NATs were identified (supplemental Fig. S1, A and B). PCA showed a clear discrepancy between HCC tumors and NATs, and higher inter-patient heterogeneity was observed among tumor samples than NATs (Fig. 2A). A total of 1492 proteins were observed to be significantly differentially expressed (fold change >1.5, p < 0.05), of which 971 were upregulated and 521 were downregulated (Fig. 2, B and C and supplemental Table S3).
Fig. 2.

Quantitative analysis of the proteome in HCC tumors and NATs.A, principal component analysis (PCA) visualization of protein expression in HCC tumors and NATs. B, the volcano plot illustrating differentially expressed proteins (fold change>1.5, p < 0.05, by two-sided Student’s t test). C, the heatmap of the identified differentially expressed proteins. D and E, KEGG pathway enrichment analysis of the upregulated (D) and downregulated (E) proteins. The analysis was conducted by clusterProfiler (version: 3.16.0). See also supplemental Fig. S1 and supplemental Table S3. HCC, hepatocellular carcinoma; KEGG, Kyoto Encyclopedia of Genes and Genomes; NATs, normal adjacent tissues.
To better understand the biological functions of the differentially expressed proteins, we performed Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis, and the results showed that they were associated with distinct pathways (Fig. 2, D and E and supplemental Table S3). For example, the upregulated proteins were most significantly enriched in pathways such as spliceosome, RNA transport, ribosome, and antigen processing and presentation (Fig. 2D). In the antigen processing and presentation pathway, MHC-I class molecule TAP binding protein (TAPBP) and HLA class I histocompatibility antigen responsible for presenting endogenous antigens and activating CD8+ T cells were upregulated (38). MHC-II class molecule HLA class II histocompatibility antigen gamma chain (CD74) involved in the formation and transport of MHC class II peptide complexes for the generation of CD4+ T cell responses was also upregulated (39). The data suggested elevated infiltration of immune cells in HCC tumors compared to NATs. In addition, we observed overexpression of multiple proteins involved in ferroptosis, a recently recognized form of cell death elicited by iron-dependent phospholipid peroxidation. For example, the long chain fatty acid CoA ligase 4 (ACSL4), a key enzyme that modulated cellular lipid composition and dictated ferroptosis sensitivity (40), was upregulated in tumors, revealing the potential vulnerability of HCC that could be exploited for ferroptosis-based therapies. Furthermore, multiple metabolism-related pathways, for example, fatty acid metabolism, carbon metabolism, biosynthesis of amino acid, were overrepresented for the downregulated proteins (Fig. 2E). GO analysis revealed that a large majority of the proteins upregulated in HCC tumors were associated with RNA processing and transport (supplemental Fig. S1C). Meanwhile, many of the downregulated proteins were metabolic enzymes (supplemental Fig. S1D). The data unveiled the global alteration of protein expression in HCC, especially related to gene expression and metabolism. The observations were consistent with an earlier large-scale study of HCC clinical samples (41).
Characterization of the K-acetylome of HCC
To explore the role of protein acetylation in the regulation of HCC, we further investigated the nonhistone K-acetylome in these eight pairs of HCC tumors and NATs. The K-acetylated peptides were isolated through immunoprecipitation with anti-K-acetylation antibody-conjugated beads (Fig. 1). Taking advantage of the high scanning speed provided by timsTOF Pro, a total of 9219 K-acetylation sites in 2625 proteins were identified, with an average of 3053 and 3972 acetylation sites identified per tumor and NAT, respectively (Fig. 3A).
Fig. 3.

Identification of the nonhistone K-acetylation sites in HCC tumors and NATs.A, the average number of K-acetylation sites and the K-acetylated proteins identified in tumor and NAT samples. Error bars represent SDs of eight samples. ns, not significant; ∗∗∗p < 0.001 by two-sided Student’s t-test. B, analysis of the amino acid sequence motifs associated with the detected acetylpeptides. The analysis was conducted by MoMo (version: 5.4.1), using shuffled input peptides as the control. C, GO analysis of the K-acetylated proteins. D, pathway enrichment of the K-acetylated proteins using the Reactome database. E, numbers of the tumor- and NAT-specific K-acetylation sites (n = 1, n = 2, n ≥ 3 out of 8 samples). F, functional enrichment analysis of proteins containing tumor- or NAT-specific acetylation sites (n ≥ 3). The GO and pathway enrichment analysis was conducted by AGOTOOL (https://agotool.org/), using the abundance-corrected proteome as the control for statistical analysis (p < 0.05 as cutoff). See also supplemental Table S4. GO, gene ontology; HCC, hepatocellular carcinoma; NATs, normal adjacent tissues.
Next, we investigated the occurrence of sequence motifs by analyzing the probabilities of amino acids from the −6 to +6 positions surrounding the identified lysine acetylation sites. The top three enriched motifs included GK, KS, and KT, accounting for 10.03%, 8.39%, and 7.01% of all the observed K-acetylated peptides, respectively (Fig. 3B).
To understand the cellular distribution and biological functions of the 2625 K-acetylated proteins, we performed UniProt keywords, cellular component (CC), biological process, and pathway enrichment analysis, using the abundance-corrected proteome as the control for statistical analysis (Fig. 3, C and D). The results showed that the acetylated proteins were associated with keywords such as alternative splicing, acetylation, and nucleus. CC analysis revealed that these proteins were mainly localized in organelle, membrane, intracellular organelle lumen, and mitochondria. Biological process analysis indicated that the K-acetylated proteins were significantly associated with cellular process and metabolic process. Pathway analysis based on the Reactome database suggested that detected K-acetylated proteins were associated with various pathways, for example, metabolism, signal transduction, gene expression (Fig. 3D).
Interestingly, we observed a number of acetylated sites specifically in tumors or NATs (Fig. 3E and supplemental Table S4). The data showed that 620 acetylation sites were repeatedly observed from NATs (n ≥ 3) but undetectable in tumors, and 196 acetylation sites were only detected in tumors (n ≥ 3). Pathway enrichment analysis revealed that proteins with NAT-specific acetylation sites were highly involved in various cellular metabolism processes, while tumor-specific acetylation sites were most significantly associated with metabolism of RNA (Fig. 3F and supplemental Table S4). CC analysis revealed that proteins with NAT-specific K-acetylation sites were mainly localized in the mitochondria and cytoplasm, while proteins containing tumor-specific K-acetylation sites were markedly enriched in the nucleus (Fig. 3F and supplemental Table S4).
Majority of the proteins with tumor-specific K-acetylation sites were upregulated in tumors at the protein level. For example, heat shock protein HSP90AB1 plays an important role in endoplasmic reticulum stress response and promotes tumor growth and metastasis (42). The proteome data showed that HSP90AB1 was overexpressed in HCC tumors (fold change = 1.66, p = 1.6 × 10−5) (Fig. 4A), and its K72 acetylation was detected specifically in the tumor samples (n = 6/8) (Fig. 4B). On the other hand, some proteins exhibited similar expression levels between tumors and NATs but were specifically acetylated in the tumors (Fig. 4A). For example, acetylation of three cytoskeleton-regulating proteins, that is, filamin A (FLNA, K865), filamin B (FLNB, K697), and cofilin 1 (CFL1, K19), were only detected in the tumor samples (Figs. 4B and S2). These three molecules play crucial roles in the regulation of actin dynamics and cell migration, a key process during tumor metastasis (43, 44, 45). Our observations suggest that the functions of these cytoskeleton-regulating molecules may be modulated through acetylation in HCC, and further investigation is required to clarify the biological implications of the identified acetylation sites.
Fig. 4.

Examples of protein K-acetylation sites specifically detected in tumors.A, comparison of the protein expression levels of HSP90AB1, FLNA, FLNB, and CFL1 between HCC tumors and NATs. B, illustration of the K-acetylation sites specifically observed in tumors from HSP90AB1 (K72), FLNA (K865), FLNB (K697), and CFL1(K19). See also supplemental Fig. S2. HCC, hepatocellular carcinoma; NATs, normal adjacent tissues.
Functions of Proteins with Differential K-acetylation in HCC
Next, we further investigated the K-acetylated proteins detected in both tumors and NATs by quantitative analysis. To rule out the impact of protein expression changes on the measured acetylation levels, we normalized the intensities of acetylpeptides with the abundance of the corresponding protein in the same sample. Overall, 5090 sites from 1610 proteins were quantified. Correlation analysis of the acetylome data showed a high degree of correlation between each sample (R > 0.8) (supplemental Fig. S3A). PCA revealed that the acetylome data from tumors and NATs formed two distinct clusters, suggesting dramatic differences of the acetylation landscape between these two types of samples (Fig. 5A). Using p < 0.05 and FC > 1.5 as the cutoff threshold, 1003 acetylation sites exhibited significant changes. Specifically, 165 sites from 129 proteins were upregulated in HCC tumor tissues, and 838 sites from 381 proteins were downregulated (Fig. 5B). The results showed that acetylation was globally suppressed in HCC tumors, which may partly be due to the impairment of TCA and the reduced level of acetyl-CoA in HCC (46). Another possibility was that tumor cells sustained a higher level of proliferation and faster protein turnover than normal cells. Weinert et al reported that growth arrest in several types of cells lead to increased acetylation likely by prolonging the exposure of proteins to acetyl-CoA (47). In a recently reported acetylome study of four paired HCC tissue samples, 792 acetylation sites in 415 proteins were identified (26). In their study, only three acetylation sites, that is, EP300 (K1550), GRHPR (K327), and GLDC (K423), were observed to be upregulated in HCC tumors. We also detected these three sites, but they were not significantly changed between tumors and NATs in our data. Next, we compared the proteins with downregulated K-acetylation, and the results showed significant overlapping between these two studies, however, many more acetylation sites were identified in our study, providing novel information regarding the K-acetylation profile in HCC (supplemental Fig. S3B).
Fig. 5.

Quantification analysis of the K-acetylome in HCC.A, principal component analysis of the identified K-acetylation sites in HCC tumors and NATs. B, the volcano plot showing the K-acetylation sites with differential levels between tumors and NATs (fold change>1.5, p < 0.05, by two-sided Student’s t test). C, reactome pathway enrichment analysis of proteins with differential K-acetylation. D and E, molecular function and cellular component enrichment analysis of proteins with K-acetylation upregulated (D) and downregulated (E) in HCC tumors. The GO and pathway enrichment analysis was conducted by AGOTOOL (https://agotool.org/), using the abundance-corrected proteome as the control for statistical analysis (p < 0.05 as cutoff). See also supplemental Fig. S3 and supplemental Table S5. GO, gene ontology; HCC, hepatocellular carcinoma; NATs, normal adjacent tissues.
To further understand the biological implications of protein K-acetylation in HCC, we performed pathway enrichment and GO analysis on the proteins exhibiting differential K-acetylation levels between tumors and NATs (Fig. 5, C–E). The results showed that the proteins with downregulated K-acetylation sites were mostly enriched in metabolism pathways associated with fatty acid, amino acid, pyruvate, and TCA cycle, while proteins with upregulated acetylation sites were related to metabolism of RNA and signaling by nuclear receptors (Fig. 5C and supplemental Table S5). Molecular function analysis showed that the differentially acetylated proteins were associated with RNA binding and catalytic activity (Fig. 5, D and E). Additionally, the proteins with upregulated K-acetylation exhibited localization enrichment in nucleus and organelle, while proteins with downregulated K-acetylation were predominantly localized in the mitochondria and cytoplasm (Fig. 5, D and E). Interestingly, we detected 16 proteins with both upregulated and downregulated acetylation sites, implying that different molecular mechanisms were involved in the regulation of their acetylation (supplemental Fig. S3C). For example, glutathione reductase catalyzes the reduction of GSSG to GSH and plays key roles in cellular redox homeostasis. Here, we observed upregulated acetylation of K110 and downregulated acetylation of K164 on glutathione reductase in HCC tumors. Taken together, the results showed that K-acetylation of nonhistone proteins may play important roles in diverse cellular processes in HCC, especially in the regulation of gene expression and cellular metabolism.
Metabolic Enzymes with Decreased K-acetylation in HCC Tumors
Our data showed the downregulated K-acetylation of metabolic enzymes in diverse metabolic pathways, for example, glycolysis, TCA cycle, fatty acid metabolism, biosynthesis of amino acid, and valine, leucine, isoleucine degradation pathways (Fig. 6A). For example, the acetylation of K12 and K73 on glucose-6-phosphate isomerase (GPI) was downregulated in HCC (Fig. 6B). In the cytoplasm, GPI catalyzes the conversion of glucose-6-phosphate to fructose-6-phosphate, the second step in glycolysis. However, how acetylation affects its activity has not been reported. In our study, we detected downregulation of the acetylation of K12 and K73 on GPI in HCC (Figs. 6B and S4A). The mitochondrial enzyme, malate dehydrogenase (MDH2), plays a key role in the TCA cycle, and the acetylation of K185, K301, K307, and K314 on MDH2 enhances its enzymatic activity (13, 47). In our study, we observed downregulated acetylation of K185, K301, K307 on MDH2, suggesting that its enzymatic activity was likely inhibited in HCC. In addition, we detected downregulated acetylation of another six lysine sites, including K78, K296, K157, K74, K324, and K203 on MDH2 (Figs. 6C and S4B). Taken together, our data highlighted the potential role of acetylation in the regulation of metabolic reprogramming in HCC.
Fig. 6.

Metabolic enzymes with decreased K-acetylation in HCC tumors.A, metabolic enzymes with downregulated K-acetylation involved in glycolysis, TCA cycle, fatty acid metabolism, biosynthesis of amino acid, and valine, leucine, isoleucine degradation. B and C, the protein structures of GPI (PDB_1JLH) (B) and MDH2 (PDB_4WLF) (C). The differentially K-acetylated sites are marked in red. The images were generated in Pymol (version: 2.5.0). See also supplemental Fig. S4. GPI, glucose-6-phosphate isomerase; HCC, hepatocellular carcinoma.
The Role of SIRT2 as a Contributing Factor in the Deacetylation of Nonhistone Proteins in HCC
Next, we sought to explore the potential molecular mechanisms regulating the observed suppressed acetylation in HCC. The acetylome in HCC tumors may be affected by multiple factors, such as nonenzymatic reaction with acetyl-CoA that targets all solvent accessible lysine residues and the differential proliferation rate and protein turnover between tumors and normal cells. Moreover, acetylation-modulating enzymes may also play a role by regulating the acetylation of specific targets. In the HCC proteome data, we detected seven enzymes associated with acetylation regulation. Histone acetyltransferase type B catalytic subunit (HAT1), histone deacetylase 1(HDAC1), histone deacetylase 2 (HDAC2), and SIRT2 were upregulated in tumors, while histone deacetylase 6 (HDAC6), NAD-dependent protein deacetylase sirtuin-3 (SIRT3), and NAD-dependent protein deacetylase sirtuin-5 (SIRT5) were downregulated (Fig. 7A). Since majority of the observed downregulated acetylation sites were from cytoplasmic proteins, we suspected that some of the observed deacetylation may be associated with the function of SIRT2 primarily localized in the cytoplasm.
Fig. 7.

Quantitative analysis of the K-acetylome changes in response to SIRT2 overexpression in HCC cells.A, differential expression of proteins involved in acetylation regulation between HCC tumors and NATs. B, PCA of the identified K-acetylation sites in SIRT2-overexpressing cells and NC cells. C, volcano plot showing the K-acetylation sites with differential levels between SIRT2-overexpressing cells and NC cells (fold change>1.5, p < 0.05, by two-sided Student’s t test). D, reactome pathway enrichment analysis of the 122 SIRT2-induced downregulated K-acetylation sites that were also decreased in HCC tumors. The analysis was conducted by AGOTOOL (https://agotool.org/) using the abundance-corrected proteome as the control for statistical analysis (p < 0.05 as cutoff). E, a schematic diagram illustrating SIRT2 overexpression-induced downregulation of K-acetylation sites on metabolic enzymes involved in glycolysis and TCA cycle. See also supplemental Figs. S5, S6, supplemental Tables S7 and S8. HCC, hepatocellular carcinoma; NATs, normal adjacent tissues; PCA, principal component analysis.
To explore the potential targets of SIRT2, we overexpressed SIRT2 in Huh7 liver cancer cells and investigated the impact on the cellular proteome and K-acetylome using the same procedure for HCC tumor tissue analysis. PCA of the proteomics data showed a clear discrepancy between SIRT2-overexpressing cells and NC cells (supplemental Fig. S5, A and B). A total of 766 proteins were observed to be significantly differentially expressed (fold change >1.5, p < 0.05), of which 551 were upregulated and 215 were downregulated (supplemental Fig. S5C and supplemental Table S6). Pathway enrichment analysis showed that the upregulated proteins were most significantly enriched in pathways such as ribosome, protein export, and nucleocytoplasmic transport (supplemental Fig. S5D and supplemental Table S6). And the downregulated proteins were most significantly enriched in proteasome, adherens junction, and carbon metabolism (supplemental Fig. S5E and supplemental Table S6).
Next, we investigated the impact of SIRT2 overexpression on the K-acetylome normalized by proteome abundance. PCA showed that the acetylome data from SIRT2-overexpressing cells and control cells formed two distinct clusters, suggesting the alteration of the K-acetylation landscape between these two types of samples (Fig. 7B). Using p < 0.05 and FC <1/1.5 as the cutoff threshold, a total of 935 K-acetyl-sites from 590 proteins were detected to be downregulated in response to SIRT2 overexpression (Fig. 7C and supplemental Table S7). In addition, 950 K-acetylation sites were repeatedly observed from NC cells (n ≥ 2 of 3 biological replicates) but not detectable in SIRT2-overexpressing cells. Pathway enrichment analysis using the Reactome database showed that the proteins with downregulated acetylation were involved in pathways such as TCA cycle, gene expression, SUMOylation, and metabolism of RNA (supplemental Fig. S6A and supplemental Table S7). These proteins were most significantly enriched in nucleoplasm, followed by nuclear lumen, intracellular organelle lumen, and cytosol (supplemental Fig. S6A and supplemental Table S7). Proteins with K-acetylation affected by SIRT2 were significantly enriched for keywords describing phosphoprotein, acetylation, cell cycle, and transcription regulation (supplemental Fig. S6B and supplemental Table S7). Intriguingly, we observed reduced acetylation of acetylation-regulating enzymes, including deacetylases HDAC1 (K31) and HDAC2 (K11, K32), and histone acetyltransferase p300 (K1001, K1769, K1554, K1590), suggesting an intertwined regulatory network among acetylation modulators.
Interestingly, 122 of the SIRT2-induced downregulated sites from 100 proteins were also observed to be decreased in HCC tissues. Reactome pathway analysis showed statistically significant enrichment in three pathways, including metabolism, fatty metabolism, pyruvate metabolism, and TCA cycle (Fig. 7D and supplemental Table S8). CC analysis showed enrichment in cytoplasm and mitochondrion (Fig. 7D and supplemental Table S8). As shown in Figure 7E, we detected four glycolytic enzymes with reduced K-acetylation in both HCC tumors and SIRT2-overexpressing cells, including GPI (K73), GAPDH (K117,) phosphoglycerate kinase (PGK1) (K91), and LDHA (K278), and five enzymes from the TCA cycle, including isocitrate dehydrogenase (K272, K180), OGDH (K869, K389), SUCLG1 (K57), SDHA (K250, K608), and MDH2 (K296). SIRT2 was previously implicated in the regulation of metabolic shift in pluripotent stem cells and T cells with different phenotypes and mechanisms (48, 49). Our data suggested that SIRT2 may also be involved in energy metabolism in HCC cells.
SIRT2 Modulates Energy Metabolism in HCC Cells
Deacetylases are being extensively investigated as anticancer drug targets (50), and there has been a great interest to explore their functions in various biological contexts. In a previous study, Hamaidi et al (49) found that knockout or inhibition of SIRT2 enhanced both glycolysis and oxidative phosphorylation in T cells and proposed that chemical inhibition of SIRT2 may enhance tumor-specific T cell responses and improve antitumor immunity by promoting metabolic reprogramming toward a profound hyper-metabolic state. However, the effects of SIRT2 on the metabolism of tumor cells is still unknown and thus information is crucial to further evaluate the role of SIRT2 as a potential therapeutic target in HCC.
To investigate the function of SIRT2 in cellular metabolism of HCC cells, we overexpressed SIRT2 in Huh7 cells and examined its impact on glycolysis and oxygen consumption. First, the overexpression of SIRT2 was confirmed by both quantitative real-time PCR and Western blotting (Fig. 8, A and B). Next, we examined the oxidative phosphorylation by measuring OCR using a Seahorse Analyzer (Fig. 8C). Huh7 cells treated with SIRT2-overexpression or empty vectors were treated sequentially with the ATP synthase inhibitor oligomycin, oxidative phosphorylation uncoupler FCCP, and electron transport chain inhibitor R/A. The results showed that SIRT2 overexpression reduced the overall oxidative phosphorylation in Huh7 cells. The values of basal respiration, maximal respiration, and spare respiratory capacity were all significantly reduced by SIRT2 overexpression. Moreover, we studied the glycolytic flux by measuring ECAR using the Seahorse Analyzer (Fig. 8D). Glucose was first added to boost the glycolysis level, and the addition of ATP synthase inhibitor oligomycin shut down oxidative phosphorylation, allowing the measurement of glycolytic capacity. The following addition of glycolysis inhibitor 2-DG inhibited glycolysis and allowed us to evaluate the glycolytic reserve. The analysis results showed that SIRT2 overexpression reduced the overall glycolytic flux in Huh7 cells. Glycolysis and glycolytic capacity were both significantly reduced by SIRT2 overexpression. Glycolytic reserve was slightly reduced but the difference was not statistically significant. In summary, the results showed that SIRT2 overexpression inhibited energy metabolism of HCC cells in both cytoplasmic glycolysis and mitochondrial respiration, likely due to the deacetylation of multiple key enzymes in both pathways.
Fig. 8.

SIRT2 overexpression inhibited both glycolysis and oxidative phosphorylation in Huh7 liver cancer cells.A, the relative expression of SIRT2 in Huh7 cells transfected with the empty vector control (pcDNA3.1) or SIRT2 (pcDNA3.1 + SIRT2) as measured by qRT-PCR. B, Western blotting analysis confirmed the overexpression of SIRT2 in cells transfected with pcDNA3.1 + SIRT2. The densities of the corresponding protein bands were measured by Image J and displayed on the right. Based on the observed molecular weights, the double bands of SIRT2 were likely formed by two alternative spliced isoforms of SIRT2, including Sirt2.1 as the canonical isoform (1–389 aa, 43 kDa) and Sirt2.2 (38–389 aa, 40 kDa). C and D, oxygen consumption rate (OCR) (C) and extracellular acidification rate (ECAR) (D) of Huh7 cells overexpressing SIRT2 and the corresponding control cells. Left panel, oligomycin, FCCP, and R/A were injected at the indicated time points. The values of basal respiration, maximal respiration, and spare respiratory capacity were calculated by the Seahorse XF24 software. Right panel, glucose, oligomycin, and 2-DG were injected at the indicated time points. The values of glycolysis, glycolytic capacity, and glycolytic reserve were calculated by the Seahorse XF24 software. Data represent mean ± s. d. of triplicate independent experiments (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant, by two-sided Student’s t test). 2-DG, 2-deoxy-glucose; FCCP, carbonyl cyanide 4-trifluoromethoxyphenylhydrazone; qRT-PCR, quantitative real-time PCR; R/A, rotenone/antimycin A.
Furthermore, we investigated the effects of SIRT2 on cell proliferation and examined cell proliferation in HCC cells by the Cell Counting Kit-8 assay. The results showed that knockdown of SIRT2 inhibited the proliferation of MHCC-97H cells (Fig. 9, A–C). Consistently, overexpression of SIRT2 enhanced cell proliferation in Huh7 cells (Fig. 9, D–F). The data indicated that SIRT2 promoted proliferation of HCC cells.
Fig. 9.

The effects of SIRT2 on the proliferation of HCC cells.A, the relative expression of SIRT2 in MHCC-97H cells transfected with siRNA was measured by qRT-PCR. B, Western blotting confirmed the knockdown of SIRT2 in cells treated with siSIRT2. The densities of the corresponding protein bands were measured by Image J and displayed on the right. C, cell proliferation of MHCC-97H cells with SIRT knockdown (siSIRT2) and the corresponding control cells (siNC) was measured by the Cell Counting Kit-8 assay at 24, 48, and 72 h. D, the relative expression of SIRT2 in Huh7 cells transfected with the empty vector control (NC) or pcDNA3.1-SIRT2 vector was measured by qRT-PCR. E, Western blotting analysis confirmed the overexpression of SIRT2. The densities of the corresponding protein bands were measured by Image J and displayed on the right. Based on the observed molecular weights, the double bands of SIRT2 were likely formed by two alternative spliced isoforms of SIRT2, including Sirt2.1 as the canonical isoform (1–389 aa, 43 kDa) and Sirt2.2 (38–389 aa, 40 kDa). F, cell proliferation of Huh7 cells with SIRT2 overexpression and the corresponding control cells was measured by the Cell Counting Kit-8 assay at 24, 48, and 72 h. Data represent mean ± s. d. of triplicate independent experiments (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by two-sided Student’s t test). HCC, hepatocellular carcinoma; qRT-PCR, quantitative real-time PCR.
Discussion
In recent years, clinical proteomics has advanced tremendously and has provided comprehensive information for the molecular features of various cancers (51). To resolve the sample complexity and achieve high coverage, prefractionation is normally performed before MS analyses, resulting in increased instrument time and limiting its applications in clinical practice for personalized medicine. Here, we employed a timsTOF Pro instrument to analyze eight HCC tissue samples using single injections, and all the MS experiments were finished within a day. The TIMS device helped to resolve the high complexity of tumor samples with an added dimension of separation in addition to HPLC and mass spectrometry. The TOF analyzer had very high acquisition rate (>100 Hz), enabling efficient sampling of ions in the sub-ms time frame. A previous study showed that TIMS-PASEF provided consistent and accurate protein quantification for whole cell lysates without prefractionation (28). In this study, 4678 proteins were identified per HCC tumor, and 4475 proteins were identified per NAT sample, comparable to the earlier large-scale HCC study, which employed a standard workflow of shot-gun proteomics with six fractions and identified 5953 proteins per tumor and 5114 proteins per nontumor tissues (41).
Acetylation is a pervasive and reversible form of PTM that regulates the activity, stability, and location of proteins (52). In this study, we detected 9219 acetylation sites from 2625 proteins from HCC tissues. Widespread downregulation of protein acetylation was observed in HCC tumors compared to NATs. Majority of the proteins with downregulated K-acetylation were localized in the mitochondria and cytoplasm, participating in various metabolic processes. For example, glycolytic enzyme LDHA that catalyzes pyruvate to lactate is frequently overexpressed in multiple tumors and plays an important role in promoting tumor progression (53, 54). It has been reported that LDHA-K5 acetylation was decreased in human pancreatic cancers, resulting in elevated LDHA enzymatic activity and enhanced cancer cell proliferation and migration (55). In our study, we observed downregulated acetylation of K126, K59, K278 of LDHA in HCC tumor tissues. Fatty acid synthase (FASN) is one of the rate-limiting enzymes in the de novo lipogenesis pathway, and the acetylation of FASN promotes its degradation and inhibits de novo lipogenesis (56). Here, we identified 29 K-acetylation sites in FASN, and two of them K2449 and K776 were downregulated in HCC. Of note, the sample size of this study is relatively small, and the observations may need to be further validated in larger sample sets. In addition, for majority of the dysregulated acetylation sites, how they are modulated and how they affect the modified proteins requires further investigation.
Our findings suggest that SIRT2 overexpression may affect some of the dysregulated K-acetylation sites in HCC tissues. Substantial evidence has implicated SIRT2 in a variety of cellular processes, such as mitosis regulation, genome integrity, cell differentiation, cell homeostasis, aging, infection, inflammation, oxidative stress, metabolism, and autophagy (57, 58, 59, 60), consistent with the widespread alterations of protein K-acetylation in SIRT2-overexpressing cells detected in this study. Moreover, our data showed that SIRT2 overexpression inhibited K-acetylation on multiple glycolytic enzymes, for example, GPI (K73), GAPDH(K117), PGK1 (K91), LDHA(K278). Previous studies have associated SIRT2 with the deacetylation of GAPDH and PGK1 (48); however, the acetylation sites remain unknown. Our data suggest that the reduced acetylation of these sites in HCC tumors may be modulated by SIRT2. In addition, SIRT2 overexpression decreased the K-acetylation on five TCA enzymes, including isocitrate dehydrogenase (K272, K180), SDHA (K250, K608), MDH2 (K296), OGDH (K869, K389), and SUCLG1 (K57). Although SIRT2 is predominately localized in the cytoplasm, several recent studies have shown that it is associated with the inner membrane of mitochondria (61) and dynamically interacts with multiple TCA enzymes (49). On the hand, the observed downregulated acetylation could be induced by the metabolic changes in SIRT2-overexpressing cells, and whether these enzymes are direct substrates of SIRT2 requires further exploration.
Several KDAC inhibitors have been tested in preclinical studies or clinical trials for their efficacy for HCC, such as vorinostat and resminostat, alone or in combination with sorafenib (62, 63). However, majority of these chemicals target class I and/or class II KDACs. The study of SIRT2 and its inhibitors in HCC is still limited. Knockdown of SIRT2 with shRNA impaired HCC cell proliferation and migration (64). A very recent study showed that SIRT1 and SIRT2 inhibitors could enhance the inhibitory effect of sorafenib, the standard of care for patients with advanced HCC (65). These studies suggest the potential of targeting SIRT2 for HCC treatment, especially in combination with sorafenib, however, more in-depth investigation is still required to explore its value as a therapeutic target in HCC.
Data Availability
The proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE (66) partner repository with the dataset identifier PXD026729 for the HCC data and PXD031636 for the Huh7 data. The annotated mass spectra for all acetylpeptides are uploaded to MS-Viewer (search key, vlovqzdx6m and xv8ot7cony).
Supplemental data
This article contains supplemental data (26).
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (21974094, 81902495), Science and Technology Project of Beijing Municipal Education Commission (KM202110025019), Top-Notch Project for Outstanding Talents in Beijing (2018000026833ZK78), Youth Top Talent Project of Beijing Municipal Education Commission (CIT&TCD201904093), and Science, Technology and Innovation Commission of Shenzhen Municipality (Grant No.: KCXFZ202002011006448). This work was also funded by the Open Project Program of the State Key Laboratory of Proteomics (SKLP-O202004) and Tianjin University Innovation Fund (2022XSU-0008).
Author contributions
J. X. and H. L. methodology; J. X., X. G., X. J., and H. L. investigation; J. X. writing–original draft; R. C. conceptualization; R. C. and Y. L. supervision; R. C. writing–review and editing; Y. L. resources; Y. L. funding acquisition.
Contributor Information
Hongyan Li, Email: hongyanli@xwhosp.org.
Ruibing Chen, Email: rbchen@tju.edu.cn.
Yinying Lu, Email: luyinying1973@163.com.
Supplemental Data
References
- 1.Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2021;7:6–34. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Au S.L., Ng I.O., Wong C.M. Epigenetic dysregulation in hepatocellular carcinoma: focus on polycomb group proteins. Front. Med. 2013;7:231–241. doi: 10.1007/s11684-013-0253-7. [DOI] [PubMed] [Google Scholar]
- 4.Tang Z.Y. Hepatocellular carcinoma—cause, treatment and metastasis. World J. Gastroenterol. 2001;7:445–454. doi: 10.3748/wjg.v7.i4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yarchoan M., Agarwal P., Villanueva A., Rao S., Dawson L.A., Llovet J.M., et al. Recent developments and therapeutic strategies against hepatocellular carcinoma. Cancer Res. 2019;79:4326–4330. doi: 10.1158/0008-5472.CAN-19-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reig M., da Fonseca L.G., Faivre S. New trials and results in systemic treatment of HCC. J. Hepatol. 2018;69:525–533. doi: 10.1016/j.jhep.2018.03.028. [DOI] [PubMed] [Google Scholar]
- 7.Bayo J., Fiore E.J., Dominguez L.M., Real A., Malvicini M., Rizzo M., et al. A comprehensive study of epigenetic alterations in hepatocellular carcinoma identifies potential therapeutic targets. J. Hepatol. 2019;71:78–90. doi: 10.1016/j.jhep.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Allfrey V.G., Faulkner R., Mirsky A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 10.Choudhary C., Weinert B.T., Nishida Y., Verdin E., Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 11.Schölz C., Weinert B.T., Wagner S.A., Beli P., Miyake Y., Qi J., et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015;33:415–423. doi: 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- 12.Narita T., Weinert B.T., Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019;20:156–174. doi: 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- 13.Zhao S., Xu W., Jiang W.Q., Yu W., Lin Y., Zhang T.F., et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernabé A.M., Balcells C., Celada J.T., Foguet C., Voillard S.B., Seve M., et al. The importance of post-translational modifications in systems biology approaches to identify therapeutic targets in cancer metabolism. Curr. Opin. Struct. Biol. 2017;3:161–169. [Google Scholar]
- 15.Lv L., Li D., Zhao D., Lin R., Chu Y., Zhang H., et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lv L., Xu Y.P., Zhao D., Li F.L., Wang W., Sasaki N.Y., et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner G.R., Payne R.M. Widespread & Enzyme-independent, Nε-Acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinert B.T., Iesmantavicius V., Moustafa T., Schölz C., Wagner S.A., Magnes C., et al. Acetylation dynamics and stoichiometry in saccharomyces cerevisiae. Mol. Syst. Biol. 2014;10:716–728. doi: 10.1002/msb.134766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y.P., Zhou L.S., Zhao Y.Z., Wang S.W., Chen L.L., Liu L.X., et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33:1304–1320. doi: 10.1002/embj.201387224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y., Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016;6:a026831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imai S., Armstrong C.M., Kaeberlein M., Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 22.Haigis M.C., Sinclair D.A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaquero A., Scher M.B., Lee D.H., Sutton A., Cheng H.L., Alt F.W., et al. SIRT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 25.de Oliveira R.M., Sarkander J., Kazantsev A.G., Outeiro T.F. SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol. 2012;3:82–91. doi: 10.3389/fphar.2012.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Q., Zhang Z., Li J., Xu F., Zhang B., Liu M., et al. Lysine acetylome study of human hepatocellular carcinoma tissues for biomarkers and therapeutic targets discovery. Front. Genet. 2020;11:572663–572677. doi: 10.3389/fgene.2020.572663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diallo I., Seve M., Cunin V., Minassian F., Poisson J.F., Michelland S., et al. Current trends in protein acetylation analysis. Exp. Rev. Proteomics. 2018;16:139–159. doi: 10.1080/14789450.2019.1559061. [DOI] [PubMed] [Google Scholar]
- 28.Meier F., Brunner A.D., Koch S., Koch H., Lubeck M., Krause M., et al. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics. 2018;17:2534–2545. doi: 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 30.Husson F., Josse J., Le S., Mazet J. FactoMineR: an R package for multivariate analysis. J. Sta. Softw. 2008;25:1–18. [Google Scholar]
- 31.Wickham H. Springer-Verlag; NY: 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 32.Yu G., Wang L.G., Han Y., He Q.Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M., Goto S. KEGG: kyoto Encyclopedia of genes and genomes. Nucl. Acids Res. 1999;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists. Nucl. Acids Res. 2019;47:191–198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schölz C., Lyon D., Refsgaard J.C., Jensen L.J., Choudhary C., Weinert B.T. Avoiding abundance bias in the functional annotation of post- translationally modified proteins. Nat. Met. 2015;12:1003–1004. doi: 10.1038/nmeth.3621. [DOI] [PubMed] [Google Scholar]
- 36.Cheng A., Grant C.E., Noble W.S., Bailey T.L. MoMo: discovery of statistically significant post-translational modification motifs. Bioinformatics. 2018;35:2774–2782. doi: 10.1093/bioinformatics/bty1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLano W.L. DeLano Scientific; Palo Alto, CA: 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 38.Garcia-Lora A., Algarra I., Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003;195:346–355. doi: 10.1002/jcp.10290. [DOI] [PubMed] [Google Scholar]
- 39.Su H., Na N., Zhang X., Zhao Y. The biological function and significance of CD74 in immune diseases. Inflamm. Res. 2017;66:209–216. doi: 10.1007/s00011-016-0995-1. [DOI] [PubMed] [Google Scholar]
- 40.Li Y., Feng D., Wang Z., Zhao Y., Sun R., Tian D., et al. Ischemia-induced ACSL4 activation contributes to ferroptosis- mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–2299. doi: 10.1038/s41418-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y., Sun A., Zhao Y., Ying W., Sun H., Yang X., et al. Chinese Human Proteome Project (CNHPP) Consortium Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567:257–261. doi: 10.1038/s41586-019-0987-8. [DOI] [PubMed] [Google Scholar]
- 42.Wang M.H., Feng L., Li P., Han N.J., Gao Y.N., Xiao T. Hsp90AB1 protein is overexpressed in non-small cell lung cancer tissues and associated with poor prognosis in lung adenocarcinoma patients. Chin. J. Lung Cancer. 2016;19:64–69. doi: 10.3779/j.issn.1009-3419.2016.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao Q.Q., Zhang T.P., Zhao W.J., Liu Z.W., You L., Zhou L., et al. Filamin A: insights into its exact role in cancers. Pathol. Oncol. Res. 2016;22:245–252. doi: 10.1007/s12253-015-9980-1. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura F., Stossel T.P., Hartwig J.H. The filamins organizers of cell structure and function. Cell Adh. Migr. 2011;5:160–169. doi: 10.4161/cam.5.2.14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W., Mouneimne G., Sidani M., Wyckoff J., Chen X., Makris A., et al. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J. Cell Biol. 2006;173:395–404. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todisco S., Convertini P., Iacobazzi V., Infantino V. TCA cycle rewiring as emerging metabolic signature of hepatocellular carcinoma. Cancers. 2019;12:68–82. doi: 10.3390/cancers12010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinert B.T., Moustafa T., Iesmantavicius V., Zechner R., Choudhary C. Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 2015;34:2620–2632. doi: 10.15252/embj.201591271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cha Y., Han M.J., Cha H.J., Zoldan J., Burkart A., Jung J.H., et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 2017;19:445–456. doi: 10.1038/ncb3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamaidi I., Zhang L., Kim N., Wang M.H., Iclozan C., Fang B., et al. Sirt2 inhibition enhances metabolic fitness and effector functions of tumor-reactive T cells. Cell Metab. 2020;32:420–436. doi: 10.1016/j.cmet.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh A.K., Bishayee A., Pandey A.K. Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients. 2018;10:731–762. doi: 10.3390/nu10060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H., Jiang N., Zhang Y., Chen R. Clinical proteomics: a driving force for cancer therapeutic target discovery and precision medicine. Cancer Biol. Med. 2019;16:623–629. doi: 10.20892/j.issn.2095-3941.2019.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu W., Li Y., Liu C., Zhao S. Protein lysine acetylation guards metabolic homeostasis to fight against cancer. Oncegene. 2013;33:2279–2285. doi: 10.1038/onc.2013.163. [DOI] [PubMed] [Google Scholar]
- 53.Feng Y., Xiong Y., Qiao T., Li X., Jia L., Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018;7:6124–6136. doi: 10.1002/cam4.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheng S.L., Liu J.J., Dai Y.H., Sun X.G., Xiong X.P., Huang G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS. J. 2012;279:3898–3910. doi: 10.1111/j.1742-4658.2012.08748.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhao D., Zou S.W., Liu Y., Zhou X., Mo Y., Wang P., et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell. 2013;23:464-476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin H.P., Cheng Z.L., He R.Y., Song L., Tian M.X., Zhou L.S., et al. Destabilization of fatty acid synthase by acetylation inhibits de novo lipogenesis and tumor cell growth. Cancer Res. 2016;76:6924–6936. doi: 10.1158/0008-5472.CAN-16-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y., Yang J.Q., Hong T.T., Chen X.J., Cui L.L. SIRT2: controversy and multiple roles in disease and physiology. Aging Res. Rev. 2019;55:100961–100976. doi: 10.1016/j.arr.2019.100961. [DOI] [PubMed] [Google Scholar]
- 58.Kim H.S., Vassilopoulos A., Wang R.H., Lahusen T., Xiao Z., Xu X., et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park S.H., Ozden O., Liu G., Song H.Y., Zhu Y., Yan Y., et al. SIRT2-mediated deacetylation and tetramerization of pyruvate kinase directs glycolysis and tumor growth. Cancer Res. 2016;76:3802–3812. doi: 10.1158/0008-5472.CAN-15-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen G.Y., Huang P., Hu C. The role of SIRT2 in cancer: a novel therapeutic target. Int. J. Cancer. 2020;147:3297–3304. doi: 10.1002/ijc.33118. [DOI] [PubMed] [Google Scholar]
- 61.Liu G., Park S.H., Imbesi M., Nathan W.J., Zou X., Zhu Y., et al. Loss of NAD-dependent protein deacetylase Sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid. Redox Signal. 2017;26:849–863. doi: 10.1089/ars.2016.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bitzer M., Horger M., Giannini E.G., Ganten T.M., Wörns M.A., Siveke J.T., et al. Resminostat in combination with sorafenib as second-line therapy of advanced hepatocellular carcinoma - the SHELTER study. J. Hepatol. 2016;65:280–288. doi: 10.1016/j.jhep.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 63.Zhang C., Yang C., Feldman M.J., Wang H., Pang Y., Maggio D.M., et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Ontotarget. 2017;8:56110–56125. doi: 10.18632/oncotarget.18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J., Anthony W.H., Chen W.Q., Zhang Z.Z., Ren J.H., Song C.L., et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3β/β-catenin signaling. Hepatology. 2013;57:2287–2298. doi: 10.1002/hep.26278. [DOI] [PubMed] [Google Scholar]
- 65.Ceballos M.P., Angel A., Delprato C.B., Livore V.I., Ferretti A.C., Lucci A., et al. Sirtuin 1 and 2 inhibitors enhance the inhibitory effect of sorafenib in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2021;892:173736–173750. doi: 10.1016/j.ejphar.2020.173736. [DOI] [PubMed] [Google Scholar]
- 66.Vizcaíno J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., et al. 2016 update of the PRIDE database and its related tools. Nucl. Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE (66) partner repository with the dataset identifier PXD026729 for the HCC data and PXD031636 for the Huh7 data. The annotated mass spectra for all acetylpeptides are uploaded to MS-Viewer (search key, vlovqzdx6m and xv8ot7cony).