

Summary
Phosphoinositides (PIPs) are low-abundant membrane lipids with critically important functions in cellular physiology. To investigate their subcellular distribution in neurons, we optimized protocols for immunofluorescence staining of intracellular or plasma membrane PIPs with commercially available antibodies. Here, we describe the preparation and transfection of primary mouse hippocampal neurons in dissociated culture, followed by immunofluorescence staining and quantitative analysis of PIP signals. In addition, we expand the application of the protocol to proteins located at the cytoplasmic leaflet of cellular membranes.
For complete details on the use and execution of this protocol, please refer to Guo et al. (2022).
Subject areas: Cell culture, Cell Membrane, Microscopy, Neuroscience
Graphical abstract
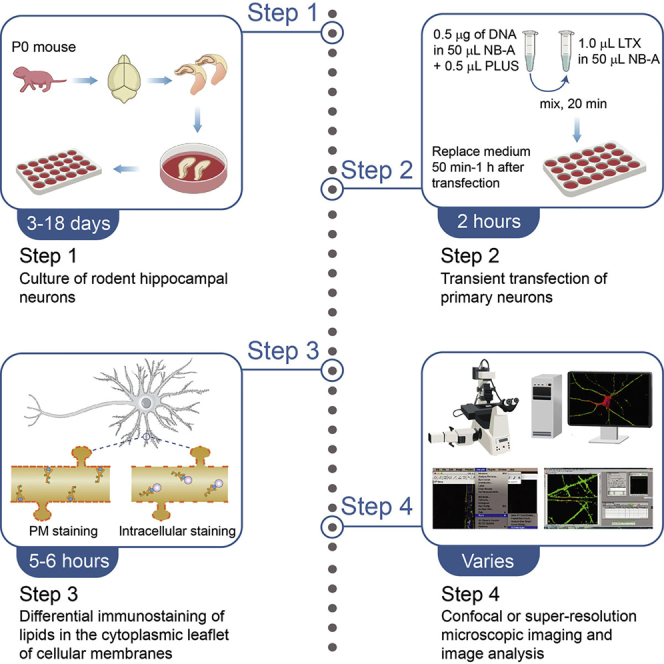
Highlights
-
•
A protocol for detecting membrane lipids in cultured mouse neurons
-
•
Culturing of neurons for analysis of the subcellular distribution of lipids
-
•
Differential immunostaining of lipids in the plasma membrane and endomembranes
-
•
Application of the protocol to proteins at the cytoplasmic leaflet of membranes
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Phosphoinositides (PIPs) are low-abundant membrane lipids with critically important functions in cellular physiology. To investigate their subcellular distribution in neurons, we optimized protocols for immunofluorescence staining of intracellular or plasma membrane PIPs with commercially available antibodies. Here, we describe the preparation and transfection of primary mouse hippocampal neurons in dissociated culture, followed by immunofluorescence staining and quantitative analysis of PIP signals. In addition, we expand the application of the protocol to proteins located at the cytoplasmic leaflet of cellular membranes.
Before you begin
This protocol is designed to provide precise details for immunofluorescence staining of PIPs in primary cultured central nervous system (CNS) neurons. The neuronal culture protocol was designed for neonatal (P0, within 12 h of birth) pups of C57BL/6J mice (wild type), but can be easily adapted to rats. The animals may be commercially obtained or involve knockout or transgenic lines. Animal experiments must receive approval from the relevant institutional review board and be conducted in strict accordance with institutional animal ethics committee guidelines. All procedures performed here were performed in compliance with the guidelines of the Animal Care and Use Committee of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (Approval code: AP2015003). P0 C57BL/6J mice were purchased from SPF (Beijing) Biotechnology Co., Ltd (Beijing, China).
The whole process of neuronal culture should be performed under sterile conditions, and all the tools and reagents used during culture should be sterile to prevent any contamination. Please note that it is optional to express green or red fluorescent proteins (e.g., mCherry) in neurons as cell fill to label the cell morphology. For transient transfection, the time length for incubation of neurons with transfection reagents should be no longer than 60 min to avoid cytotoxicity. In order to improve the reproducibility of experimental results, the antibodies and membrane-permeabilizing detergents (digitonin and saponin) should be stored at −20°C and avoid repeated freeze–thaw. The protocol below describes the specific steps for primary cultured hippocampal neurons. We have also applied this protocol to HeLa cells, NIH/3T3 and Neuro-2a cells (Guo et al., 2022).
Before starting the experiment several chemicals, buffers, and media should be prepared (For detailed information please see key resources table and materials and equipment section).
Institutional permissions
All procedures performed here were approved by an Institutional Animal Care and Use Committee.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-PI3P, IgG (1:50) | Echelon Biosciences | Cat# Z-P003, RRID: AB_427221 |
| Mouse anti-PI4P, IgM (1:100 -1:200) | Echelon Biosciences | Cat# Z-P004, RRID: AB_11127796 |
| Mouse anti-PI(4,5)P2, IgM (1:50 - 1:150) | Echelon Biosciences | Cat# Z-A045, RRID: AB_427211 |
| Rabbit anti-Rab8, IgG (1:150) | Sigma-Aldrich | Cat# R5530, RRID: AB_2175306 |
| Rabbit anti-TGN46, IgG (1:200) | Abcam | Cat# ab50595, RRID: AB_2203289 |
| Rabbit anti-endophilin A1, IgG (1:100) | Synaptic Systems | Cat# 159002, RRID: AB_887757 |
| Alexa Fluor 647 donkey anti-rabbit, IgG (1:1500) | Invitrogen | Cat# A-31573, RRID: AB_2536183 |
| Alexa Fluor 488 goat anti-mouse, IgG (1:1500) | Invitrogen | Cat# A-11001, RRID: AB_2534069 |
| Alexa Fluor 488 goat anti-mouse, IgM (1:1500) | Invitrogen | Cat# A-21042, RRID: AB_141357 |
| Chemicals, peptides, and recombinant proteins | ||
| Concentrated sulfuric acid | Sinopharm Chemical Reagent Co., Ltd | Cat# SJ01006 |
| Potassium dichromate | Sinopharm Chemical Reagent Co., Ltd | Cat# SJ01003 |
| Poly-D-Lysine | Sigma-Aldrich | Cat# P6407 |
| HBSS (without Ca2+, Mg2+, Phenol Red) | Gibco | Cat# 14175095 |
| HEPES (1 M) | Gibco | Cat# 15630080 |
| Penicillin-Streptomycin (100×) | HyClone | Cat# SV30010 |
| DMEM with sodium pyruvate | HyClone | Cat# SH30243.01 |
| Fetal Bovine Serum | Gibco | Cat# 16000044 |
| Ham’s F-12 Nutrient Mix | Gibco | Cat# 11765054 |
| Trypsin | HyClone | Cat# SH30042.02 |
| Neurobasal-A Medium | Gibco | Cat# 10888022 |
| B-27 Supplement (50×) | Gibco | Cat# 17504044 |
| GlutaMAX | Gibco | Cat# 35050061 |
| Na2HPO4 | Sigma-Aldrich | Cat# S5136 |
| KH2PO4 | Sigma-Aldrich | Cat# P5655 |
| NaCl | Sigma-Aldrich | Cat# S6191 |
| KCl | Sigma-Aldrich | Cat# P5405 |
| Formaldehyde | Thermo Scientific | Cat# 28906 |
| Sucrose | BBI | Cat# A610498 |
| NH4Cl | Sigma-Aldrich | Cat# 213330 |
| PIPES | AMRESCO | Cat# 0488 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| DMSO | Sigma-Aldrich | Cat# D5879 |
| Normal Goat Serum | ZSGB-BIO | Cat# ZLI-9022 |
| DAPI | Roche | Cat# 10236276001 |
| Propylgallate | Sigma-Aldrich | Cat# 3130 |
| Glycerol | Sigma-Aldrich | Cat# v900122 |
| Glutaraldehyde | Electron Microscopy Sciences | Cat# 16020 |
| Saponin | Sigma-Aldrich | Cat# S7900 |
| Critical commercial assays | ||
| Lipofectamine® LTX Reagent and PLUS™ Reagent | Invitrogen | Cat# 15338100 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (P0, within 12 h of birth; male and female) | SPF (Beijing) Biotechnology Co., Ltd. | Cat# B204 |
| Recombinant DNA | ||
| pAOV-CAMKIIα-mCherry-2A-3Flag | OBiO Technology (Shanghai) Corp. Ltd. | Cat# AKD001 |
| Software and algorithms | ||
| Fiji-ImageJ | NIH | Fiji ImageJ http://fiji.sc, RRID: SCR_002285 |
| NIS-Elements AR 3.1 | Nikon Corporation |
https://www.nikoninstruments.com/Products/Software RRID: SCR_014329 |
| Graphpad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/, RRID: SCR_002798 |
| Other | ||
| Fisherbrand® Microscope Cover Glass | Fisher Scientific | Cat# 12-545-80 |
| 150 mm glass petri dish | Beijing Beibo Keyi Commerce & Trade Co., Ltd. | Cat# BL02006 |
| 90 mm glass petri dish | Beijing Beibo Keyi Commerce & Trade Co., Ltd. | Cat# BL02003 |
| 24-well cell culture plates | Costar | Cat# 3524 |
| Straight ophthalmic forceps | Beijing Beibo Keyi Commerce & Trade Co., Ltd. | Cat# XYN-ZC |
| Fine smooth straight forceps | RWD | Cat# F11020-11 |
| Mayo spring scissors | RWD | Cat# S11007-12 |
| Sharp straight delicate scissors | Beijing Beibo Keyi Commerce & Trade Co., Ltd. | Cat# YYJ-ZJ |
| Small spatula | Fisher Scientific | Cat# 21-401-25A |
| 100 mm cell culture petri dish | In Vitro Scientific | Cat# 010100 |
| 1.5 mL centrifuge tube | Eppendorf | Cat# 30108116 |
| 50 mL polypropylene conical Tube | Falcon | Cat# 352070 |
| 15 mL polypropylene conical Tube | Falcon | Cat# 352086 |
| 20 mL syringe | ZYMM | Cat# GCA10313 |
| 0.22 μm syringe filter | Millipore | Cat# SLGPR33RB |
| Hemocytometer | Qiujing | Cat# HY-00570 |
| Stereomicroscope | Beijing Tech Instrument Co., LTD. | Cat# XTS20 |
| Microscope for cell counting | Olympus | Cat# CKX53SF |
| Confocal microscope | Nikon Corporation | Cat# 537353 |
Materials and equipment
-
•
Preparation of cleaning solution.
| Reagent | Final concentration | Amount |
|---|---|---|
| Concentrated sulfuric acid | 10% (v/v) | 20 mL |
| Potassium dichromate | 10% (w/v) | 20 g |
| ddH2O | n/a | 180 mL |
| Total | n/a | 200 mL |
CRITICAL: Concentrated sulfuric acid is highly corrosive. Use personal protective equipment as required, such as protective gloves, face protection, and protective clothing. Prepare the cleaning solution under chemical fume hood by adding 20 mL of concentrated sulfuric acid to 180 mL of 11.1% (w/v) potassium dichromate solution slowly while stirring constantly. Do not add aqueous solution into concentrated sulfuric acid.
Note: Store at 21°C–25°C. The cleaning solution could be used repeatedly until it turns green.
-
•
Preparation of the Poly-D-Lysine (PDL) solution (0.1 mg/mL).
| Reagent | Final concentration | Amount |
|---|---|---|
| PDL | 0.1 mg/mL | 5 mg |
| ddH2O | n/a | 50 mL |
| Total | n/a | 50 mL |
Note: Store at 4°C and away from light for up to 3 months.
-
•
Preparation of dissection buffer.
| Reagent | Final concentration | Amount |
|---|---|---|
| HBSS (without Ca2+ or Mg2+) | n/a | 490 mL |
| 1 M HEPES | 10 mM | 5 mL |
| Penicillin-Streptomycin (100×) | 100 U/mL Penicillin 100 μg/mL Streptomycin |
5 mL |
| Total | n/a | 500 mL |
Note: Store at 4°C for up to 1 month.
-
•
Preparation of the DMEM culture medium.
| Reagent | Final concentration | Amount |
|---|---|---|
| High glucose DMEM (with L-Glutamine and Sodium Pyruvate) | n/a | 40 mL |
| Fetal Bovine Serum (FBS) | 10% (v/v) | 5 mL |
| Ham’s F-12 Nutrient Mix | 10% (v/v) | 5 mL |
| Total | n/a | 50 mL |
Note: Prepare the medium just before use.
-
•
Preparation of the Neurobasal-A (NB-A) culture medium.
| Reagent | Final concentration | Amount |
|---|---|---|
| Neurobasal-A | n/a | 48.5 mL |
| B-27 (50×) | 2% (v/v) | 1 mL |
| GlutaMAX (100×) | 1% (v/v) | 500 μL |
| Total | n/a | 50 mL |
Note: Prepare the medium just before use and store at 4°C for up to 1 week.
-
•
Preparation of 10×PBS solution (pH = 7.4).
| Reagent | Final concentration | Amount |
|---|---|---|
| Na2HPO4 | 100 mM | 14.2 g |
| KH2PO4 | 18 mM | 2.4 g |
| NaCl | 1.4 M | 81.8 g |
| KCl | 27 mM | 2 g |
| ddH2O | n/a | 1 L |
| Total | n/a | 1 L |
Note: Store at 21°C–25°C for up to 1 year.
-
•
Preparation of PBS containing 4% (v/v) formaldehyde (FA).
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% FA | 4% | 250 μL |
| 10×PBS | 1× | 100 μL |
| ddH2O | n/a | 650 μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use, and keep away from light.
-
•
Preparation of 60% (w/v) sucrose.
| Reagent | Final concentration | Amount |
|---|---|---|
| Sucrose | 60% | 6 g |
| ddH2O | n/a | 10 mL |
| Total | n/a | 10 mL |
Note: Store at 21°C–25°C for up to 1 year.
-
•
Preparation of PBS containing 2% FA and 4% sucrose.
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% FA | 2% | 125 μL |
| 10×PBS | 1× | 100 μL |
| 60% sucrose | 4% | 66.7 μL |
| ddH2O | n/a | 708.3 μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use, and keep away from light.
-
•
Preparation of 5 M NH4Cl solution.
| Reagent | Final concentration | Amount |
|---|---|---|
| NH4Cl | 5 M | 13.4 g |
| ddH2O | n/a | 50 mL |
| Total | n/a | 50 mL |
Note: Store at 21°C–25°C for up to 1 year.
-
•
Preparation of PBS containing 50 mM NH4Cl.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NH4Cl | 50 mM | 500 μL |
| 10×PBS | 1× | 5 mL |
| ddH2O | n/a | 44.5 mL |
| Total | n/a | 50 mL |
Note: Store at 4°C for up to 1 week.
-
•
Preparation of 2×buffer A (pH 6.8).
| Reagent | Final concentration | Amount |
|---|---|---|
| PIPES | 40 mM | 0.6 g |
| NaCl | 274 mM | 0.8 g |
| KCl | 5.4 mM | 20.1 mg |
| ddH2O | n/a | 50 mL |
| Total | n/a | 50 mL |
Note: 2×buffer A can be stored at −20°C for up to one year and should be stored at 4°C and used within one week once thawed.
-
•
Preparation of 25 mg/mL digitonin.
| Reagent | Final concentration | Amount |
|---|---|---|
| Digitonin | 25 mg/mL | 12.5 mg |
| DMSO | n/a | 500 μL |
| Total | n/a | 500 μL |
Note: Store at −20°C for up to 1 year and avoid repeated freeze–thaw.
-
•
Preparation of buffer A containing 25 μg/mL digitonin.
| Reagent | Final concentration | Amount |
|---|---|---|
| 25 mg/mL digitonin | 25 μg/mL | 1 μL |
| 2×buffer A | 1× | 500 μL |
| ddH2O | n/a | 499 μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use.
-
•
Preparation of buffer A (pH 6.8).
| Reagent | Final concentration | Amount |
|---|---|---|
| 2×buffer A | 1× | 25 mL |
| ddH2O | n/a | 25 mL |
| Total | n/a | 50 mL |
Note: Store at 4°C for up to 1 week.
-
•
Preparation of buffer A containing 5% Normal Goat Serum (NGS) and 50 mM NH4Cl.
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% NGS | 5% | 500 μL |
| 5 M NH4Cl | 50 mM | 10 μL |
| 2×buffer A | 1× | 500 μL |
| Total | n/a | 1010 μL |
Note: Store at 4°C for up to 1 week.
-
•
Preparation of buffer A containing 5% NGS.
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% NGS | 5% | 500 μL |
| 2×buffer A | 1× | 500 μL |
| Total | n/a | 1 mL |
Note: Store at 4°C for up to 1 week.
-
•
Preparation of PBS containing 2% FA.
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% FA | 2% | 125 μL |
| 10×PBS | 1× | 100 μL |
| ddH2O | n/a | 775 μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use, and keep away from light.
-
•
Preparation of DAPI solution for nuclear staining.
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 mg/mL DAPI | 1 μg/mL | 1 μL |
| PBS | n/a | 1 mL |
| Total | n/a | 1 mL |
Note: Store at 4°C for up to 1 day and keep away from light.
-
•
Preparation of mounting medium.
| Reagent | Final concentration | Amount |
|---|---|---|
| Propylgallate | 4% | 2 g |
| Glycerol | 80% (v/v) | 40 mL |
| 10×PBS | 1× | 5 mL |
| ddH2O | n/a | 5 mL |
| Total | n/a | 50 mL |
Note: Mounting medium should be stored away from light at 4°C for up to 1 year.
-
•
Preparation of PBS containing 8% FA and 0.4% Glutaraldehyde (GA).
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% FA | 8% | 500 μL |
| 8% GA | 0.4% | 50 μL |
| 10×PBS | 1× | 100 μL |
| ddH2O | n/a | 350 μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use, and keep away from light.
-
•
Preparation of PBS containing 4% FA, 0.2% GA and 4% sucrose.
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% FA | 4% | 250 μL |
| 8% GA | 0.2% | 25 μL |
| 60% sucrose | 4% | 66.7 μL |
| 10×PBS | 1× | 100 μL |
| ddH2O | n/a | 558.3μL |
| Total | n/a | 1 mL |
Note: Prepare the solution immediately before use, and keep away from light.
-
•
Preparation of 10% (w/v) saponin.
| Reagent | Final concentration | Amount |
|---|---|---|
| Saponin | 10% | 100 mg |
| Buffer A | n/a | 1 mL |
| Total | n/a | 1 mL |
Note: Store at −20°C for up to 1 year and avoid repeated freeze–thaw.
-
•
Preparation of buffer A containing 5% NGS, 50 mM NH4Cl and 0.5% saponin.
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% NGS | 5% | 500 μL |
| 5 M NH4Cl | 50 mM | 10 μL |
| 10% saponin in buffer A | 0.5% | 50 μL |
| 2×buffer A | 1× | 500 μL |
| Total | n/a | 1060 μL |
Note: Prepare the solution immediately before use.
-
•
Preparation of buffer A containing 5% NGS and 0.1% saponin.
| Reagent | Final concentration | Amount |
|---|---|---|
| 10% NGS | 5% | 500 μL |
| 10% saponin in buffer A | 0.1% | 10 μL |
| 2×buffer A | 1× | 500 μL |
| Total | n/a | 1010 μL |
Note: Prepare the solution immediately before use.
Step-by-step method details
Preparation one: Culturing primary mouse hippocampal neurons
The major steps for hippocampal neuron culture are shown in Figure 1.
Figure 1.

Culturing of mouse hippocampal neurons
(A) Dissecting microscope and surgical instruments used for hippocampal dissection.
(B) Dissect whole brain from P0 C57BL/6J mouse. Arrows indicate direction of cuts.
(C) Mouse brain before and after stripping away meninges.
(D) Isolate the hippocampus from the cortex.
(E) Trypsinize hippocampi, rinse with the DMEM culture medium and collect tissue for trituration. Shown are hippocampi before and after trypsinization.
(F) Rinse PDL-coated coverslips with autoclaved distilled water.
(G) Plate dissociated hippocampal neurons on coverslips. Replenish culture medium regularly Scale bars: 1 mm.
Preparation of coverslips for dissociated culture of primary hippocampal neurons
Timing: 3–7 days
-
1.
Soak coverslips in cleaning solution for 12–16 h or longer in 150 mm glass petri plates on a flat rotator.
-
2.
Remove cleaning solution, rinse coverslips sequentially with distilled water (dH2O) and double distilled water (ddH2O) for more than ten times each on a flat rotator, change water every 30 min or longer.
Note: Concentrated sulfuric acid in cleaning solution removes inorganic and organic dirt on the surface of coverslips and produces a large number of hydroxyl, amino, sulfhydryl groups and other reactive groups by dissociating the sodium, potassium and calcium cations, which renders coverslips highly hydrophilic. As sulfuric acid is harmful to cells, it is imperative to rinse the coverslips thoroughly before coating and plating of cells.
-
3.
Rinse coverslips twice with absolute alcohol for 1 h or longer on a flat rotator.
-
4.
Autoclave sterilize the coverslips in 90 mm glass petri dish.
-
5.
Store and use the coverslips in 3 months.
Coating of coverslips with Poly-D-Lysine (PDL) in a tissue culture hood
-
6.
Add coverslips into 24-well cell culture plate, one coverslip per well.
-
7.
Coat coverslips with 300 μL PDL solution (0.1 mg/mL) for at least 2 h before culturing of primary neurons. Proceed to step 15 during trypsin treatment of hippocampal cells.
Preparation of surgical instruments
Sterilize surgical instruments (Dissection tools were purchased from manufacturers in China. Equivalents can be found at https://www.dumonttweezers.com/) by ultraviolet light irradiation or soaking in 75% ethanol for more than 20 min and air dry in a tissue culture hood before use (Figure 1A).
Dissection of mouse hippocampi
-
8.
Add 15 mL dissection buffer to a sterile 100 mm cell culture petri dish for dissection and 1 mL dissection buffer to a 1.5 mL sterile Eppendorf tube for collection of hippocampi. Keep the 1.5 mL sterile Eppendorf tube with dissection buffer on ice.
-
9.
Sanitize P0 C57BL/6J mouse by quick immersion in 75% ethanol, transfer the pup to a sterile petri dish, and decapitate mouse quickly with sharp straight delicate scissors (Figure 1B).
-
10.
Fix the head with straight ophthalmic forceps, cut the skull along the medulla oblongata with mayo spring scissors, dissect out mouse brain and transfer to precooled dissection buffer in 100 mm petri dish with a small spatula.
-
11.
Remove the olfactory bulb, epencephalon and brainstem, thoroughly strip away meninges under a dissecting microscope (stereomicroscope) with a pair of fine smooth forceps (Figure 1C). For more detailed description of brain tissue preparation, please refer to Güler et al. (Güler et al., 2021).
-
12.
Reverse the brain, fix it gently on the periphery of the cortex with fine forceps, cut off one hippocampus from the cortex, clear up the meninges and remove extra tissue from the hippocampus with fine smooth forceps (Figure 1D). Isolate the other hippocampus from the contralateral side of the cortex.
-
13.
Transfer hippocampus to the precooled dissection buffer in 1.5 mL sterile Eppendorf tube, keep the tube on ice until all hippocampi are collected. Put no more than 6 hippocampi in one tube. Transfer to a 15 mL conical centrifuge tube for later steps if more than 6 hippocampi are collected.
Dissociation and plating of hippocampal neurons
-
14.
Remove the dissection buffer, add 0.25% trypsin (# hippocampi ≤ 6: 1.5 mL, ˃ 6 : 2–5 mL) and keep the tube in a humidified incubator (5% CO2, 37°C) for 15–17 min, gently reverse the centrifuge tube 3–4 times every 5 min (Figure 1E).
-
15.
During trypsinization of hippocampal tissue, aspirate off PDL from coverslips, add 500 μL autoclaved ddH2O to wells, shake the plate gently and keep coverslips in water for 5 min or shorter (Figure 1F).
-
16.
Repeat step 15 at least twice.
-
17.
Aspirate off water, position coverslips in the center of wells, and let them air dry for less than 10 min in the hood, then keep the plate in a humidified incubator (Figure 1F).
-
18.
Remove 0.25% trypsin, rinse the hippocampi with equal volume of DMEM culture medium by reversing the tube 6–8 times gently and let stand for 3 min at 21°C–25°C or until the hippocampi settle at the bottom (Figure 1E).
-
19.
Remove the culture medium and repeat rinsing of hippocampi twice.
-
20.
Add 0.5 mL DMEM culture medium, transfer hippocampi to a fresh 1.5 mL Eppendorf tube, and gently triturate the hippocampal tissue 8 times with P1000 pipette. Let the tube stand still for ∼ 3 min, transfer the supernatant (dissociated cells in suspension) to a fresh 1.5 mL Eppendorf tube.
-
21.
Add 0.5 mL DMEM culture medium to the pellet and repeat gentle trituration with P1000 pipette for 8 times to dissociate more cells. Let the tube stand still for ∼ 3 min, collect the supernatant and discard the pellet.
-
22.
Combine the supernatants (cell suspensions) collected from steps 20 and 21.
-
23.
Count the number of cells in suspension with a hemocytometer.
Note: If the cell suspension is too diluted, concentrate by centrifugation at 300×g for 10 min, remove the supernatant and resuspend cells with appropriate volume of DMEM culture medium.
-
24.
Add hippocampal neurons in 60–70 μL DMEM culture medium on air-dried PDL-coated coverslip carefully at a density of 2–3×104 cells/well, keep the tissue culture plate in humidified incubator (5% CO2, 37°C) for 5–10 min. Then add 450 μL DMEM culture medium into each well, incubate in humidified incubator (5% CO2, 37°C) for 4 h (Figure 1G).
-
25.
Replace DMEM with 500 μL Neurobasal-A culture medium pre-warmed to 37°C.
-
26.
Replenish the dissociated culture by adding 500 μL of fresh Neurobasal-A culture medium to each well on Day 3 in vitro (DIV3) and every three days afterward.
Preparation two (optional): Transient transfection to express green or red fluorescent protein as morphological marker, or overexpress/silence gene of interest
This section describes transfection of mouse hippocampal neurons in dissociated culture with the Lipofectamine® LTX & PLUS™ Reagents (http://tools.thermofisher.com/content/sfs/manuals/LipofectamineLTX_PLUS_Reag_protocol.pdf).
-
27.
Dilute 0.5 μg DNA in 50 μL Neurobasal-A Medium, then add 0.5 μL PLUS™ Reagent and mix gently by pipetting up and down. Incubate for 5 min at 21°C–25°C.
-
28.
Dilute 1 μL Lipofectamine® LTX Reagent in 50 μL Neurobasal-A Medium and mix gently. Incubate for 5 min at 21°C–25°C.
-
29.
Add the mixture of diluted DNA and PLUS™ Reagent to diluted Lipofectamine® LTX Reagent and mix gently. Incubate for 20 min at 21°C–25°C.
-
30.
Replace the culture medium in each well of the 24-well culture plate with 400 μL Neurobasal-A Medium (pre-warmed to 37°C). Save the culture medium in a new centrifuge tube for later use. To achieve high transfection efficiency, the transfection is usually performed on DIV 3–12.
-
31.
Add the DNA-lipid complex to neurons cultured in 24-well culture plate drop by drop. Shake the plate gently back and forth to uniformly disperse the DNA-lipid complex.
-
32.
Incubate neurons for 50–60 min at 37°C, and replace the DNA-lipid complex with culture medium (mixture of old and fresh Neurobasal-A culture medium, and the fresh medium accounts for 1/3–1/2 of the total volume).
-
33.
Incubate neurons at 37°C in a CO2 incubator for 72 h or longer to express mCherry in neurons as cell fill (morphological marker).
Intracellular PIP staining of primary cultured mouse hippocampal neurons
For intracellular PIP staining of primary cultured mouse hippocampal neurons, all steps were performed at 21°C–25°C. This protocol was modified from a previously published protocol described by Hammond et al. (Hammond et al., 2009).
-
34.For intracellular PIP staining without any manipulation before fixation, please refer to step a. For staining with manipulation, such as inducing long-term potentiation (LTP) before fixation (Guo et al., 2022), please refer to step b.Note: Keeping growth medium at the fixation step can protect neuronal cells from shrinking caused by hypo-osmotic shock. Likewise, 4% sucrose is added to the fixing solution to maintain osmolality when neurons are incubated in medium-free buffer for drug treatments such as LTP induction. There is no apparent difference between the two fixation methods in terms of final output.
-
a.Fix neurons in 200 μL medium by adding 200 μL PBS containing 4% FA (pre-warmed to 21°C–25°C) along the sidewall of the culture plate to a final concentration of 2%, and incubate for 15 min at 21°C–25°C.Note: Compared with FA, GA is a much stronger fixative that irreversibly crosslinks proteins. GA preserves membrane effectively but may alter protein configuration to mask target epitopes, and prevents penetration of antibodies into the cell interior by crosslinking proteins in the cytoplasm, so it is only used for immunostaining of PM-distributed antigens. For immunostaining of intracellular PIPs and proteins, mild fixation of cells with 2% FA allows accession of antibodies to epitopes located at the cytosolic side of endomembranes.
-
b.Remove buffer or medium and fix neurons rapidly by adding 400 μL PBS containing 2% FA and 4% sucrose (pre-warmed to 21°C–25°C) along the sidewall of the culture plate and incubate for 15 min at 21°C–25°C.
-
a.
-
35.
Rinse neurons three times with PBS containing 50 mM NH4Cl, 5 min each.
-
36.
Permeabilize neurons with buffer A containing 25 μg/mL digitonin for 5–10 min.
Note: Digitonin is a weak nonionic detergent, which at low concentrations selectively renders the plasma membrane permeable but leaves intracellular organelles intact, so it is used for immunodetection of antigens located at the cytosolic side of endomemebranes.
-
37.
Rinse neurons immediately for three times with buffer A, 5 min each.
-
38.
Block neurons with buffer A containing 5% (v/v) NGS and 50 mM NH4Cl for 45 min.
-
39.
Rinse neurons once with buffer A.
-
40.
Incubate neurons with primary antibodies (PI3P, Z-P003, Echelon, 1:50, PI4P, Z-P004, Echelon, 1:200, or PI(4,5)P2, Z-A045, Echelon, 1:150 and Rab8, R5530, Sigma-Aldrich, 1:150 or TGN46, ab50595, Abcam, 1:200) in buffer A containing 5% NGS for 1 h.
-
41.
Rinse neurons for three times with buffer A, 5 min each.
-
42.
Incubate neurons with secondary antibodies (Alexa Fluor 488 goat anti-mouse, IgG, A-11001, 1:1500 or Alexa Fluor 488 goat anti-mouse, IgM, A-21042, 1:1500 and Alexa Fluor 647 donkey anti-rabbit, IgG, A-31573, 1:1500) in buffer A containing 5% NGS for 45 min away from light. Do keep neurons away from light for the rest of the procedure.
-
43.
Rinse neurons for three times with buffer A, 5 min each.
-
44.
Postfix neurons with 2% FA in PBS for 5 min.
-
45.
Rinse neurons for three times with PBS containing 50 mM NH4Cl to remove the residual FA.
-
46.
Incubate neurons with PBS containing DAPI for 5 min.
-
47.
Mount coverslips on glass slide with mounting medium after one rinse with PBS and then with distilled ddH2O.
Using the procedures described above, we performed costaining of intracellular PIP (PI3P, PI4P or PI(4,5)P2) and Rab8 (protein marker for secretory endosome) or TGN46 (protein marker for the trans-Golgi network) in neurons. The intracellular PIPs and Rab8/TGN46 were detected easily with the intracellular staining procedure (Figure 2). We can see in Figure 2 that the distribution of PI3P, PI4P and PI(4,5)P2 in neurons are different. Overall, both the cell body and dendrites are enriched in PI3P and PI4P, whereas PI(4,5)P2 signals in the cell body are scarce. Different PIPs show differential distribution indicated by their extent of colocalization with Rab8 and TGN46. Specifically, colocalization of PI4P with Rab8 is slightly higher than PI3P and PI(4,5)P2 (Figures 2A–2C, 5I, and 5J).
Figure 2.

Immunofluorescence staining of intracellular PIPs and Rab8/TGN46 in mouse hippocampal neurons
Mouse hippocampal neurons were fixed on DIV12 and co-immnunostained for intracellular PI3P, PI4P or PI(4,5)P2 and Rab8 or TGN46. DAPI-stained cell nuclei are shown in blue.
(A–F) Shown are representative confocal microscopy images of intracellular PI3P and Rab8 (A), intracellular PI4P and Rab8 (B), intracellular PI(4,5)P2 and Rab8 (C), intracellular PI3P and TGN46 (D), intracellular PI4P and TGN46 (E), intracellular PI(4,5)P2 and TGN46 (F). Rab8: protein marker for secretory endosomes, TGN46: protein marker for the trans-Golgi network. Lower panels are magnifications of outlined regions in the top panels. Scale bars: 5 μm.
Figure 5.

Colocalization analysis of PIPs and Rab8 of dendrites
(A) Open an image with the NIS-Elements software.
(B) Open LUTs and amplify fluorescence signal of PI4P to overexposed to identify the background region within the dendrite.
(C) Draw ROI on one background regions within the dendrite, and view the intensity of PI4P within ROI.
(D) Set appropriate values to subtract image intensity.
(E) Subtract the intensity of PI4P within ROI to zero.
(F) Subtract the intensity of Rab8 within ROI to zero.
(G) Draw ROI of the dendrite and analyze the colocalization of PI4P and Rab8.
(H) Colocalization parameters from (G).
(I) Quantification of colocalization between PI3P and Rab8 (Mander’s overlap coefficient). n = 10, 19 dendrite fragments. Data represent mean ± SEM.
(J) Quantification of colocalization between PI4P and Rab8 (Mander’s overlap coefficient). n = 10, 19 dendrite fragments. Data represent mean ± SEM.
Plasma membrane (PM) PIP staining of primary cultured mouse hippocampal neurons
For PM PIP staining of primary cultured mouse hippocampal neurons, all steps were performed on ice with all buffers and solutions pre-chilled after removing the fixing reagent with rinsing buffer. This protocol was also modified from a previously published protocol described by Hammond et al. (Hammond et al., 2009).
-
48.For PM PIP staining without any manipulation before fixation, please refer to step a. For staining with manipulation, such as inducing LTP in neurons before fixation (Guo et al., 2022), please refer to step b.
-
a.Fix neurons rapidly in 200 μL medium by adding 200 μL PBS containing 8% FA and 0.4% GA (pre-warmed to 21°C–25°C) along the sidewall of the culture plate to a final concentration of 4% FA and 0.2% GA for 15 min at 21°C–25°C.
-
b.Remove buffer or medium and fix neurons rapidly with 400 μL PBS containing 4% FA, 0.2% GA and 4% sucrose (pre-warmed to 21°C–25°C) along the sidewall of the culture plate for 15 min at 21°C–25°C.
-
a.
-
49.
Rinse neurons three times with PBS containing 50 mM NH4Cl at 21°C–25°C, 5 min each.
-
50.
Place coverslips in ice bath for 4 min to pre-chill samples before blocking.
-
51.
Block neurons with buffer A containing 5% (v/v) NGS, 50 mM NH4Cl and 0.5% saponin for 45 min.
Note: Saponin can punch holes in the cell membrane by specifically dissolving cholesterol in the cell membrane, thus allowing access of antibodies to antigens located at the cytoplasmic leaflet of the plasma membrane. Because saponin-mediated cell permeabilization is reversible, it is critical to keep the cells in the presence of low concentration saponin (0.1%) during PM immunostaining.
-
52.
Rinse neurons once with buffer A.
-
53.
Incubate neurons with primary antibodies (PI3P, Z-P003, Echelon, 1:50, PI4P, Z-P004, Echelon, 1:100, or PI(4,5)P2, Z-A045, Echelon, 1:50 and endophilin A1, 159002, Synaptic Systems, 1:100) in buffer A containing 5% NGS and 0.1% saponin for 1 h.
-
54.
Rinse neurons twice with buffer A, 5 min each.
-
55.
Incubate neurons with secondary antibodies (Alexa Fluor 488 goat anti-mouse, IgG, A-11001, 1:1500 or Alexa Fluor 488 goat anti-mouse, IgM, A-21042, 1:1500 and Alexa Fluor 647 donkey anti-rabbit, IgG, A-31573, 1:1500) in buffer A containing 5% NGS and 0.1% saponin for 45 min away from light. Do keep neurons away from light for the rest of the procedure.
-
56.
Rinse neurons for three times with buffer A, 5 min each.
-
57.
Postfix neurons with 2% FA in PBS on ice for 10 min before warming to 21°C–25°C for an additional 5 min.
-
58.
Rinse neurons for three times with PBS containing 50 mM NH4Cl to remove the residual FA.
-
59.
Incubate neurons with PBS containing DAPI for 5 min.
-
60.
Mount coverslips on glass slide with mounting medium after one rinse with PBS and then with distilled ddH2O.
Following the protocol described above, we performed the PM PIP (PI3P, PI4P and PI(4,5)P2) staining (Figures 3A–3C), and the costaining of PI(4,5)P2 and endophilin A1 (EEN1, a peripheral membrane protein (Yang et al., 2021) in neurons (Figure 3D). The staining results show that PI4P and PI(4,5)P2 distribute in the PM of neurons with different patterns (Figures 3B and 3C).
Figure 3.

Immunofluorescence staining of PM PIPs and EEN1 in mouse hippocampal neurons
Mouse hippocampal neurons were transfected with pAOV-CaMKIIα-mCherry-2A-3Flag on DIV5 to express mCherry as volume marker (Note: the CaMKIIα promoter drives gene expression specifically in excitatory neurons but not inhibitory neurons) (Kohara et al., 2020), fixed on DIV8 and immnunostained for PM PI3P, PI4P or PI(4,5)P2. DAPI-stained cell nuclei are shown in blue.
(A–D) Shown are representative confocal microscopy images of PM PI3P (A), PM PI4P (B), PM PI(4,5)P2 (C), and costaining of PM PI(4,5)P2 and PM EEN1 (D). Lower panels are magnifications of boxed regions in the top panels. Scale bars: 5 μm.
Of note, consistent with previous studies, PI3P is not detected in the PM of neurons (Figure 3A). Furthermore, using PI(4,5)P2 as a control for antigens located in the cytoplasmic leaflet of the PM, PM-associated endophilin A1 was labeled successfully (Figure 3D) (Yang et al., 2021), demonstrating the applicability of the PM staining method to proteins localized at the inner leaflet of the PM.
Quantitative analysis of fluorescent images
To measure the signal intensity of PIPs or proteins in neurons, we used different methods to quantitatively analyze confocal images of intracellular and PM PIP staining. First, we analyzed the intensity of intracellular PIPs or Rabs (for example, intracellular PI3P and Rab8 in dendrites of neurons expressing mCherry (Figure 4)) with the Fiji-ImageJ software (NIH).
-
61.
Open an image with the Fiji-ImageJ software (view stack with: Hyperstack; color mode: Composite; Memory management: Use virtual stack) (Figure 4A).
-
62.
Convert image format to grayscale images (Image / Type / 16-bit) (Figure 4B).
-
63.
Adjust Brightness/Contrast of the fluorescent signals of the volume marker (mCherry: Channel 2. Image / Color / Channels Tool, Image / Adjust / Brightness/Contrast) to view the morphology of dendrites clearly.
-
64.
Draw a region of interest (ROI) around the dendrite in the mCherry channel with Freehand Selections (Figure 4C). Open ROI Manager (Analyze / Tools / ROI Manager) and press the Add button to select the region specified (Figure 4D).
-
65.
Adjust Designate fluorescence intensity measurement using Set Measurements (Analyze / Set Measurements / Select mean gray value).
-
66.
Switch to channel 1 (PI3P) with Channels Tool (Channels Tool / Color / Channel 1) (Figure 4E), press M or click Analyze/Measure to measure fluorescence mean intensity of PI3P in the dendrite selected at step 64 (Figure 4G).
-
67.
Switch to channel 3 (Rab8) with Channels Tool (Channels Tool / Color / Channel 3) (Figure 4F), press M or click Analyze/Measure to measure fluorescence mean intensity of Rab8 in ROI (Figure 4G).
-
68.
Transfer the data to Excel, analyze the mean intensity of PI3P or Rab8 of more than 10 neurons. Perform statistical analysis (t-tests or One-way ANOVA) using the GraphPad Prism software.
Figure 4.

Quantitative analysis of intracellular PIPs or Rab signals in dendrites
(A) Open an image with the Fiji-ImageJ software.
(B) Convert image format to 16-bit.
(C) Draw ROI around the dendrite outlined by adjusting the Brightness/Contrast of the fluorescent signals of mCherry the volume marker.
(D) Record ROI from Step C with the ROI Manager.
(E) Measure mean intensity of PI3P in ROI.
(F) Measure mean intensity of Rab8 in ROI.
(G) Record mean intensity of PI3P or Rab8 signals in ROI.
From the above results, we can see that intracellular PIP staining method can be employed to investigate distribution of PIPs in protein marker-labeled organelles or vesicular structures. Next, we performed colocalization analysis of PIPs and Rab proteins with the NIS-Elements AR software (Nikon, Tokyo, Japan) (alternatively, use Fiji-ImageJ), for example, the colocalization between PI4P and the Rab8-positive secretory endosome (Figure 5).
-
69.
Open an image (PI4P-Rab8 co-staining of the dendrite) with the NIS-Elements software (Figure 5A).
-
70.
Select one channel, e.g., the PI4P channel. Open LUTs (View / View LUTs), and amplify fluorescence signal to overexposed to identify the background region within the dendrite (Figure 5B).
-
71.
Draw ROI on one or more of the background regions within the dendrite, and open Automated Measurement Results (View / Analysis Controls / Automated Measurement Results) to view the intensity of PI4P within ROI. (Figure 5C).
-
72.
Open Image Intensity (Image / Adjust Image / Intensity Transformation) and subtract the intensity of ROI to zero to clear background (Figures 5D and 5E).
-
73.
Perform the same manipulation (steps 70–72) for the Rab8 channel and subtract the intensity of ROI to zero in the same way (Figure 5F).
-
74.
Draw ROI on the entire dendrite and open Colocalization (View / Analysis Controls / Colocalization) to view the results of colocalization analysis (Figures 5G and 5H).
-
75.
Export data to Excel, analyze the mean value of Mander’s overlap coefficient of PI4P and Rab8 from 10 neurons (Figures 5I and 5J).
To investigate the distribution of PIPs in the PM, we analyzed the signal intensity of PM PIPs (for example, PM PI4P in dendrite (Figure 6)) with the NIS-Elements AR software (Nikon, Tokyo, Japan). Quantitative analysis of signal intensity in the PM can also be done with Fiji-ImageJ (NIH).
-
76.
Open an image with the NIS-Elements software, perform analysis on the 515/30 channel (PI4P signals) (Figure 6A).
-
77.
Open LUTs (View / View LUTs), amplify fluorescence signal to overexposed so that we can identify the background area within dendrite.
-
78.
Select a region within dendrite where there is no positive signal using the “area” tool (View / Analysis Controls / Annotations and Measurements / area), area and mean fluorescence intensity of the region will appear in the table below (Figure 6B).
-
79.
Subtract background using constant to ensure mean intensity of the selected background region < 3. (Image / Background / Subtract Background Using Constant) (Figure 6B).
-
80.
Adjust designate fluorescence intensity measurement using Object Features (Measure / Object Features), add or remove features for measure as acquired. MeanGreen is selected to measure fluorescence intensity of PM PI4P.
-
81.
Open Automated Measurement Results (View / Analysis Controls / Automated Measurement Results), select ‘Current ROI’ and click ‘Keep updating measurement ON’.
-
82.
Draw ROI around the PM of a dendritic segment. (Turn ROI On/Off / Draw Polygonal ROI) (Figure 6C).
-
83.
Export data generated from “Automated Measurement Results” to Excel (Figure 6D), analyze mean intensity of PM PI4P from more than 10 neurons. Perform statistical analysis (t-tests or One-way ANOVA) using the GraphPad Prism software.
Figure 6.

Steps of PM PIP analysis routine
(A) Open an image with the NIS-Elements AR software.
(B) Subtract background using constant.
(C) Draw ROI around plasma membrane of a dendritic segment.
(D) Export data to Excel for further analysis.
Expected outcomes
Immunofluorescence staining of PIPs was modified from a previously published protocol developed by Hammond et al. (Hammond et al., 2009). Hammond et al. performed immunofluorescence staining of PIPs in different cell lines, such as CHO (Chinese-hamster ovary), HEK (human embryonic kidney cells)-293, and HeLa cells, etc. Our protocols are applied to mammalian hippocampal neurons from primary culture with modifications to preserve the delicate morphology of neurites and enhance accessibility of antibodies. Compared with TritonX-100, digitonin and saponin are mild detergents and can specifically remove cholesterol in lipid bilayers, allowing gentle permeabilization of cellular membranes while maintaining their structural integrity to preserve lipid or protein antigens better. Notably, we costained PIPs (PI3P, PI4P and PI(4,5)P2) with protein markers for intracellular structures (Figure 2), which facilitated study of the subcellular localization and function of PIPs (Guo et al., 2022), and avoided the artifact and toxicity of lipid probe expression that might interfere with cellular physiology (Wills et al., 2018). Moreover, using the protocols described above, we not only analyzed the distribution of PI4P in various organelle membranes, but also monitored dynamic changes in the levels and subcellular distribution of PI4P and PI(4,5)P2 in dendrites of rodent hippocampal neurons during LTP induction and expression, which was not detected by lipidomic analysis of whole cells (Guo et al., 2022). Further, we applied the PM staining protocol to investigate membrane-associated proteins, and found that endophilin A1 and PI4KIIIα redistribute from the cytoplasm to the plasma membrane in dendrites of primary mouse hippocampal neurons undergoing LTP (Guo et al., 2022; Yang et al., 2021). Our studies demonstrated that combined with high-resolution microscopy and quantification of fluorescent images (Figures 4, 5, and 6), immunostaining of PIPs and proteins localized to the cytosolic side of cellular membranes provides a fast and cost-effective method to detect and analyze their dynamics in CNS neurons at the subcellular level.
Limitations
As we have shown in this paper, the intracellular and PM lipid staining methods work well to detect PIPs in the cytoplasm and PM of CNS neurons, respectively. The protocols can also be employed to detect membrane-localized protein in cells. Unfortunately, detection of PM and intracellular PIPs in the same cell does not work with the sequential application of the two protocols. We have tried different concentrations (0.2%, 0.1%, 0.05%, 0.025%, 0%) of glutaraldehyde (GA, a strong fixative to stabilize membranes) for immunostaining and the results showed that the PM PIP signals were lost without GA and the intracellular PIPs cannot be detected even with the lowest concentration of GA (0.025%) tested. In addition, our attempts to stain PM PIPs in mature neurons expressing volume marker (GFP or mCherry) failed for unknown reasons (Guo et al., 2022). Therefore, we recommend PM staining of non-transfected mature neurons (DIV14-18) and developing neurons (DIV3-9) transiently transfected with fluorescent protein as cell fill.
Troubleshooting
Problem 1
After triturating the hippocampi with 1 mL pipette tip, only a small amount of cells dissociated from the brain tissue (steps 20 and 21).
Potential solution
Poor digestion leads to pieces of tissue remaining after triturating. The activity of 0.25% Trypsin is unstable and declines over time. To keep the enzyme activity, we store 0.25% Trypsin at -20°C for short-term storage and at -80°C for long-term storage. Thaw 0.25% Trypsin just before use. Another possible reason for inadequate digestion is the residual cortex around the hippocampus after dissection. For high quality culture, remove extra brain tissue from the hippocampus.
Problem 2
Neurons die within several hours after DMEM is replaced with the Neurobasal-A culture medium (step 25).
Potential solution
The B-27 supplement is crucial for neuron growth and survival. Make sure B-27 is properly stored (at -20°C, avoid repeated freeze/thaw) and not expired.
Problem 3
The cell bodies of several neurons start to aggregate on DIV1-3, and the neurites are curly. (step 26).
Potential solution
Aggregation of cell bodies is an indication that cells are not healthy. It is imperative to thoroughly remove cleaning solution by intensive rinses with ddH2O when preparing coverslips. The frequency and strength of triturating is very important, as excessive trituration can lead to cell damage and even death. Do perform the trituration gently. Avoid generation of air bubbles during trituration and avoid repeated trituration for individual cells that have been dissociated.
Problem 4
There are many glial cells in neuronal culture, which interfere with immunolabeling of intracellular PIP in neurons (step 26).
Potential solution
Too many glia cells would interfere with immunostaining of intracellular PIP in neurons. FBS is beneficial to the proliferation of glial cells, so the incubation time for DMEM culture medium should be no longer than 4 h after plating and the Neurobasal-A culture medium is not supplemented with FBS.
Problem 5
Neurons grown on some areas of the coverslip die after transfection for one day or more (step 33).
Potential solution
The possible reason may be the DNA-lipid complex is not evenly distributed in the well, which leads to local concentration of transfection reagents and hence cytotoxicity or even cell death. Add DNA-lipid complex drop-by-drop with 200 μL pipette tip to multiple sites in each well, and avoid adding the DNA-lipid complex alongside the well wall. Then shake the plate gently back and forth to disperse the DNA-lipid complex.
Problem 6
For intracellular PIP staining, no signal or nonspecific signals in cells (steps 34–47).
Potential solution
For intracellular PIP staining, cell density should not be too high to achieve thorough fixation. Neurons should be plated on coverslips in 24-well plates at a density of ≤3×104 cells/well (step 24).
Problem 7
For intracellular PIP staining, the fluorescent signals are weak or unevenly distributed, especially in the cell body there is no signal or signals are confined to some areas (steps 34–47).
Potential solution
Incubation of cells with detergent is key to successful staining. Buffer A containing digitonin should be evenly distributed to ensure efficient permeabilization of cultured neurons. We generally use digitonin at 25 μg/mL to permeabilize neurons for 10 min at 21°C–25°C, but for different batches of digitonin, the time needed for membrane permeabilization should be tested within the range of 5–10 min (step 36).
Problem 8
The PM PIP fluorescent signals are not distributed throughout the dendrites (steps 48–60).
Potential solution
The PM PIP staining protocol is quite reliable. We occasionally encountered the problem that the PM PIP fluorescent signals were not distributed throughout the dendrites. We checked the procedure and reagents, and found that the glutaraldehyde we used was expired. To avoid the problem, we only use it within one month once the vial is opened and store it at 4°C (step 48).
Problem 9
High background fluorescence after PIP staining due to non-specific staining (steps 40, 42, 53 and 55).
Potential solution
Keep coverslips in humidity chambers while incubating with antibodies, and avoid prolonged exposure to the air. Place the coverslips face down on parafilm on top of two layers of wet filter paper in a humidity chamber. Spin down aggregates from secondary antibodies to the bottom of the tube before use.
Problem 10
Immunostaining fails with routine manipulation.
Potential solution
One of the probable reasons is antibody inactivation due to inappropriate storage. Positive control is recommended to confirm the activity of the antibody. Antibodies are stored at 2°C–8°C for up to 30 days, −20°C for greater than 30 days. Do not freeze thaw multiple times.
Problem 11
The PIP staining signal becomes weaker over time.
Potential solution
For PIP staining, slides should be stored at 4°C for up to 1 week, and −20°C for 1–2 month away from light. Collect data as soon as possible to avoid gradual decay of fluorescent signals.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, [Jia-Jia Liu (jjliu@genetics.ac.cn)].
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by funding from the National Natural Science Foundation of China (31530039, 31921002, 91954126, and 32070785) and the State Key Laboratory of Molecular Developmental Biology (2022-MDB-KF-13).
Author contributions
Z.G., C.T., and Y.Y. adapted and optimized the protocols. Z.G., C.T., Y.Y., and J.-J.L. wrote the manuscript. Z.G. and C.T. performed and analyzed the experiments. J.-J.L. revised and edited the manuscript. J.-J.L. supervised the project.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate new datasets.
References
- Güler B.E., Krzysko J., Wolfrum U. Isolation and culturing of primary mouse astrocytes for the analysis of focal adhesion dynamics. STAR Protoc. 2021;2:100954. doi: 10.1016/j.xpro.2021.100954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Jiang C.-H., Tong C., Yang Y., Wang Z., Lam S.M., Wang D., Li R., Shui G., Shi Y.S., Liu J.J. Activity-dependent PI4P synthesis by PI4KIIIα regulates long-term synaptic potentiation. Cell Rep. 2022;38:110452. doi: 10.1016/j.celrep.2022.110452. [DOI] [PubMed] [Google Scholar]
- Hammond G.R., Schiavo G., Irvine R.F. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4, 5)P(2) Biochem. J. 2009;422:23–35. doi: 10.1042/bj20090428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K., Inoue A., Nakano Y., Hirai H., Kobayashi T., Maruyama M., Baba R., Kawashima C. BATTLE: genetically engineered strategies for split-tunable allocation of multiple transgenes in the nervous system. iScience. 2020;23:101248. doi: 10.1016/j.isci.2020.101248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills R.C., Goulden B.D., Hammond G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell. 2018;29:1526–1532. doi: 10.1091/mbc.e17-12-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.R., Chen J., Chen X., Li D., He J.F., Wang S., Zhao S., Yang X.Y., Deng S.K., Tong C.F., et al. Endophilin A1 drives acute structural plasticity of dendritic spines in response to Ca2+/calmodulin. J. Cell Biol. 2021;220:e202007172. doi: 10.1083/jcb.202007172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new datasets.