

Abstract
Advances in whole-genome sequencing (WGS) promise to enable the accurate and comprehensive structural variant (SV) discovery. Dissecting SVs from WGS data presents a substantial number of challenges and a plethora of SV detection methods have been developed. Currently, evidence that investigators can use to select appropriate SV detection tools is lacking. In this article, we have evaluated the performance of SV detection tools on mouse and human WGS data using a comprehensive polymerase chain reaction-confirmed gold standard set of SVs and the genome-in-a-bottle variant set, respectively. In contrast to the previous benchmarking studies, our gold standard dataset included a complete set of SVs allowing us to report both precision and sensitivity rates of the SV detection methods. Our study investigates the ability of the methods to detect deletions, thus providing an optimistic estimate of SV detection performance as the SV detection methods that fail to detect deletions are likely to miss more complex SVs. We found that SV detection tools varied widely in their performance, with several methods providing a good balance between sensitivity and precision. Additionally, we have determined the SV callers best suited for low- and ultralow-pass sequencing data as well as for different deletion length categories.
Keywords: Variant calling, Structural Variant, Bioinformatics
Introduction
Structural variants (SVs) are genomic regions that contain an altered DNA sequence due to deletion, duplication, insertion, or inversion [1]. SVs are present in approximately 1.5% of the human genome [1, 2], but this small subset of genetic variations has been implicated in the pathogenesis of psoriasis [3], Crohn’s disease [4] and other autoimmune disorders [5], autism spectrum and other neurodevelopmental disorders [6–9] and schizophrenia [10–13]. Specialized computational methods—often referred to as SV callers—are capable of detecting SVs directly from sequencing data. At present, although several benchmarking studies have been previously carried out [14, 15, 16], our study is the first to utilize a complete polymerase chain reaction (PCR)-validated gold standard with respect to the alignment file. We benchmarked currently available whole-genome sequencing (WGS)-based SV callers to determine the efficacy of available tools and find methods with a good balance between sensitivity and precision.
The lack of comprehensive benchmarking makes it impossible to adequately compare the performance of SV callers. In the absence of benchmarking, biomedical studies rely on the consensus of several SV callers [17, 18]. In order to compare SV callers given the current lack of a comprehensive gold standard dataset, a recent study [19] used long read technologies to define a ground truth in order to evaluate a large number of currently available tools [20, 21]. However, a comprehensive gold standard dataset is still needed; current long read technologies are prone to producing high error rates, which confounds efforts to detect SVs at single-base pair resolution. In response to the pressing need for a comprehensive gold standard dataset, our article presents a rigorous assessment of the sensitivity and precision of SV detection tools when applied to both mouse and human WGS data.
Results
Preparing the mouse gold standard data and WGS data
Over the last decade, a plethora of SV detection methods have been developed (Table 1 and Supplemental Table S1), but the relative performance of these tools is unknown [22–28]. In order to assess the precision and accuracy of the currently available SV callers, we simplified the problem presented to the SV callers using a set of homozygous deletions present in inbred mouse chromosomes. More specifically, we chose to limit our analysis to mouse chr19 as it is the smallest. We used a PCR-validated set of deletions, in which the mouse deletions were manually curated, and targeted PCR amplification of the breakpoints and sequencing were used to resolve the ends of each deletion to the base pair [45, 46]. The same read alignment file which was used for the manual curation of the deletions was used as an input to the SV callers, making our gold standard complete and containing all possible true deletions [true positives (TPs)]. To ensure that our gold standard is complete with respect to the alignment file, we first examined all possible deletions manually, and then validated each deletion by PCR. Thus, although our gold standard may not be universally complete, it was complete with respect to the alignment files which were provided to the SV callers as all deletions which could be possibly detected from the alignment were recorded and further examined using PCR. Details about the preparation of the gold standard are provided in the supplementary material.
Table 1.
Overview of SV-detection methods included in this study
| Software tool | Version | Under lying algorithm | Published year | Tool’s webpage | Bioconda version | Format |
|---|---|---|---|---|---|---|
| GASV [29] | 1.4 | RP | 2009 | http://compbio.cs.brown.edu/projects/GASV/ | No | Custom |
| Pindel [30] | 0.2. 5b9 | RP + S R SR (2015, 0.2.5 b9) | 2009 | http://gmt.genome.wustl.edu/packages/pindel/ | Yes | Custom |
| RDXplorer [31] | 3.2 | RD | 2009 | http://RDXplorer.sourceforge.net/ | No | Custom |
| CLEVER [32] | 2.4 | RP | 2011 | https://bitbucket.org/tobiasmarschall/CLEVER-toolkit/wiki/Home | Yes | Custom |
| DELLY [33] | 0.8.2 | RP + SR | 2012 | https://github.com/DELLYtools/DELLY | Yes | Custom |
| BreakDancer [34] | 1.3.6 | RP | 2012 | https://github.com/genome/BreakDancer | Yes | Custom |
| indelMINER [35] | N/A | RP + SR | 2014 | https://github.com/aakrosh/indelMINER | No | VCF |
| GRIDSS [30] | 2.5.1 | RP + SR | 2015 | https://github.com/PapenfussLab/GRIDSS | Yes | VCF |
| MiStrVar [4] | N/A | N/A | 2015 | https://bitbucket.org/compbio/MiStrVar | No | VCF |
| LUMPY [36] | 0.2.4 | RP, SR, RD | 2016 | https://github.com/brentp/smoove | Yes | VCF |
| PopDel [37] | 1.1.3 | RP | 2017 | https://github.com/kehrlab/PopDel | Yes | VCF |
| CREST [38] | 1.0 | SR | 2017 | https://www.stjuderesearch.org/site/lab/zhang | No | Custom |
| Manta [39] | 1.6.0 | SR | 2017 | https://github.com/Illumina/manta | Yes | VCF |
| Genome STRiP [40] | 2.0 | RP+SR+RD | 2017 | http://software.broadinstitute.org/software/genomestrip/ | Yes | VCF |
| Ocotopus [41] | 0.7.4 | SR | 2018 | https://luntergroup.github.io/octopus/ | Yes | VCF |
| Deep Variant [42] | 1.2.0 | N/A | 2018 | https://github.com/google/deepvariant | Yes | VCF |
| Tardis [43] | 1.04 | RP + RD + SR | 2019 | https://github.com/BilkentCompGen/tardis | Yes | VCF |
| GROM [44] | 1.0.3 | RD | 2021 | https://osf.io/6rtws/ | No | VCF |
Surveyed SV detection methods sorted by their year of publication from 2009 to 2018 are listed along with their underlying algorithm: read-depth (RC), read-pair algorithms (RP), split-read approaches (SR), discordant pairs (DP) or a combination of algorithms. We documented the version of the software tool used in the study (‘Version’), the year the software tool was published (‘Published year’), the webpage where each SV detection method is hosted (‘Tool’s webpage’) and whether or not the Bioconda package of the software was available (‘Bioconda version’), Geometric Analysis of Structural Variants (GASV), clique-enumerating variant finder (CLEVER), Clipping REveals STructure (CREST), Genome Rearrangement OmniMapper (GROM), Variant caller format (VCF).
The set of deletions we used among seven inbred strains, called with reference to C57BL/6J, is shown in Figure 1A and listed in Supplemental Table S2 [45]. We filtered out deletions shorter than 50 bp as such genomic events are usually detected by indel callers rather than SV callers. In total, we obtained 3710 deletions with lengths ranging from 50 to 239 572 base pairs (Supplemental Figure S1 and Table S2). Almost half of the deletions were in the range of 100–500 bp. Almost 30% of deletions were larger than 1000 bp (Supplemental Figure S1). High-coverage sequence data were used as an input to the SV callers in the form of aligned reads. Reads were mapped to the mouse genome (GRCm38 Mouse Build) using BWA with the - a option. In total, we obtained 5.2 billion 2 × 100 bp paired-end reads across seven mouse strains. The average depth of coverage was 50.75× (Supplemental Table S3). Details regarding the gold standard and raw data preparation and analysis are presented in the supplementary materials.
Figure 1.

Comparison of inferred deletions across SV callers on mouse data. (A) Length distribution of molecularly confirmed deletions from chromosome 19 across seven strains of mice. (B) Number of molecularly confirmed deletions (‘true deletions’ black color) and number of deletions detected by SV callers. (C) Bar plot depicting the total number of TP calls across all error thresholds for each SV caller. (D) Scatter plot depicting the number of correctly detected deletions (TP: true positives) by the number of incorrectly detected deletions (FP: false positives) at the 100 bp threshold. Deletion is considered to be correctly predicted if the distance of right and left coordinates are within the given threshold from the coordinates of true deletion. An SV caller was considered to detect a given nondeletion if no deletions were reported in a given region.
Preparing the human gold standard data and WGS data
We used public benchmark data for the Ashkenazi Jewish Trio son (NA24385/HG002) from the genome-in-a-bottle (GIAB) consortium. The alignment files were publicly available from the GIAB website and were used as an input to the variant callers. The average depth of coverage was 45×, and the reads were 2 × 250 bp paired-end reads. We used the GIAB preliminary variant set containing deletions in HG002 as our gold standard. The preliminary HG002 deletion set available is the first reference set that is near complete within defined high-confidence regions of the genome defined by a bed file and hence allowed us to systematically benchmark the performance of variant callers within those high-confidence regions. The set contained 37 412 deletions, out of which 10 159 deletions remained after extracting the high-confidence regions. Similar to mouse data, we filtered out deletions shorter than 50 bp. Almost 30% of the deletions were in the range of 100–500 bp. Around 8% of deletions were larger than 1000 bp (Figure 6). The complete details of how the human gold standard was prepared are provided in the Methods section.
Figure 6.

Comparison of inferred deletions across SV callers on human data. (A) Length distribution of molecularly confirmed deletions from the human whole genome. (B) Number of molecularly confirmed deletions (‘true deletions’ black color) and number of deletions detected by SV callers. (C) Bar plot depicting the total number of TP calls across all error thresholds for each SV caller. (D) Scatter plot depicting the number of correctly detected deletions (TP: true positives) by the number of incorrectly detected deletions (FP: false positives) at the 100 bp threshold.
Choice of SV callers
For this benchmarking study, we selected methods capable of detecting SVs from aligned WGS reads. SV detection algorithms typically use information about the coverage profile in addition to the alignment patterns of abnormal reads. We excluded tools that were designed to detect SVs in tumor-normal samples (e.g. Patchwork [47], COpy number using Paired Samples (COPS) [48], recursive Smith-Waterman-seq (rSW-seq) [49], bic-seq [39], seqCBS [51]) and tools designed to detect only small (less than 50 bp in length) SVs (e.g. GATK [52, 53], Platypus [54], Varscan [37]). Some tools were not suitable for inclusion in our dataset as they were unable to process aligned WGS data (e.g. Magnolya [55]). Other tools were designed solely for long reads (e.g. Sniffles [56, 57]). The complete list of tools excluded from our analysis is provided in Supplemental Table S4. In total, we identified 61 suitable SV methods capable of detecting deletions from WGS data (Table 1 and Supplemental Table S1).
Our benchmarking study produced an analysis of the results generated by 15 SV detection tools for mouse data and 14 tools for human data (Table 1). We were able to internally install and run all tools. The remaining 42 tools could not be installed and were not included in this study. Supplemental Table S4 presents detailed information about the issues that prevented us from installing these software tools. Commands to install the tools and details of the installation process are provided in the supplementary materials.
Comparing the performance of SV callers on mouse WGS data
We compared the performance of 15 SV callers with respect to inferring deletions. The number of deletions detected varied from 899 (indelMINER [35]) to 82 225 (GASV [37]). In all 53% of the methods reported fewer deletions than are known to be present in the sample (Figure 1B). We allowed deviation in the coordinates of the detected deletions and compared deviations with the coordinates of the true deletions. Even at a relaxed stringency, the best method correctly detected the breakpoints of only 20% of known deletions in our curated dataset.
The majority of SV callers typically detect deletions whose coordinates differ from the correct positions by up to 100 bp. Figure 1C shows the TP rates for the SV callers at four different resolution values. The total number of false negative (FN) and false positive (FP) calls decreased with an increase in the threshold (Supplemental Figure S2). The FP rate for pindel Popdel [52] was more susceptible to changes in the threshold as compared with Pindel [30] and GASV [29]. In general, the length distribution of the detected deletions varied across tools and was substantially different from the distribution of true deletions across multiple SV detection methods (Figure 2 and Supplementary Table S2). Deletions detected by BreakDancer [34] were the closest to the true median deletion length whereas 7 out of 15 SV callers overestimated the deletion lengths (Figure 2).
Figure 2.
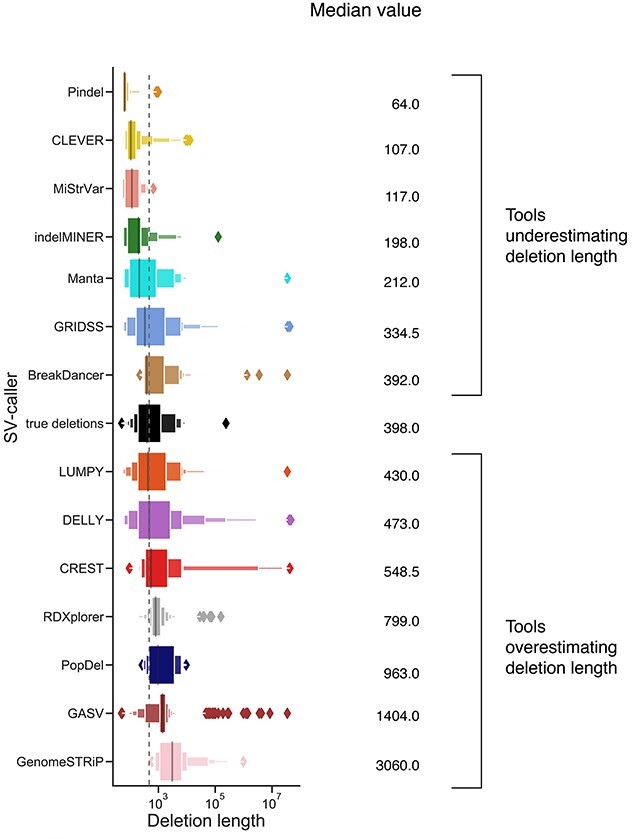
Length distribution of deletions detected by each SV caller for mouse data. True deletions are indicated in black. Tools were sorted in increasing order based on their median deletion length. The vertical dashed line corresponds to the median value of true deletions.
Increasing the resolution threshold increases the number of deletions detected by the SV callers (Figure 1C). Several methods detected all deletions in the sample at 10 000 bp resolution but overpredicted deletions leading to a precision close to zero (Figure 3B). We used the harmonic mean between precision and sensitivity (F-score) rates to determine the method with the best balance between sensitivity and precision. Several methods (e.g. Manta [39], LUMPY [36]) offered the highest F-score for deletion detection, consistently between 100 and 10 000 bp resolution across all the mouse strains (Supplemental Figure S3). For a resolution of 10 bp, the method with the best performance for all the samples was LUMPY [36] while at higher resolutions the best performing method was Manta [39](Supplemental Figure S3). The method with the best precision for a threshold of 100–1000 bp was PopDel [37], but the sensitivity rate of PopDel [37] did not exceed 50% (Figure 3A, Supplemental Figures S4 and S5).
Figure 3.
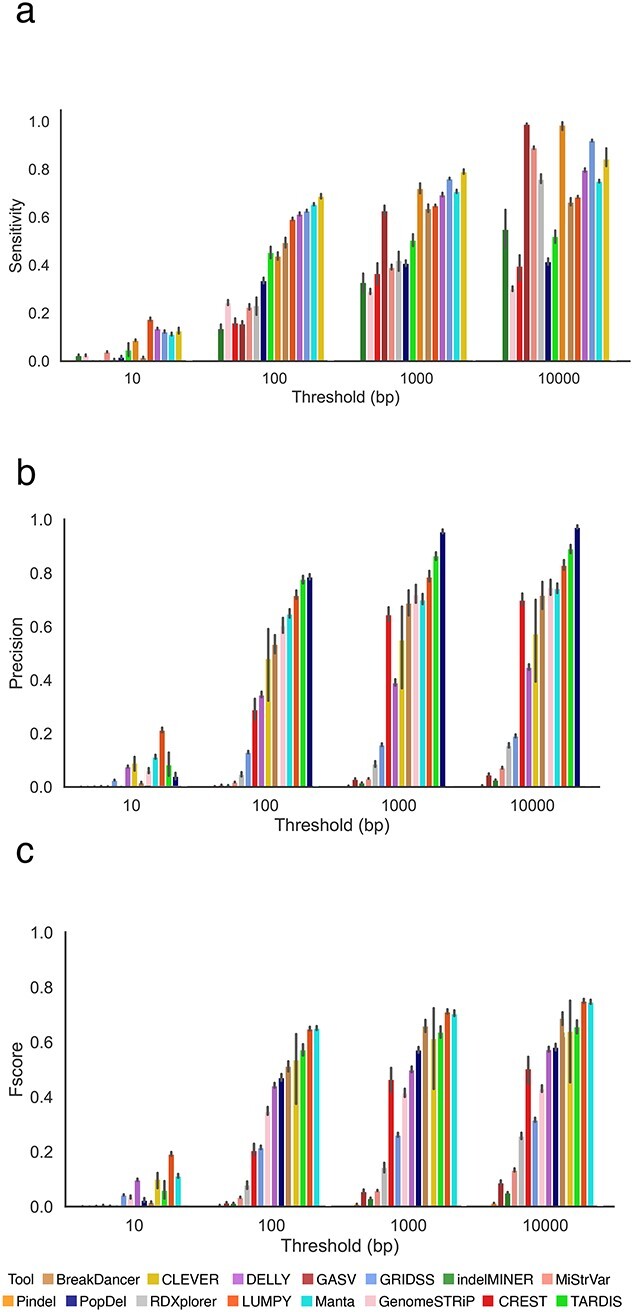
Comparing the performance of SV callers based on WGS data across seven inbred mouse strains. A deletion is considered to be correctly predicted if the distance of right and left coordinates are within the threshold τ from the coordinates of a true deletion. (A) Sensitivity of SV callers at different thresholds. (B) Precision of SV callers at different thresholds. (C) F-score of SV callers at different thresholds. Figures (A–C) are sorted in increasing order based on their performance at the 100 bp threshold. Results for other thresholds are presented in Supplemental Figure S6.
Methods that produced a higher F-score are the most balanced in precision and sensitivity; few methods skewed towards just one of the metrics (Supplemental Figures S7 and S29). Manta [39], LUMPY [36] and CLEVER [32] were the only methods able to successfully balance precision and sensitivity, with rates above 50% for each metric (Figure 3E and Supplemental Figure S7). CLEVER [32] was able to achieve the highest sensitivity rate at the majority of thresholds (Figure 3A and Supplemental Figure S5). The most precise method we observed was PopDel [37], with rates exceeding 80% for thresholds 1000 bp onwards, but the sensitivity of this method was two times lower than the majority of other tools (Figure 3B).
We examined whether the SV callers included in this study maintained similar SV detection accuracy across the different mouse strains. We compared results from each tool to study how consistent the results were across the samples. Among the tools with a sensitivity rate above 10%, LUMPY [36] maintained the most consistent sensitivity rate across samples with the highest rate of 60% when applied to both C3H/HeJ and CBA/J strains. The lowest sensitivity rate achieved by LUMPY [36] was 58% for A/J and DBA/2J strains. Sensitivity rates were the most stable across the seven different strains (Supplemental Figure S5). Precision shows the second highest variability across the strains, with the most stable results provided by Pindel [30] and indelMINER [35] (Supplemental Figure S4).
We have also compared CPU time and the maximum amount of RAM used by each of the tools. Across all tools, GASV [29] required the highest amount of RAM whereas PopDel [37] required the lowest amount of RAM to run the analysis. CREST [38] required the longest time to perform the analysis. Breakdancer [34] was the fastest tool. We have also compared the computational resources and speed of SV callers based on datasets with full coverage and those with ultralow coverage (Supplemental Figure S9).
Performance of SV detection tools on low- and ultralow- coverage mouse data
We assessed the performance of SV callers at different coverage depths generated by downsampling the original WGS data. The simulated coverage ranged from 32× to 0.1×, and 10 subsamples were generated for each coverage range. For each method, the number of correctly detected deletions generally decreased as the coverage depth decreased (Supplemental Figure S10). Some of the methods were able to call deletions from ultralow coverage (≤0.5×) data. Although tools like Manta [39] reached a precision of 82%, the overall sensitivity and F-score values were less than 8% for all tools. None of the methods were able to detect deletions from 0.1× coverage.
As suggested by other studies [51], most tools reached a maximum precision at an intermediate coverage (Figure 4B). Both the sensitivity rate and the F-score improved as the coverage increased (Figure 4A and C). Overall, DELLY [33] showed the highest F-score for coverage below 4× (Figure 4C). For coverage between 8 and 32×, Manta [39] showed the best performance. LUMPY [36] was the only tool to attain precision above 90% for coverage 1× to 4×. However, a decreased sensitivity in coverage below 4× led to a decreased F-score when compared with DELLY [33]. Precision in results from DELLY [33] for ultralow-coverage data was above 90% when the threshold was set at 1000 bp, but changing the threshold had no effect on LUMPY [36] (Supplemental Figure S11).
Figure 4.
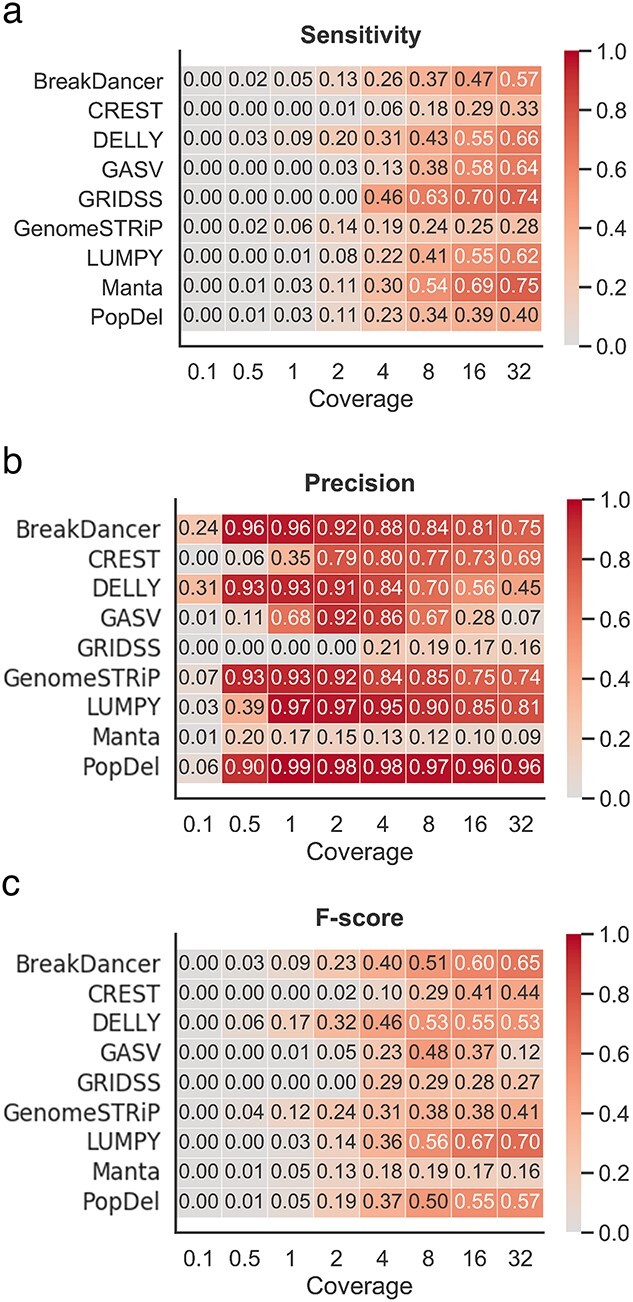
Performance of SV detection tools on low- and ultralow-coverage mouse data. (A) Heatmap depicting the sensitivity based on the 100 bp threshold across various levels of coverage. (B) Heatmap depicting the precision based on the 100 bp threshold across various levels of coverage. (C) Heatmap depicting the F-score based on the 100 bp threshold across various levels of coverage.
Length of deletions impacts the performance of the SV callers
As different variant callers are designed for different deletion length categories, we separately assessed the effect of deletion length on the accuracy of detection for four categories of deletions (Figure 5). The performance of the SV callers was significantly affected by deletion length. For example, for deletions shorter than 100 bp, precision and F-score values were typically below 40%, regardless of the tool (Figure 5B and C and Supplemental Figures S12, S14 and S15) whereas sensitivity values were above 50% for several tools (Figure 5A) (Supplemental Figure S13). For deletions longer than 100 bp, the best-performing tool in terms of sensitivity and precision significantly varied depending on the deletion length (Figure 5A and B). CLEVER [32] provided a sensitivity of above 60% for deletions less than 500 bp; however DELLY [33] provided the highest sensitivity for deletions longer than 500 bp (Figure 5A and Supplemental Figures S17, S21 and S25). LUMPY [36] delivered the best precision for deletion lengths from 50 to 500 bp, and CLEVER [32] performed well for longer deletion lengths (Figure 5B and Supplemental Figures S14, S18, S22 and S26). indelMINER [35] provided the high precision rate of detection of deletions in the range of 100–500 bp and when longer than 1000 bp, but the precision of detecting deletion in the 500–1000 bp range was lower than that of other tools (Figure 5B). In general, Manta [39] and LUMPY [36] were the only methods able to deliver an F-score above 30% across all categories (Figure 5D and Supplemental Figures S15, S19, S23 and S27).
Figure 5.
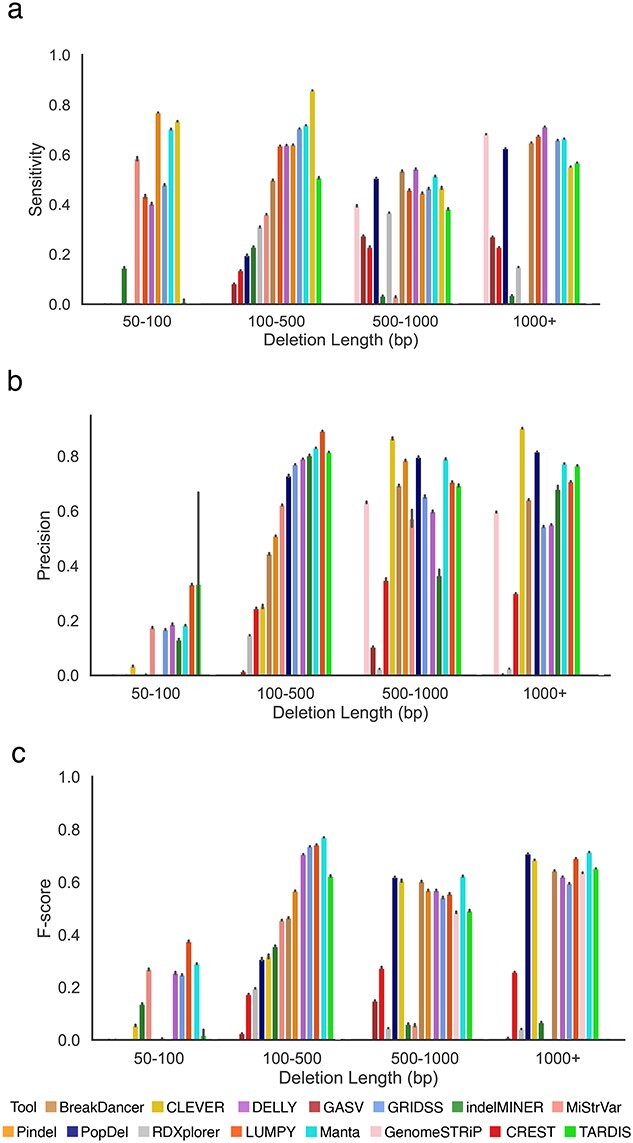
Comparing the performance of SV callers across various deletions lengths on mouse data. (A) Sensitivity of SV callers at the 100 bp threshold across deletion length categories. (B) Precision of SV callers at the 100 bp threshold across deletion length categories. (C) F-score of SV callers at the 100 bp threshold across deletion length categories.
Comparing the performance of SV callers on human WGS data
We compared the performance of 14 SV callers with respect to inferring deletions. The number of deletions detected varied from 342 (LUMPY [36]) to 1 371 466 (CLEVER [32]). Although tools like BreakDancer [34], GenomeSTRiP [40], LUMPY [36] and PopDel [37] reported fewer deletions than the gold standard and gasv, rdxplorer and clever reported higher deletions than the gold standard, consistently for both mouse and human data, a reverse trend was observed for CREST [32], Manta [39] and DELLY [33] (Figure 6).
We analyzed the TP rates for the SV callers at four different resolution thresholds (Figure 6). In general, the length distribution of detected deletions varied across tools and was substantially different from the distribution of true deletions across multiple SV detection methods (Supplemental Figure S28). Deletions detected by Manta [39] were the closest to the true median deletion length. In contrast to mouse data, a majority of the SV callers overestimated deletion lengths, with only 3 out of 14 callers underestimating deletions. A possible explanation for this could be the large number of FP calls by the tools, which could skew the distribution towards a longer median deletion length (Supplemental Figure S28).
Similar to mouse data, we analyzed the ability of the tools to balance precision and sensitivity (Figure 7). Although Manta was able to achieve the highest sensitivity for thresholds less than 100 bp, CLEVER [32] was the best performing tool for 10 000 bp threshold (Figure 7A). Octopus [41] was able to maintain a high sensitivity rate across the majority of thresholds at the cost of decreased precision. Similarly, Manta [39] achieved the highest precision for thresholds less than 100 bp, and PopDel [37] for thresholds 1000 and 10 000 bp (Figure 7B). In concordance with the results on mouse data, Manta [39] was the top-performing tool in terms of F-score, achieving the best balance between sensitivity and precision (Figure 7C). Other tools achieving a high F-score on human data, consistently across all thresholds were DELLY [33] and GROM [44] (Supplemental Figure S30).
Figure 7.
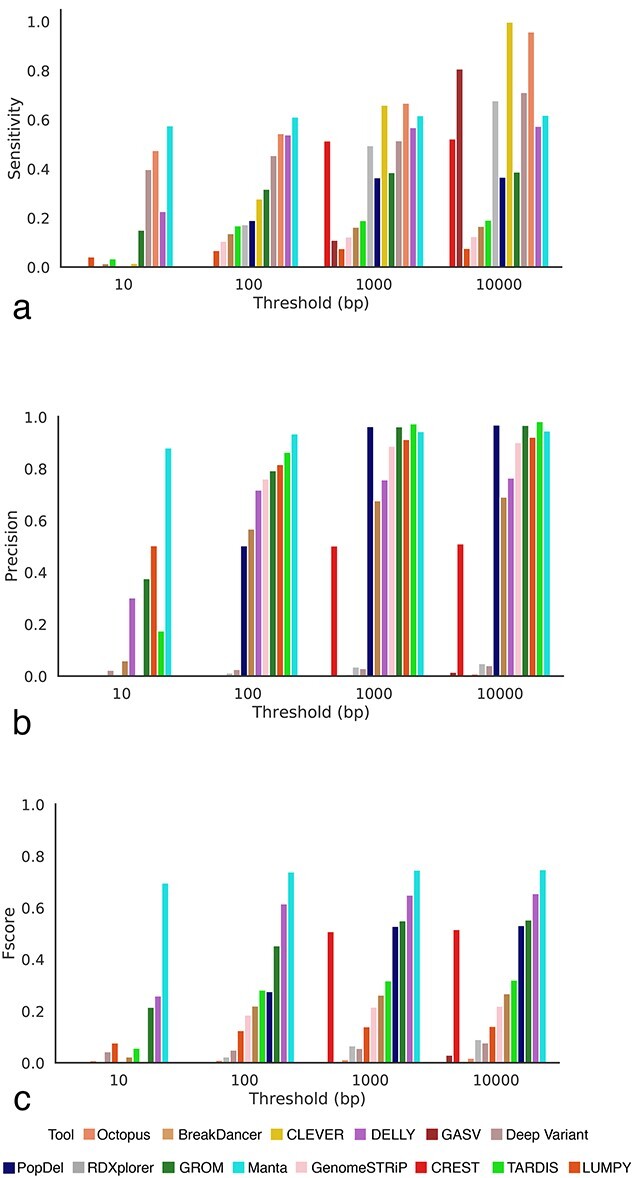
Comparing the performance of SV callers on human data. A deletion is considered to be correctly predicted if the distance of right and left coordinates are within the threshold τ from the coordinates of a true deletion. (A) Sensitivity of SV callers at different thresholds. (B) Precision of SV callers at different thresholds. (C) F-score of SV callers at different thresholds.
Consistent with results on mouse data, the sensitivity of the SV callers on human data was substantially affected by deletion length (Figure 8). For example, Octopus [41] was able to achieve the highest sensitivity of 0.745 in the 50–100 bp category; however its sensitivity dropped to 0.009852 in the 500–1000 bp category (Figure 8A). Manta [39] had the highest sensitivity for 100–500 bp, DELLY [33] for 500–1000 bp and GenomeSTRiP [40] for deletions larger than 1000 bp. While Manta [39] consistently provided the highest values of precision for 100–500 bp and greater than 1000 bp, Octopus [41] was the highest performing tool for the 500–1000 bp category (Figure 8B). For shorter deletions less than 100 bp, GROM [44] achieved the highest precision. Manta [39] achieved the highest F-score for the 100–500 bp category (Figure 8B).
Figure 8.
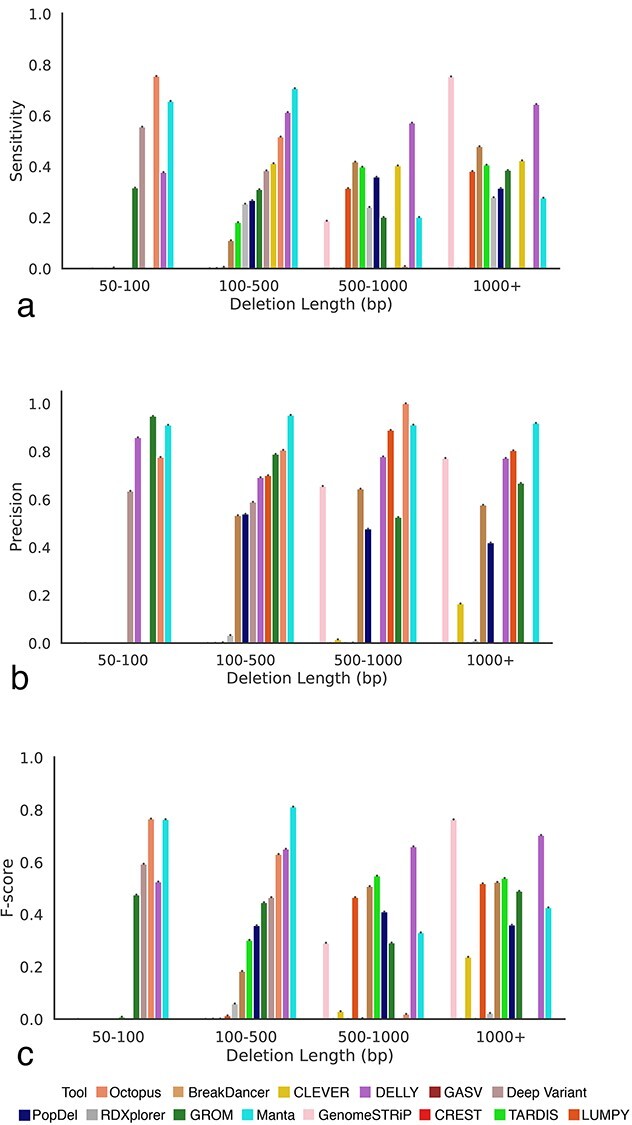
Comparing the performance of SV callers across various deletions lengths on human data. (A) Sensitivity of SV callers at the 100 bp threshold across deletion length categories. (B) Precision of SV callers at the 100 bp threshold across deletion length categories. (C) F-score of SV callers at the 100 bp threshold across deletion length categories.
Discussion
In this article, we performed a systematic benchmarking of algorithms to identify SVs from WGS data. In contrast to methods that are used to identify single nucleotide polymorphisms and have coalesced around a small number of approaches, there is currently no consensus on the best way to detect SVs in mammalian genomes. Indeed, we were able to find 61 different methods, each claiming relatively high sensitivity rates in the original publication.
In comparison to previous benchmarking efforts based on simulated data [22, 24, 28, 58], we obtained and employed a set of molecularly defined deletions for which breakpoints are known at base pair resolution. Other benchmarking studies have employed long–read-based gold standard datasets with approximate coordinates of deletions [19]. Our benchmarking method, using a gold standard set of molecular-defined deletions, overcomes the limitations of simulated data and incomplete characterization. Thus, our benchmarking study represents a robust assessment of the performance of the currently available SV detection methods when applied to a representative dataset.
In order to assess the precision and accuracy of the currently available SV callers, we simplified the problem presented to the detectors by using a set of homozygous deletions present in inbred mouse chromosomes. Although homozygous strains do not fully reflect the biology of real data, we use it in our mouse model as an easy-to-detect simple baseline. SV callers rely on the presence of the deletion allele and/or the read pairs affected by the deletion. A homozygous deletion, having only the deletion allele, can therefore be expected to have a stronger signal for split reads and discordant read pairs [44]. A stronger signal makes a homozygous variant easier to detect than a heterozygous variant. Methods failing to detect homozygous variants are unlikely to detect heterozygous variants. Among SVs, we limit our analysis to the detection of deletions. We exclude other types of SVs because of the lack of a PCR-validated gold standard. We excluded insertions because of the poor reliability of variant calling methods to detect them. Medium-sized indels, such as the FLT3-ITD, have proved difficult to detect by most methods [60].
When installing the majority of SV callers, we noticed significant difficulties due to inadequate software implementation and technical factors [61]. Deprecated dependencies and segmentation faults were the most common reasons preventing successful tool installation [62]. The majority of the tools have a consensus on the output format to be used (Supplemental Table S6), but the requirements for the format varied among tools. The lack of documentation about format requirements may further limit the use of SV callers. In our benchmarking study, we only considered methods with a novel variant detection algorithm. Benchmarking of consensus-based algorithms such as parliment2 [63], svclassify [64, 65] and SURVIVOR [66] that combine individual tools has already been performed [67] and were excluded from our analysis. Although we were able to run 15 tools on mouse data, we had to exclude indelminer and pindel for our analysis on human data. This is because of exceptionally high computational resources required by these tools, both in terms of running time and memory, on the human data.
In contrast to existing benchmarking studies, we did not filter the output of the SV callers. Filtering is extremely context specific and may vary significantly for different experiments, and good post-hoc filtering would require individual consideration of each tool’s quality metrics and thresholds to get comparable results. Although it may be acceptable to tolerate a few FPs in studies in which it is vital to detect as many true variants as possible, others may be very conservative require only high-quality variants. As different tools have different variant quality cut-offs, keeping the cutoff too low could result in many potential FPs. We do not exclude any variants based on the filter field and leave it up to the end users to decide the feasible filtering cut-offs which are best for their study. Additionally, we intentionally did not apply any quality control measures such as filtering on genotype and genotype quality to ensure all the tools evaluated in the study were restricted to homozygous deletions as choosing settings appropriate for homozygous deletions would have advantaged those tools that allow this option. Our aim was to perform a fair comparison of the performance of the tools in situations where the genotypes are unknown. As the detection of homozygous deletions is an easier task as compared with the detection of heterozygous deletions, we believe this provided for a fair comparison. We used Truvari (https://github.com/spiralgenetics/truvari), a tool widely adopted by the community for the evaluation of SV calls to compute the metrics of this study.
We identified a series of factors that determined the performance of the SV caller methods. The most important factors were the size of deletions and the coverage of WGS data. For example, BreakDancer [34] only detected deletions larger than 100 bp. Some tools achieved excellent sensitivity with the caveat that their precision was close to zero. For example, Pindel [34] achieved the highest sensitivity rate among all the tools with a precision rate of less than 0.1%. Other tools (e.g. PopDel [37]) employ a more conservative SV detection approach, resulting in higher precision at the cost of decreased sensitivity for smaller deletion events. A few tools were able to maintain a good balance between precision and sensitivity. For example, Manta [39], CLEVER [32], LUMPY [36] and BreakDancer [34] maintained both precision and sensitivity rates above 40%. In addition to differences in the accuracy of SV detection, we observed substantial differences in run times and required computational resources (Supplemental Figure S9). We studied the effect of the SV detection algorithm on performance, including split read, read depth and read pair. We found that most tools were a combination of different algorithms (Table 1). We did not find any correlation between the algorithm used and the performance of the tool. Although we have explored the effect of coverage, resolution threshold, organism and deletion length on variant detection, a study of the distribution of the variants location wise across the genome, the copy number of the variants and the different subcategories within deletions is an important area that needs to be explored by future studies.
Our reported top-performing variant caller-based on the F-score, Manta [39], is consistent with prior studies [66, 68]. Although other studies [68] use concordance as a comparison metric and do not adjust for the incompleteness of the human gold standard, we chose to make the gold standard complete by extracting the high-confidence regions defined in the bed file, allowing us to report metrics like sensitivity and precision.
We envision that future SV caller methods should enable the detection of deletions with precise coordinates. The inability of current methods to precisely detect breakpoints was related to the issue of the majority of tools underestimating the true size of SVs. Given the variation in the performance of the callers based on deletion length, coverage and organism, the results in this analysis can be combined to create an integrated SV calling method that has the potential to outperform individual callers. We acknowledge the existence of numerous implementations of an ensemble calling approach, such as Parliament2 [63] and FusorSV [69]. Although it has been shown to be easy to improve the sensitivity of the set of SVs by taking either the intersect or union of the calls created by the various callers, we note it is a nontrivial task to find universal thresholds and rules for the integration of the set of SVs into an ensemble set that maximizes both precision and recall at the same time. We hope that the results reported in this benchmarking study can help researchers choose appropriate variant calling tools based on the organism, data coverage and deletion length.
Methods
Run SV detection tools
Commands required to run each of the tools and the installation details are available in Supplemental Table S5. LUMPY [36] was run using smoove as recommended by the developers of smoove. The diploidSV vcf files were used for Manta based on the recommendation of the developers.
Convert the output of the SV detection tool to a universal format
We have adopted the VCF format proposed by VCFv4.2 as the universal format used in this study. Custom formats of the SV detection tools were converted to VCFv4.2. The description of custom formats is provided in Supplemental Table S6. The scripts to convert custom formats of SV detection tools to VCFv4.2 are available at https://github.com/Mangul-Lab-USC/benchmarking_SV
Generate high-confidence vcf files for human data
The human high-confidence vcf file for the gold standard was generated using a Python script. The script directly compares the high-confidence bed file to the csv file, by chromosome number. Then, high confidence vcf deletions were extracted if the deletion was fully contained within a high confidence region in the bed file, i.e., if the start position of the bed file was smaller than the start position of the vcf file and the end positions of the bed file was larger than the end position of the vcf file. These VCF files contained regions where the gold standard was high confidence and complete, meaning that it contained all possible deletions. Similar to the gold standard, the tool’s high-confidence vcf files were produced to extract regions that were fully contained within the high-confidence regions defined by the bed file. These true high-confidence files were then used as a gold standard to estimate the accuracy of the SV callers. The script and true high-confidence vcf file for the HG002 sample are available at: https://github.com/Mangul-Lab-USC/benchmarking_SV
Compare deletion inferred from WGS data with the gold standard
We compared the deletions inferred from SV callers from WGS data (inferred deletions) with the molecular-based gold standard (true deletions) using Truvari. Start and end positions of the deletion were considered when comparing true deletions and inferred deletions. Inferred deletion was considered to be correctly predicted if the distance of right and left coordinates are within the resolution threshold τ from the coordinates of true deletion. We consider the following values for resolution threshold τ: 0, 10, 100, 1000 and 10 000 bp. As most tools had zero matches when the threshold was kept at 0 bp, the starting threshold in the figures is kept as 10 bp. TPs were correctly predicted deletions, and were defined as deletions reported by the SV caller that were also present in the gold standard. In case an inferred deletion matches several true deletions, we randomly choose one of them. Similarly, in case a true deletion matches several inferred deletions, we choose the first deletion that matches. The deletion predicted by the SV caller but not present in the golden standard was defined as a FP. Similarly, each deletion present in the gold standard was matched with only one deletion predicted by the software. The SVs that were not predicted by the SV caller were defined as FN. SV detection accuracy was assessed using various detection thresholds (τ). The accuracy at threshold τ is defined as the percentage of SVs with an absolute error of deletion coordinates smaller or equal to τ. We have used the following measures to compare the accuracy of SV- callers:
● Sensitivity = TP/(TP + FN).
● Precision = TP/(TP + FP).
● F-score = 2×Sensitivity×Precision/(Sensitivity + Precision).
Compare computational performance of SV callers
The CPU time and RAM of each tool were measured to determine its computational performance. The statistics were measured for 1× coverage and full-coverage bam files, with sample A/J and BALB/cJ for mouse data. The CPU time was computed using either the GNU time program that is inbuilt in make bash terminals or the Hoffman2 Cluster qsub command. For GNU time, we used this specific command /usr/bin/time -f ‘%e\t%U\t%S\t%M’ which we had to run either manually on an interactive qsub session or through another method that was not a qsub. This GNU time command would output one line containing Wallclock time in seconds, user-time in seconds, kernel-space time in seconds, and peak memory consumption of the process in kilobytes. The CPU time was calculated by adding user-time and kernel-space time. RAM usage was equivalent to peak memory consumption in the case of this command. For qsubs on the Hoffman2 Cluster, we used the command qsub -m e which would email the user a full list of records when the tool finished running. This list included CPU-time and Max mem which was designated as RAM usage for each tool.
Downsample the Wgs Samples
We have used a custom script to downsample the full coverage bam file to desired coverage. Existing tools (e.g. samtools) are not suitable for this purpose as they treat each read from a read pair independently, resulting in singletons reads in the downsample bam file.
Data Availability
WGS mouse strains for the samples A/J, AKR/J, BALB/cJ, C3H/HeJ, DBA/2 J and LP/J used for benchmarking of the SV callers are available under the following accession numbers in the European Nucleotide Archive: ERP000038, ERP000037, ERP000039, ERP000040, ERP000044 and ERP000045. The output VCFs produced by the tools, the gold standard VCFs, the analysis scripts, figures and log files are available at https://github.com/Mangul-Lab-USC/benchmarking_SV. The human high-confidence bed file can be found here https://www.nist.gov/programs-projects/genome-bottle
The novoaligned bam data for the HG002_NA24385 son genome were downloaded from https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/
Code Availability
The source code to compare SV detection methods and to produce the figures contained within this text is open source and free to use under the Massachusetts Institute of Technology (MIT) license. All code required to produce the figures and analysis performed in this article are freely available at https://github.com/Mangul-Lab-USC/benchmarking_SV
Authors’ Contributions
V.S. created scripts for running and evaluating the software tools. M.K, N.R, A.S, E.W., J.C., M.G.D., N.D.-M., R.A., R.C., R.L., S.C. and V.S. contributed to installing, running and evaluating software tools. S.N. applied GRIDSS [30], LUMPY [36], Manta [39] and PopDel [37] to the mouse data and discussed evaluation metrics. R.A. curated data and prepared the mouse data for evaluation with the software tools. M.K, A.S, A.L., R.A. and V.S. generated the figures; R.A. and V.S. generating the tables. E.E., J.F., M.G.D., S.M., S.N. and V.S. wrote, reviewed and edited the manuscript. J.F. and S.M. led the project.
Supplementary Material
Acknowledgements
We thank Dr Lana Martin for the helpful discussions and comments on the manuscript. We thank the authors of the tools surveyed in this work—Cenk Sahinalp, Ryan Layer, Ira Hall, Tony Papenfuss, Gerton Lunter, Michael Schatz, Alexander Schoenhuth, Ken Chen, Aakrosh Ratan and Tobias Rausch—for providing helpful feedback and verifying the information related to their tool.
Author Biographies
Varuni Sarwal is a Computer Science PhD student at UCLA.
Serghei Mangul is an assistant professor at the USC School of Pharmacy.
Contributor Information
Varuni Sarwal, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA; Indian Institute of Technology Delhi, Hauz Khas, New Delhi, Delhi 110016, India.
Sebastian Niehus, Berlin Institute of Health (BIH), Anna-Louisa-Karsch-Str. 2, 10178 Berlin, Germany; Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Charitéplatz 1, 10117 Berlin, Germany.
Ram Ayyala, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Minyoung Kim, Department of Quantitative and Computational Biology, University of Southern California, 1050 Childs Way, Los Angeles, CA 90089.
Aditya Sarkar, School of Computing and Electrical Engineering, Indian Institute of Technology Mandi, Kamand, Mandi, Himachal Pradesh 175001, India.
Sei Chang, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Angela Lu, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Neha Rajkumar, Department of Bioengineering, Department of Bioengineering, University of California Los Angeles, Los Angeles, CA, 90095.
Nicholas Darci-Maher, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Russell Littman, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Karishma Chhugani, Department of Clinical Pharmacy, School of Pharmacy, University of Southern California 1985 Zonal Avenue Los Angeles, CA 90089-9121.
Arda Soylev, Department of Computer Engineering, Konya Food and Agriculture University, Konya, Turkey.
Zoia Comarova, Department Civil and Environmental Engineering, University of Southern California, Los Angeles, CA, United States.
Emily Wesel, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Jacqueline Castellanos, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Rahul Chikka, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Margaret G Distler, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA.
Eleazar Eskin, Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA; Department of Human Genetics, David Geffen School of Medicine at UCLA, 695 Charles E. Young Drive South, Box 708822, Los Angeles, CA, 90095, USA; Department of Computational Medicine, David Geffen School of Medicine at UCLA, 73-235 CHS, Los Angeles, CA, 90095, USA.
Jonathan Flint, Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior, University of California Los Angeles, 760 Westwood Plaza, Los Angeles, CA 90095, USA.
Serghei Mangul, Department of Clinical Pharmacy, School of Pharmacy, University of Southern California 1985 Zonal Avenue Los Angeles, CA 90089-9121.
Funding
S.N. is funded by the The Federal Ministry of Education and Research (BMBF) Grant #031 L0180 from the German Federal Ministry for Education and Research. SM is supported by the National Science Foundation grants 2041984 and 2135954. J.F. is supported by NIH grant R01MH122569. E.E. is supported by the National Science Foundation grants 1705197 and 1910885. The authors acknowledge the Quantitative and Computational Biology Institute and the Department of Computational Medicine at University of California Los Angeles (UCLA) for their support for the Bruins in Genomics Summer Program. Ram Ayyala, Nicholas Darci-Maher, Sei Chang, Emily Wesel and Jacqueline Castellanos received funding from The National Science Foundation (NSF) grant 1705197 and National Institute of Health (NIH) grant R25MH109172.
References
- 1. Feuk L, et al. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet 2006;79:965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pang AW, et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol 2010;11:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollox EJ, Barber JCK, Brookes AJ, et al. Defensins and the dynamic genome: what we can learn from structural variation at human chromosome band 8p23.1. Genome Res 2008;18:1686–97. [DOI] [PubMed] [Google Scholar]
- 4. McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet 2008;40:1107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fanciulli M, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet 2007;39:721–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Girirajan S, Eichler EE. De novo CNVs in bipolar disorder: recurrent themes or new directions? Neuron 2011;72:885–7. [DOI] [PubMed] [Google Scholar]
- 7. Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010;466:368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011;70:863–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elia J, et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 2011;44:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012;17:142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008;455:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walsh T. et al., Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008;320:539–43. [DOI] [PubMed] [Google Scholar]
- 13. Marshall CR, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 2017;49:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudmant PH, et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015;526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hehir-Kwa JY, et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat Commun 2016;7:12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kosugi S, et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 2019;20(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collins RL, et al. An open resource of structural variation for medical and population genetics. bioRxiv 2019;578674. 10.1101/578674. [Google Scholar]
- 18. Werling DM, et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet 2018;50:727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kosugi S, et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 2019;20:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao X, et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. The American Journal of Human Genetics 2021;108(5):919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao X, et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am J Hum Genet 2021;108(5):919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alkodsi A, Louhimo R, Hautaniemi S. Comparative analysis of methods for identifying somatic copy number alterations from deep sequencing data. Brief Bioinform 2015;16:242–54. [DOI] [PubMed] [Google Scholar]
- 23. Pabinger S, Rödiger S, Kriegner A, et al. A survey of tools for the analysis of quantitative PCR (qPCR) data. Biomol Detect Quantif 2014;1:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duan J, Zhang J-G, Deng H-W, et al. Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS One 2013;8:e59128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mills RE, et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011;470:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Legault M-A, Girard S, Lemieux Perreault L-P, et al. Comparison of sequencing based CNV discovery methods using monozygotic twin quartets. PLoS One 2015;10:e0122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hasan MS, Wu X, Zhang L. Performance evaluation of indel calling tools using real short-read data. Hum Genomics 2015;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neuman JA, Isakov O, Shomron N. Analysis of insertion-deletion from deep-sequencing data: software evaluation for optimal detection. Brief Bioinform 2013;14:46–55. [DOI] [PubMed] [Google Scholar]
- 29. Sindi S, Helman E, Bashir A, et al. A geometric approach for classification and comparison of structural variants. Bioinformatics 2009;25:i222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye K, Schulz MH, Long Q, et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoon S, et al. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 2009;19(9):1586–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marschall T, et al. CLEVER: clique-enumerating variant finder. Bioinformatics 2012;28:2875–82. [DOI] [PubMed] [Google Scholar]
- 33. Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan X, Abbott TE, Larson D, et al. BreakDancer: identification of genomic structural variation from paired-end read mapping. Curr Protoc Bioinformatics 2014;45:15.6.1–15.6.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ratan A, Olson TL, Loughran TP, Jr, et al. Identification of indels in next-generation sequencing data. BMC Bioinform 2015;16:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Layer RM, et al. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014;15(6):R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niehus S, et al. PopDel identifies medium-size deletions jointly in tens of thousands of genomes. bioRxiv 2020;740225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8:652–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen X, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016;32:1220–2. [DOI] [PubMed] [Google Scholar]
- 40. Noll AC, et al. Clinical detection of deletion structural variants in whole-genome sequences. NPJ Genom Med 2016;1:16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cooke DP, Wedge DC, Lunter G. A unified haplotype-based method for accurate and comprehensive variant calling. Nat Biotechnol 2021;39(7):885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poplin R, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 2018;36:983–7. [DOI] [PubMed] [Google Scholar]
- 43. Soylev A, et al. Toolkit for automated and rapid discovery of structural variants. Methods 2017;129:307. [DOI] [PubMed] [Google Scholar]
- 44. Smith SD, et al. Lightning-fast genome variant detection with GROM. GigaScience 2017;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Keane TM, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011;477:289–94 s 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yalcin B, et al. Sequence-based characterization of structural variation in the mouse genome. Nature 2011;477(7364):326–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mayrhofer M, DiLorenzo S, Isaksson A. Patchwork: allele-specific copy number analysis of whole-genome sequenced tumor tissue. Genome Biol 2013;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krishnan NM, Gaur P, Chaudhary R, et al. COPS: a sensitive and accurate tool for detecting somatic copy number alterations using short-read sequence data from paired samples. PLoS One 2012;7:e47812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim T-M, Luquette LJ, Xi R, et al. rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinform 2010;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xi R, Lee S, Xia Y, et al. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res 2016;44:6274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Bell JM, Zavala NA, et al. R, Allele-specific copy number profiling by next-generation DNA sequencing. Nucleic Acids Res 2015;43:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McKenna A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rimmer A, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014;46:912–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nijkamp JF, et al. De novo detection of copy number variation by co-assembly. Bioinformatics 2012;28:3195–202. [DOI] [PubMed] [Google Scholar]
- 56. Sedlazeck FJ, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 2018;15:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sedlazeck FJ, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 2018;15(6):461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guan P, Sung W-K. Structural variation detection using next-generation sequencing data: A comparative technical review. Methods 2016;102:36–49. [DOI] [PubMed] [Google Scholar]
- 59. Cameron DL, Di Stefano L, Papenfuss AT. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat Commun 2019;10:3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spencer DH, Abel HJ, Lockwood CM, et al. Detection of FLT3 internal tandem duplication in targeted, short-read-length, next generation sequencing data. J Mol Diagn 2012;15:81–93. [DOI] [PubMed] [Google Scholar]
- 61. Mangul S, Martin LS, Eskin E, et al. Improving the usability and archival stability of bioinformatics software. Genome Biol 2019;20:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mangul S, et al. Challenges and recommendations to improve the installability and archival stability of omics computational tools. PLoS Biol 2019;17:e3000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zarate S, et al. Parliament2: accurate structural variant calling at scale. GigaScience 2020;9(12):giaa145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zook JM, et al. A robust benchmark for detection of germline large deletions and insertions. Nat Biotechnol 2020;38(11):1347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Parikh H, et al. svclassify: a method to establish benchmark structural variant calls. BMC Genomics 2016;17(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jeffares DC, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 2017;8(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Collins RL, et al. A structural variation reference for medical and population genetics. Nature 2020;581(7809):444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H. FermiKit: assembly-based variant calling for Illumina resequencing data. Bioinformatics 2015;31:3694–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Becker T, et al. FusorSV: an algorithm for optimally combining data from multiple structural variation detection methods. Genome Biol 2018;19(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soylev, A, et al. Toolkit for automated and rapid discovery of structural variants. Methods 2017;129:3–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WGS mouse strains for the samples A/J, AKR/J, BALB/cJ, C3H/HeJ, DBA/2 J and LP/J used for benchmarking of the SV callers are available under the following accession numbers in the European Nucleotide Archive: ERP000038, ERP000037, ERP000039, ERP000040, ERP000044 and ERP000045. The output VCFs produced by the tools, the gold standard VCFs, the analysis scripts, figures and log files are available at https://github.com/Mangul-Lab-USC/benchmarking_SV. The human high-confidence bed file can be found here https://www.nist.gov/programs-projects/genome-bottle
The novoaligned bam data for the HG002_NA24385 son genome were downloaded from https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/