

Abstract
Recent technological advances have led to an exponential expansion of biological sequence data and extraction of meaningful information through Machine Learning (ML) algorithms. This knowledge has improved the understanding of mechanisms related to several fatal diseases, e.g. Cancer and coronavirus disease 2019, helping to develop innovative solutions, such as CRISPR-based gene editing, coronavirus vaccine and precision medicine. These advances benefit our society and economy, directly impacting people’s lives in various areas, such as health care, drug discovery, forensic analysis and food processing. Nevertheless, ML-based approaches to biological data require representative, quantitative and informative features. Many ML algorithms can handle only numerical data, and therefore sequences need to be translated into a numerical feature vector. This process, known as feature extraction, is a fundamental step for developing high-quality ML-based models in bioinformatics, by allowing the feature engineering stage, with design and selection of suitable features. Feature engineering, ML algorithm selection and hyperparameter tuning are often manual and time-consuming processes, requiring extensive domain knowledge. To deal with this problem, we present a new package: BioAutoML. BioAutoML automatically runs an end-to-end ML pipeline, extracting numerical and informative features from biological sequence databases, using the MathFeature package, and automating the feature selection, ML algorithm(s) recommendation and tuning of the selected algorithm(s) hyperparameters, using Automated ML (AutoML). BioAutoML has two components, divided into four modules: (1) automated feature engineering (feature extraction and selection modules) and (2) Metalearning (algorithm recommendation and hyper-parameter tuning modules). We experimentally evaluate BioAutoML in two different scenarios: (i) prediction of the three main classes of noncoding RNAs (ncRNAs) and (ii) prediction of the eight categories of ncRNAs in bacteria, including housekeeping and regulatory types. To assess BioAutoML predictive performance, it is experimentally compared with two other AutoML tools (RECIPE and TPOT). According to the experimental results, BioAutoML can accelerate new studies, reducing the cost of feature engineering processing and either keeping or improving predictive performance. BioAutoML is freely available at https://github.com/Bonidia/BioAutoML.
Background
Considering advances in sequencing, an increasing number of biological sequences have been generated [1, 2]. With the expansion in volume and complexity of biological data, machine learning (ML) algorithms have been successfully applied to their analysis [3–5]. ML algorithms can extract new and useful knowledge from biological data [6], allowing complex analyses, speeding up new findings and reducing research costs [7]. These advances bring important social and economical benefits, such as improving diagnosis, treatment and the design of new medications [7–9], e.g. coronavirus disease 2019 [8, 10], cancer diagnosis [11] and CRISPR/Cas9-based gene-editing technology [12, 13].
Moreover, with the advancement of next-generation sequencing technologies and multi-omics analysis [14], studies have focused on discovering and characterizing small noncoding RNAs (sRNAs) in bacteria and archaea, expanding the understanding of gene regulation and elucidating new biological mechanisms [15]. Moreover, Noncoding RNAs (ncRNAs) have distinct classes with specific functions, depending on their spatial structure, sequence composition and length [16]. Regarding genome annotation, the identification of protein-coding and non-protein-coding sequences is the first and most crucial step [17]. In addition, sRNAs control gene expression in prokaryotes, regulating processes, e.g. stress responses, nutrient acquisition, virulence and biofilm formation [18]. According to [19], there is a large number of regulatory ncRNAs, highlighting their potential links to bacterial pathogenesis.
Nevertheless, one of the main difficulties for applying ML algorithms to ncRNAs and other sequences is the categorical and unstructured nature of biological sequence data. A frequent alternative to deal with this problem is to apply, in a feature engineering process, feature extraction techniques (e.g. DNA sequences: A, C, T, G), to transform biological sequences into numerical data (e.g. GC content and k-mers) with a structured format. Feature extraction techniques based on various aspects have been proposed to extract numbers from these sequences, including physicochemical, biological and mathematical features [6, 9]. No matter the aspect, the features must capture the relevant information present in the biological sequence, as the predictive performance of the model induced by an ML algorithm strongly depends on the representativeness of the input feature vector [20]. A common approach to increase the representativeness of the features is to select, among the extracted features, the subset that leads to the best predictive performance of a model induced by an ML algorithm. This approach, known as feature selection using wrappers, is also a part of the feature engineering process.
The feature engineering process often requires extensive domain knowledge, performed manually by a human expert, and is one of the most time-consuming steps in the ML pipeline [20]. Furthermore, according to [21], ncRNAs are divided into categories based on their cellular functionality and their sequential, thermodynamic and structural properties, assuming that their sequence can provide robust discriminative features. However, the same sequences can act as more than one type of ncRNAs, e.g. mature micro RNA (miRNA) can also be transfer RNA (tRNA) fragments. Consequently, most computational approaches can predict only the presence of ncRNAs. Even those designed to classify more than one type (class) of ncRNAs do not work well with more than three types [21, 22].
These limitations motivated the development of a novel open-source software package, called BioAutoML, that can extract features based on different aspects, and automate the feature selection, algorithm(s) recommendation and algorithm(s) tuning steps for multi-class classification of biological data. BioAutoML is an end-to-end Automated Machine Learning (AutoML) tool for experiments using biological sequences; BioAutoML is able to deal with different categories of ncRNA in bacteria, such as small RNA (sRNA), tRNA, ribosomal RNA (rRNA), precursor-microRNA (pre-miRNA), miRNA, small nucleolar RNA (snoRNA), small nuclear RNA (snRNA) and transfer-messenger RNA (tmRNA) [21, 23]. According to [24, 25], AutoML has a proposal similar to the area of hyper-heuristics, automatically recommending pipelines, algorithms or hyper-parameters for specific tasks, reducing dependence on user knowledge. These tasks can include different ways of preprocessing or feature engineering, as well as algorithms and optimization of its parameters (hyper-parameter tuning) [24–26]. In this study, BioAutoML calls the MathFeature package [27, 28] to extract feature descriptors representing relevant numerical information from ncRNA sequences (Feature Extraction module). After receiving the feature values, BioAutoML, automatically recommends, using Bayesian Optimization [29], the best pair of selected features and predictive model.
To select the features (Feature Selection module), BioAutoML follows the wrapper approach, using a predictive model to assess how good a feature set is. The feature extraction and feature selection model are part of the Feature Engineering process. To recommend the best model, BioAutoML recommends the best number of predictive models, and the ML algorithm to be used for the induction of each model (Algorithm Recommendation module). These two tasks are carried simultaneously, whereby one feeds information to the other in the Bayesian optimization process, until the predictive performance obtained by the pair is not further improved. It is important to point out that the predictive model used in the wrapper and induced by the recommended algorithm can be an ensemble of predictive models.
This occurs when the Algorithm recommendation model recommends more than one ML algorithm. In this case, each recommended algorithm induces a predictive model, and the induced models are combined in an ensemble. As an ensemble of predictive models is, by itself, a predictive model, for the sake of simplicity, we will name the ensemble also a predictive model. Having finished the Feature Engineering module and the Algorithm Recommendation module, BioAutoML goes to the fourth module, which uses AutoML to fine tune the hyper-parameters of the recommended ML algorithms [24–26], aiming to improve the predictive performance of the model (Tuning and Combination module). If the predictive model is an ensemble, the algorithm that induces each model in the ensemble has its hyper-parameters tuned. Afterwards, the predictive models induced by the tuned algorithms are combined in the ensemble.
To make the role clearer of each module in the whole process, the part of the pipeline with the Feature Extraction module and the Feature Selection module, as they mainly work at the feature level, is named here Feature Engineering. The other part of the pipeline, with the Algorithm Recommendation module and the Tuning and Combination module, as their work at the ML level, is called henceforth Metalearning. Thus, BioAutoML creates an automated pipeline working at the data feature and algorithm level. In this research, we have investigated several insights to support our hypothesis, as follows:
Hypothesis: Automated feature engineering and metalearning provides an efficient mechanism to extract features based on different aspects, and automates the feature selection, algorithm(s) recommendation and tuning steps, and hence, a high-quality prediction of categories of ncRNAs in bacteria.
The approach will help us to answer our Research Question (RQ), and consequently be used to confirm or deny the hypothesis, described as follows:
RQ: Is it possible to predict different categories of bacterial ncRNAs using automated feature engineering and metalearning pipelines?
Finally, to support our hypothesis and research question, we have validated BioAutoML into three different case studies using seven bacterial phyla.
Feature engineering
According to Chou’s five-step rule [3, 30], numerically representing biological sequences with an efficient and adequate mathematical expression is one of the most relevant steps to establish an effective statistical predictor for a biological system. In ML, biological sequences must be represented by a fixed number of features (e.g. binary, categorical or continuous), transforming originally unstructured data into a structured format. Feature extraction or feature encoding is a key step in the construction of high-quality ML-based models, determining the effectiveness of trained models in bioinformatics applications, such as biological sequence classification [5, 31, 32]. Nevertheless, the feature engineering process is a time-intensive step and requires domain knowledge of experts [20, 33, 34], which is a complex exercise [34]. Therefore, to develop our proposal and answer our research question, we define the automated feature engineering task formally explained as follows:
Given a set of sequence data,
 , divided into train (
, divided into train (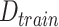 ) and test (
) and test (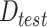 ), a set of feature descriptors,
), a set of feature descriptors,  , where
, where 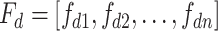 , our aim is to select the best numerical representation, that is, the feature vector (
, our aim is to select the best numerical representation, that is, the feature vector (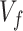 ), combining different feature descriptors in the training set (
), combining different feature descriptors in the training set ( ), using an objective function that considers the most important feature descriptor (
), using an objective function that considers the most important feature descriptor ( ) to evaluate the best
) to evaluate the best  .
.
Metalearning
One of the main difficulties in applying ML algorithms to a new dataset is selecting the most adequate algorithm for this dataset. Each ML algorithm has an inductive bias, which can be defined by the way it searches for a robust model, e.g. starting with simple models and gradually increasing the complexity of the models, until a robust model is found, and the format adopted to represent the models, e.g. a model represented by a decision tree. Although it can be seen as a limitation, the bias is necessary for learning to occur. As a consequence, each algorithm fits better datasets with particular conformations. Thus, there is no champion ML algorithm that performs better than all the others in every situation, but each ML algorithm performs better than the others on some datasets, which are not known beforehand [35]. A good alternative to select the best ML algorithm for a new dataset is to use previous knowledge regarding the performance of a set of algorithms in previous learning experiences. This is the idea behind a particular approach for metalearning, defined in [36] as learning to learn. According to the authors, metelarning is a research area that investigates how to recommend the most suitable algorithm, or set of algorithms, for a new task. In this study, we use metalearning to do the following:
Given a set of selected features, recommend the ML algorithm(s) able to induce the best predictive model, which can be a set of algorithms, each one inducing a model, and combine these models into an ensemble (
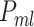 ), recommending the best algorithm. Ensemble methods can boost the performance of simple classifiers (e.g. using multiple prediction models for solving the same problem) and has proven its effectiveness in bioinformatics [37–39].
), recommending the best algorithm. Ensemble methods can boost the performance of simple classifiers (e.g. using multiple prediction models for solving the same problem) and has proven its effectiveness in bioinformatics [37–39].
Related works
After a carrying out systematic literature review, we found 14 related studies proposing packages that use feature engineering (feature extraction and selection) and ML algorithms for biological sequence classification: PseAAC [40], propy [41], PseKNC-General [42], SPiCE [43], Pse-in-One [3], repDNA [44], Rcpi [45], BioSeq-Analysis [46], PyFeat [31], iLearn [5], iLearnPlus [6], BioSeq-BLM [47], autoBioSeqpy [48] and AutoGenome [49]. For each package, we checked if it uses AutoML for feature engineering, ML algorithms and, when they use these algorithms, tune their hyper-parameters. Table 1 summarizes our findings.
Table 1.
Use of AutoML for feature engineering, recommendation of ML algorithm and hyper-parameter tuning
| Study | Feature Engineering | ML algorithm | Tuning |
|---|---|---|---|
| PseAAC | − | − | |
| propy | − | − | − |
| PseKNC-General | − | − | − |
| SPiCE | − | − | − |
| Pse-in-One | − | − | − |
| repDNA | − | − | − |
| Rcpi | − | − | − |
| BioSeq-Analysis | − | − | |
| PyFeat | − | − | − |
| iLearn | − | V | V |
| iLearnPlus | − | V | V |
| BioSeq-BLM | − | − | − |
| autoBioSeqpy | − | V | V |
| AutoGenome | − | V | V |
| BioAutoML | V | V | V |
The most similar packages to BioAutoML are iLearn, iLearnPlus, autoBioSeqpy and AutoGenome, which apply AutoML to recommend ML algorithms, but they do not use automated feature engineering. The most similar package to our proposal, iLearn, requires an initial configuration file (choosing descriptors and classifiers), which needs domain knowledge from a human expert. Even in its most sophisticated version, iLearnPlus, a file needs to be inserted with the extracted features, instead of automatic feature engineering. The autoBioSeqpy and AutoGenome packages focus on recommending the best deep learning architecture. Thus, to the best of our knowledge, BioAutoML automates the longest pipeline for biological sequence analysis, encompassing feature engineering, ML algorithm recommendation and hyper-parameter tuning. Furthermore, BioAutoML is a user-friendly tool for non-experts.
Looking at more general applications of AutoML, we can cite RECIPE [24] and TPOT [50]. Tree-based Pipeline Optimization Tool (TPOT) is an AutoML tool that optimizes ML pipelines using genetic programming. REsilient ClassifIcation Pipeline Evolution (RECIPE) is an AutoML framework with grammar-based genetic programming. One of the most notable methods of RECIPE is how it uses grammar to organize the knowledge acquired from the literature [24]. RECIPE can also be an alternative to TPOT, as TPOT can create ML pipelines that are arbitrary, failing to solve a classification problem, therefore leading to a waste of computational resources [24]. The major difference compared with BioAutoML is the lack of a feature extraction module for biological sequences. These two packages are for any application domain, requiring a previously selected feature vector.
Prediction techniques of ncRNAs in bacteria
Many ML-based techniques have been proposed to identify ncRNAs in bacteria [51–56]. In [55], the authors compare the predictive performance of different techniques for RNAs classes, such as tRNAs, rRNAs and mRNAs. For such, they use normalized minimum free energy of folding, motif frequency and several RNA-folding parameters, such as base-pairing propensity, Shannon entropy and base-pair distance. The model induced by the Random Forest algorithm presented 89.5% of predictive accuracy. Another related study [51] constructed ML models to discriminate bona fide sRNAs applying five ML algorithms to random genomic sequences from five bacterial species. Seven features were used, including secondary structure. In [53] the support vector machine (SVM) algorithm was applied to a Non-Coding DNA (ncDNA) benchmark dataset, collected from Saccharomyces cerevisiae. SVM was also used in [57] to identify sRNAs in bacteria, particularly Salmonella Typhimurium LT2, Escherichia coli (E. coli) K-12 and Salmonella Typhi (S. Typhi). Some features are combined to achieve better results with accuracy of 81.25% and 88.82% for E. coli K-12 and S. Typhi Ty2. Unlike BioAutoML, these approaches did not apply an end-to-end ML pipeline.
BioAutoML package
BioAutoML is a user-friendly multi-class and binary classification package that allows the use of automated feature engineering and metalearning, as illustrated by Figure 1. Its use does not require specialized human assistance. BioAutoML only needs a training dataset of biological sequences (FASTA files) to perform an end-to-end ML experiment, from the feature engineering to generating of the predictive model induced by tuned ML algorithms. Nevertheless, the modules implemented in the BioAutoML package can be run independently, i.e. users can just generate the best numerical representation and send it to another ML model generation package, or they can use features extracted from other packages to generate a predictive model. For such, BioAutoML has two components with two modules each: (1) automated feature engineering (feature extraction and selection) and (2) Metalearning (algorithm recommendation and hyperparameters tuning). In the next sections, we briefly describe each component and module.
Figure 1.

Components implemented in the BioAutoML package: (1) Automated Feature Engineering (feature extraction and selection) and (2) Metalearning (algorithm recommendation and hyper-parameters tuning).
Feature extraction
This module, which is the first feature engineering stage, extracts feature descriptors using the MathFeature package [28], e.g. Mathematical descriptors (Fourier, Shannon, Tsallis, among others) and Conventional descriptors [Nucleic Acid Composition (NAC), dinucleotide composition (DNC), trinucleotide composition (TNC), ORF Features, Xmer k-Spaced Ymer composition frequency (kGap), Fickett score, among others]. As a result, more than 15 feature extraction techniques can numerically represent information found in biological sequences.
BioAutoML - selection and recommendation
The second module carries out automated feature engineering, selecting the best feature vector and ML algorithm to induce predictive models, which can be an ensemble of predictive models, as shown in Figure 2. For such, it uses the Bayesian optimization technique [29]. We use this technique because there is a large number of alternatives for the types and number of feature descriptors, characterizing an NP-hard problem. This module receives the following as input:
Figure 2.

Illustration of how BioAutoML works: selection and recommendation module.
All feature descriptors extracted by the first module;
An objective function, e.g. in our case, balanced accuracy for binary problems and F1-score (weighted) for multi-class problems;
ML algorithms (CatBoost [58], AdaBoost [59], Random Forest [60] and LightGBM [61]). These classifiers are responsible for analyzing the potential of the selected features. These algorithms are used for the wrapper-based feature selection, using different feature subsets as input. We chose these ML algorithms because they have good predictive performance and induce interpretable predictive models, allowing the understanding of the internal decision-making process [62]. The algorithms are widely adopted in the bioinformatics literature [37–39].
To represent the search space (for selecting feature descriptors and recommending ML algorithms), we use a partially binary input vector, e.g. [1, 0, 1, 0, 0, 1, [2]], when the last position can be a value from the set 0, 1, 2, 3, representing each of the four ML algorithms. In the other position, value 0 means that the feature descriptor was not selected for the subset to be evaluated, and value 1 that was selected. Next, using Bayesian optimization (Hyperopt library - Tree of Parzen Estimators [63]), BioAutoML selects a quasi-optimal feature vector, regarding the predictive performance of the model used in the wrapper. We chose Bayesian optimization based on studies in the literature [29, 64, 65], which demonstrate that it saves time and improves performance, presenting benefits over random search [66]. As can be seen in Figure 2, BioAutoML generates combinations of features and ML algorithm(s) until it finds the best pair (selected feature set, recommended ML algorithm) to send to the fourth module, hyper-parameter tuning. We adopted as stopping criterion for the optimization when the predictive performance reaches a plateau or after assessing 50 pairs (this number can be changed by the user). Let us remember that this module can recommend one ML algorithm or a set of ML algorithms (when an ensemble model induced by ML algorithms is recommended).
ML algorithm(s) hyper-parameter tuning
The last module is tuning, where users can generate a predictive model using the recommendation of the feature vector and ML algorithms (among those whose implementation is available at BioAutoML). These algorithms will use the feature vector to induce a set of classification models, whose output will be combined using an ensemble-based approach. The quality of the classification models will be affected by the hyper-parameter values used for the recommended ML algorithms. In this work, Bayesian optimization is used to tune their hyper-parameters. For such, we separate part of the training set. The hyper-parameters tuned for each algorithm are defined by their official documentation, e.g. Random Forest (n_estimators, max_features, criterion, max_depth, min_samples_split, min_samples_leaf and bootstrap). The optimization stops when the predictive performance reaches a plateau or after assessing 100 possible sets of values. In addition, this module generates important performance analysis files as outputs, e.g. best features, performance results, trained model and feature importance, among others.
Experimental results
The main purpose of this article was to provide a user-friendly and open-access package that allows automated feature engineering and metalearning for the analysis of biological sequences. To assess the relevance of the proposed package, we evaluate its predictive performance in three case studies, described in the following sections: (1) Case Study I - Genomic Pipeline, (2) Case Study II - Pipeline with Annotated Bacterial Sequences and (3) Case Study III - BioAutoML versus other proposed packages for automated experiments.
Case study I—genomic pipeline
We designed an experiment to classify ncRNA families in bacteria, using three known types of bacterial RNA: sRNA, tRNA and rRNA. These RNAs are often considered and studied to analyze ncRNA sequences, e.g. (1) tRNAs and rRNA can contaminate sRNA samples isolated from cytoplasmic total RNA extracts [67], and (2) sRNAs in bacteria, key actors in transcriptional and posttranscriptional regulation [55], emphasizing the importance of accurate prediction of these sequences. To further demonstrate the usefulness of our package, we generated our dataset using a standard bioinformatics pipeline, as shown in Figure 3, extracting sequences from genomes and then applying ML algorithms to predictive models. Our aim is to demonstrate that non-experts can easily connect their pipeline to BioAutoML.
Figure 3.

Case Study I - Genomic Pipeline.
To collect the RNAs from genomes, we used the Infernal application [68]. First, in our genomic pipeline, we accessed the Rfam Public MySQL Database obtaining a list of families for each RNA type [69], using the Rfam database in its 14.7 version. Next, with the lists and the complete Rfam Covariance Model (CM), we generated three CM files using cmfetch, i.e. one for each RNA type. We use cmsearch considering the gathering cutoff (GA) selected by the Rfam curators to extract the sequences for the RNA types [70], given the CM files and a genome. Once the sequences are extracted, they are passed as input to BioAutoML. Thereby, we selected Escherichia coli K-12 genome for an initial experiment with the genomic pipeline. In Table 2, we show the sequences generated for training and testing (Hold-out 80% training and 20% test).
Table 2.
Number of sequences from E. coli K-12 used for training and testing
| RNA type | Samples | Training | Testing |
|---|---|---|---|
| sRNA | 166 | 133 | 33 |
| tRNA | 50 | 40 | 10 |
| rRNA | 40 | 32 | 8 |
BioAutoML returned a combination of three feature descriptors that considered to better numerically represent this dataset, kGap, Fourier and Tsallis entropy. After automated feature engineering, our package selects a feature vector and recommended an ML algorithm to be finely tuned. The final results are shown in Table 3 and Figure 4-A. As our problem is multi-class, we report the main results using precision (Macro and Weighted), recall (Macro and Weighted), F1-score (Macro and Weighted) and confusion matrix [71].
Table 3.
Results: E. coli K-12—case study I
| Precision | Recall | F1-Score | |
|---|---|---|---|
| sRNA | 1.00 | 0.97 | 0.98 |
| tRNA | 1.00 | 1.00 | 1.00 |
| rRNA | 0.89 | 1.00 | 0.94 |
| Macro Average | 0.96 | 0.99 | 0.98 |
| Weighted Average | 0.98 | 0.98 | 0.98 |
Figure 4.

Confusion matrix of the experiments. (A) Case Study I—E. Coli K-12. (B) Case Study I—Bacterial phyla. (C) Case Study II—Annotated Bacterial Sequences.
BioAutoML was performed between 0.96 and 0.98 (macro and weighted average) in this initial experiment, showing a robust numerical representation for the input genome. Next, we used the recommended algorithm to induce a predictive model to classify new unknown sequences. To test the potential of our package, we did a more complex experiment using bacterial phyla, as shown in Table 4. We analyzed the generalization potential for the classification of new bacterial genomes as new organisms will not be in the training set, e.g. training with Chloracidobacterium and classifying a new genome as Terriglobus roseus. Moreover, according to [72], the different GC content skew patterns throughout bacterial phylogenetic groups could change relevant characteristics of the sequences’ primary structure used for the generation of descriptors. The bacterial phyla used for this experiment are shown in Table 4 and Figure 4-B.
Table 4.
Number of sequences for bacterial phyla by RNA type
| Phylum | Bacteria | sRNA | tRNA | rRNA | Gram | NCBI Taxonomy ID | Used as |
|---|---|---|---|---|---|---|---|
| Acidobacteria | Chloracidobacterium | 21 | 50 | 7 | Negative | 2821542 | Train |
| Terriglobus roseus | 4 | 47 | 11 | Negative | 926566 | Test | |
| Actinobacteria | Corynebacterium diphtheriae | − | 43 | 14 | Positive | 1450520 | Train |
| Mycobacterium tuberculosis | 11 | 45 | 8 | Positive | 83332 | Test | |
| Bacteroidota | Flavobacterium sediminis | − | 41 | 19 | Negative | 2201181 | Train |
| Mucilaginibacter gossypii | − | 46 | 20 | Negative | 551996 | Test | |
| Cyanobacteria | Oscillatoria acuminata | 9 | 65 | 19 | Negative | 56110 | Train |
| Prochlorococcus marinus | 2 | 39 | 7 | Negative | 167539 | Test | |
| Firmicutes | Staphylococcus aureus | 84 | 35 | 31 | Positive | 93061 | Train |
| Staphylococcus epidermidis | 44 | 38 | 11 | Positive | 1282 | Test | |
| Proteobacteria | Escherichia coli | 166 | 50 | 40 | Negative | 83333 | Train |
| Salmonella enterica | 118 | 52 | 39 | Negative | 99287 | Test | |
| Verrucomicrobia | Akkermansia glycaniphila | − | 44 | 7 | Negative | 1679444 | Test |
| Luteolibacter ambystomatis | 29 | 46 | 14 | Negative | 2824561 | Train |
We randomly selected one type of bacteria from each phylum for a fair split, as shown in Table 4. We used seven bacteria for training and testing. The number of sequences generated by RNA type is also presented. The sequences were extracted using the same pipeline exposed in Figure 3. The performance metrics can be seen in Table 5.
Table 5.
Results: Bacterial phyla—case study I
| Precision | Recall | F1-Score | |
|---|---|---|---|
| sRNA | 0.97 | 0.97 | 0.97 |
| tRNA | 0.98 | 1.00 | 0.99 |
| rRNA | 0.99 | 0.95 | 0.97 |
| Macro Average | 0.98 | 0.97 | 0.98 |
| Weighted Average | 0.98 | 0.98 | 0.98 |
Our package recommended six feature descriptors that best represent this new scenario: NAC, TNC, kGap, Fourier and ORF. Two of these descriptors were in the initial experiment (kGap and Fourier). Again, BioAutoML showed good predictive results, between 0.97 and 0.98 (macro and weighted average). However, we were classifying new bacterial sequences that were not in training, indicating the package’s ability to recommend robust feature descriptors for the input problem.
Case study II—pipeline with annotated bacterial sequences
For this case, we extracted annotated bacterial sequences from databases, standard pipeline in several studies [37, 48, 49]. We used eight classes for this analysis: pre-miRNA, miRNA, snoRNA, snRNA, tmRNA, tRNA, rRNA and mRNA. Compared with case study I, we worked with specific types of sRNAs to study BioAutoML capacity for dealing with more classes. In addition, we used mRNA as a counterpoint by containing coding regions compared with the ncRNAs. These classes can be separated into regulatory, and housekeeping ncRNAs [73]. We also demonstrate the performance metrics for the application considering recurrent problems such as the classification of pre-miRNA between miRNA [21, 74, 75], and the prediction of miRNA by itself [76]. There are few studies related to the prediction of miRNAs in bacteria [77] as the number of these annotated sequences is still small [78].
We collected ncRNA sequences from RNAcentral, a database of ncRNA sequences that provide a single access point to at least 44 RNA resources in its last version [23]. We accessed the RNAcentral Public Postgres database running SQL queries to filter active cross-reference sequences by type, limited to 1000 sequences, and restricting them to bacterial organisms. With the results from the queries, FASTA files for each class were created. Considering how collecting from diverse databases could bring some redundancy, we used CD-HIT Est [79] to cluster the sequences, removing redundancy at 95% similarity. The same preprocessing pipeline was applied for mRNA, but we collected the sequences from GenBank [80], filtering for bacterial organisms. In Table 6, we show the numbers of examples collected from RNAcentral and GenBank, the numbers after applying CD-HIT Est with the preprocessing method used in BioAutoML, and how many of these sequences are used for training and testing (Hold-out 80% training and 20% test). The results generated by BioAutoML are presented in Table 7 and Figure 4-C.
Table 6.
Number of sequences used for training and testing—case study II
| RNA type | Samples | Preprocessing | Training | Testing |
|---|---|---|---|---|
| pre-miRNA | 327 | 253 | 203 | 50 |
| miRNA | 464 | 263 | 211 | 52 |
| snoRNA | 331 | 178 | 143 | 35 |
| snRNA | 176 | 113 | 91 | 22 |
| tmRNA | 1000 | 350 | 280 | 70 |
| tRNA | 1000 | 445 | 356 | 89 |
| rRNA | 1000 | 687 | 549 | 138 |
| mRNA | 1000 | 702 | 514 | 188 |
Table 7.
Results generated by BioAutoML in the case study II
| Precision | Recall | F1-Score | |
|---|---|---|---|
| pre-miRNA | 0.69 | 0.76 | 0.72 |
| miRNA | 0.63 | 0.58 | 0.60 |
| snoRNA | 0.60 | 0.60 | 0.60 |
| snRNA | 0.65 | 0.50 | 0.56 |
| tmRNA | 0.99 | 0.96 | 0.97 |
| tRNA | 0.95 | 0.99 | 0.97 |
| rRNA | 0.95 | 0.98 | 0.96 |
| mRNA | 0.98 | 0.97 | 0.97 |
| Macro Average | 0.80 | 0.79 | 0.80 |
| Weighted Average | 0.89 | 0.89 | 0.89 |
Again, our package presented robust performance, even for eight classes, ranging from 0.79 to 0.89 (macro and weighted average). By analyzing each class individually, we observed a better performance for rRNA (F1-Score: 0.96), tRNA (F1-Score: 0.97), tmRNA (F1-Score: 0.97), pre-miRNA (F1-Score: 0.72) and mRNA (F1-Score: 0.97), but lower performance for miRNA, snoRNA and snRNA (F1-Score: both around 0.60). However, multi-class classification problems present more challenges than binary classification problems, e.g. an imbalanced dataset. Even so, BioAutoML recommended a good feature vector formed by the descriptors NAC, DNC, TNC, kGAP, ORF feature, Fourier and Tsallis entropy.
Case study III—comparing BioAutoML with other AutoML packages
In this last case study, we compared BioAutoML with well-known AutoML packages used in different classification tasks [81, 82]. In our literature review, we did not find any tool for biological sequence classification with automatic feature engineering, characterizing the innovative nature of BioAutoML. To allow the experimental comparison, we chose two packages using AutoML: RECIPE [24] and TPOT [50]. The major difference compared with BioAutoML is the lack of a feature extraction module for biological sequences.
Similar to BioAutoML, they include feature selection, algorithm recommendation and hyper-parameter tuning. These two packages are for any application domain, requiring a previously selected feature vector. For a fair comparison, we used the same output from the feature extraction module in the AutoML packages (all feature descriptors), feature descriptors recommended by BioAutoML and datasets from the previous case studies. These experiments assess whether BioAutoML can build predictive models with recommended feature vectors and ML algorithms as robust as RECIPE and TPOT, which are well known for the quality of their pipelines [81, 82]. The BioAutoML results shown in Table 8 are the average of 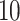 runs. All experiments, package configurations (default parameters) and datasets can be consulted in our repository (https://github.com/Bonidia/BioAutoML--Case Studies). We performed the experiments using a machine with Intel Core i3-9100F CPU (3.60GHz), 16GB memory and running in Debian GNU/Linux 10.
runs. All experiments, package configurations (default parameters) and datasets can be consulted in our repository (https://github.com/Bonidia/BioAutoML--Case Studies). We performed the experiments using a machine with Intel Core i3-9100F CPU (3.60GHz), 16GB memory and running in Debian GNU/Linux 10.
Table 8.
Case study III—BioAutoML versus other AutoML packages
| Dataset | Version | Precision (Weighted) | Recall (Weighted) | F1-Score (Weighted) | Time (min) |
|---|---|---|---|---|---|
| CS-I-phyla | BioAutoML | 0.97 | 0.97 | 0.97 | 16.34 |
| Recommended Feature Descriptors | RECIPE | 0.97 | 0.97 | 0.97 | 32.48 |
| Recommended Feature Descriptors | TPOT | 0.99 | 0.99 | 0.99 | 72.41 |
| All Feature Descriptors | RECIPE | 0.96 | 0.96 | 0.96 | 30.46 |
| All Feature Descriptors | TPOT | 0.98 | 0.98 | 0.98 | 46.39 |
| CS-II | BioAutoML | 0.88 | 0.88 | 0.88 | 85.02 |
| Recommended Feature Descriptors | RECIPE | 0.87 | 0.61 | 0.68 | 272.46 |
| Recommended Feature Descriptors | TPOT | 0.89 | 0.89 | 0.89 | 416.28 |
| All Feature Descriptors | RECIPE | 0.77 | 0.36 | 0.38 | 151.12 |
| All Feature Descriptors | TPOT | 0.90 | 0.89 | 0.89 | 338.55 |
As can be seen, we observed similar performance between BioAutoML and other tools (TPOT and RECIPE) in CS-I and CS-II, considering two different types of experiments: (i) with all feature descriptors, and (ii) with the vector recommended by BioAutoML. We also noted the improvement prediction of TPOT when the input was provided by vector recommended from BioAutoML (gain of 2% and 1% for CS-I and CS-II, respectively). Another interesting result is related to computational time to generate an ML model when both TPOT and RECIPE spent a huge computational effort (416.26 and 272.46 min, respectively in CS-II), while BioAutoML spent 85.02 min. BioAutoML also recommends the best vector be extracted automatically. It is important to highlight that both TPOT and RECIPE do not have any mechanism to recommend the best vector to be automatically extracted for biological sequences. Finally, the statistical significance was applied in this case study (difference in F1-Score (Weighted)), using Friedman’s test, indicating that there is no statistical significance in performance ( , using
, using 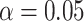 ), suggesting that our proposal is as robust as known methods in the literature.
), suggesting that our proposal is as robust as known methods in the literature.
Discussion
We assessed BioAutoML in three case studies with ncRNA sequences. We consider different ncRNA categories for multi-class classification tasks using ncRNA bacteria data. For case study I, we used Infernal, which builds statistical models of RNA secondary structure and sequence consensus called Covariance Models (CMs) [68]. Infernal is still widely used for genome annotation, especially for detecting ncRNA [83, 84]. However, one of its limitations for creating CM is the need for a secondary structure model for the RNA families. The experimental results from this case study shows the success of BioAutoML in using only primary structure features to predict what we found with Infernal.
For case study II, we considered eight classes, including miRNAs. Although some studies in the literature consider that prokaryotes do not have true miRNA as in eukaryotes [85, 86], recently, many similarities between the noncoding sequences were observed, indicating miRNA-like mechanisms in prokaryotes, which resulted in the annotated sequences used in our study [86, 87]. Prokaryotic miRNAs can also accumulate in the nucleolus as pre-miRNAs, and mature miRNAs [87], emphasizing the challenge for an accurate classification in these two classes. Other classes used, such as snoRNA [88] and snRNA [89], are also relatively rare in prokaryote organisms. Nevertheless, it is relevant to discover more of these noncoding sequences in bacteria with the advancements in RNA sequencing technology and ML-based algorithms.
Finally, in case study III, we observed the robust predictive performance of BioAutoML when compared with AutoML tools found in the literature, mainly due to the quality of their pipelines [81, 82]. The experimental results indicated the efficiency of the feature extraction module, which can extract features based on different aspects, automated feature selection, algorithm(s) recommendation and tuning steps. Together, they predicted the categories of ncRNAs in bacteria with high predictive accuracy, even when the number of classes was increased.
Conclusion
In this article, we propose and experimentally evaluate a new package, BioAutoML, to classify biological sequences. BioAutoML uses AutoML to select the best feature vector from a set of descriptors extracted by the MathFeature package, to recommend the best ML algorithms, and tune the hyper-parameters of the recommended algorithm. For such, it initially performs automated feature engineering and metalearning for noncoding sequences in bacteria, which has the potential to accelerate new studies in bioinformatics. We develop a package that does not require specialized human assistance, supporting research on challenging problems in biological sequence analysis. Our findings support our hypothesis, showing the benefits of using automated feature engineering and metalearning. Although in this study, BioAutoML is applied only to ncRNA sequences in bacteria, it can be used in other DNA/RNA sequence scenarios. We focused exclusively on bacteria, due to the biotechnological potential existing in the investigated strains. Nevertheless, the first module of BioAutoML is an important task for providing feature descriptors for different types of sequences (nucleotides or proteins, i.e. prediction of structural features along the primary sequence of amino acids). We also used our previous framework, MathFeature, to extract features for BioAutoML. BioAutoML can be used for binary and multi-class classification problems, allowing its integration with many existing packages. Finally, in future work, we intend to expand the BioAutoML to proteins and add new feature extraction packages, e.g. iLearn, BioSeq-Analysis and BioSeq-BLM, testing other feature selection methods such as combining Bayesian Optimization and Lipschitz Optimization, Genetic Algorithm and Genetic programming.
Key Points
The first study to propose an automated feature engineering and metalearning pipeline for ncRNA sequences in bacteria;
BioAutoML can be used in multi-class and binary problems;
BioAutoML can be employed in other DNA/RNA sequences scenarios;
BioAutoML can accelerate new bioinformatics studies, reducing the feature engineering time-consuming stage and improving the design and performance of ML pipelines;
BioAutoML reduces requirement of human expert assistance.
Acknowledgments
The authors would like to thank USP, CAPES, Google (LARA - 2021), CNPq, and FAPESP for the financial support for this research.
Funding
This project was supported by the Coordenacâo de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) - Finance Code 001, Google (LARA - 2021), Universidade de São Paulo (USP) and São Paulo Research Foundation (FAPESP) - grants #2013/07375-0, #2021/08561-8.
Author Biographies
Robson P. Bonidia received the M.Sc. degree in bioinformatics from the Federal University of Technology - Paraná (UTFPR), Brazil. He is currently pursuing the Ph.D. degree in computer science and computational mathematics with the University of São Paulo - USP. His main research topics are in computational biology and pattern recognition, feature extraction and selection, meta- heuristics, and sports data mining.
Anderson P. Avila Santos received M.Sc. degree in Computer Science from the University of Londrina - Paraná (UEL), Brazil. He is currently pursuing the Ph.D. degree in computer science and computational mathematics with the University of São Paulo-USP. His main research topics are in computational biology and pattern recognition, data mining, machine learning, evolutionary algorithms, bioinformatics.
Breno L. S. de Almeida is a Computer Science undergraduate student at the Institute of Mathematics and Computer Sciences - University of São Paulo (USP), where also pursued Certificate Programs in Data Science and Data Engineering. His main research interests are in Machine Learning, Feature Extraction, and Computational Biology.
Peter F. Stadler received a PhD in Chemistry from U. Vienna in 1990, where he then worked as Assistant and Associate Professor for Theoretical Chemistry. Since 2002 he is Full Professor for Bioinformatics at U. Leipzig. He is External Professor at the Santa Fe Institute, External Scientific Member of the Max Planck Society, Corresponding Member Abroad of the Austrian Academy of Sciences, and Honorary Professor of the Universidad Nacional de Colombia.
Ulisses N. da Rocha received a PhD in Microbial Ecology from the University of Groningen (The Netherlands) in 2010. In 2017, he joined the Department of Environmental Microbiology at the Helmholtz Centre for Environmental Research - UFZ (Leipzig, Germany) as a junior group leader. In 2021, he became a senior scientist at the UFZ. Further, he leads the Microbial Data Science group at the UFZ since 2017 where he works at the intersection of Microbial Ecology, Bioinformatics, and Computational Biology.
Danilo S. Sanches received the Ph.D. degree in electrical engineering from the University of Sao Paulo, in 2013. He is currently an Associate Professor with the Computer Science Department, Federal University of Technology - Paraná (UTFPR), Brazil. His research includes data mining, machine learning, evolutionary algorithms, bioinformatics, and pattern recognition approaches.
André C.P.L.F. de Carvalho is a full professor at the Department of Computer Science, University of São Paulo. He is the Vice Dean of the Mathematics and Computer Science Institute of the University of São Paulo, ICMC-USP, Vice Director of the Center for Mathematical Sciences Applied to Industry, USP, and Vice President of the Brazilian Computer Society, SBC. His research interests are in machine learning, data mining, and data science.
Contributor Information
Robson P Bonidia, Institute of Mathematics and Computer Sciences, University of São Paulo, São Carlos 13566-590, Brazil.
Anderson P Avila Santos, Institute of Mathematics and Computer Sciences, University of São Paulo, São Carlos 13566-590, Brazil; Department of Environmental Microbiology, Helmholtz Centre for Environmental Research-UFZ GmbH, Leipzig, Saxony, Germany.
Breno L S de Almeida, Institute of Mathematics and Computer Sciences, University of São Paulo, São Carlos 13566-590, Brazil.
Peter F Stadler, Department of Computer Science and Interdisciplinary Center of Bioinformatics, University of Leipzig, Leipzig, Saxony, Germany.
Ulisses N da Rocha, Department of Environmental Microbiology, Helmholtz Centre for Environmental Research-UFZ GmbH, Leipzig, Saxony, Germany.
Danilo S Sanches, Department of Computer Science, Federal University of Technology - Paraná, UTFPR, Cornélio Procópio 86300-000, Brazil.
André C P L F de Carvalho, Institute of Mathematics and Computer Sciences, University of São Paulo, São Carlos 13566-590, Brazil.
Availability of data and materials
BioAutoML, documentation and datasets are available in the Github repository: https://github.com/Bonidia/BioAutoML.
References
- 1. Hashemi FSG, Ismail MR, Yusop MR, et al. Intelligent mining of large-scale bio-data: Bioinformatics applications. Biotechnology & Biotechnological Equipment 2018;32(1):10–29. [Google Scholar]
- 2. Lou H, Schwartz M, Bruck J, et al. Evolution of k-mer frequencies and entropy in duplication and substitution mutation systems. IEEE Transactions on Information Theory 2020;66:3171–3186.. [Google Scholar]
- 3. Liu B, Liu F, Wang X, et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 05 2015;43(W1):W65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greener JG, Kandathil SM, Moffat L, et al. A guide to machine learning for biologists. Nat Rev Mol Cell Biol 2021;1–16. [DOI] [PubMed] [Google Scholar]
- 5. Chen Z, Zhao P, Li F, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 04 2019;21(3):1047–57. [DOI] [PubMed] [Google Scholar]
- 6. Chen Z, Zhao P, Li C, et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res 02 2021;gkab122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharma M, et al. Emerging trends of bioinformatics in health informatics. In: Computational Intelligence in Healthcare. Springer, 2021, 343–67. [Google Scholar]
- 8. Cannataro M, Harrison A. Bioinformatics helping to mitigate the impact of COVID-19- Editorial. Brief Bioinform 03 2021;22(2):613–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghannam RB, Techtmann SM. Machine learning applications in microbial ecology, human microbiome studies, and environmental monitoring. Comput Struct Biotechnol J 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Randhawa GS, Soltysiak MPM, Roz, et al. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: Covid-19 case study. Plos one 2020;15(4):e0232391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maros ME, Capper D, Jones DTW, et al. (eds). Machine learning workflows to estimate class probabilities for precision cancer diagnostics on dna methylation microarray data. Nat Protoc 2020;1–34. [DOI] [PubMed] [Google Scholar]
- 12. Li VR, Zhang Z, Troyanskaya OG. CROTON: an automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics 07 2021;37:i342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitrofanov A, Alkhnbashi OS, Shmakov SA, et al. CRISPRidentify: identification of CRISPR arrays using machine learning approach. Nucleic Acids Res 12 2020;49(4):e20–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turner AW, Wong D, Khan MD, et al. Multi-Omics Approaches to Study Long Non-coding RNA Function in Atherosclerosis. Frontiers in Cardiovascular Medicine feb 2019;6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shira Stav, Ruben M. Atilho, Gayan Mirihana Arachchilage, Giahoa Nguyen, Gadareth Higgs, and Ronald R. Breaker. Genome-wide discovery of structured noncoding RNAs in bacteria. BMC Microbiol, 19(1):1–18, mar 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costa MCSF, de Araújo Oliveira JV, Silva WMC, et al. Machine learning studies of non-coding rnas based on artificially constructed training data. In: BIOINFORMATICS, 2021, 176–83.33812384
- 17. Stefan Washietl, Sven Findeiß, Stephan A. Müller, Stefan Kalkhof, Martin Von Bergen, Ivo L. Hofacker, Peter F. Stadler, and Nick Goldman. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA, 17(4):578–94, apr 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dar D, Sorek R. Bacterial noncoding RNAs excised from within protein-coding transcripts. MBio sep 2018;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waqas Ahmed, Ke Zheng, and Zheng Fei Liu. Small non-coding RNAs: New insights in modulation of host immune response by intracellular bacterial pathogens. Front Immunol, 7(OCT):431, oct 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waring J, Lindvall C, Umeton R. Automated machine learning: Review of the state-of-the-art and opportunities for healthcare. Artif Intell Med 2020;104:101822. [DOI] [PubMed] [Google Scholar]
- 21. Stavridis M, Korfiati A, Sakellaropoulos G, et al. Non-coding rna sequences identification and classification using a multi-class and multi-label ensemble technique. In: IFIP International Conference on Artificial Intelligence Applications and Innovations. Springer, 2018, 179–88. [Google Scholar]
- 22. Chen C-C, Qian X, Yoon B-J. RNAdetect: efficient computational detection of novel non-coding RNAs. Bioinformatics 08 2018;35(7):1133–41. [DOI] [PubMed] [Google Scholar]
- 23. RNAcentral Consortium . RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res 10 2020;49(D1):D212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Sá AGC, Walter José GS, Pinto LO, et al. Recipe: a grammar-based framework for automatically evolving classification pipelines. In: European Conference on Genetic Programming. Springer, 2017, 246–61. [Google Scholar]
- 25. He X, Zhao K, Chu X. Automl: A survey of the state-of-the-art. Knowledge-Based Systems 2021;212:106622. [Google Scholar]
- 26. Santos A, Castelo S, Felix C, et al. (eds). Visus: An interactive system for automatic machine learning model building and curation. In: Proceedings of the Workshop on Human-In-the-Loop Data Analytics, 2019, 1–7.
- 27. Robson Parmezan Bonidia . Feature extraction approaches for biological sequences: A comparative study of mathematical features. https://github.com/Bonidia/FeatureExtraction_BiologicalSequences, 2020. [DOI] [PubMed]
- 28. Bonidia RP, Domingues DS, Sanches DS, et al. Mathfeature: feature extraction package for dna, rna and protein sequences based on mathematical descriptors. Brief Bioinform 2022;23(1):bbab434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frazier PI. A tutorial on bayesian optimization arXiv preprint arXiv:1807.02811. 2018.
- 30. Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 2011;273(1):236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muhammod R, Ahmed S, Farid DM, et al. PyFeat: a Python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics 03 2019;35(19):3831–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khatun MS, Hasan MM, Shoombuatong W, et al. Proin-fuse: improved and robust prediction of proinflammatory peptides by fusing of multiple feature representations. J Comput Aided Mol Des 2020;34(12):1229–36. [DOI] [PubMed] [Google Scholar]
- 33. Khurana U, Turaga D, Samulowitz H, et al. Cognito: Automated feature engineering for supervised learning. In: 2016 IEEE 16th International Conference on Data Mining Workshops (ICDMW), 2016, 1304–7.
- 34. Chen X, Lin Q, Luo C, et al. (eds). Neural feature search: A neural architecture for automated feature engineering. In: 2019 IEEE International Conference on Data Mining (ICDM). IEEE, 2019, 71–80. [Google Scholar]
- 35. Wolpert DH, Macready WG. No free lunch theorems for optimization. IEEE transactions on evolutionary computation 1997;1(1):67–82. [Google Scholar]
- 36. Brazdil PB, van Rijn JN, Soares C, et al. Metalearning: Applications to Automated Machine Learning and Data Mining. Leiden University, Institute of Advanced Computer Science, 2022. [Google Scholar]
- 37. Liu Y, Zhaomin Y, Cheng Chen Y, et al. Prediction of protein crotonylation sites through lightgbm classifier based on smote and elastic net. Anal Biochem 2020;609:113903. [DOI] [PubMed] [Google Scholar]
- 38. Hancock J, Khoshgoftaar TM. Catboost for big data: an interdisciplinary review. Research Square 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He S, Dou L, Li X, et al. Review of bioinformatics in azheimer’s disease research. Comput Biol Med 2022;143:105269. [DOI] [PubMed] [Google Scholar]
- 40. Shen H-B, Chou K-C. Pseaac: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 2008;373(2):386–8. [DOI] [PubMed] [Google Scholar]
- 41. Cao D-S, Xu Q-S, Liang Y-Z. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 02 2013;29(7):960–2. [DOI] [PubMed] [Google Scholar]
- 42. Chen W, Zhang X, Brooker J, et al. PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 09 2014;31(1):119–20. [DOI] [PubMed] [Google Scholar]
- 43. van den Berg BA, Reinders MJT, Roubos JA, et al. Spice: a web-based tool for sequence-based protein classification and exploration. BMC bioinformatics 2014;15(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bin Liu, Fule Liu, Longyun Fang, Xiaolong Wang, and Kuo-Chen Chou. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics, 31(8):1307–9, 12 2014. [DOI] [PubMed] [Google Scholar]
- 45. Chiu T-P, Comoglio F, Zhou T, et al. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics 12 2015;32(8):1211–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu B. Bioseq-analysis: a platform for dna, rna and protein sequence analysis based on machine learning approaches. Brief Bioinform 2017;20(4):1280–94. [DOI] [PubMed] [Google Scholar]
- 47. Li H-L, Pang Y-H, Liu B. Bioseq-blm: a platform for analyzing dna, rna and protein sequences based on biological language models. Nucleic Acids Res 2021;49(22):e129–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jing R, Li Y, Xue L, et al. autobioseqpy: a deep learning tool for the classification of biological sequences. J Chem Inf Model 2020;60(8):3755–64. [DOI] [PubMed] [Google Scholar]
- 49. Liu D, Chi X, He W, et al. Autogenome: an automl tool for genomic research. Artificial Intelligence in the Life Sciences 2021;1:100017. [Google Scholar]
- 50. Le TT, Weixuan F, Moore JH. Scaling tree-based automated machine learning to biomedical big data with a feature set selector. Bioinformatics 2020;36(1):250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eppenhof EJJ, Peña-Castillo L. Prioritizing bona fide bacterial small rnas with machine learning classifiers. PeerJ 2019;7:e6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Almeida BLS, Queiroz AP, Santos APA, et al. Feature importance analysis of non-coding dna/rna sequences based on machine learning approaches. In: Brazilian Symposium on Bioinformatics. Springer, 2021, 81–92. [Google Scholar]
- 53. He W, Ying J, Zeng X, et al. Sc-ncdnapred: a sequence-based predictor for identifying non-coding dna in saccharomyces cerevisiae. Front Microbiol 2018;9:2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie J, Zhang L, Xiao M. A review of artificial intelligence applications in bacterial genomics. In: 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE, 2020, 1870–6. [Google Scholar]
- 55. Barik A, Das S. A comparative study of sequence-and structure-based features of small rnas and other rnas of bacteria. RNA Biol 2018;15(1):95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bar A, Argaman L, Altuvia Y, et al. Prediction of novel bacterial small rnas from ril-seq rna–rna interaction data. Front Microbiol 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barman RK, Mukhopadhyay A, Das S. An improved method for identification of small non-coding rnas in bacteria using support vector machine. Sci Rep 2017;7(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prokhorenkova L, Gusev G, Vorobev A, et al. Catboost: unbiased boosting with categorical features. In: Advances in neural information processing systems, 2018, 6638–48.
- 59. Schapire RE. Explaining adaboost. In: Empirical inference. Springer, 2013, 37–52. [Google Scholar]
- 60. Liaw A, Wiener M. Classification and regression by randomforest. R news 2002;2(3):18–22. [Google Scholar]
- 61. Ke G, Meng Q, Finley T, et al. Lightgbm: A highly efficient gradient boosting decision tree. Advances in neural information processing systems 2017;30. [Google Scholar]
- 62. Bonidia RP, Machida JS, Negri TC, et al. A novel decomposing model with evolutionary algorithms for feature selection in long non-coding rnas. IEEE Access 2020;8:181683–97. [Google Scholar]
- 63. Bergstra J, Yamins D, Cox DD. Hyperopt: A python library for optimizing the hyperparameters of machine learning algorithms. In: Proceedings of the 12th Python in science conference, Vol. 13. Citeseer, 2013, 20. [Google Scholar]
- 64. Helen Victoria A, Maragatham G. Automatic tuning of hyperparameters using bayesian optimization. Evolving Systems 2021;12(1):217–23. [Google Scholar]
- 65. Elsayad AM, Nassef AM, Al-Dhaifallah M. Bayesian optimization of multiclass svm for efficient diagnosis of erythemato-squamous diseases. Biomedical Signal Processing and Control 2022;71:103223. [Google Scholar]
- 66. Turner R, Eriksson D, McCourt M, et al. (eds). Bayesian optimization is superior to random search for machine learning hyperparameter tuning: Analysis of the black-box optimization challenge 2020. In: NeurIPS 2020 Competition and Demonstration Track. PMLR, 2021, 3–26. [Google Scholar]
- 67. Kwang SNG, Loong and Santosh K Mishra. Unique folding of precursor micrornas: quantitative evidence and implications for de novo identification. RNA 2007;13(2):170–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster rna homology searches. Bioinformatics 2013;29(22):2933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kalvari I, Nawrocki EP, Ontiveros-Palacios N, et al. Rfam 14: expanded coverage of metagenomic, viral and microrna families. Nucleic Acids Res 2021;49(D1):D192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ioanna Kalvari, Eric P Nawrocki, Joanna Argasinska, Natalia Quinones-Olvera, Robert D Finn, Alex Bateman, and Anton I Petrov. Non-coding rna analysis using the rfam database. Current protocols in bioinformatics, 62(1):e51, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grandini M, Bagli E, Visani G. Metrics for multi-class classification: an overview arXiv preprint arXiv:2008.05756. 2020.
- 72. Jennifer L, Salzberg SL. Skewit: The skew index test for large-scale gc skew analysis of bacterial genomes. PLoS Comput Biol 2020;16(12):e1008439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang P, Wenyi W, Chen Q, et al. Non-coding rnas and their integrated networks. J Integr Bioinform 2019;16(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tasdelen A, Sen B. A hybrid cnn-lstm model for pre-mirna classification. Sci Rep 2021;11(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xiangzheng F, Zhu W, Cai L, et al. Improved pre-mirnas identification through mutual information of pre-mirna sequences and structures. Front Genet 2019;10:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang D, Zhang Y, Zhao Y. Lightgbm: an effective mirna classification method in breast cancer patients. In: Proceedings of the 2017 International Conference on Computational Biology and Bioinformatics, 2017, 7–11.
- 77. Dang THY, Tyagi S, D’Cunha G, et al. Computational prediction of micrornas in marine bacteria of the genus thalassospira. PloS one 2019;14(3):e0212996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cardin S-E, Borchert GM. Viral micrornas, host micrornas regulating viruses, and bacterial microrna-like rnas. Bioinformatics in MicroRNA Research 2017;39–56. [DOI] [PubMed] [Google Scholar]
- 79. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22(13):1658–9. [DOI] [PubMed] [Google Scholar]
- 80. Sayers EW, Cavanaugh M, Clark K, et al. Genbank. Nucleic Acids Res 2019;47(D1):D94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Balaji A, Allen A. Benchmarking automatic machine learning frameworks arXiv preprint arXiv:1808.06492. 2018.
- 82. Zöller M-A, Huber MF. Benchmark and survey of automated machine learning frameworks. Journal of artificial intelligence research 2021;70:409–72. [Google Scholar]
- 83. Li J-Y, Li W-X, Wang A-T, et al. Mitoflex: an efficient, high-performance toolkit for animal mitogenome assembly, annotation and visualization. Bioinformatics 2021;37(18):3001–3. [DOI] [PubMed] [Google Scholar]
- 84. Chan PP, Lin BY, Mak AJ, et al. trnascan-se 2.0: improved detection and functional classification of transfer rna genes. Nucleic Acids Res 2021;49(16):9077–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. David P. Clark, Nanette J. Pazdernik, and Michelle R. McGehee. Chapter 19 - noncoding rna. In David P. Clark, Nanette J. Pazdernik, and Michelle R. McGehee, editors, Molecular Biology (Third Edition), pages 604–21. Academic Cell, third edition edition, 2019. [Google Scholar]
- 86. Watkins D, Arya DP. Regulatory roles of small rnas in prokaryotes: Parallels and contrast with eukaryotic mirna. Non-coding RNA Investig 2019;3:28. [Google Scholar]
- 87. Soltani-Fard E, Taghvimi S, Kichi ZA, et al. Insights into the function of regulatory rnas in bacteria and archaea. International Journal of Translational Medicine 2021;1(3):403–23. [Google Scholar]
- 88. Streit D, Shanmugam T, Garbelyanski A, et al. The existence and localization of nuclear snornas in arabidopsis thaliana revisited. Plan Theory 2020;9(8):1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lindsay MA, Griffiths-Jones S, Valadkhan S, et al. Role of small nuclear rnas in eukaryotic gene expression. Essays Biochem 2013;54:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
BioAutoML, documentation and datasets are available in the Github repository: https://github.com/Bonidia/BioAutoML.