

Abstract
Single-cell RNA sequencing trades read-depth for dimensionality, often leading to loss of critical signaling gene information that is typically present in bulk data sets. We introduce DURIAN (Deconvolution and mUltitask-Regression-based ImputAtioN), an integrative method for recovery of gene expression in single-cell data. Through systematic benchmarking, we demonstrate the accuracy, robustness and empirical convergence of DURIAN using both synthetic and published data sets. We show that use of DURIAN improves single-cell clustering, low-dimensional embedding, and recovery of intercellular signaling networks. Our study resolves several inconsistent results of cell–cell communication analysis using single-cell or bulk data independently. The method has broad application in biomarker discovery and cell signaling analysis using single-cell transcriptomics data sets.
Keywords: single-cell RNA-seq, cell-signaling, imputation, deconvolution, ADMM, LDA
Introduction
Single-cell RNA sequencing (scRNA-seq) provides insights into the diversity of tissue composition and regulation in multi-cellular organisms [1, 2]. It can also dissect cellular development process using lineage inference [3, 4] or transcriptional dynamics [5–7]. More recently, cell–cell communication analysis with scRNA-seq measurements has drawn increasing attention to reveal the complex multiscale signaling regulations during cell development or disease [8, 9]. These advances were made possible using existing sequencing technologies by embracing a fundamental trade-off between sequencing depth and sequencing breadth [2, 10].
In single-cell data, the possibility of undetected transcripts (commonly referred as the ‘dropout’ phenomenon), which results from various factors such as biological noise, limited capture efficiency or amplification bias [2, 10, 11], can pose serious threats for the performance and validity of downstream analysis. For instance, current analytical tools for cell–cell communication commonly quantify the strengths of intercellular communications through the gene expression values of ligand and receptor pairs in corresponding sender or receiver cells [8, 12, 13]. Therefore, dropout of key signaling ligand genes [8, 14] in single-cell data sets may affect the accurate inference of cellular interaction relations. Historically, confirmation of these dropout events involves analysis of traditional bulk RNA sequencing data sets, where the measurements of important genes are likely to remain, due to greater read depth and immunity to biological sources of dropout such as stochastic gene expression because of averaging effects [2, 15]. On the other hand, bulk data lack the resolution to distinguish the specific celltypes in tissues. An important strategy called deconvolution [16–19] is necessary to resolve the heterogenous celltypes in bulk data sets. With the increasing availability of data sets containing both measurements of single-cell and bulk sequencing in clinical studies [20, 21], it is also necessary to develop computational methods to integrate the single-cell and bulk data sets efficiently and effectively.
To deal with dropouts and impute single-cell data, many algorithms have been developed. The pooling approach implemented scran R package used a hierarchical model of sparsity to address biological sources of dropout at the normalization stage [22]. The SCDE and ZINB-WAVE packages explicitly account for dropout at the differential-expression stage [15, 23]. ZIFA [24], URSM [25], CIDR [26] and scVI [27] represent dropout within a dimensionality-reduction framework. Methods such as ScImpute [28], VIPER [29] and SAVER [30] fit a sparse regression model whose response is the observed expression value.
In addition, McImpute [31] and PBLR [32] exploit the low-rank structure of single-cell data to perform imputation of dropout reads. However, these algorithms focus solely on the single-cell data, lacking the ability to enforce consistency with bulk data sets.
Although we present DURIAN (Deconvolution and mUltitask-Regression-based ImputAtioN) primarily as a tool to enhance downstream analysis via dropout imputation, the simultaneous deconvolution of bulk data by DURIAN is essential to this task. Existing approaches for bulk data deconvolution rely either on marker-gene specification to constrain the output of an unsupervised method or on additional data sets to allow supervised learning. Semi-supervised methods such as CellMix utilize specific markers for non-negative matrix factorization [33, 34], whereas mixture model-based methods such as RNA-Sieve and BayICE aim to perform variable selection implicitly [35, 36]. Supervised methods such as MuSiC [37] and Bisque [38] use weighted non-negative least-squares (NNLS) regression to estimate the bulk celltype proportions by incorporating multiple single-cell data sets with gene-specific weighting or corrections, and SCDC [39] proposes the ensemble approach for deconvolution across multiple single-cell references. High-confidence prior knowledge about marker genes or multiple single-cell reference data sets are important to the robust performance of current deconvolution algorithms [19].
Recently, new methods have been proposed to impute single-cell data by utilizing bulk measurements. URSM [25] fits a unified generative model of single-cell and bulk measurements, which involves specific assumptions about dropout mechanisms and counts distributions, yet to be justified for data sets generated from different sequencing platforms or biological systems. SCRABBLE [40] utilizes the data-driven approach of low-rank matrix completion, constraining the imputation by integrating bulk and single-cell data sets to enforce consistency between the average imputed expression of each gene and its bulk-sequencing counterpart, though the heterogeneous celltype fractions in bulk samples have not been fully accounted for. Thus, an efficient and flexible computational framework to simultaneously and explicitly accommodate single-cell imputation and bulk deconvolution is still lacking.
Here, we introduce the modular, iterative learning framework DURIAN, which imputes single-cell data with more efficient use of bulk data by taking deconvoluted celltype profiles into consideration. To facilitate this, DURIAN alternates between the deconvolution of bulk data sets and the imputation of single-cell data sets (Figure 1). In the deconvolution step, the bulk celltype fractions are estimated with respect to imputed single-cell biomarkers; whereas in the imputation step, the sparse single-cell expression matrix is further refined under the guidance of deconvoluted celltype fractions and expression values in bulk data sets. We designed the benchmarking on both synthetic and downsampled real data to reveal the accuracy and robustness of DURIAN and inspected the empirical convergence of the iterative algorithm. We then applied DURIAN to clinical and experimental data sets, demonstrating its capability to improve signals in single-cell data and highlighting its unique integration of single-cell and bulk data sets for cellular communication analysis.
Figure 1.

Schematic of DURIAN method. (A) Input: single-cell expression data (m × n). (B) Input: bulk expression data (m × w). (C) Deconvolution of bulk data, yielding a matrix of celltype proportions (w × k). (D) Construct deconvolution map (m × w) where each column corresponds to the pseudobulk average of the current imputation result according to the celltype ratios calculated in (C). (E) Update the imputation objective with the current deconvolution map. Perform imputation to replace zeros in the single-cell expression data (m × n) with estimated counts. Finally, update the deconvolution model with the imputation estimate. (F) Output: final deconvolution matrix of celltype proportions (w × k). (G) Output: final estimate of single-cell data with dropout replaced by imputed reads. (H) Application: use imputed single-cell data to directly compute cell-signaling patterns.
With respect to our benchmarking of DURIAN against existing imputation methods, we focus on approaches which encompass both similar and distinct approaches to our own: smoothing approaches that perform naive clusterin on the data to generate a reference profile (DrImptue), convex rank-minimization methods (SCRABBLE, DURIAN, CMF-Impute), hierarchical generative methods that simulate missing reads from a posterior predictive distribution, based on some causal hypothesis about the data (URSM, DURIAN-dsLDA), methods that utilize bulk data to regularize imputation estimates (SCRABBLE, URSM, DURIAN), iterative methods that involve structure-recovery of paired bulk data to aid dropout-detection (DURIAN, URSM) and graph-based smoothing methods (G2S3).
Results
Overview of DURIAN
Input for DURIAN includes a single-cell data set and a second bulk (or pseudobulk) data set (Figure 1A and B). At each iteration, DURIAN first computes the deconvolution, an estimate of the percentage of each celltype in the bulk data (Figure 1C). Either Gibbs sampling or non-negative regression may be used for deconvolution, depending on the research priority and available metadata. The scheme under this modular approach is outlined in Algorithms 1 and 2 (Supplementary Text), following notation provided in Supplemental Methods. Regardless of algorithm selection, deconvolution of the bulk data is supervised by the original single-cell data during the first iteration and by the current single-cell imputation estimate during all subsequent iterations. Following deconvolution, the deconvolution map is constructed (Figure 1D). The deconvolution map converts the current imputation estimate to a pseudobulk reference for the observed bulk data via the deconvolution estimate (Supplemental Methods). Next, the imputation objective is updated based on the deconvolution map and the minimum-rank imputation estimate is computed (Figure 1E). This imputation is constrained by the difference between the continuously updated pseudobulk reference and the observed bulk data (Supplemental Methods). The next DURIAN iteration then begins with deconvolution of the bulk (Figure 1C), supervised by the newly imputed single-cell data. Stopping criteria are satisfied when the optimized objectives of two successive DURIAN iterations are within tolerance, or a maximum number of iterations has been performed. Empirically, this scheme leads to correlated improvement in both imputation and deconvolution as DURIAN converges (Figure S1). The final output includes both the final deconvolution of the bulk data and the imputed single-cell data (Figure 1F and G). Direct applications of this output include inference of intercellular communication networks (Figure 1H), based on complete data.
Iterative imputation scheme
Imputation of single-cell data in DURIAN fits a convex optimization objective utilizing a tissue-matched bulk data set to improve accuracy. Here 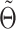 represents the deconvolution of the bulk data
represents the deconvolution of the bulk data 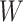 (Equation 1), which is initialized using raw single-cell data, and based on the imputation estimate
(Equation 1), which is initialized using raw single-cell data, and based on the imputation estimate 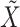 during subsequent iterations. Once
during subsequent iterations. Once 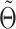 is estimated, we update the deconvolution map
is estimated, we update the deconvolution map 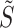 , a matrix whose individual columns represent the pseudobulk reference for the corresponding bulk sample in
, a matrix whose individual columns represent the pseudobulk reference for the corresponding bulk sample in 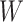 (Equation 2). Finally, dropout reads in
(Equation 2). Finally, dropout reads in 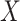 are imputed based on the singular-value thresholding scheme described in [41], which is closely related to the ADMM implementation described in [40]. The 1st term in Equation 3 (
are imputed based on the singular-value thresholding scheme described in [41], which is closely related to the ADMM implementation described in [40]. The 1st term in Equation 3 (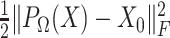 ) penalizes the imputation
) penalizes the imputation 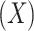 of nonzero entries recorded in the original unimputed data (
of nonzero entries recorded in the original unimputed data (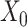 ) by projecting
) by projecting 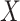 onto
onto 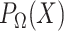 . The projection operator
. The projection operator 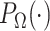 sets any entry in the argument to zero which was zero in the
sets any entry in the argument to zero which was zero in the 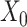 before imputation. Thus, if any nonzero entry in the original data
before imputation. Thus, if any nonzero entry in the original data 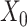 is altered, term 1 of Equation 3 will be positive and zero otherwise. In contrast to SCRABBLE, we utilize a deconvolution map to pull information from celltype-specific gene expression signatures in the bulk data via the 3rd term in Equation 3. In particular, the parameter
is altered, term 1 of Equation 3 will be positive and zero otherwise. In contrast to SCRABBLE, we utilize a deconvolution map to pull information from celltype-specific gene expression signatures in the bulk data via the 3rd term in Equation 3. In particular, the parameter 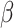 (Equation 3) penalizes the discrepancy between the expression levels
(Equation 3) penalizes the discrepancy between the expression levels 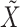 estimated by imputation and observed bulk data
estimated by imputation and observed bulk data 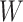 . We also include the standard parameters for convex matrix completion [40, 41] in Equation 3: the parameter
. We also include the standard parameters for convex matrix completion [40, 41] in Equation 3: the parameter 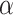 penalizes the rank of the imputed data
penalizes the rank of the imputed data  , and the parameter
, and the parameter 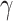 controls the ADMM learning rate. Mathematical details of the two deconvolution approaches we address under this scheme (including our novel topic model), in addition to pseudocode for DURIAN under these two approaches, are provided in the Supplemental Methods.
controls the ADMM learning rate. Mathematical details of the two deconvolution approaches we address under this scheme (including our novel topic model), in addition to pseudocode for DURIAN under these two approaches, are provided in the Supplemental Methods.
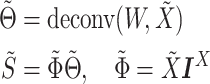 |
(1) |
 |
(2) |
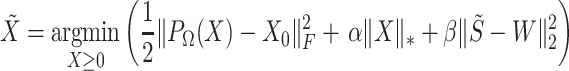 |
(3) |
Imputation benchmarks for downsampled and synthetic data
We compared the performance of DURIAN to existing imputation methods using two standard benchmarking approaches: downsampled scRNA-seq data to introduce zeros at known locations [28, 40] and simulating single-cell data via the Splatter method [42]. For each benchmarking strategy we compare the performance of six algorithms: DrImpute [43], SCRABBLE [40], URSM [25] (scaled posterior celltype distributions as described in Methods), mtSCRABBLE (a single iteration of DURIAN with a deconvolution map based on the known celltype percentages in the bulk), DURIAN [MuSiC] and DURIAN [dsLDA]. The performance of both DURIAN and SCRABBLE is dependent upon the optimization parameters 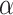 ,
,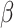 and
and 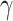 (see Methods). In both methods, the
(see Methods). In both methods, the 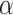 parameter governs the relative importance of low-rank, which favors smaller numbers of implicit cell clusters, and
parameter governs the relative importance of low-rank, which favors smaller numbers of implicit cell clusters, and 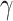 determines the relative rate of convergence (ADMM step-size). In DURIAN, the
determines the relative rate of convergence (ADMM step-size). In DURIAN, the 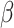 parameter uniquely controls the relative importance of the deconvolution in the imputation estimate (Figure S4). For each benchmarking task below, we provide results for DURIAN, mtSCRABBLE and SCRABBLE that utilize a moderate
parameter uniquely controls the relative importance of the deconvolution in the imputation estimate (Figure S4). For each benchmarking task below, we provide results for DURIAN, mtSCRABBLE and SCRABBLE that utilize a moderate 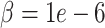 value and a high
value and a high 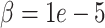 value. The high value forces the imputation to lean more on celltype information present in the bulk data.
value. The high value forces the imputation to lean more on celltype information present in the bulk data.
Benchmarking with downsampling
Downsampling is a common strategy for imputation benchmarking that takes real single-cell expression data as input and subsequently sets individual entries to zero [28, 40]. Previous downsampling benchmarks utilized a constant gene-wise dropout probability across single-cell donors, which does not reproduce the batch-related dropout effects introduced via the separate sequencing of biological replicates, which has previously been reported as a common source of technical bias [44]. We resolve this issue by utilizing donor-specific dropout rates rather than imposing a single, gene-wise dropout probability. Our approach sets the observed value to zero with probability 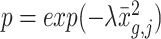 where
where 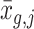 is the mean expression of gene
is the mean expression of gene 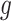 in single-cell donor
in single-cell donor 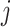 . Here
. Here 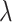 represents the dropout rate, given the single-cell sample, where lower values of
represents the dropout rate, given the single-cell sample, where lower values of 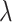 correspond to higher dropout rates. For each of three dropout rates (
correspond to higher dropout rates. For each of three dropout rates (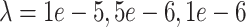 ), we ran imputation on 50 data replicates each composed of 1000 randomly selected genes and 800 randomly selected islet (alpha/beta/gamma/delta/epsilon) cells taken from published single-cell data from healthy adult human pancreatic tissue [45]. In this tissue, the differentiated celltypes provide a strong foothold for deconvolution [37], and this represents the best-case test for DURIAN’s performance compared with other methods. We present the cell-wise mean imputation error (log RMSE), and the L2 imputation error in Figures 2A and 3A, respectively. In each case, the average sparsity of the the three tested
), we ran imputation on 50 data replicates each composed of 1000 randomly selected genes and 800 randomly selected islet (alpha/beta/gamma/delta/epsilon) cells taken from published single-cell data from healthy adult human pancreatic tissue [45]. In this tissue, the differentiated celltypes provide a strong foothold for deconvolution [37], and this represents the best-case test for DURIAN’s performance compared with other methods. We present the cell-wise mean imputation error (log RMSE), and the L2 imputation error in Figures 2A and 3A, respectively. In each case, the average sparsity of the the three tested 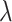 values is shown in the 2nd-level horizontal facet on the right.
values is shown in the 2nd-level horizontal facet on the right.
Figure 2.
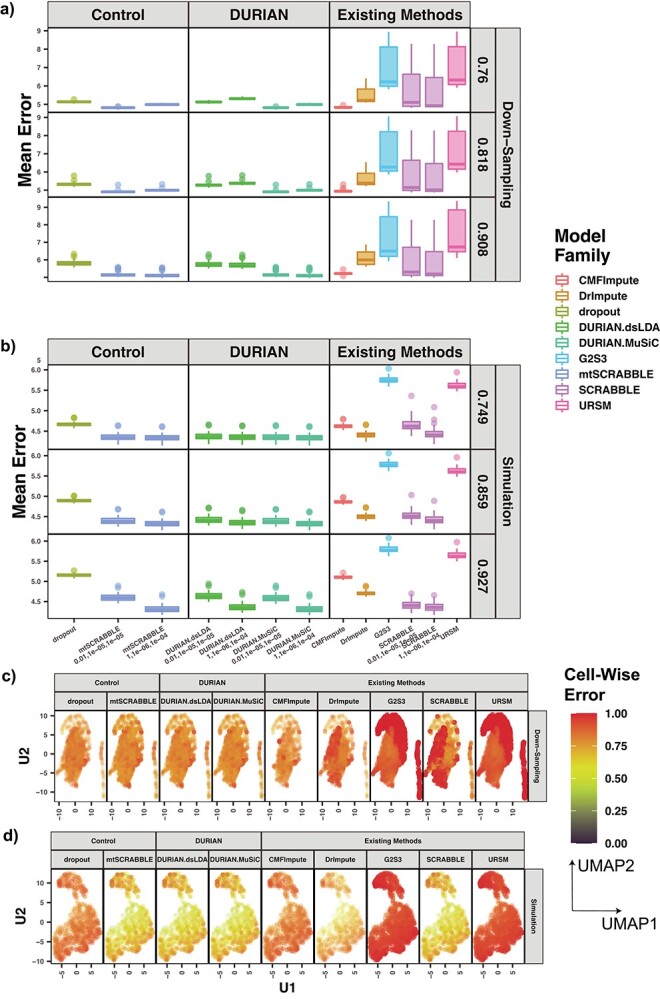
Mean error of imputation benchmarks. (A-B) Mean error (log RMSE) of imputation for both synthetic data strategies: downsampling and simulation. For each strategy, the mean sparsity (percentage of zeros in the unimputed data) of all replicates corresponding to each of three values for the strategy-specific dropout parameter is shown in the 2nd level vertical label: 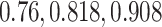 (downsampling),
(downsampling), 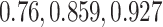 (simulation). The x-axis is arranged into three method categories (shown in top label): control, DURIAN, existing methods. Control methods include dropout: unimputed data and mtSCRABBLE: the DURIAN algorithm where the deconvolution map is permanently set according to the true bulk celltype percentages. Both dsLDA and NNLS (MuSiC) deconvolution approaches are included for DURIAN benchmarks. Two sets of values for the ADMM parameters of the SCRABBLE/DURIAN objective are provided for DURIAN, SCRABBLE and mtSCRABBLE:
(simulation). The x-axis is arranged into three method categories (shown in top label): control, DURIAN, existing methods. Control methods include dropout: unimputed data and mtSCRABBLE: the DURIAN algorithm where the deconvolution map is permanently set according to the true bulk celltype percentages. Both dsLDA and NNLS (MuSiC) deconvolution approaches are included for DURIAN benchmarks. Two sets of values for the ADMM parameters of the SCRABBLE/DURIAN objective are provided for DURIAN, SCRABBLE and mtSCRABBLE: 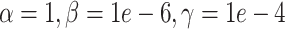 and
and 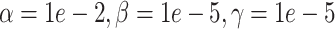 . (C) Scatter plots of a UMAP embedding for a replicate of the downsampling strategy at sparsity
. (C) Scatter plots of a UMAP embedding for a replicate of the downsampling strategy at sparsity 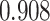 , color corresponds to the centered and scaled logistic transformation of cellwise RMSE. (D) Scatter plots of a UMAP embedding for a replicate from the simulation strategy at mean sparsity
, color corresponds to the centered and scaled logistic transformation of cellwise RMSE. (D) Scatter plots of a UMAP embedding for a replicate from the simulation strategy at mean sparsity 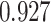 . DURIAN parameters for C–D are
. DURIAN parameters for C–D are 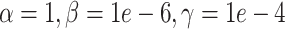 .
.
For down-sampled data, DURIAN (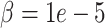 ) achieves significantly-lower mean error than other methods at the two lower dropout levels (Tables S13–16). For very sparse down-sampled data, DURIAN (
) achieves significantly-lower mean error than other methods at the two lower dropout levels (Tables S13–16). For very sparse down-sampled data, DURIAN (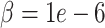 ) still acheives lower mean error than other methods but is not significantly lower than CMFImpute (Tables S17–18). Each data point in Figure 2A represents the log mean error across the entire data set. To better see how the error may relate to the underlying celltype, the mean across all genes in each individual cell for a single, representative downsampled replicate is depicted graphically in Figure 2C. Here, a single, unimputed replicate from the maximum dropout group (
) still acheives lower mean error than other methods but is not significantly lower than CMFImpute (Tables S17–18). Each data point in Figure 2A represents the log mean error across the entire data set. To better see how the error may relate to the underlying celltype, the mean across all genes in each individual cell for a single, representative downsampled replicate is depicted graphically in Figure 2C. Here, a single, unimputed replicate from the maximum dropout group (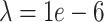 , mean sparsity
, mean sparsity 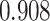 ) was embedded via UMAP [46] and each cell is colored according to the error resulting from each of the imputation methods. In Figure 2C we can see that DURIAN is more resistant than other methods to celltype related bias in dropout, as depicted by the relatively uniform coloring of the UMAP embedding. In contrast, both DrImpute and SCRABBLE show much higher celltype dependent error in the left-most lobe of the UMAP embedding, whereas URSM shows higher celltype dependent error in the right-most lobe of the embedding.
) was embedded via UMAP [46] and each cell is colored according to the error resulting from each of the imputation methods. In Figure 2C we can see that DURIAN is more resistant than other methods to celltype related bias in dropout, as depicted by the relatively uniform coloring of the UMAP embedding. In contrast, both DrImpute and SCRABBLE show much higher celltype dependent error in the left-most lobe of the UMAP embedding, whereas URSM shows higher celltype dependent error in the right-most lobe of the embedding.
In contrast to the average error, which provides a broad picture of how well imputation performs regardless of underlying celltypes or the individual genes, the L2 error (the largest singular value of the error matrix) shows how well the imputed data match the true data when considering the imputed data as a low-rank approximation to the true data [47]. We found that DURIAN’s measured improvement with respect to the other approaches, across multiple benchmarking strategies, was statistically significant (Tables S13 and S14). For downsampled data in particular, we found that for individual dropout levels DURIAN achieves significantly lower L2 error when the 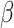 parameter is increased from
parameter is increased from 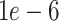 to
to 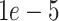 (Figure 3A and Tables S15 and S16). For very sparse data (corresponding to
(Figure 3A and Tables S15 and S16). For very sparse data (corresponding to 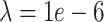 ) DURIAN’s L2 error is significantly lower than all other methods when
) DURIAN’s L2 error is significantly lower than all other methods when 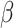 is increased forcing the imputation estimate to rely more heavily on celltype information present in the bulk, which DURIAN obtains via deconvolution (Tables S21 and S22). For the lowest dropout level, the L2 error of DURIAN is slightly higher than the tested smoothing methods on down-sampled data (Tables S17–20). These results also show that our novel topic-model based deconvolution approach (dsLDA) provides a slight advantage in L2 error rate at lower dropout levels. A complementary perspective to the cell-specific L2 error across genes, is the gene-specific average error across cells via an adaptation of the MA plot for bulk sequencing [48]. Each of the facets of Figure 3C show the gene-wise error of the true versus imputed version of a single downsampled replicate for the respective method. For each method, log ratio of true versus imputed gene-wise average over the log average of true and imputed gene-wise counts is plotted, with overestimates highlighted in red and underestimates highlighted in blue, with error threshold of
is increased forcing the imputation estimate to rely more heavily on celltype information present in the bulk, which DURIAN obtains via deconvolution (Tables S21 and S22). For the lowest dropout level, the L2 error of DURIAN is slightly higher than the tested smoothing methods on down-sampled data (Tables S17–20). These results also show that our novel topic-model based deconvolution approach (dsLDA) provides a slight advantage in L2 error rate at lower dropout levels. A complementary perspective to the cell-specific L2 error across genes, is the gene-specific average error across cells via an adaptation of the MA plot for bulk sequencing [48]. Each of the facets of Figure 3C show the gene-wise error of the true versus imputed version of a single downsampled replicate for the respective method. For each method, log ratio of true versus imputed gene-wise average over the log average of true and imputed gene-wise counts is plotted, with overestimates highlighted in red and underestimates highlighted in blue, with error threshold of 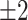 . These plots clearly show that DURIAN’s use of celltype expression profiles in the bulk prevent overestimation observed in SCRABBLE, a trend which was also reported previously [40].
. These plots clearly show that DURIAN’s use of celltype expression profiles in the bulk prevent overestimation observed in SCRABBLE, a trend which was also reported previously [40].
Figure 3.
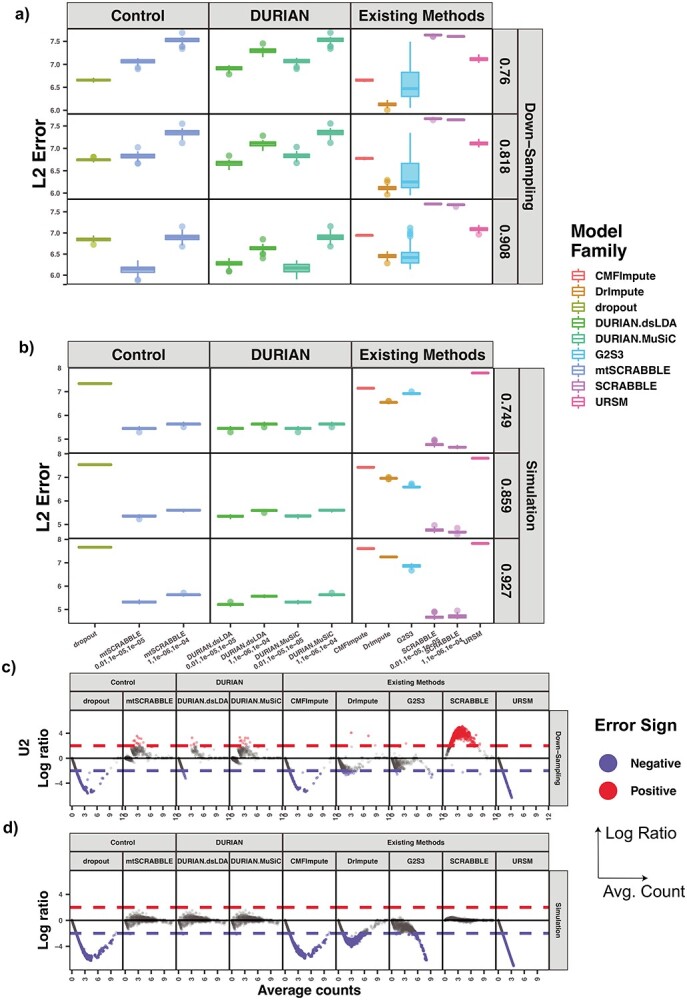
L2 error of Imputation Benchmarks. (A-B) The y-axis is the log L2 norm of difference between the imputed data and the true data, for both synthetic data strategies: downsampling and simulation. For each strategy, the mean sparsity (percentage of zeros in the unimputed data) of all replicates corresponding to each of three values for the strategy-specific dropout parameter is shown in the 2nd level vertical label: 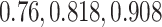 (downsampling),
(downsampling), 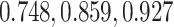 (simulation). The x-axis is arranged into three method categories (shown in top label): Control, DURIAN, Existing Methods. Control methods include dropout: unimputed data, and mtSCRABBLE: the DURIAN algorithm where the deconvolution map is permanently set according to the true bulk celltype percentages. Both dsLDA and NNLS (MuSiC) deconvolution approaches are included for DURIAN benchmarks. Two sets of values for the SCRABBLE objective are provided for DURIAN, SCRABBLE and mtSCRABBLE:
(simulation). The x-axis is arranged into three method categories (shown in top label): Control, DURIAN, Existing Methods. Control methods include dropout: unimputed data, and mtSCRABBLE: the DURIAN algorithm where the deconvolution map is permanently set according to the true bulk celltype percentages. Both dsLDA and NNLS (MuSiC) deconvolution approaches are included for DURIAN benchmarks. Two sets of values for the SCRABBLE objective are provided for DURIAN, SCRABBLE and mtSCRABBLE: 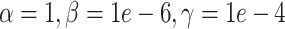 and
and 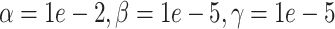 . (C) MA plots for a replicate of the downsampling strategy at sparsity
. (C) MA plots for a replicate of the downsampling strategy at sparsity 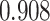 . The y-axis is the log ratio of true vs imputed gene-wise average. The x-axis is the log average over true and imputed gene-wise counts. (D) MA plots for a replicate of the simulation strategy at mean sparsity
. The y-axis is the log ratio of true vs imputed gene-wise average. The x-axis is the log average over true and imputed gene-wise counts. (D) MA plots for a replicate of the simulation strategy at mean sparsity 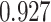 . DURIAN parameters for B–C are
. DURIAN parameters for B–C are  .
.
Benchmarking on synthetic data
In contrast to the downsampled pancreatic data above, a continuum of very closely related celltypes, in addition to batch effects provides a worst-case example of DURIAN’s performance relative to other methods. In this benchmark, batch effects separate simulated single-cell samples as well as the simulated bulk samples. These challenges are compounded by the decreased distinction between individual celltypes based on a poorly differentiated lineage. The splatter R package [42] implements a zero-inflated generative model of single-cell library simulation. This model is a standard platform for assessing the effects of biological and technical noise that has been repeatedly validated [49–51]. We used splatter to simulate 50 single-cell data sets, each composed of 1600 cells and 1000 genes, from four celltypes in a simulated lineage, subject to dropout rates defined by the logistic midpoint parameter (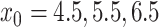 ), where higher values of
), where higher values of 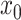 correspond to higher dropout rates. We used splatter’s batch effect model to generate four batches for each simulation and reserved two batches each for the single-cell and pseudobulk data. For each simulated replicate, the two pseudobulk samples were constructed by taking the mean across all cells bearing their respective batch IDs. Like the downsampling approach above, we present the cell-wise mean imputation error (log RMSE) in Figure 2B and the corresponding L2 error in Figure 3B. In each case, the average sparsity of the three tested
correspond to higher dropout rates. We used splatter’s batch effect model to generate four batches for each simulation and reserved two batches each for the single-cell and pseudobulk data. For each simulated replicate, the two pseudobulk samples were constructed by taking the mean across all cells bearing their respective batch IDs. Like the downsampling approach above, we present the cell-wise mean imputation error (log RMSE) in Figure 2B and the corresponding L2 error in Figure 3B. In each case, the average sparsity of the three tested 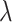 values is shown in the 2nd-level horizontal facet of Figures 2 and 3.
values is shown in the 2nd-level horizontal facet of Figures 2 and 3.
For simulated data, non-parametric tests over the combined data at all dropout levels reveal that DURIAN has significantly lower mean error than existing methods (Tables S11 and S12). This favorable performance gap appears to widen when 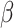 is decreased to
is decreased to 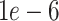 , enforcing a weaker reliance on the bulk data.
, enforcing a weaker reliance on the bulk data.
Furthermore, even when a closer match to the deconvolution is enforced at 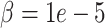 , only SCRABBLE achieves a lower mean error than DURIAN (Figure 2B and Tables S11 and S12). The graphical representation of mean error for simulated data in Figure 2D clearly shows that splatter simulation is less challenging to other methods, relative to DURIAN than the downsampling approach based on real data above. More importantly, the UMAP embedding plotted in Figure 2D shows that globally, the four simulated celltypes are more closely related and less distinct than the adult islet cell populations in the downsampled real data. Therefore, it is expected that DURIAN’s utilization of population structure detected in the bulk data will be less important when imputing data where the global distinction between celltypes is lost.
, only SCRABBLE achieves a lower mean error than DURIAN (Figure 2B and Tables S11 and S12). The graphical representation of mean error for simulated data in Figure 2D clearly shows that splatter simulation is less challenging to other methods, relative to DURIAN than the downsampling approach based on real data above. More importantly, the UMAP embedding plotted in Figure 2D shows that globally, the four simulated celltypes are more closely related and less distinct than the adult islet cell populations in the downsampled real data. Therefore, it is expected that DURIAN’s utilization of population structure detected in the bulk data will be less important when imputing data where the global distinction between celltypes is lost.
Even under this more challenging scenario, DURIAN's L2 norm error is not significantly (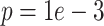 ) lower than SCRABBLE and significantly lower than other methods (Tables S23 and S24). This is consistent with the fact that SCRABBLE, while unable to use celltype-specific gene expression to its advantage when such global information is rich, is also immune to very challenging deconvolution tasks. The overall trends shown in the MA plots (error ratio over average expression) of splatter simulated data in Figure 3D mirror those for downsampled data in Figure 3D above, except that SCRABBLE does not overestimate the counts as badly in this plot. We attribute this difference to the fact that SCRABBLE’s regularizer based on the average bulk counts is more effective for very closely related celltypes in this simulated lineage.
) lower than SCRABBLE and significantly lower than other methods (Tables S23 and S24). This is consistent with the fact that SCRABBLE, while unable to use celltype-specific gene expression to its advantage when such global information is rich, is also immune to very challenging deconvolution tasks. The overall trends shown in the MA plots (error ratio over average expression) of splatter simulated data in Figure 3D mirror those for downsampled data in Figure 3D above, except that SCRABBLE does not overestimate the counts as badly in this plot. We attribute this difference to the fact that SCRABBLE’s regularizer based on the average bulk counts is more effective for very closely related celltypes in this simulated lineage.
Overall, benchmarking of DURIAN on both the best- and worst-case scenarios suggests that the method can accurately address the simultaneous tasks of single-cell imputation and bulk-deconvolution and is robust to concrete selection of deconvolution schemes. Furthermore, we found that when results were pooled across simulation strategies and dropout rates, DURIAN mean and L2 error were significantly lower 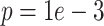 than the the other methods (Tables S1,S2 and S13,S14, respectively). This is due to DURIAN’s more consistent performance across downsampled and simulated benchmarks, especially in contrast to SCRABBLE (Figure 3A-B). Finally, we note that because Splatter’s ZINB generative model is distinct from the logistic midpoint thresholding employed by downsampling, we have empirically determined the values of the parameters for both of these strategies such that approximately the same three empirical dropout levels are observed.
than the the other methods (Tables S1,S2 and S13,S14, respectively). This is due to DURIAN’s more consistent performance across downsampled and simulated benchmarks, especially in contrast to SCRABBLE (Figure 3A-B). Finally, we note that because Splatter’s ZINB generative model is distinct from the logistic midpoint thresholding employed by downsampling, we have empirically determined the values of the parameters for both of these strategies such that approximately the same three empirical dropout levels are observed.
DURIAN improves clustering quality for real data
The downsampling approach described above directly measures imputation performance on data that is as close as possible to real-world cases. Meanwhile, imputation is also expected to improve the score of raw single-cell data for some standard cluster-quality metrics. In Figure 4a, we present five standard quality metrics (described below) [52] for DURIAN, SCRABBLE and DrImpute applied to adult pancreas (healthy and diabetic), adult skin (healthy and eczema) and embryonic (E13.5) mouse skin. The intracluster spanning-tree widest gap (cwidegap) takes the spanning tree computed for each cluster and calculates the longest distance between two cells in the tree. The cluster diameter is the longest distance between any two cells in a single cluster. For both of these metrics, DURIAN and DrImpute are equivalent on both of the pancreatic data sets, whereas DURIAN has the largest clustering quality on two of the three remaining data sets. The Dunn index (dunn) is the minimum separation over the maximum diameter for the data set. For this metric, DURIAN is equivalent to DrImpute on both pancreatic data sets, and on diseased skin, in the meantime yields better clustering quality for both healthy skin and embryonic skin. The silhouette index (sindex) is the mean of the bottom 10% of the distances from every cell in a cluster to the closest cell not in the same cluster. DURIAN is consistent with DrImpute for both pancreatic data sets, as well as healthy skin, and achieves the improved clustering quality for both diseased skin and embryonic skin. The sum-of-squares distance (within.cluster.ss) is half the sum of the squared distances for each pair of cells in the cluster divided by the cluster size. For this metric DURIAN is equivalent to DrImpute for both pancreatic tissue and embryonic skin, and achieved satisfactory results for both adult skin data sets.
Figure 4.

Quantitation of cluster-quality for imputated single-cell data (A) Single-cell data from adult human pancreas (healthy and diabetic) and skin (healthy and eczema), and mouse embryonic skin was imputed via DrImpute, SCRABBLE and DURIAN. The y-axis corresponds to the log of the following cluster statistics: ‘cwidegap’ (lower is better): the longest edge in the within-cluster spanning tree of each cluster (each data point corresponds to a single cluster), ‘diameter’ (lower is better) the longest distance between two cells of a single cluster (each data point corresponds to a single cluster), ‘dunn’ (higher is better): each point represents the Dunn index corresponding to the minimum separation verus maximum diameter for two (possibly the same) clusters in the corresponding data set, ‘sindex’ (higher is better): the Silhouette Index corresponding to the mean of the bottom 10% of the distances from every cell in a cluster to the closest cell not in the same cluster (each data point corresponds to a single cluster), ‘within.cluster.ss’ (lower is better): half the sum of the squared distances for each pair of cells in the cluster divided by the cluster size (each data point corresponds to a single cluster). (B–C) Scatter plots of the imputed result from each data set, colored by the published label.
In addition to the quantitative cluster-quality metrics above, we also compared the robustness of DURIAN’s output with respect to the global structure of the data by inspecting the low-dimensional embeddings of imputed data sets. Figures 4B and C show scatter plots for the UMAP embeddings of each imputation method on the human skin and pancreatic data analyzed in Figure 4A. From these figures, it is obvious that a fair amount of global heterogeneity already exists between mature islet cells in the raw data (shown as ‘dropout’ method) for pancreatic tissue (Figure 4B). In this case, we qualitatively measure the success of imputation based on how well it preserves this separation in the low-dimensional embedding, which could easily be obscured if error was introduced through overestimation. Conversely, the skin data set (Figure 4C) is composed of many celltypes, some of which are not well separated and were identified via clustering on a subset of the data in the original report [14]. Here, we can see that both DURIAN and DrImpute improve the global separation between celltypes, including closely related fibroblasts that were found to express APOE and FBN1 in a mutually exclusive fashion [8, 53], in the cluster approximately centered at (0,-5) in the common UMAP embedding of Figure 4C. Overall, DURIAN improves the quality of unsupervised clustering on single-cell data sets, but can also reveal separation between distinct celltypes not present in the original data.
DURIAN enhances intracellular ligand recovery in embryonic skin
Intercellular signaling relies on the presentation of one or more ‘sender’ molecules (ligands) by one cell, the presentation of one or more ‘receiver’ molecules (receptors and co-receptors) by another cell. The expression of these molecules in general, and ligands in particular, is vital to the cell communication analysis using single-cell data. For instance, CellChat determines significant intercellular interactions via a resampling approach across potential signaling partners such that all possible signaling events between two cells, which are recapitulated by less than a threshold percentage of randomized samples (default is 5%) are declared significant [8]. However, the expression of signaling genes is notoriously difficult to detect in single-cell data from tissues where bulk sequencing and other molecular assays have shown activation of their respective pathways [8, 54]. We reason that DURIAN’s approach to single-cell data imputation using bulk data deconvolution can resolve the challenge and inconsistency in cell communication analysis. We demonstrate the application of imputation to a popular biological domain: embryonic development and highlight specific pathway findings for ncWNT.
We first compared the cell signaling analysis performance using the imputed single-cell data by DURIAN and DrImpute (as baseline control without bulk data) from mouse E13.5 embryonic skin [14]. In Figure 5A, each row of the bar plot shows a pathway for which the relative abundance of predicted interactions for both DURIAN and DrImpute is depicted as a percentage of all recovered interactions for that pathway for the respective method. DURIAN dominates the recovery of signaling interactions in all 26 pathways except three (including MK and PTN, for which the two methods are tied). The remaining 23 pathways that highlighted DURIAN’s recovery include noncanonical WNT (ncWNT) signaling, which is broadly implicated in skin pattern formation at E13.5 [55, 56].
Figure 5.
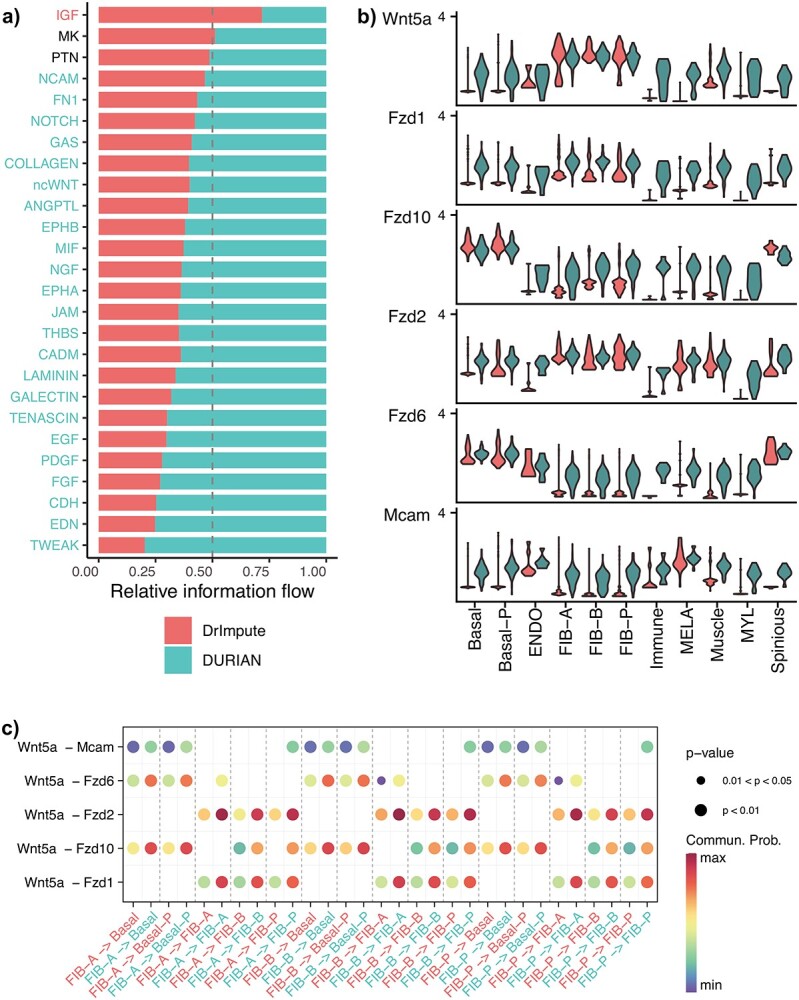
Quantitative comparison of imputation via DURIAN and DrImpute on mouse embryonic skin . (A–C) CellChat comparative analysis of signaling in mouse E13.5 embryonic skin. (A) Relative information flow (higher is better) on each pathway (for which significant interactions were detected), defined as the sum of communication probability among all pairs of cell groups in the inferred network. (B) Violin plots for markers in the noncanonical Wnt pathway after imputation by either DrImpute (in red) or DURIAN (in green), units of expression are log(counts+1). (C) Bubble plots showing the communication probability from individual fibroblast clusters to individual basal cell clusters. Colors on the x-axis labels denote imputation by either DrImpute (in red) or DURIAN (in green). For each row, adjacent bubble pairs within a set of vertical dashed lines represent signaling detected by DrImpute versus DURIAN.
Next, we explored the imputation effects of key markers in important pathways. The CellChat database contains a partial list of ligands, receptors, co-receptors and other molecular species for each available pathway. Figure 5B shows the relative imputed expression of all ncWNT pathway markers involved in predicted interactions for the data set. Here we can see that DURIAN recovers ligand expression more broadly and at higher magnitude than Dr Impute, and provides the enhancement of all ligands expression in several celltypes (immune, myeloid, melanocyte) for which DrImpute was unable to recover as robustly as DURIAN.
Interestingly, formation of the dermal placode at E13.5–14.5 relies on communication between the fibroblasts of the nascent dermal condensate and the cells of the basal epithelium [14]. We therefore examined the relative intensity of predicted signaling interactions between these two celltypes, based on imputation with DURIAN versus DrImpute in Figure 5C. Here, we can see that DURIAN detects previously hidden interactions between members of the multicellular lineage of differentiated (FIB-A,FIB-B) and proliferating (FIB-P) fibroblasts. In summary, DURIAN can be utilized to enhance signaling analysis in single-cell data with the recovery of intracellular ligand expression.
Discussion
Single-cell transcriptomics trade sensitivity for resolution, while maintaining the high dimensionality of bulk sequencing [2]. For applications such as embedding and certain lineage inference tasks, this trade-off introduces few compromises and can even aid feature selection [57, 58]. In contrast, additional steps may be required in applications which seek to identify rare celltypes or rely on the recovery of specific markers that are prone to dropout [8, 53]. In particular, inference of cell–cell communication based on kinetic representation of signaling events cannot make use of implicit dropout correction and are less tolerant to noise, since individual markers must be robustly detected in subsets of the population [8, 59]. Here we present DURIAN, an iterative learning framework that achieves accurate and robust bulk-data deconvolution and single-cell data imputation simultaneously (Figure S1), which is useful to enhance single-cell signals, and particularly suitable for the integrative analysis of cellular communications using both single-cell and bulk data sets.
Three key features distinguish our approach from existing methods for joint analysis of both single-cell and bulk data such as URSM [25]. First, DURIAN serves as the first fully supervised imputation method to learn and utilize the deconvolution of bulk data for improved performance, while simultaneously utilizing prior knowledge about celltypes. This is in contrast to the generative, semi-supervised method employed by URSM [25], relies on latent celltype signatures during both deconvolution and imputation. Second, in contrast to URSM, which comprises a unified graphical model, our approach is modular and permits the use of diverse deconvolution strategies. Finally, though both URSM and DURIAN utilize deconvolution of bulk data to supervise imputation, URSM treats the single-cell signature matrix of the model as latent, whereas we utilize prior labeling of the cells. This allows us to utilize not only the output of existing unsupervised clustering pipelines [59–61], but also the celltype signatures associated with labels that are impossible to derive from genetic data alone, such as labeling revealed by functional assays [62, 63].
Compared with other imputation methods involving bulk data, DURIAN can make use of celltype-specific gene expression patterns in the bulk expression data to reduce its error rate [40]. By iteratively sharing information between its imputation and deconvolution stages, DURIAN improves the usefulness of both the bulk (via the deconvolution) and single-cell data during the imputation task. The performance of our supervised rank-minimization approach measurably improves in the presence of increasing global structure in the bulk data (Figure 2A). In contrast to methods that only provide supervision via global gene expression averages, our analysis suggests that deconvoluted bulk data provide critical information about expression at the single-cell level (Figure 2A).
Because deconvolution is critical DURIAN’s use of bulk data to supervise imputation, we directly measured imputation accuracy against several existing methods both in a realistic scenario (downsampling) and a scenario in which poorly differentiated celltypes are further obscured by high-magnitude batch effects (Figures 2A and 3A). These experiments suggest that mis-representation of specific sub-populations (L2 error) is the most critical limitation of all methods, highlighting the improved consistency of our approach (Figures 3A and 3B). Like other optimization-based approaches, parameter-selection is critical to DURIAN’s performance [40] and our benchmarking procedure could be adapted for parameter selection to prevent overestimation in practice. In particular, the error could be empirically minimized on downsampled versions of the target single-cell data matrix, while adjusting the learning rate (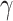 ).
).
Our synthetic benchmarks (Figures 3B and 3D) in combination with reported rates of signaling ligand recovery (Figure 5B) highlight the contrast between DURIAN and methods like DrImpute that rely on single-cell data alone to predict dropout [43]. Because DrImpute utilizes the average gene expression from unsupervised clustering of the input data to estimate missing reads, it is less prone to overestimating reads and to adding reads to nonzero entries in the original data, especially when the celltypes in the original data are well differentiated (Figure 3A) [43]. However, as suggested by the relative increase of predicted signaling rates in embryonic skin (Figure 5b), the rare expression of some genes within a given cell subpopulation makes it more likely that DrImpute will underestimate these reads, which may be critical to inference of cell-signaling events.
Though DURIAN shows promise on a wide variety of data, several refinements of our approach could increase performance. Preprocessing of single-cell data is essential to downstream applications [64]. In the experiments above, all methods were provided with the same input data, including all genes expressed by over 5% of cells. While this eliminated dimensionality as a variable in our benchmarking, it is likely that more rigorous gene selection could improve deconvolution for data sets where the maximum expression of celltype markers is lower. Additionally, it is known that NNLS methods like MuSiC are significantly less accurate when dropout in the single-cell reference is above 40%. Therefore, initial imputation could be performed by SCRABBLE (at a reduced learning rate), until the sparsity of the single-cell data reaches the threshold [37]. With regard to the impact of imputation on differentially expressed genes, we regard this as a critical area of future study, especially with respect to the impact of preprocessing on downstream performance. Finally, we note that one major advantage of DURIAN is that it can recover a gene that is completely missing in the single-cell data through the deconvolution, which is not the case for some other methods such as DrImpute or G2S3. On the other hand, while SCRABBLE can recover such missing genes, the cell cluster information is lost.
We present a new method for correcting drop-out in single-cell expression data that utilizes deconvoluted bulk expression data to supervise imputation. Novel features of this approach include its integrative and iterative schemes combining data imputation and bulk deconvolution, with modular design allowing deconvolution using either NNLS or a novel topic-model network featuring a distributed MCMC sampler. DURIAN is accurate and robust in benchmarks and can improve quality of unsupervised clustering and embedding of single-cell data, as well as enhance the recovery of signaling pathway markers and facilitate cell communication analysis combining single-cell and bulk data sets. DURIAN will be useful to dissect the multiscale characterization [8] of intercellular interactions and study the diversity and regulation of multicellular lineages [1, 65].
Key Points
A novel mathematical framework is presented for single-cell and bulk data integration with detailed empirical analysis of convergence.
Superior performance is demonstrated under a unique modular design allowing flexible incorporation of various choices in deconvolution, such as best-in-class NNLS regression or a newly proposed Bayesian network.
Imputation is fully supervised via the latent population structure of bulk expression data.
Methods
Package website and code availability
Package examples and source code are provided at https://mkarikom.github.io/DURIAN. A docker image providing a working installation of the DURIAN package and its vignette library at the time of publication is available at 10.5281/zenodo.6544669. Algorithmic details of the method, including deconvolution modules are presented in the Supplementary Materials.
Supplementary Material
Acknowledgements
We extend special thanks to Imam Toufiq and Nick Santucci UCI Research Cyberinfrastructure Center (RCIC) for computing support. M.K., Q.N. and P.Z. conceived the study. M.K. and P.Z. designed the algorithm. M.K. implemented the algorithm, developed the software and performed data analysis. Q.N. supervised the research. M.K. wrote the manuscript draft and supplementary materials. All authors read, wrote and approved the final manuscript.
Funding
National Institutes of Health (grants U01AR073159, R01DE030565); National Science Foundation (grants DMS1763272, MCB2028424); Simons Foundation (grant 594598).
Author Biographies
Matthew Karikomi is a PhD student in the MCSB program at University of California, Irvine, USA.
Peijie Zhou is a visiting assistant professor at the Mathematics Department, University of California, Irvine, USA.
Qing Nie is a Chancellor's Professor of Mathematics and Developmental & Cell Biology, University of California, Irvine, USA.
Contributor Information
Matthew Karikomi, Department of Mathematics.
Peijie Zhou, Department of Cell and Developmental Biology.
Qing Nie, NSF-Simons Center for Multiscale Cell Fate Research, University of California, Irvine.
References
- 1. MacLean AL, Hong T, Nie Q. Exploring intermediate cell states through the lens of single cells. Curr Opin Syst Biol June 2018;9:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol November 2016;34(11):1145–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol April 2014;32(4):381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wolf FA, Hamey FK, Plass M, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol March 2019;20(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manno, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature August 2018;560(7719):494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergen V, Lange M, Peidli S, et al. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol December 2020;38(12):1408–14. [DOI] [PubMed] [Google Scholar]
- 7. Zhou P, Wang S, Li T, et al. Dissecting transition cells from single-cell transcriptome data through multiscale stochastic dynamics. Nat Commun 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun February 2021;12(1):1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sha Y, Wang S, Bocci F, et al. Inference of intercellular communications and multilayer gene-regulations of epithelial–mesenchymal transition from single-cell transcriptomic data. Front Genet 2021;11:1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang MJ, Ntranos V, Tse D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat Commun February 2020;11(1):774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang R, Sun T, Song D, et al. Statistics or biology: the zero-inflation controversy about scRNA-seq data. Genome Biol 2022;23(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Armingol E, Officer A, Harismendy O, et al. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet 2021;22(2):71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuxuan H, Peng T, Gao L, et al. Cytotalk: de novo construction of signal transduction networks using single-cell transcriptomic data. Sci Adv 2021;7(16):eabf1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta K, Levinsohn J, Linderman G, et al. Single-cell analysis reveals a hair follicle dermal niche molecular differentiation trajectory that begins prior to morphogenesis. Dev Cell January 2019;48(1):17–31.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods July 2014;11(7):740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu P, Nakorchevskiy A, Marcotte EM. Expression deconvolution: a reinterpretation of DNA microarray data reveals dynamic changes in cell populations. Proc Natl Acad Sci 2003;100(18):10370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang K, Meng Q, Shats I, et al. Cdseq: a novel complete deconvolution method for dissecting heterogeneous samples using gene expression data. PLoS Comput Biol 2019;15(12):e1007510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cobos FA, Alquicira-Hernandez J, Powell JE, et al. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Commun November 2020;11(1):5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen GM, Chen C, Das RK, et al. Integrative bulk and single-cell profiling of premanufacture t-cell populations reveals factors mediating long-term persistence of car t-cell therapy. Cancer Discov 2021;11(9):2186–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Zou D, Zhu T, et al. Gene expression nebulas (gen): a comprehensive data portal integrating transcriptomic profiles across multiple species at both bulk and single-cell levels. Nucleic Acids Res 2022;50(D1):D1016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lun ATL, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol April 2016;17(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van den Berge, Perraudeau F, Soneson C, et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol February 2018;19(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pierson E, Yau C. ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol November 2015;16(1):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu L, Lei J, Devlin B, et al. A unified statistical framework for single cell and bulk RNA sequencing data. Ann Appl Stat March 2018;12(1):609–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin P, Troup M, Ho JWK. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol March 2017;18(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez R, Regier J, Cole MB, et al. Deep generative modeling for single-cell transcriptomics. Nat Methods December 2018;15(12):1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun March 2018;9(1):997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen M, Zhou X. VIPER: variability-preserving imputation for accurate gene expression recovery in single-cell RNA sequencing studies. Genome Biol November 2018;19(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang M, Wang J, Torre E, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods July 2018;15(7):539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mongia A, Sengupta D, Majumdar A. McImpute: matrix completion based imputation for single cell RNA-seq data. Front Genet 2019;10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Zhang S. Imputing single-cell RNA-seq data by considering cell heterogeneity and prior expression of dropouts. J Mol Cell Biol 2021;13(1):29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gaujoux R, Seoighe C. Semi-supervised nonnegative matrix factorization for gene expression deconvolution: a case study. Infect Genet Evol July 2012;12(5):913–21. [DOI] [PubMed] [Google Scholar]
- 34. Gaujoux R, Seoighe C. CellMix: a comprehensive toolbox for gene expression deconvolution. Bioinformatics September 2013;29(17):2211–2. [DOI] [PubMed] [Google Scholar]
- 35. Tai A-S, Tseng GC, Hsieh W-P. Bayice: a Bayesian hierarchical model for semireference-based deconvolution of bulk transcriptomic data. Ann Appl Stat 2021;15(1):391–411. [Google Scholar]
- 36. Erdmann-Pham DD, Fischer J, Hong J, et al. Likelihood-based deconvolution of bulk gene expression data using single-cell references. Genome Res 2021;31(10):1794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Park J, Susztak K, et al. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun January 2019a;10(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jew B, Alvarez M, Rahmani E, et al. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat Commun 2020;11(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong M, Thennavan A, Urrutia E, et al. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief Bioinform January 2021;22(1):416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peng T, Zhu Q, Yin P, et al. SCRABBLE: single-cell RNA-seq imputation constrained by bulk RNA-seq data. Genome Biol May 2019;20(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai J-F, Candés EJ, Shen Z. A singular value thresholding algorithm for matrix completion. SIAM J Optim January 2010;20(4):1956–82. [Google Scholar]
- 42. Zappia L, Phipson B, Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol September 2017;18(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gong W, Kwak I-Y, Pota P, et al. DrImpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinf June 2018;19(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hicks SC, William Townes F, Teng M, et al. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2018;19(4):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baron M, Veres A, Wolock SL, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst October 2016;3(4):346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McInnes L, Healy J, Melville J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426. 2018.
- 47. Markovsky I. Low Rank Approximation: Algorithms, Implementation, Applications, Vol. 906. Springer, 2012. [Google Scholar]
- 48. Robinson MD, McCarthy DJ, Smyth GK. edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lähnemann D, Köster J, Szczurek E, et al. Eleven grand challenges in single-cell data science. Genome Biol 2020;21(1):1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saelens W, Cannoodt R, Todorov H, et al. A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019;37(5):547–54. [DOI] [PubMed] [Google Scholar]
- 51. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 2019;15(6):e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hennig C. FPC: Flexible Procedures for Clustering, 2020, R package version 2.2-9.
- 53. He H, Suryawanshi H, Morozov P, et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol June 2020;145(6):1615–28. [DOI] [PubMed] [Google Scholar]
- 54. Kumar MP, Du J, Lagoudas G, et al. Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep November 2018;25(6):1458–1468.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Andl T, Reddy ST, Gaddapara T, et al. Wnt signals are required for the initiation of hair follicle development. Dev Cell 2002;2(5):643–53. [DOI] [PubMed] [Google Scholar]
- 56. Reddy S, Andl T, Bagasra A, et al. Characterization of wnt gene expression in developing and postnatal hair follicles and identification of wnt5a as a target of sonic hedgehog in hair follicle morphogenesis. Mech Dev 2001;107(1-2):69–82. [DOI] [PubMed] [Google Scholar]
- 57. Qiu P. Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 2020;11(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moon KR, van Dijk, Wang Z, et al. Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol 2019;37(12):1482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang S, Karikomi M, MacLean AL, et al. Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res June 2019b;47(11):e66–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol February 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol December 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yankaskas CL, Thompson KN, Paul CD, et al. A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat Biomed Eng June 2019;3(6):452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Y-C, Humphries B, Brien R, et al. Functional isolation of tumor-initiating cells using microfluidic-based migration identifies phosphatidylserine decarboxylase as a key regulator. Sci Rep January 2018;8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lueken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol June 2019;15(6):e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lander AD, Gokoffski KK, Wan FYM, et al. Cell lineages and the logic of proliferative control. PLoS Biol January 2009;7(1): e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.