

Abstract
Recently reported to be effective in patients with lung cancer, KRASG12C inhibitors bind only to the inactive, or guanosine diphosphate-bound, state of the oncoprotein and require guanosine triphosphate (GTP) hydrolysis for inhibition. Yet, KRAS mutations prevent the catalytic arginine of GTPase-activating proteins (GAPs) from enhancing an otherwise slow hydrolysis rate. If KRAS mutants are indeed insensitive to GAPs, it is unclear how KRASG12C hydrolyzes sufficient GTP to allow inactive state selective inhibition. Here we show that RGS3, a GAP previously known for regulating G-protein coupled receptors, can also enhance the GTPase activity of mutant and wild-type KRAS proteins. Our study reveals an unexpected mechanism that inactivates KRAS and explains the vulnerability to emerging clinically effective therapeutics.
One Sentence Summary:
RGS3 inactivates common oncogenic KRAS mutants by enhancing their guanosine triphosphate activity.
KRAS hydrolyzes guanosine triphosphate (GTP) to guanosine diphosphate (GDP) and controls diverse cellular functions by cycling between an active (GTP-bound) and an inactive (GDP-bound) conformation (1–4). Its weak intrinsic GTPase activity is enhanced by GTPase-activating proteins (GAPs) and the exchange of GDP for GTP is enhanced by guanine-nucleotide exchange factors (5–7). KRAS mutations are among the most common activating alterations found in cancer (4, 8). Most of these mutations (~80%) result in single amino acid substitutions of glycine 12 (for example, G12C/D/V/etc.) and prevent the catalytic arginine residue in GAPs from enhancing GTP-hydrolysis (3, 9, 10). Insensitivity to GAPs is thought to render KRAS oncoproteins constitutively active and independent of upstream input for activation.
Emerging evidence suggests that some KRAS oncoproteins require nucleotide exchange for activation. Perhaps the strongest evidence is provided by KRASG12C inhibitors (11–17), which selectively target the inactive state of the oncoprotein to prevent its activation by nucleotide exchange. We have previously shown that these drugs induce responses in ~1/3 of patients with lung cancer (18) and that inhibition requires an intact GTPase activity by KRASG12C (12, 17). If KRAS mutants are insensitive to GAPs, it is unclear how KRASG12C hydrolyzes sufficient GTP to allow inactive state selective inhibition.
Intrinsic hydrolysis alone may be sufficient to enable inactive state selective inhibition. To this end, we compared the rate of inhibition in cancer cells to the rate of intrinsic GTP hydrolysis by KRASG12C. The time required for maximal inhibition of KRAS in cells treated with a potent and selective G12Ci (MRTX1257, fig. S1A) ranged from ~20 min in serum-deprived medium, a condition that suppresses nucleotide exchange (half-life ~ 2 min, Fig. 1A), to ~60 min in medium with 10% serum (half-life ~ 10 min, fig. S1B). Similar cellular inhibition kinetics were observed with other potent inhibitors (kinac/KI ≥ 10 mM−1 s−1), such as MRTX849 and AMG510 (Fig. 1A).
Fig. 1. Cancer cell extracts enhance the GTPase activity of KRASG12C.

(A) KRASG12C-mutant cell lines were serum-deprived for 16 h and treated as shown. The level of active KRAS (GTP-bound) was determined by RAS-binding domain (RBD)-pulldown and quantified by densitometry. Bottom: The potency of drug binding to G12C (kinac/KI) and the half-life for cellular inhibition (mean from the indicated number of cell lines). (B) KRASG12C was loaded with GTP[ɣ32P] and its intrinsic GTPase activity was assayed over time, ensuring sufficient time for the reaction to reach completion (mean ± s.e.m, n=2). Also shown is the effect of MRTX1257 treatment on the ratio of active to total cellular KRAS. (C) GTP[ɣ32P] hydrolysis by KRASG12C was assayed in the absence or presence of whole-cell extracts (WCE) from H358 lung cancer cells (mean ± s.e.m, n=3, two-tailed p value). WCE were either added directly or after fractionation with a 3 kDa molecular cut-off column. (D) KRAS splice variants 4A or 4B, either WT or G12C mutant, were loaded with GTP[ɣ32P] and assayed for GTP-hydrolysis over time. (E) Thin layer chromatography (TLC) was used to separate α32P-labeled nucleotides that were eluted from KRASG12C incubated with the NF1 GAP-related domain (GRD) or H358 cellular extracts. Shown are the fold increase in the absolute GDP[α32P] signal and the percent α32P-labeled nucleotide relative to the total signal per sample. FC: fold change. A representative of at least two experimental repeats is shown in D and E.
By comparison, purified KRASG12C hydrolyzed GTP[ɣ32P] at a much slower rate (half-life 120–300 min, Fig. 1B and fig. S1C). The intrinsic hydrolysis half-life for KRAS proteins varies across publications (fig. S1D, range: 25–200 min). Regardless of this variation, the rate of cellular KRASG12C inhibition with potent inhibitors was quicker than even the fastest intrinsic hydrolysis estimate (19) reported in the literature (fig. S1E).
The discrepancy between the rate of cellular inactivation and the rate of the intrinsic GTPase reaction suggests the presence of cellular factors that enhance GTP-hydrolysis by KRASG12C. To test this possibility, purified KRAS was loaded with GTP[ɣ32P] and incubated with whole-cell extracts (WCE) from lung cancer models. Because cytoplasmic RAS-GAPs, such as NF1, enhance the GTPase activity of wild-type (WT) KRAS, the latter was used as a positive control. The KRAS-nucleotide complex was immobilized and the remaining GTP[ɣ32P] was quantified by autoradiography (hereafter referred to as the ɣ-phosphate assay, fig. S1C). As shown in Fig. 1C and fig. S2A, little intrinsic GTP[ɣ32P]-hydrolysis was detected for KRASG12C at 1 h of incubation. In the presence of cellular extracts, however, there was a near complete hydrolysis of GTP[ɣ32P] (Fig. 1C and fig. S2A). Under these conditions, the reaction reached a steady-state maximum within 2 min of incubation (Fig. 1D and fig. S2B). Cellular extracts did not affect KRAS protein stability in these experiments (fig. S2C and D). The activity was greatly reduced in the flow-through from a 3 kDa molecular cut-off fractionation column (Fig. 1C and fig. S2A), suggesting that a cellular protein is responsible.
It has been reported that cellular extracts enhance hydrolysis by wild-type but not by mutant (G12V/D) NRAS (5). If the same holds true for KRASG12C has not been tested in the literature. In agreement with previous work, our data show that the GTPase activity of KRASWT was enhanced in a concentration dependent manner and that the effect on KRASWT was greater than on KRASG12C (fig. S2D). However, rather than being completely insensitive to cellular extracts, KRASG12C was also responsive, with activity detected at higher lysate concentrations (fig. S2D and E).
It is possible that the KRASG12C effect reflects an enhanced rate of nucleotide dissociation or the exchange of GTP for GDP. To address these possibilities, the GTPase activity of KRASG12C was assayed by loading it with GTP[α32P] and subsequent incubation with cellular extracts (hereafter referred to as the α-phosphate assay, fig. S2F). Following immobilization of KRAS, the bound nucleotides were eluted and subjected to separation by thin-layer chromatography (TLC). As expected, after incubation with the GAP-related domain (GRD) of NF1, the predominant [α32P]-labeled nucleotide bound to KRASG12C was GTP (Fig. 1E). By comparison, the predominant [α32P]-labeled nucleotide bound to KRASG12C incubated with cell extracts was GDP. If nucleotide exchange (or nucleotide dissociation) was responsible, then GTP[α32P] would be replaced with cold nucleotide and no GDP[α32P] signal would be detected. Because an increase in GDP[α32P] was indeed detected (Fig. 1E), the data support the presence of a cellular protein that accelerates GTP-hydrolysis by KRASG12C. Moreover, the GTPase enhancing effect of cellular extracts was attenuated when A59G — a transition state mutation that blocks GTPase activity (20) — was introduced alongside KRASG12C (fig. S2G).
To isolate the protein responsible for enhancing the GTPase activity of KRASG12C, we devised a purification scheme wherein KRASG12C-mutant cancer cell extracts (see Materials and Methods) were subjected to two rounds of size-exclusion chromatography (SEC, steps 1 and 2), desalting (step 3), anion-exchange chromatography (AEC, step 4) and mass spectrometry (MS, step 5) to identify proteins classified as GAPs (Fig. 2A). The eluted fractions from each chromatographic step were assayed for their optical density and their ability to enhance KRASG12C hydrolysis. Fractions that retained activity were pooled and carried forward to the next step. SEC step 1 elution volumes ranging from 52 to 72 mL had KRASG12C-directed GAP activity (fig. S3A). Four of these were independently subjected to SEC step 2, in order to confirm the activity and to identify which step 2 fractions to carry forward (Fig. 2B). SEC step 2 elution volumes ranging from 12 to 15 mL were pooled, desalted and analyzed by AEC with a linear salt-gradient (fig. S3B). The latter identified five peaks with KRASG12C-directed GAP activity (Fig. 2C).
Fig. 2. Identification of a protein that enhances the GTPase activity of KRASG12C.

(A) The purification schema included size-exclusion chromatography (SEC, steps 1 and 2), desalting (step 3), anion-exchange chromatography (AEC, step 4) and mass spectrometry (MS, step 5). (B) H358 cell extracts (2 g) were subjected to SEC and eluted fractions (1 mL each) were incubated with GTP[ɣ32P]-loaded KRASG12C for 1 h and subjected to the ɣ-phosphate assay. The G12C-GTP[ɣ32P] signal in each fraction from step 2 is shown. See fig. S3A for step 1 analysis. (C) Active fractions from step 2 (12–17 mL) were pooled, desalted and subjected to AEC using a linear salt-gradient. The effect of each fraction on GTPase activity is shown. See also fig. S3B. (D) The elution of RGS3 p75 and p25 variants in fractions from peak 1 (P1) and the effect on KRASG12C GTPase activity.
The active fractions eluted in step 4 were pooled, acid-precipitated and subjected to MS in order to identify proteins annotated as GAP (fig. S3C). One of these, RGS3, is a GAP for the Gαi/q subunit of heterotrimeric G-protein coupled receptors (21–23). Many of the RGS3 peptides identified by mass spectrometry localized in the RGS domain of the protein (RGSD, the GAP domain, fig. S3D–E). RGS3 lacks the catalytic arginine (R) residue present in canonical RAS-GAPs (24, 25). We thus reasoned that RGS3 might not be impeded by G12 mutations and decided to study it further. The broader RGS family contains 20 members (fig. S3F), which often have alternatively spliced variants. At least 9 variants of RGS3 have been reported. As expected, the 75 kDa (p75) and/or the 25 kDa (p25) RGS3 isoforms (both of which contain the RGS GAP domain) eluted in one or more fractions that enhanced GTP[ɣ32P]-hydrolysis by KRASG12C (Fig. 2D).
In beginning to validate the effect of RGS3, we asked if it interacts with KRASG12C in cells. To this end, we relied on the ability of the G12Ci to selectively displace effector proteins from KRASG12C, while sparing those bound to wild-type KRAS, NRAS and HRAS (12, 13). Co-immunoprecipitation (IP) studies in three KRASG12C-mutant cell lines showed an interaction between endogenous KRASG12C and RGS3; an interaction that was diminished upon G12Ci-treatment (fig. S4A). GST-pulldown experiments confirmed that RGS3 interacts with either KRASG12C 4A or 4B splice variants (fig. S4B), an interaction that was less pronounced with KRASWT (fig. S4C). Direct binding assays with purified proteins suggested a preference for the active (or GTPɣS-loaded) conformation of KRASG12C, as compared to the inactive (GDP-loaded) state (fig. S4D).
To directly test if RGS3 acts as a GAP for KRASG12C, we carried out GTP-hydrolysis assays with purified KRAS in the presence of RGS3 variants. In addition to the radioactive ɣ- and α-phosphate assays (figs. S1C and S2F), we also used a non-radioactive hydrolysis assay that enables continuous measurement of phosphate release over time (fig. S5A). As expected, RGS3 enhanced GTP-hydrolysis by KRASG12C (Fig. 3A–B and fig. S5B), leading to a concentration-dependent increase in the rate constant (fig. S5C and D). RGS3 was more effective at enhancing KRASG12C hydrolysis than canonical RAS-GAPs, such as NF1 and RASA1 (fig. S5E and F). Whereas no measurable effect was detected for RASA1, some activity was observed for NF1 (Fig. 3A–B and fig. S5E and F). The latter, however, was not significantly attenuated when the catalytic arginine of NF1 was mutated to alanine (R1276A), supporting our suspicion that KRASG12C-directed GAP activity occurs independently of the arginine residue, which is also known as the “R-finger”.
Fig. 3. Mechanistic basis for RGS3-assisted GTP-hydrolysis by KRASG12C.

(A, B) GTP[ɣ32P]-loaded KRAS variants were incubated with buffer, NF1 GAP-related domain (GRD) or RGS3 (p75) for the indicated times, followed by determination of GTPase activity by using the ɣ-phosphate assay (A) or the α-phosphate assay (B). (C) As in A, except that the hydrolysis transition-state mutation A59G was engineered alongside G12C. (D) Molecular model of the interaction of KRAS-GMPPNP with the GAP-domain of RGS3 (RGSD). The catalytic arginine residue of RASA1 (R) is superimposed as a reference point (not part of the modeling). (E, F) KRASG12C was loaded with GTP[ɣ32P] (E) or GTP[α32P] (F) and then incubated with WT or asparagine-to-histidine (NH) mutant RGS3 followed by determination of GTPase activity using the ɣ-phosphate (E) or the α-phosphate (F) assay. A representative of at least two experimental repeats is shown in A-C, E and F. (G) Schematic of the mechanism that enables mutant KRAS-inclusive GAP activity.
Rather than having an idiosyncratic effect on KRASG12C, RGS3 also enhanced GTP-hydrolysis by G12D/V and G13C/D mutant KRAS (fig. S6A–C), which together comprise ~95% of KRAS mutations found in cancer. RGS3 also enhanced the GTPase activity of KRASWT, but in this setting RGS3 was much less effective than NF1 or RASA1 (Fig. 3A–B and fig. S5G).
RGS3 had little effect when the transition state mutation A59G was engineered alongside G12C (Fig. 3C). This suggests that RGS3 enhances hydrolysis by helping the reaction progress past the transition state. To better understand the mechanism, we constructed a molecular model using crystal structures of KRAS-GMPPNP (6OB2, ref. 26), RGS3 GAP domain (2OJ4, ref. 27) and Gaia1-GDP·AlF4:RGS4 (1AGR, ref. 24). The model identified an asparagine (N) residue in the GAP domain of RGS3 as potentially involved in the hydrolysis reaction (Fig. 3D). This residue was predicted to orient away from the P-loop and, therefore, less likely to be impeded by G12 substitutions. Also, asparagine residues serve a catalytic function in other (non RAS-specific) GAPs, including those enhancing GTP-hydrolysis by RAP1 and RHEB GTPases (28, 29).
We thus tested if the asparagine residue in RGS3 was necessary for the GTPase-activating effect on KRASG12C. A histidine (H) substitution on N460 in p75 (RGS3) or on N147 in p25 (RGSD) attenuated the effect of RGS3 proteins on GTP-hydrolysis by KRASG12C, as evidenced by the ɣ-phosphate (Fig. 3E) and the α-phosphate (Fig. 3F) hydrolysis assays. Because RGS3 NH mutants had some residual activity against KRASG12C, it is likely that other residues also contribute to the GAP activity. This is in agreement with reports indicating that multiple RGS residues (besides asparagine) participate in enhancing GTP hydrolysis by Gα (24). The asparagine residue is conserved between RGS isoforms, suggesting that other members of the family also enhance the KRAS GTPase activity. Indeed, this was the case for RGS4 (Fig. 3E). Together, the biochemical data suggest that RGS3 enhances the GTPase activity of KRAS in a mutant-inclusive manner (Fig. 3G).
If RGS3 enhances KRASG12C-hydrolysis it may inactivate this oncoprotein in cells. As expected, sgRNA-mediated deletion of RGS3 in KRASG12C-mutant lung cancer cells led to an increase in KRAS activation (Fig. 4A and fig. S7A). Loss of RGS3 also enhanced proliferation in culture and accelerated tumor growth in xenograft studies (Fig. 4B and fig. S7B). The proliferative advantage of RGS3−/− cells was largely dependent on KRASG12C, as evidenced by a previously validated (17, 30) KRASG12C-specific siRNA (fig. S7C). In agreement, siRNA-mediated knockdown of RGS3 increased proliferation in cells expressing KRASG12C but not in cells expressing KRASG12C/A59G (fig. S7D), which is insensitive to the GTPase-enhancing effect of WCE (fig. S2G) or RGS3 (Fig. 3C).
Fig. 4. RGS3 diminishes KRASG12C activation in cancer cells.
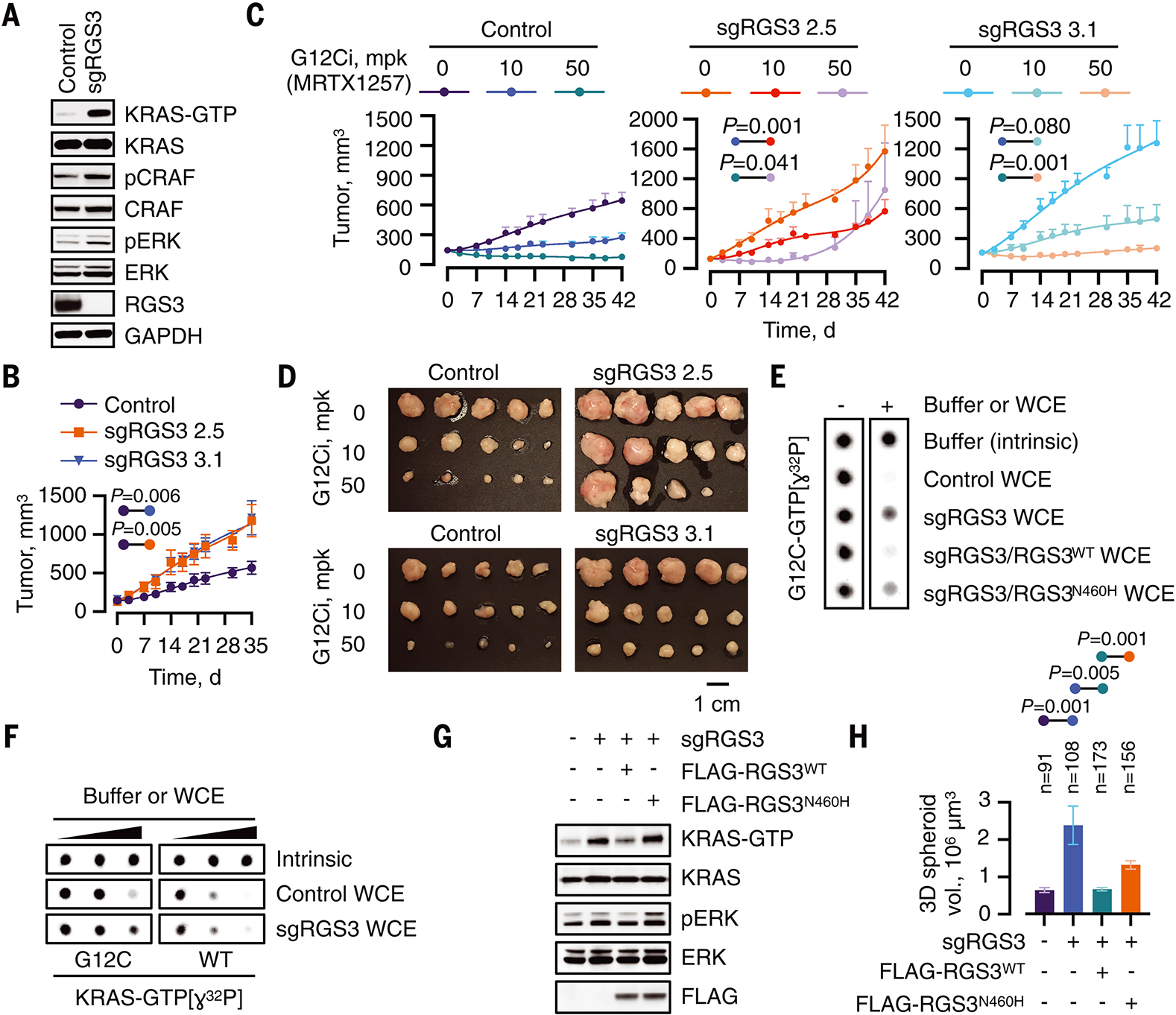
(A) WCE from control or RGS3-null H358 cells were immunoblotted to determine the effect on the indicated signaling intermediates. A representative of at least three experimental repeats is shown. (B) Control or RGS3−/− clones were implanted in athymic mice to determine the effect on tumor growth (n=10 for control and n=5 for each sgRGS3 clone). Mean ± s.e.m and two-tailed p values are shown. (C, D) Mice bearing control or RGS3−/− xenografts were treated as shown to determine the effect on tumor growth (n=10 for the control and n=5 for each sgRGS3 clone). Two-tailed p values (C) and endpoint tumor volumes (D) are shown. (E) Purified KRASG12C was loaded with GTP[ɣ32P] and assayed for GTP-hydrolysis in the absence or presence of extracts (0.4 μg/μL) from control and RGS3−/− cells as well as extracts from RGS3−/− cells engineered to re-express WT or N460H mutant RGS3. (F) Purified KRASWT or KRASG12C proteins were loaded with GTP[ɣ32P] and assayed for hydrolysis in the absence (intrinsic) or in the presence of extracts (WCE) from control or RGS3−/− cells (0, 0.1, 0.2 μg/μL). (G) WCE from the indicated cell lines were evaluated by RBD-pulldown to determine the amount of active KRAS. (H) The cells were cultured as 3D tumor spheroids in collagen I-containing medium for 17 days. The spheroid volumes were determined from representative images.
To determine if RGS3 expression inversely correlated with KRAS activation in patients with lung cancer, we established a mutant KRAS-dependent transcriptional output score (fig. S8A and B) and experimentally validated its ability to detect changes in KRASG12C-driven signaling following G12Ci-treatment (fig. S8C). As shown in fig. S8D and E, a higher RGS3 expression was associated with lower mutant KRAS output in lung cancers harboring G12C or any KRAS mutation. The negative correlation was observed with distinct KRAS up-regulated or down-regulated gene expression signatures (fig. S8F and G). Little to no correlation was noted in KRASWT lung cancers.
RGS3−/− cells had an attenuated response to G12Ci-treatment, as compared to their isogenic RGS3 wild-type cells. This was evidenced by: 1) a diminished inhibition of KRAS-GTP levels and downstream signaling (fig. S9A, B), 2) a diminished antiproliferative effect (fig. S9C, D), and 3) a less potent antitumor effect in vivo (Fig. 4C, D and fig. S9E, F). Moreover, RGS3 expression correlated with susceptibility to G12Ci-treatment, in a panel of 9 lung cancer patient-derived xenograft (PDX) models. The PDX were established from 7 patients and their treatment response profile (fig. S10A) was similar to that reported in a recent clinical study (18). RNA sequencing (fig. S10B) and immunohistochemistry with an RGS3-specific antibody (fig. S10C, D) revealed that tumors with higher RGS3 expression had a larger magnitude of inhibition.
To provide additional evidence that RGS3 functions as a mutant KRAS-inclusive GAP, we tested the ability of RGS3-depleted extracts to directly enhance GTP[ɣ32P]-hydrolysis by KRAS. As shown in Fig. 4E, almost complete KRASG12C hydrolysis was observed in the presence of control extracts (row 2 vs. row 1); an activity that was attenuated in RGS3−/− extracts (row 3). By comparison, the activity was restored in extracts from RGS3−/− cells expressing RGS3WT (row 4) and diminished again in those expressing RGS3N460H (row 5). RGS3−/− extracts did not significantly affect GTP[ɣ32P] hydrolysis by KRASWT (Fig. 4F). The latter agrees with the notion that RGS3−/− cells still express conventional RAS-GAPs (such as NF1), which greatly enhance hydrolysis by KRASWT (Fig. 3B and fig. S5G: NF1>>RGS3), but only modestly that by KRASG12C (Fig. 3B and fig. S5E: RGS3>NF1). Lastly, the increase in cellular KRAS activation (Fig. 4G) and proliferation (Fig. 4H) conferred by the deletion of RGS3 were reversed by re-expression of RGS3WT, but much less so by RGS3N460H.
Here we report that the GTPase activity of KRAS is enhanced by non arginine-finger dependent GAPs, such as RGS3 (see also supplementary text). As evidenced above, RGS3 enhanced GTP hydrolysis in mutant KRAS-inclusive manner, affecting both wild-type and several oncogenic mutants (including G12C, G12D, G12V, G13C and G13D). This effect was dependent on a key asparagine residue in the GAP domain of RGS3. KRAS mutations impede the catalytic arginine (or R-finger) of canonical RAS-GAPs (such as NF1 or RASA1) from enhancing an otherwise slow GTP hydrolysis rate. If, however, KRAS mutants were completely insensitive to GAPs, then inactive state selective KRASG12C inhibitors would require a long time to take effect, when considering that the time to inhibition is limited by the rate of hydrolysis. While inhibitor potency might have hindered initial observations (13), highly-potent G12Ci inactivate the oncoprotein with minutes; quicker than even the fastest estimate of intrinsic hydrolysis rate in the literature (19). RGS3-assisted hydrolysis by KRASG12C was approximately 1–2 orders of magnitude slower than NF1- or RASA1-stimulated hydrolysis by KRASWT, suggesting that RGS3 is unlikely to inactivate the entire cellular pool of KRASG12C; that is, not to the same extent as canonical R-finger GAPs are able to do for KRASWT. As a result, KRASG12C has a longer residency time in its active (GTP-bound) conformation than KRASWT, allowing the oncoprotein to drive proliferation despite its susceptibility to ‘atypical-for-RAS’ GAPs (Fig. 3G). Collectively, our experiments uncover an unexpected regulatory mechanism with important implications for understanding and therapeutically targeting KRAS oncoprotein-driven cancers.
Supplementary Material
Acknowledgments:
The authors thank Megan Mroczkowski for discussing this work throughout its stages and for reviewing the manuscript. The authors thank Rajesh Soni from the Columbia University mass spectrometry core facility for helping with protein identification, James Christensen from Mirati Therapeutics for graciously providing MRTX1257 and MRTX849 and Russell Lipford from Amgen for graciously providing AMG510.
Funding:
This work has been supported in part by the NIH/NCI (1R01CA230745-01 to P.L., 1R01CA230267-01A1 to P.L., K08CA191082-01A1 to P.L. and F30CA232549-01 to J.X.), the MSKCC Josie Robertson Investigator Program and the MSKCC Support Grant-Core Grant program (P30 CA008748). P.L. is also supported in part by The Pew Charitable Trusts and the Damon Runyon Cancer Research Foundation.
Footnotes
Competing interests:
P.L. reports research grants to his institution from Mirati, Revolution Medicines and Strategia. P.L. is an inventor on patents filed by MSKCC regarding treatment of KRAS or BRAF mutant cancers. P.L. has received scientific advisory fees from Revolution Medicines and Black Diamod Therapeutics. The other authors have no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials.
List of Supplementary Materials:
References and Notes:
- 1.Malumbres M, Barbacid M, RAS oncogenes: the first 30 years. Nature reviews. Cancer 3, 459–465 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Papke B, Der CJ, Drugging RAS: Know the enemy. Science 355, 1158–1163 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Simanshu DK, Nissley DV, McCormick F, RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S, Balmain A, Counter CM, A model for RAS mutation patterns in cancers: finding the sweet spot. Nature reviews. Cancer 18, 767–777 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Trahey M, McCormick F, A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542–545 (1987). [DOI] [PubMed] [Google Scholar]
- 6.Margarit SM et al. , Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell 112, 685–695 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Bos JL, Rehmann H, Wittinghofer A, GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D, RAS oncogenes: weaving a tumorigenic web. Nature reviews. Cancer 11, 761–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollag G, McCormick F, Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 351, 576–579 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Scheffzek K et al. , The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lito P, Solomon M, Li LS, Hansen R, Rosen N, Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patricelli MP et al. , Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 6, 316–329 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Janes MR et al. , Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Canon J et al. , The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Hallin J et al. , The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue JY et al. , Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong DS et al. , KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 383, 1207–1217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter JC et al. , Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13, 1325–1335 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Hall BE, Bar-Sagi D, Nassar N, The structural basis for the transition from Ras-GTP to Ras-GDP. Proc Natl Acad Sci U S A 99, 12138–12142 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ, RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature 383, 172–175 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Ross EM, Wilkie TM, GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69, 795–827 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Neubig RR, Siderovski DP, Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov 1, 187–197 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Tesmer JJ, Berman DM, Gilman AG, Sprang SR, Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell 89, 251–261 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Srinivasa SP, Watson N, Overton MC, Blumer KJ, Mechanism of RGS4, a GTPase-activating protein for G protein alpha subunits. J Biol Chem 273, 1529–1533 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Rabara D et al. , KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc Natl Acad Sci U S A 116, 22122–22131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezabkova L et al. , 14–3-3 protein interacts with and affects the structure of RGS domain of regulator of G protein signaling 3 (RGS3). J Struct Biol 170, 451–461 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Daumke O, Weyand M, Chakrabarti PP, Vetter IR, Wittinghofer A, The GTPase-activating protein Rap1GAP uses a catalytic asparagine. Nature 429, 197–201 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Inoki K, Guan KL, Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol 24, 7965–7975 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunaga N et al. , Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther 10, 336–346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs JB, Schaber MD, Allard WJ, Sigal IS, Scolnick EM, Purification of ras GTPase activating protein from bovine brain. Proc Natl Acad Sci U S A 85, 5026–5030 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webb MR, A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci U S A 89, 4884–4887 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lito P et al. , Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 25, 697–710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waters AM et al. , Evaluation of the selectivity and sensitivity of isoform- and mutation-specific RAS antibodies. Sci Signal 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bankhead P et al. , QuPath: Open source software for digital pathology image analysis. Sci Rep 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue Y et al. , An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 23, 929–937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmann MH et al. , BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov 11, 142–157 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.