

Abstract
B-cell activating factor (BAFF) is an essential cytokine for B-cell maturation, differentiation and survival, and excess BAFF induces aggressive or neoplastic B-cell disorders and contributes to development of autoimmune diseases. Metformin, an anti-diabetic drug, has recently garnered a great attention due to its anti-proliferative and immune-modulatory features. However, little is known regarding the effect of metformin on BAFF-stimulated B cells. Here, we show that metformin attenuated human soluble BAFF (hsBAFF)-induced cell proliferation and survival by blocking the Erk1/2 pathway in normal and B-lymphoid (Raji) cells. Pretreatment with U0126, knockdown of Erk1/2, or expression of dominant negative MKK1 strengthened metformin’s inhibition of hsBAFF-activated Erk1/2 and B-cell proliferation/viability, whereas expression of constitutively active MKK1 rendered high resistance to metformin. Further investigation found that overexpression of wild type PTEN or ectopic expression of dominant negative Akt potentiated metformin’s suppression of hsBAFF-induced Erk1/2 activation and proliferation/viability in Raji cells, implying a PTEN/Akt-dependent mechanism involved. Furthermore, we noticed that metformin hindered hsBAFF-activated mTOR pathway in B cells. Inhibition of mTOR with rapamycin or knockdown of mTOR enhanced metformin’s suppression of hsBAFF-induced phosphorylation of S6K1, PTEN, Akt, and Erk1/2, as well as B-cell proliferation/viability. These results indicate that metformin prevents BAFF activation of Erk1/2 from cell proliferation and survival by impeding mTOR-PTEN/Akt signaling pathway in normal and neoplastic B-lymphoid cells. Our findings support that metformin has a great potential for prevention of excessive BAFF-induced aggressive B-cell malignancies and autoimmune diseases.
Keywords: Metformin, B-cell activating factor, Extracellular signal-related kinases 1/2, B cells, Survival
1. Introduction
The B-cell activating factor (BAFF), also known as TALL-1, THANK, BLyS, and zTNF4, is one of TNF ligand family members displaying typical features of type II transmembrane proteins [1–5]. BAFF can bind to three receptors, B-cell activating factor receptor (BAFF-R), transmembrane activator and CAML interactor (TACI) and B-cell maturation antigen (BCMA), which are expressed predominantly on the cell membrane of B cells, and plays a critical role in B-cell survival, proliferation, and differentiation [6,7]. Elevated circulating levels of BAFF have been observed in patients with autoimmune disorders including systemic lupus erythematosus (SLE), SjÖgren’s syndrome (SS), and multiple sclerosis (MS) [8–12]. So, overexpression of BAFF is closely related to autoimmunity risk, and circulating BAFF level can serve as a biomarker of disease severity and progression in autoimmune disorders. The pathophysiological role of BAFF in autoimmune diseases has been well established, but it remains a great challenge how to target BAFF signaling to control aggressive or neoplastic B-cell disorders.
Metformin has been demonstrated to be a safe and effective oral hypoglycemic agent that is widely used in the treatment of type 2 diabetes (T2D) [13]. Recent studies in humans have revealed novel pleiotropic actions of metformin, spanning from its consolidated role in T2D management to various regulatory properties, including cardio-, nephro- and neuro-protection, as well as antiproliferative, antifibrotic, antiapoptotic and antioxidant effects [14–18]. Of note, emerging evidence suggests that metformin also has immune-modulatory function [19–23]. Numerous studies have shown that the best-characterized effect of metformin relies on activation of AMP-activated protein kinase (AMPK), a major sensor that modulates lipid and glucose metabolism, although AMPK-independent mechanisms have been recently disclosed [24]. In fact, downstream consequences of AMPK activation may account for many effects of metformin on immune homeostasis. The mammalian target of rapamycin (mTOR), which is negatively regulated by AMPK, senses environmental cues (growth factors, nutrients, and energy/oxidative stress) and controls protein synthesis, cell growth, metabolism, and autophagy [25]. Especially, mTOR plays a critical role in B cell function. It has been described that conditional deletion of MTOR gene in B cells strongly impairs B cell proliferation and germinal center (GC) differentiation [26]. Inhibition of mTOR with rapamycin markedly inhibits cell proliferation and antibody responses in mouse and human B cells [26,27]. We have recently identified that excessive human soluble BAFF (hsBAFF) activates mTOR pathway contributing to proliferation and survival in cultured B lymphocytes [27,28]. Based on these findings, we postulated that metformin might be an effective agent against BAFF-induced aggressive B-cell malignancies and autoimmune disorders.
The phosphatase and tensin homologue deleted on chromosome 10 (PTEN), a dual-specificity lipid and protein phosphatase, acts as a tumor suppressor and is frequently found deficient in patients with tumors [29]. PTEN negatively regulates both phosphatidylinositol 3′-kinase (PI3K)-Akt-dependent and -independent signaling pathways, such as the mitogen-activated protein kinase (MAPK) pathway [30]. The PI3K/Akt/mTOR and Ras/extracellular signal-regulated kinases 1/2 (Erk1/2) signaling pathways are crucial in controlling cell growth, survival, proliferation, migration, and differentiation [31,32]. Previous studies have reported that loss of PTEN function elicits increased PI3K and MAPK signaling, thereby leading to hyperplasia and tumorigenesis [29,30,32]. Consistently, our group has demonstrated that hsBAFF stimulates cell proliferation and survival by activating Ca2+-CaMKII-dependent PTEN/Akt-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells [33]. However, it is unknown whether metformin inhibits BAFF-activated cell proliferation and survival in B cells by mediating a crosstalk between the PTEN, Akt, mTOR and Erk1/2 pathways.
Here we show that metformin inhibits hsBAFF-stimulated cell proliferation and survival by activation of PTEN and inactivation of Akt, thereby resulting in inhibition of Erk1/2 pathway in normal and neoplastic B-lymphoid cells. Furthermore, we demonstrate that the effect of metformin on the PTEN/Akt-Erk1/2 pathway is dependent on inhibition of mTOR in the cells. Our data highlight that metformin is a potential agent for prevention of excessive BAFF-induced aggressive B-cell malignancies and autoimmune diseases.
2. Materials and methods
2.1. Materials
Anti-CD19 magnetic fluorobeads-B was purchased from One Lambda (Canoga Park, CA, USA). Metformin, U0126 and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA), whereas Akt inhibitor X were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Rapamycin was from ALEXIS (San Diego, CA, USA). Refolded human soluble BAFF (hsBAFF) was a recombinant form of the extracellular domain of the BAFF produced in Escherichia coli from this group [34]. RPMI 1640 medium and fetal bovine serum (FBS) were from Gibco (Rockville, MD, USA). CellTiter 96®AQueous One Solution Cell Proliferation Assay kit was from Promega (Madison, WI, USA). Enhanced chemiluminescence solution was supplied by Sciben Biotech Company (Nanjing, China). The following primary antibodies were used: phosphorylated Akt (p-Akt) (Ser473), p-S6K1 (Thr389), p-S6 (Ser235/236) (Cell Signaling Technology, Danvers, MA, USA), S6K1, S6, p-Erk1/2 (Thr202/Tyr204), Erk2 (Santa Cruz Biotechnology), p-PTEN (Thr366), PTEN (Epitomics, Burlingame, CA, USA), MKK1, mTOR (Sigma), Erk1/2, survivin, Akt, β-actin, FLAG, HA (Sciben Biotech Company); Goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Sciben Biotech Company). Other chemicals used in this work were purchased from local commercial sources and were of analytical grade.
2.2. Cell culture
Neoplastic B-lymphoid Raji cell line (American Type Culture Collection, Manassas, VA, USA) was maintained in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin and incubated at 37 °C in a humidified incubator containing 5% CO2. To verify the results from Raji cell line, murine primary B lymphocytes were purified from fresh splenic cells of healthy mice using anti-CD19 magnetic fluorobeads-B and cultured as described previously [28]. All procedures for this study were approved by the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
2.3. Recombinant adenoviral constructs and infection of cells
The recombinant adenovirus encoding HA-tagged dominant negative Akt (Ad-dn-Akt, T308A/S473A) was generously provided from Dr. Kenneth Walsh (Boston University, Boston, MA). The recombinant adenoviruses expressing FLAG-tagged constitutively active MKK1 (Ad-MKK1-R4F), FLAG-tagged dominant negative MKK1 (Ad-MKK1-K97M), wild-type human PTEN (Ad-PTEN), and the control adenovirus expressing the green fluorescent protein (GFP) (Ad-GFP) were described previously [35,36]. For experiments, Raji cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Expression of HA-tagged dn-Akt and FLAG-tagged MKK1 was detected by Western blotting with antibodies to HA and FLAG, respectively.
2.4. Lentiviral shRNA cloning, production and infection
Lentiviral shRNAs to Erk1/2, mTOR, and GFP (for control) were generated and used as described [37,38]. For use, Raji cells, when grown to about 70% confluence, were infected with above lentivirus containing supernatants in the presence of 8 μg/ml polybrene for 12 h twice at an interval of 6 h. Uninfected cells were eliminated by exposure to 2 μg/ml puromycin for 48 h before use. After 5 days of culture, cells were used for experiments.
2.5. Assays for cell proliferation, cell viability, and live cell number
Purified mouse splenic B lymphocytes and Raji cells, or Raji cells infected with Ad-MKK1-R4F, Ad-MKK1-K97M, Ad-PTEN, Ad-dn-Akt and Ad-GFP, respectively, or Raji cells infected with lentiviral shRNAs to Erk1/2, mTOR, and GFP, respectively, were seeded in 24-well plates (3 × 105 cells/well, for cell proliferation assay and live cell assay) or 96-well plates (3 × 104 cells/well, for cell viability assay) and cultured for overnight in a humidified incubator of 5% CO2 at 37 °C. Next day, cells were treated with metformin (0–10 mM) for 48 h, or pretreated with/without metformin (1–5 mM or 1.5 mM) for 4 h, or pretreated with/without metformin (1.5 mM) for 4 h and then with/without rapamycin (100 ng/ml) for 2 h, or Akt inhibitor X (20 μM) or U0126 (5 μM) for 1 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 48 h with 5 replicates of each treatment. Subsequently, cell proliferation was assessed using a Coulter Counter (Beckman Coulter, Fullerton, CA, USA), and the viability of the cells, after incubation with MTS reagent (one solution reagent) (20 μl/well) for 4 h, was detected with a Victor X3 Light Plate Reader (PerkinElmer, Waltham, MA, USA), as described previously [39]. Live cells were estimated by counting viable cells using trypan blue exclusion.
2.6. Western blot analysis
After treatments, Western blotting was performed as described [37]. The blots for detected proteins were semi-quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA) and were normalized using β-actin as an internal control.
2.7. Statistical analysis
All results were presented as mean values ± standard error (mean ± SE). The Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. Significance was accepted at P < 0.05.
3. Results
3.1. Metformin attenuates hsBAFF-induced cell proliferation and survival through blocking the Erk1/2 pathway in normal and lymphoma B cells
It has been described that Erk1/2 play a vital role in proliferation and survival of different types of cells [40]. Our recent study has identified that hsBAFF-stimulated cell proliferation and survival is dependent on activation of Erk1/2 [41]. Additionally, multiple studies have documented that metformin may activate or inhibit Erk1/2, depending on the concentrations of metformin or distinct cell lines used [42–45]. Here we wondered whether metformin attenuates hsBAFF-stimulated cell proliferation and survival by targeting Erk1/2. Firstly, Raji cells and purified mouse splenic B lymphocytes were treated with metformin (0–10 mM) for 12 h or 48 h, showing that metformin reduced the levels of p-Erk1/2 and survivin (Fig. 1A and B), and evoked cell viability reduction (Fig. 1C) in a dose-dependent manner. Based on the reports from others [21,46,47], subsequently, the concentrations of 1–5 mM metformin were chosen for the experiments co-treated with/without hsBAFF. As predicted, after the indicated cells were pretreated with/without metformin (1–5 mM) for 4 h and then stimulated with/without hsBAFF (2.5 μg/ml) for 12 h or 48 h, Western blot analysis showed that metformin effectively inhibited hsBAFF-induced expressions of p-Erk1/2 and survivin dose-dependently (Fig. 1D and E). Interestingly, metformin also potently reduced hsBAFF-stimulated cell proliferation and survival, as evaluated by cell counting (Fig. 1F), MTS assay (Fig. 1G), and trypan blue exclusion (Fig. 1H), respectively. The results suggest that metformin inhibits hsBAFF-induced B-cell proliferation and survival, possibly associated with inhibition of the Erk1/2 pathway. As 1.5 mM of metformin was able to inhibit the cell proliferation/viability nearly to the basal level, this concentration was selected for further studies, as described below.
Fig. 1.

Metformin attenuates hsBAFF-induced phosphorylation of Erk1/2 as well as proliferation and survival in normal and lymphoma B cells. Raji cells and purified mouse splenic B lymphocytes were treated with metformin (0–10 mM) for 12 h or 48 h, or with/without metformin (1–5 mM) for 4 h and then stimulated with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and/or viability assay). (A and D) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and E) The relative densities for p-Erk1/2 and survivin to β-actin were semi-quantified using NIH image J. (F) Cell proliferation was estimated by cell counting. (C and G) Cell viability was evaluated by MTS assay. (H) Relative number of live cells was detected by trypan blue exclusion assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs normal control group; #P < .05, difference vs 2.5 μg/ml hsBAFF group.
To substantiate the above finding, Raji cells and purified mouse splenic B lymphocytes were pre-incubated with/without U0126 (5 μM) (a selective inhibitor of MKK1/2, upstream of Erk1/2) alone, or in combination with metformin (1.5 mM). We found that U0126 or metformin alone obviously suppressed the basal and hsBAFF-stimulated expression of p-Erk1/2/survivin and cell proliferation/viability, whereas co-treatment with metformin/U0126 exhibited the inhibitory effects more potently than treatment with metformin or U0126 alone in the cells (Fig. 2A–D).
Fig. 2.

Inhibition of Erk1/2 potentiates metformin’s suppression of hsBAFF-stimulated proliferation and viability in B cells. Raji cells and purified mouse splenic B lymphocytes, or Raji cells, infected with lentiviral shRNA to Erk1/2 or GFP (as control), respectively, were pretreated with/without metformin (1.5 mM) for 4 h and then with/without U0126 (5 μM) for 1 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation/viability assay). (A and E) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and F) The relative densities for p-Erk1/2 and survivin to β-actin were semi-quantified using NIH image J. (C and G) Cell proliferation was estimated by cell counting. (D and H) Cell viability was evaluated by MTS assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs 0 μg/ml hsBAFF group; #P < .05, difference vs 2.5 μg/ml hsBAFF group; $P < .05, difference vs hsBAFF/Metformin group or hsBAFF/U0126 group; †P < .05, Erk1/2 shRNA group vs GFP shRNA group.
To further validate the vital role of Erk1/2 in metformin’s inhibition of hsBAFF-induced B-cell proliferation/viability, we extended our studies using gene silencing or overexpression experiments. As shown in Fig. 2E, lentiviral shRNA to Erk1/2, but not to GFP, silenced the expression of Erk1/2 protein by ~90% in Raji cells. Knockdown of Erk1/2 obviously reduced the basal or hsBAFF-induced expression of p-Erk1/2 and survivin, as detected by Western blotting (Fig. 2E and F). Coincidently, silencing Erk1/2 also resulted in a significant decrease of the basal or hsBAFF-stimulated proliferation/viability in Raji cells (Fig. 2G and H), which was strengthened by addition of metformin (Fig. 2E–H).
In addition, we employed recombinant adenoviruses encoding FLAG-tagged constitutively active MKK1 (Ad-MKK1-R4F) and dominant negative MKK1 (Ad-MKK1-K97M), respectively. Infection of Raji cells with Ad-MKK1-R4F and Ad-MKK1-K97M, but not Ad-GFP (as control), resulted in high expression of FLAG-tagged MKK1 mutants (Fig. 3A). Expression of MKK1-R4F triggered robust expression of p-Erk1/2 and survivin even without stimulation with hsBAFF, whereas expression of MKK1-K97M led to suppression of the basal and hsBAFF-stimulated expression of p-Erk1/2 and survivin (Fig. 3A and B), indicating that the MKK1 mutants function in the cells as expected. Notably, expression of MKK1-R4F in Raji cells remarkably elevated the basal or hsBAFF-stimulated cell proliferation/viability, and conferred profound resistance to metformin’s inhibitory effects (Fig. 3C and D). On the contrary, expression of MKK1-K97M in the cells markedly repressed the basal or hsBAFF-stimulated cell proliferation/viability, which was strengthened by addition of metformin (Fig. 3C and D). Taken together, these results strongly support the notion that metformin attenuates hsBAFF-induced cell proliferation and survival through blocking the Erk1/2 pathway in normal and lymphoma B cells.
Fig. 3.
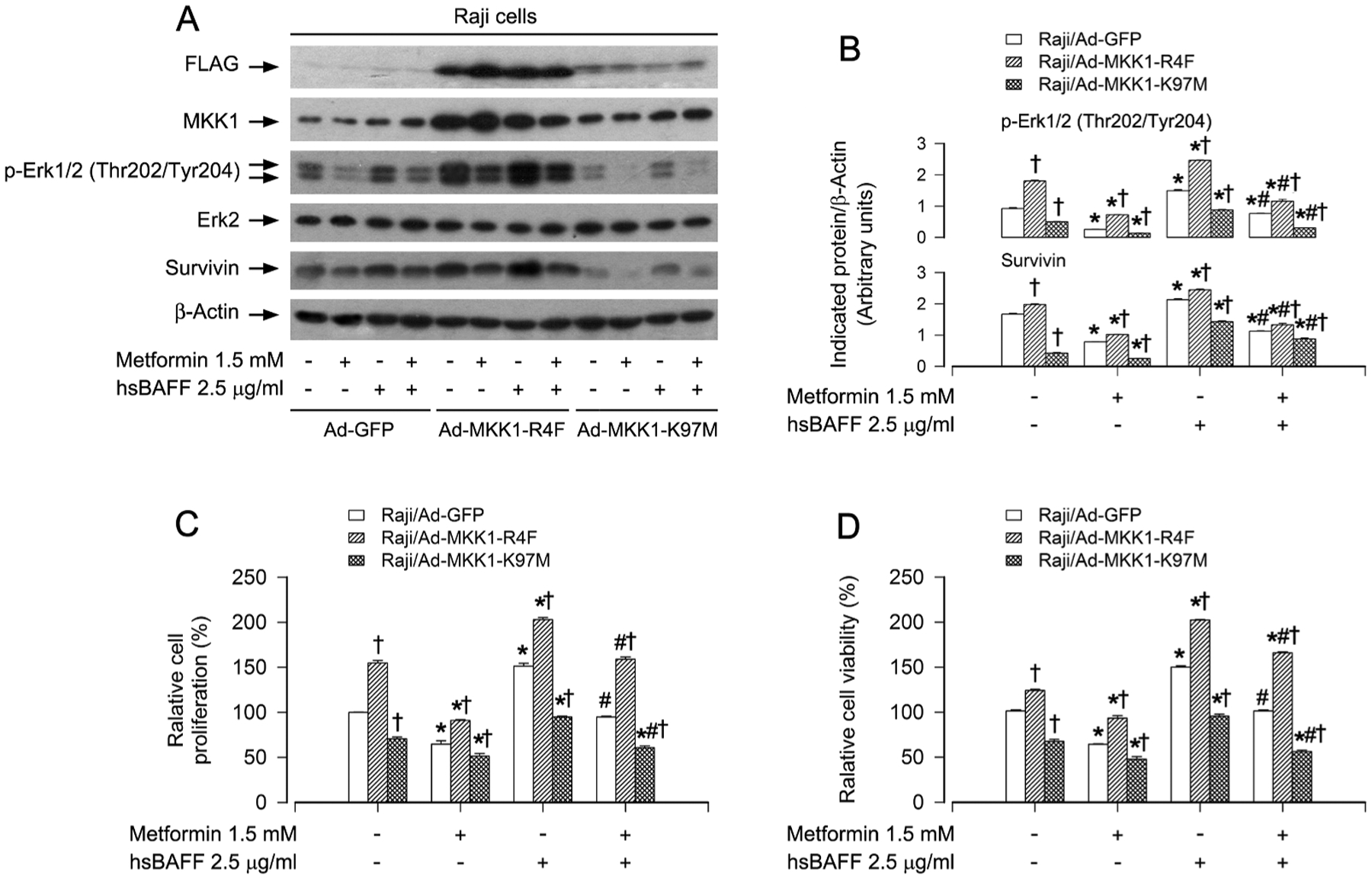
Expression of constitutively active or dominant negative MKK1 affects metformin’s inhibition of hsBAFF-induced Erk1/2 activation and proliferation/viability in B cells. Raji cells, infected with Ad-MKK1-R4F, Ad-MKK1-K97M and Ad-GFP (as control), respectively, were pretreated with/without metformin (1.5 mM) for 4 h and then stimulated with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation/viability assay). (A) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B) The relative densities for p-Erk1/2 and survivin to β-actin were semi-quantified using NIH image J. (C) Cell proliferation was estimated by cell counting. (D) Cell viability was evaluated by MTS assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs 0 μg/ml hsBAFF group; #P < .05, difference vs 2.5 μg/ml hsBAFF group; †P < .05, Ad-MKK1-R4F group or Ad-MKK1-K97M group vs Ad-GFP group.
3.2. Metformin suppresses hsBAFF-induced Erk1/2 activation and proliferation/viability via a PTEN/Akt-dependent mechanism in B cells
It is well known that PTEN negatively regulates the PI3K/Akt signaling [48]. Recent studies have shown that metformin reduces HaCat cell viability, and that metformin synergistically enhances the anticancer activity of cisplatin in gallbladder cancer, both by inhibiting the PI3K/Akt pathway [44,49]. This prompted us to examine the role of PTEN/Akt in metformin’s inhibition of hsBAFF-stimulated B-cell proliferation and survival. Our results showed that metformin obviously suppressed hsBAFF-induced phosphorylation of PTEN (Thr366) and Akt (Ser473) dose-dependently in Raji cells and purified mouse splenic B lymphocytes (Fig. 4A and B), implying that metformin may block hsBAFF-induced inactivation of PTEN and activation of Akt, thus hindering hsBAFF-stimulated B-cell proliferation/viability.
Fig. 4.

Metformin suppresses hsBAFF-induced Erk1/2 activation and proliferation/viability via a PTEN/Akt-dependent mechanism in B cells. Raji cells and purified mouse splenic B lymphocytes were pretreated with/without metformin (1–5 mM) for 4 h and then stimulated with/without hsBAFF (2.5 μg/ml) for 12 h or Raji cells, infected with Ad-PTEN, Ad-dn-Akt and Ad-GFP (as control), respectively, were pretreated with/without metformin (1.5 mM) for 4 h and then with/without Akt inhibitor X (20 μM) or U0126 (5 μM) for 1 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation/viability assay). (A, C, G) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B, D, H) The relative densities for p-PTEN, p-Akt, p-Erk1/2, and survivin to β-actin were semi-quantified using NIH image J. (E and I) Cell proliferation was estimated by cell counting. (F and J) Cell viability was evaluated by MTS assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs 0 μg/ml hsBAFF group; #P < .05, difference vs 2.5 μg/ml hsBAFF group; †P < .05, Ad-PTEN group or Ad-dn-Akt group vs Ad-GFP group.
Since PTEN can negatively regulate the Erk1/2 pathway in several malignancies, and Akt can activate Erk1/2 through PKC [30], next, we wondered whether metformin blocks hsBAFF-induced Erk1/2 activation and proliferation/viability by modulating PTEN/Akt signaling in B cells. To this end, Raji cells, infected with recombinant adenovirus expressing wild-type human PTEN (Ad-PTEN) or Ad-GFP (as control), were pretreated with/without metformin (1.5 mM) for 4 h or Akt inhibitor X (20 μM) for 1 h, followed by stimulation with hsBAFF (2.5 μg/ml) for 12 h or 48 h. The results showed that infection with Ad-PTEN increased the expression of PTEN and depressed the basal and hsBAFF-elicited expression of p-Akt, p-Erk1/2, and survivin (Fig. 4C and D). Overexpression of PTEN reinforced the inhibitory effects of metformin or Akt inhibitor X on hsBAFF-induced expression of p-Akt, p-Erk1/2, and survivin (Fig. 4C and D). Of importance, overexpression of PTEN also potentiated metformin’s or Akt inhibitor X’s suppression on hsBAFF-stimulated B-cell proliferation/viability (Fig. 4E and F). The findings support the idea that metformin inhibits hsBAFF-triggered activation of Erk1/2 and consequential B-cell proliferation/viability, by blocking hsBAFF-induced inactivation of PTEN and activation of Akt.
To further validate the role of Akt in metformin’s blockage of hsBAFF-induced Erk1/2 activation and proliferation/viability in B cells, Raji cells were infected with recombinant adenovirus expressing HA-tagged dominant-negative Akt (Ad-dn-Akt). As shown in Fig. 4G, there exited a high expression of HA-tagged Akt mutant in Ad-dn-Akt-infected cells, but not in Ad-GFP-infected cells. Ectopic expression of dn-Akt markedly repressed the basal and hsBAFF-evoked expression of p-Akt, p-Erk1/2 and survivin in the cells (Fig. 4G and H). Meanwhile, we also observed that both metformin and U0126 substantially suppressed the basal and hsBAFF-induced expression of p-Akt, p-Erk1/2, and survivin (Fig. 4G and H). Of note, overexpression of dn-Akt was able to reinforce the inhibitory effects of metformin or U0126 on hsBAFF-stimulated expression of p-Akt, p-Erk1/2, and survivin (Fig. 4G and H), as well as proliferation/viability (Fig. 4I and J) in the cells. Collectively, these data indicate that metformin suppresses hsBAFF-induced Erk1/2 activation and proliferation/viability via a PTEN/Akt-dependent mechanism in B cells.
3.3. Inhibition of mTOR is critical for metformin’s regulation of PTEN/Akt signaling and consequential suppression of Erk1/2 and cell proliferation/viability in hsBAFF-stimulated B cells
AMPK is a negative regulator of mTOR, a central controller of cell growth, proliferation and survival [25,50]. Metformin can activate AMPK, thus inhibiting mTOR signaling both in vivo and in vitro [21,51]. Our recent studies have demonstrated that excess hsBAFF activates the mTOR pathway promoting B-cell proliferation and survival [27,28]. Therefore, we asked whether metformin exerts the inhibitory effects on hsBAFF-induced B-cell proliferation/viability through hampering the mTOR pathway. As shown in Fig. 5A and B, metformin significantly suppressed hsBAFF-induced phosphorylation of mTOR, S6K1 and S6 dose-dependently in Raji cells and purified mouse splenic B lymphocytes, suggesting that metformin indeed inhibits hsBAFF-activated mTOR pathway in B cells.
Fig. 5.

Metformin inhibits mTOR, thereby blocking hsBAFF-induced inactivation of PTEN and activation of Akt, leading to suppression of Erk1/2 and proliferation/viability in B cells. Raji cells and purified mouse splenic B lymphocytes were pretreated with/without metformin (1–5 mM) for 4 h, or pretreated with/without metformin (1.5 mM) for 4 h and then with/without rapamycin (100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation/viability assay). (A and C) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and D) The relative densities for p-mTOR, p-S6K1, p-S6, p-PTEN, p-Akt, p-Erk1/2, and survivin to β-actin were semi-quantified using NIH image J. (E) Cell proliferation was estimated by cell counting. (F) Cell viability was evaluated by MTS assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs 0 μg/ml hsBAFF group; #P < .05, difference vs 2.5 μg/ml hsBAFF group; $P < .05, difference vs hsBAFF/Metformin group or hsBAFF/Rapamycin group.
Next, we test whether metformin’s inhibition of mTOR pathway is related to metformin’s suppression of hsBAFF-induced inactivation of PTEN and activation of Akt, leading to repression of Erk1/2 and cell proliferation/viability in B cells. For this, Raji cells and purified mouse splenic B lymphocytes were pretreated with/without metformin (1.5 mM) for 4 h and then with/without rapamycin (100 ng/ml, a specific mTOR inhibitor) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h or 48 h. We found that rapamycin or metformin alone obviously suppressed the basal and hsBAFF-stimulated expression of p-mTOR, p-S6K1, p-PTEN, p-Akt, p-Erk1/2 and survivin in the cells, which was strengthened by co-treatment with metformin/rapamycin (Fig. 5C and D). In line with this, co-treatment with metformin/rapamycin also exhibited more potent inhibition on hsBAFF-triggered B-cell proliferation/viability than treatment with metformin or rapamycin alone, as evidenced by cell counting and MTS assay (Fig. 5E and F).
In agreement with the above findings, knockdown of mTOR (by ~90%) in Raji cells (Fig. 6A) resulted in suppression of the basal or hsBAFF-induced activation of mTOR signaling, as p-mTOR (S2448) and p-S6K1 (T389) (an indicator of mTOR kinase activity) was almost non-detectable by Western blot analysis (Fig. 6B and C). Of importance, knockdown of mTOR drastically blocked hsBAFF-induced expression of p-PTEN, p-Akt, p-Erk1/2 and survivin in the cells even without pretreatment with metformin (Fig. 6B and C). In addition, as expected, knockdown of mTOR obviously suppressed the basal and hsBAFF-induced cell proliferation/viability, and potentiated the inhibitory effect of metformin (Fig. 6D and E). Taken together, our data underscore the concept that inhibition of mTOR is critical for metformin’s regulation of PTEN/Akt signaling and consequential suppression of Erk1/2 and cell proliferation/viability in hsBAFF-stimulated B cells.
Fig. 6.

Knockdown of mTOR strengthens metformin’s inhibition of hsBAFF-induced inactivation of PTEN, activation of Ak and Erk1/2, and proliferation/viability in B cells. Raji cells, infected with lentiviral shRNA to mTOR or GFP (as control), were pretreated with/without metformin (1.5 mM) for 4 h and then stimulated with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation/viability assay). (A and B) Total cell lysates were subjected to Western blotting with indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (C) The relative densities for p-mTOR, p-S6K1, p-PTEN, p-Akt, p-Erk1/2, and survivin to β-actin were semi-quantified using NIH image J. (D) Cell proliferation was estimated by cell counting. (E) Cell viability was evaluated by MTS assay. The results were expressed as mean ± SE (n = 3–5). *P < .05, difference vs 0 μg/ml hsBAFF group; #P < .05, difference vs 2.5 μg/ml hsBAFF group; †P < .05, mTOR shRNA group vs GFP shRNA group.
4. Discussion
BAFF, a member of the TNF family, is a fundamental factor for B-cell maturation, differentiation and survival [52]. Recently, it has been reported that BAFF can attenuate oxidative stress-induced cell death by regulation of mitochondria membrane potential in B lymphoblasts [53]. However, increased levels of BAFF have been associated with autoimmune diseases, including SLE, SS and rheumatoid arthritis (RA), as well as multiple myeloma, non-Hodgkin’s lymphoma, B-lineage lymphomas and Hodgkin’s lymphoma [10,11,54,55]. The role of BAFF in aggressive B-cell malignancies and autoimmune diseases has been well established, but how to target the BAFF signaling to control these diseases remains a great challenge. Metformin, the most commonly prescribed medicine for type 2 diabetes (T2D), has recently garnered more attention because of its pleiotropic effects [17,18,22,56]. Metformin is known to affect cell energy metabolism and suppress cell cycle [46,47,57,58]. For example, metformin induces activation of the energy-sensor AMPK in various cells [58]; Metformin causes cell cycle arrest in the G1 phase in pre-clinical and clinical diffuse large B-cell lymphoma [47]; Metformin inhibits cell prolferation and growth in B-/T-lymphoma cells and SKM-1 cells via AMPK-mediated cell cycle arrest, and metformin-induced AMPK activation leads to inhibition of the mTOR signaling contributing to the cell growth inhibition [46,57]. In this study, for the first time, we provide evidence that metformin is able to suppress BAFF-stimulated cell proliferation and survival by activating PTEN and inactivating Akt, thereby leading to inhibition of Erk1/2 pathway in normal and neoplastic B-lymphoid cells. Further, we demonstrate that the effect of metformin on the PTEN/Akt-Erk1/2 pathway is dependent on the blockage of mTOR in the cells.
It has been shown that metformin impacts diverse cellular functions by regulating the Erk1/2 signaling pathway in distinct cell lines [42–45]. Our previous studies have identified that hsBAFF activates the Erk1/2 pathway promoting cell proliferation and survival in normal and neoplastic B-lymphoid cells [41]. In this study, we found that metformin attenuated the basal and hsBAFF-stimulated expression of p-Erk1/2 and survivin, as well as proliferation and survival in Raji cells and purified mouse splenic B lymphocytes in a dose-dependent manner (Fig. 1). Further investigation revealed that metformin inhibited hsBAFF-induced B-cell proliferation/viability by inhibition of the Erk1/2 pathway, as pharmacological inhibition of Erk1/2 with U0126, or depletion of Erk1/2 by RNA interference strengthened metformin’s suppression of hsBAFF-evoked proliferation/viability in the cells (Fig. 2). Furthermore, we also demonstrated that ectopic expression of dominant negative MKK1 (MKK1-K97M) enhanced metformin’s inhibition of hsBAFF-stimulated p-Erk1/2 and cell proliferation/viability in Raji cells, whereas expression of constitutively active MKK1 (MKK1-R4F) had opposite effects (Fig. 3). Taken together, these findings support that metformin attenuates hsBAFF-induced cell proliferation and survival through blocking the Erk1/2 pathway in normal and lymphoma B cells.
PTEN, a dual-specificity lipid and protein phosphatase, negatively regulates PI3K/Akt signaling [29]. In the current study, we observed that metformin elevated PTEN activity and repressed Akt activity, as metformin reduced hsBAFF-induced p-PTEN and p-Akt in Raji cells and purified mouse splenic B lymphocytes (Fig. 4A and B). Emerging studies have documented that PTEN can also negatively regulate Erk1/2 pathway, and Akt can activate Erk1/2 through PKC in several malignancies [30,32]. Therefore, we postulated that metformin might mediate a crosstalk between PTEN, Akt and Erk1/2 pathways in the B cells in response to BAFF. Here, we unveiled that metformin inhibited Erk1/2-dependent B-cell proliferation/viability by preventing hsBAFF from inactivation of PTEN and activation of Akt. This is strongly supported by the observations that overexpression of wild type PTEN or dominant negative Akt, or pretreatment with Akt inhibitor X or U0126 remarkably potentiated metformin’s suppression of hsBAFF-induced Erk1/2 activation and proliferation/viability in Raji cells (Fig. 4C–J). Collectively, our results highlight that metformin hinders hsBAFF-induced Erk1/2 activation and proliferation/viability through a PTEN/Akt-dependent mechanism in normal and lymphoma B cells.
mTOR senses and integrates a variety of environmental cues to regulate organismal growth and homeostasis, and is negatively regulated by AMPK [25,50]. It has been demonstrated that the major molecular targets of metformin include AMPK and mTOR complex 1 (mTORC1) [18,59]. mTOR plays a critical role in B-cell differentiation, proliferation/growth and antibody responses [26,27]. Our recent studies have shown that BAFF promotes B-cell proliferation and survival via activation of mTORC1/2 signaling [27]. Accordingly, we reasoned that metformin inhibits B-cell proliferation/viability via impeding the PTEN/Akt-Erk1/2 signaling pathway, which may be associated with inhibition of mTOR. In this study, as expected, we observed that metformin indeed blocked hsBAFF-activated mTOR pathway in B cells (Fig. 5A and B). Of importance, inhibition of mTOR with rapamycin potentiated metformin’s inhibitory effects on p-PTEN, p-Akt, p-Erk1/2 and B-cell proliferation/viability (Fig. 5C–F). This is further supported by the findings that silencing mTOR mimicked rapamycin’s effects in Raji cells in response to metformin and/or hsBAFF (Fig. 6). These results indicate that metformin exerts the inhibitory effects on hsBAFF-stimulated cell proliferation/viability by impeding mTOR-dependent PTEN/Akt-Erk1/2 signaling pathway in B cells.
In summary, here we have identified that metformin prevents BAFF activation of Erk1/2 from cell proliferation and survival by impeding mTOR-PTEN/Akt signaling pathway in normal and neoplastic B-lymphoid cells (Fig. 7). Our findings support that metformin has a great potential for prevention of excessive BAFF-induced aggressive B-cell malignancies and autoimmune diseases.
Fig. 7.

Potential mechanisms of how metformin inhibits hsBAFF-stimulated B-cell proliferation and survival. Metformin prevents BAFF activation of Erk1/2 from cell proliferation and survival by impeding mTOR-PTEN/Akt signaling pathway in normal and neoplastic B-lymphoid cells.
Acknowledgements
This work was supported in part by the grants from National Natural Science Foundation of China (No.31172083), National Institutes of Health (CA115414), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010), and American Cancer Society (RSG-08-135-01-CNE).
Abbreviations:
- Akt
protein kinase B (PKB)
- AMPK
AMP-activated protein kinase
- BAFF
B-cell activating factor
- BLyS
B lymphocyte stimulator
- BCMA
B cell maturation antigen
- Erk1/2
extracellular signal-related kinases 1/2
- FBS
fetal bovine serum
- GC
germinal center
- GFP
green fluorescent protein
- hsBAFF
human soluble BAFF
- MAPK
mitogen-activated protein kinase
- MKK
mitogen-activated protein kinase kinase
- MS
multiple sclerosis
- mTOR
mammalian target of rapamycin
- mTORC1/2
mTOR complexes 1/2
- PKC
protein kinase C
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- PBS
phosphate buffered saline
- PI3K
phosphatidylinositol 3′-kinase
- RA
rheumatoid arthritis
- S6K1
ribosomal protein S6 kinase 1
- SLE
systemic lupus erythematosus
- SS
Sjögren’s syndrome
- TACI
transmembrane activator and cyclophilin ligand interactor
- TALL-1
TNF and apoptosis ligand-related leukocyte-expressed ligand 1
- THANK
TNF homologue that activates apoptosis, nuclear factor κB, and c-Jun NH2-terminal kinase
- TRPML1
transient receptor potential mucolipin 1
- T2D
type 2 diabetes
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The data used to support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Shu H-B, Hu W-H, Johnson H, TALL-1 is a novel member of the TNF family that is down-regulated by mitogens, J. Leukoc. Biol 65 (5) (1999) 680–683. [PubMed] [Google Scholar]
- [2].Mukhopadhyay A, Ni J, Zhai Y, et al. , Identification and characterization of a novel cytokine, THANK, a TNF homologue that activates apoptosis, nuclear factor-kappaB, and c-Jun NH2-terminal kinase, J. Biol. Chem 274 (23) (1999) 15978–15981. [DOI] [PubMed] [Google Scholar]
- [3].Moore PA, Belvedere O, Orr A, et al. , BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator, Science 285 (5425) (1999) 260–263. [DOI] [PubMed] [Google Scholar]
- [4].Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH, TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease, Nature 404 (6781) (2000) 995–999. [DOI] [PubMed] [Google Scholar]
- [5].Mackay F, Schneider P, Rennert P, et al. , BAFF AND APRIL: a tutorial on B cell survival, Annu. Rev. Immunol 21 (2003) 231–264. [DOI] [PubMed] [Google Scholar]
- [6].Jackson SW, Davidson A, BAFF inhibition in SLE-Is tolerance restored? Immunol. Rev 292 (1) (2019) 102–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dekaban GA, Thawer S, Pathogenic antibodies are active participants in spinal cord injury, J. Clin. Invest 119 (10) (2009) 2881–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mackay F, Schneider P, Cracking the BAFF code, Nat. Rev. Immunol 9 (7) (2009) 491–502. [DOI] [PubMed] [Google Scholar]
- [9].Battaglino RA, Nguyen N, Summers M, Morse LR, B cell-activating factor is associated with testosterone and smoking status in non-ambulatory men with chronic spinal cord injury, J. Neurotrauma 36 (24) (2019) 3332–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vadacca M, Margiotta D, Sambataro D, et al. , BAFF/APRIL pathway in Sjogren syndrome and systemic lupus erythematosus: relationship with chronic inflammation and disease activity, Reumatismo 62 (4) (2010) 259–265. [DOI] [PubMed] [Google Scholar]
- [11].Steri M, Orru V, Idda ML, et al. , Overexpression of the cytokine BAFF and autoimmunity risk, N. Engl. J. Med 376 (17) (2017) 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Marin-Rosales M, Cruz A, Salazar-Camarena DC, et al. , High BAFF expression associated with active disease in systemic lupus erythematosus and relationship with rs9514828C>T polymorphism in TNFSF13B gene, Clin. Exp. Med 19 (2) (2019) 183–190. [DOI] [PubMed] [Google Scholar]
- [13].Rojas LB, Gomes MB, Metformin: an old but still the best treatment for type 2 diabetes, Diabetol. Metab. Syndr 5 (1) (2013) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA, Metformin as a tool to target aging, Cell Metab. 23 (6) (2016) 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cunha V, Cotrim HP, Rocha R, Carvalho K, Lins-Kusterer L, Metformin in the prevention of hepatocellular carcinoma in diabetic patients: A systematic review, Ann. Hepatol 19 (3) (2020) 232–237. [DOI] [PubMed] [Google Scholar]
- [16].Gao J, Yuan J, Wang Q, et al. , Metformin protects against PM2.5-induced lung injury and cardiac dysfunction independent of AMP-activated protein kinase alpha2, Redox Biol. 28 (2020), 101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Glossmann HH, Lutz OMD, Metformin and aging: A review, Gerontology 65 (6) (2019) 581–590. [DOI] [PubMed] [Google Scholar]
- [18].Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I, Metformin as an Anticancer Agent, Trends Pharmacol. Sci 39 (10) (2018) 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cui Y, Chang L, Wang C, et al. , Metformin attenuates autoimmune disease of the neuromotor system in animal models of myasthenia gravis, Int. Immunopharmacol 75 (2019), 105822. [DOI] [PubMed] [Google Scholar]
- [20].Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D, Metformin improves in vivo and in vitro B cell function in individuals with obesity and Type-2 Diabetes, Vaccine 35 (20) (2017) 2694–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee SY, Moon SJ, Kim EK, et al. , Metformin suppresses systemic autoimmunity in Roquinsan/san mice through inhibiting B cell differentiation into plasma cells via regulation of AMPK/mTOR/STAT3, J. Immunol 198 (7) (2017) 2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ursini F, Russo E, Pellino G, et al. , Metformin and autoimmunity: A “New Deal” of an old drug, Front. Immunol 9 (2018) 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tomczynska M, Bijak M, Saluk J, Metformin - The drug for the treatment of autoimmune diseases; A new use of a known anti-diabetic drug, Curr. Top. Med. Chem 16 (19) (2016) 2223–2230. [DOI] [PubMed] [Google Scholar]
- [24].Rena G, Hardie DG, Pearson ER, The mechanisms of action of metformin, Diabetologia 60 (9) (2017) 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu GY, Sabatini DM, mTOR at the nexus of nutrition, growth, ageing and disease, Nat. Rev. Mol. Cell Biol 21 (4) (2020) 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K.e., Patel R, Hoover S, Simpson RM, Simmons J, Gary J, Snapper CM, Casellas R, Mock BA, B cell-specific deficiencies in mTOR limit humoral immune responses, J. Immunol 191 (4) (2013) 1692–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zeng Q, Qin S, Zhang H, Liu B, Qin J, Wang X, Zhang R, Liu C, Dong X, Zhang S, Huang S, Chen L, Rapamycin attenuates BAFF-extended proliferation and survival via disruption of mTORC1/2 signaling in normal and neoplastic B-lymphoid cells, J. Cell. Physiol 233 (1) (2018) 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ke Z, Liang D, Zeng Q, Ren Q, Ma H, Gui L, Chen S, Guo M, Xu Y, Gao W, Zhang S, Chen L, hsBAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway, Cytokine 62 (2) (2013) 310–321. [DOI] [PubMed] [Google Scholar]
- [29].Chalhoub N, Baker SJ, PTEN and the PI3-kinase pathway in cancer, Annu. Rev. Pathol 4 (1) (2009) 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chetram MA, Hinton CV, PTEN regulation of ERK1/2 signaling in cancer, J. Recept. Signal Transduct. Res 32 (4) (2012) 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Song MS, Salmena L, Pandolfi PP, The functions and regulation of the PTEN tumour suppressor, Nat. Rev. Mol. Cell Biol 13 (5) (2012) 283–296. [DOI] [PubMed] [Google Scholar]
- [32].Xing Y, Wang R, Li C, Minoo P, PTEN regulates lung endodermal morphogenesis through MEK/ERK pathway, Dev. Biol 408 (1) (2015) 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zeng Q, Zhou Z, Qin S, et al. , Rapamycin inhibits B-cell activating factor (BAFF)-stimulated cell proliferation and survival by suppressing Ca2+-CaMKII-dependent PTEN/Akt-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells, Cell Calcium 87 (2020), 102171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cao P, Mei JJ, Diao ZY, Zhang SQ, Expression, refolding, and characterization of human soluble BAFF synthesized in Escherichia coli, Protein Expr. Purif 41 (1) (2005) 199–206. [DOI] [PubMed] [Google Scholar]
- [35].Liu L, Chen L, Luo Y, Chen W, Zhou H, Xu B, Han X, Shen T, Huang S, Lau ATY, Rapamycin inhibits IGF-1 stimulated cell motility through PP2A pathway, PLoS One 5 (5) (2010) e10578, 10.1371/journal.pone.0010578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Findley CM, Cudmore MJ, Ahmed A, Kontos CD, VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling, Arterioscler. Thromb. Vasc. Biol 27 (12) (2007) 2619–2626. [DOI] [PubMed] [Google Scholar]
- [37].Chen L, Liu L, Luo Y, Huang S, MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis, J. Neurochem 105 (1) (2008) 251–261. [DOI] [PubMed] [Google Scholar]
- [38].Chen L, Xu B, Liu L, Liu C, Luo Y, Chen X, Barzegar M, Chung J, Huang S, Both mTORC1 and mTORC2 are involved in the regulation of cell adhesion, Oncotarget 6 (9) (2015) 7136–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zeng Q, Zhang H, Qin J, Xu Z, Gui L, Liu B, Liu C, Xu C, Liu W, Zhang S, Huang S, Chen L, Rapamycin inhibits BAFF-stimulated cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells, Cell. Mol. Life Sci 72 (24) (2015) 4867–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roskoski R Jr., ERK1/2 MAP kinases: structure, function, and regulation, Pharmacol. Res 66 (2) (2012) 105–143. [DOI] [PubMed] [Google Scholar]
- [41].Liang D, Zeng Q, Xu Z, Zhang H, Gui L, Xu C, Chen S, Zhang S, Huang S, Chen L, BAFF activates Erk1/2 promoting cell proliferation and survival by Ca2+-CaMKII-dependent inhibition of PP2A in normal and neoplastic B-lymphoid cells, Biochem. Pharmacol 87 (2) (2014) 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Malekpour-Dehkordi Z, Teimourian S, Nourbakhsh M, et al. , Metformin reduces fibrosis factors in insulin resistant and hypertrophied adipocyte via integrin/ERK, collagen VI, apoptosis, and necrosis reduction, Life Sci. 233 (2019), 116682. [DOI] [PubMed] [Google Scholar]
- [43].Yue H, Hu B, Luo Z, et al. , Metformin protects against sevoflurane-induced neuronal apoptosis through the S1P1 and ERK signaling pathways, Exp. Ther. Med 17 (2) (2019) 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bi T, Zhu A.o., Yang X, Qiao H, Tang J, Liu Y, Lv R, Metformin synergistically enhances antitumor activity of cisplatin in gallbladder cancer via the PI3K/AKT/ERK pathway, Cytotechnology 70 (1) (2018) 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu C-H, Hua N.a., Fu X.i., Pan Y-L, Li B, Li X-D, Metformin regulates atrial SK2 and SK3 expression through inhibiting the PKC/ERK signaling pathway in type 2 diabetic rats, BMC Cardiovasc. Disord 18 (1) (2018) 236, 10.1186/s12872-018-0950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shi W-Y, Xiao D, Wang L, Dong L-H, Yan Z-X, Shen Z-X, Chen S-J, Chen Y, Zhao W-L, Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy, Cell Death Dis. 3 (3) (2012) e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Singh AR, Gu JJ, Zhang Q, et al. , Metformin sensitizes therapeutic agents and improves outcome in pre-clinical and clinical diffuse large B-cell lymphoma, Cancer Metab. 8 (2020) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Polak P, Hall MN, mTOR and the control of whole body metabolism, Curr. Opin. Cell Biol 21 (2) (2009) 209–218. [DOI] [PubMed] [Google Scholar]
- [49].Wang Z, Guo J, Han X, et al. , Metformin represses the pathophysiology of AAA by suppressing the activation of PI3K/AKT/mTOR/autophagy pathway in ApoE−/− mice, Cell Biosci. 9 (2019) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mossmann D, Park S, Hall MN, mTOR signalling and cellular metabolism are mutual determinants in cancer, Nat. Rev. Cancer 18 (12) (2018) 744–757. [DOI] [PubMed] [Google Scholar]
- [51].Fan H, Yu X, Zou Z, Zheng W, Deng X, Guo L, Jiang W, Zhan Q, Lu S-H, Metformin suppresses the esophageal carcinogenesis in rats treated with NMBzA through inhibiting AMPK/mTOR signaling pathway, Carcinogenesis 40 (5) (2019) 669–679. [DOI] [PubMed] [Google Scholar]
- [52].Carrillo-Ballesteros FJ, Oregon-Romero E, Franco-Topete RA, et al. , B-cell activating factor receptor expression is associated with germinal center B-cell maintenance, Exp. Ther. Med 17(3) (2019) 2053–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Park S, Jang JW, Moon EY, BAFF attenuates oxidative stress-induced cell death by the regulation of mitochondria membrane potential via Syk activation in WiL2-NS B lymphoblasts, Sci. Rep 10 (1) (2020) 11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Saidoune F, Even G, Lamri Y, Chezel J, Gaston A-T, Escoubet B, Papo T, Charles N, Nicoletti A, Sacre K, Effects of BAFF neutralization on atherosclerosis associated with systemic lupus erythematosus, Arthritis Rheumatol. 73 (2) (2021) 255–264. [DOI] [PubMed] [Google Scholar]
- [55].Panzer SE, Anti-BAFF therapy: A new tool to target B cells in antibody-mediated rejection? Transplantation 104 (1) (2020) e3–e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jia L, Xiong Y, Zhang W, Ma X, Xu X, Metformin promotes osteogenic differentiation and protects against oxidative stress-induced damage in periodontal ligament stem cells via activation of the Akt/Nrf2 signaling pathway, Exp. Cell Res 386 (2) (2020) 111717, 10.1016/j.yexcr.2019.111717. [DOI] [PubMed] [Google Scholar]
- [57].Zhou X, Kuang Y, Liang S, Wang L.i., Metformin inhibits cell proliferation in SKM-1 cells via AMPK-mediated cell cycle arrest, J. Pharmacol. Sci 141 (4) (2019) 146–152. [DOI] [PubMed] [Google Scholar]
- [58].Foretz M, Guigas B, Bertrand L, et al. , Metformin: from mechanisms of action to therapies, Cell Metab. 20 (6) (2014) 953–966. [DOI] [PubMed] [Google Scholar]
- [59].Wang Y.u., An H, Liu T, Qin C, Sesaki H, Guo S, Radovick S, Hussain M, Maheshwari A, Wondisford FE, O’Rourke B, He L, Metformin improves mitochondrial respiratory activity through activation of AMPK, Cell Rep. 29 (6) (2019) 1511–1523.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon reasonable request.