

Abstract
Autophagy, a process for self-degradation of intracellular components and dysfunctional organelles, is closely related with neurodegenerative diseases. It has been shown that cadmium (Cd) induces neurotoxicity partly by impairing autophagy. However, the underlying mechanism is not fully elucidated. In this study, we show that Cd induced expansion of autophagosomes with a concomitant abnormal expression of autophagy-related (Atg) proteins in PC12 cells and primary murine neurons. 3-MA, a classical inhibitor of autophagy, attenuated Cd-induced expansion of autophagosomes and apoptosis in the cells. Further investigation demonstrated that Cd activated JNK pathway contributing to autophagosome expansion-dependent neuronal apoptosis. This is supported by the findings that pharmacological inhibition of JNK with SP600125 or expression of dominant negative c-Jun markedly attenuated Cd-induced expansion of autophagosomes and abnormal expression of Atg proteins, as well as apoptosis in PC12 cells and/or primary neurons. Furthermore, we noticed that chelating intracellular free Ca2+ ([Ca2+]i) with BAPTA/AM profoundly blocked Cd-elicited activation of JNK pathway and consequential expansion of autophagosomes, abnormal expression of Atg proteins, and apoptosis in the neuronal cells. Similar events were also seen following prevention of [Ca2+]i elevation with EGTA or 2-APB, implying a Ca2+-dependent mechanism involved. Taken together, the results indicate that Cd impairs autophagy leading to apoptosis by Ca2+-dependent activation of JNK signaling pathway in neuronal cells. Our findings highlight that manipulation of intracellular Ca2+ level and/or JNK activity to ameliorate autophagy may be a promising intervention against Cd-induced neurotoxicity and neurodegeneration.
Keywords: Cadmium, Autophagy, Calcium ion, JNK, Neuronal cells, Apoptosis
Introduction
Cadmium (Cd), a widespread industrial and environmental pollutant in air, water, and soil, easily enters the human body through the contaminated food chain and accumulates in diverse organs with a long half-life (15–20 years) [1]. As a very toxic heavy metal, Cd has toxic effects on multiple organs/systems, such as kidney, liver, lung, testis, bones and brain [1, 2]. In terms of neurotoxicity, Cd can cause headache and vertigo, olfactory dysfunction, slowing of vasomotor functioning, peripheral neuropathy, decreased equilibrium, neurobehavioral defects in attention, psychomotor speed, and learning disabilities [1, 2]. Of note, Cd poisoning has been documented as a possible pathogenesis for several major neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS) [1, 3–7].
Autophagy is a multistep process involving double membrane formation, elongation, vesicle maturation, and final delivery of the targeted materials to the lysosome, resulting in orderly degradation and recycling of cellular components [8–11]. There are three general types of autophagy, including macroautophagy, microautophagy and chaperone-mediated autophagy, each owning different mechanisms of substrate delivery to lysosome, and the term “autophagy” usually indicates macroautophagy unless otherwise specified [10–12]. Overwhelming evidence has unequivocally unraveled that in the context of neurodegenerative disorders, autophagosome is abundant in neurons, which confers neuroprotection or promotes neuronal cell death, and has been recognized as an arbiter that decides neuronal survival and death [9, 13]. Substantial efforts have demonstrated that autophagy plays context-dependent roles at various stages of neurodegenerative diseases, and that defects of autophagy and the resulting accumulation of protein aggregates represent a common pathobiological feature of neurodegenerative disorders [10, 11, 14].
It has been well-known that a series of autophagy-related (Atg) proteins regulate autophagosome initiation and formation [11, 15]. For example, the microtuble-associated protein 1 light chain 3 (LC3), a mammalian homologue of the yeast protein Atg8, contains two molecular forms with LC3-I and LC3-II. LC3-I is the unconjugated form in the cytosol, whereas LC3-II is the phosphatidylethanolamine-conjugated form localizing to autophagosomal membranes through an enzymatic cascade involving Atg7 (as an E1-like enzyme), Atg3 (as an E2-like enzyme) and Atg5-Atg12-Atg16 complex [11, 15–17]. As LC3-II directly correlates with the number of autophagosomes, the expression of LC3-II or green fluorescent protein (GFP)-LC3-II is monitored as a marker for evaluating the status of autophagy [16, 17]. Beclin 1, Atg14 and Ambra 1 are members of class III phosphatidylinositol 3’-kinase (PI3K) complex [11, 15]. Our recent work has shown that Cd evokes autophagosome accumulation-dependent apoptosis in neuronal cells [18]. However, the relationship between the manifestation of Atg proteins and accumulation of autophagosomes-dependent apoptosis in neuronal cells induced by Cd, as well as the underlying mechanism have not been addressed.
The mitogen-activated protein kinases (MAPKs), including the c-Jun N-terminal kinase (JNK), the extracellular signal-regulated kinases 1/2 (Erk1/2) and/or the p38 MAPK (p38), exert a critical role in autophagic cell survival and death under various conditions [19–21], and especially underscoring the relationship between JNK and autophagy [15, 22, 23]. Calcium ion (Ca2+) is a versatile and dynamic second messenger responsible for regulating numerous cellular processes, such as proliferation/growth, differentiation, survival and apoptosis including autophagy [24, 25]. It has been described that Disruption of intracellular free Ca2+ ([Ca2+]i) homeostasis by Cd contributes to autophagy and apoptosis in skin epidermal cells [26], hepatic cells [27], mesangial cells [28, 29], and thymocytes [30]. Our previous studies have shown that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 implicate neuronal apoptosis in response to Cd [31, 32], and unveiled that Cd elevates [Ca2+]i-dependent activation of MAPK pathway leading to neuronal apoptosis [33]. This prompted us to study whether Cd induces autophagosome accumulation-mediated neuronal apoptosis by Ca2+-dependent MAPK signaling pathway.
Here, we show that Cd induces apoptosis by evoking expansion of autophagosomes with a concomitant abnormal expression of Atg proteins in neuronal cells, by Ca+-dependent activation of JNK pathway. These findings highlight that manipulation of intracellular Ca2+ level and/or JNK activity to ameliorate autophagy may be a promising intervention against Cd-induced neurotoxicity and neurodegeneration.
Materials and methods
Materials
Cadmium chloride, poly-D-lysine (PDL), monodansylcadaverine (MDC), 4’,6-diamidino-2-phenylindole (DAPI), 3-methyladenine (3-MA), SP600125, U0126, PD169136, ethylene glycol tetra-acetic acid (EGTA), and protease inhibitor cocktail were purchased from Sigma (St. Louis, MO, USA). 1,2-bis (o-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid tetra (acetoxymethyl) ester (BAPTA/AM) and 2-aminoethoxydiphenyl borane (2-APB) were purchased from Calbiochem (San Diego, CA, USA). z-Val-Ala-Asp-CH2F (zVAD-fmk) were acquired from ALEXIS Biochemicals Corporation (San Diego, CA). CellTiter 96®AQueous One Solution Cell Proliferation Assay kit was from Promega (Madison, WI, USA). Enhanced chemiluminescence solution was from Sciben Biotech Company (Nanjing, China). Dulbecco’s modified Eagle medium (DMEM), 0.05% Trypsin–EDTA, NEUROBASAL™ Media, and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
Cell Culture
Rat pheochromocytoma (PC12) cell line (American Type Culture Collection, Manassas, VA, USA), which was used for no more than 10 passages, were seeded in a PDL (0.2 μg/ml)-coated 6-well or 96-well plate and grown in antibiotic-free DMEM supplemented with 10% horse serum and 5% FBS. To verify the data obtained from PC12 cells, primary murine neurons were also used in this study. For this, primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in female ICR mice (being pregnant) as described [34], and sowed in a PDL (10 μg/ml)-coated 6-well or 96-well plate for experiments after 6 days of culture. The cells were maintained in a humidified incubator of 5% CO2 atmosphere at 37 °C. All procedures used in this study were approved by the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Recombinant Adenoviral Constructs and Infection of Cells
The recombinant adenoviruses expressing FLAG-tagged dominant negative c-Jun (FLAG-△169) (Ad-dn-c-Jun) was a gift from Dr Jonathan Whitfield (Eisai London Research Laboratories, University College London, London, UK) [35], and the control virus expressing β-galacftosidase (Ad-LacZ) were described previously [36]. Adenovirus expressing green fluorescence protein (GFP)-LC3 fusion protein (Ad-GFP-LC3) was purchased from Sciben Biotech Company (Nanjing, China). For experiments, PC12 cells and/or primary neurons were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Afterwards, cells were used for experiments. Cells infected with Ad-LacZ served as a control. Expression of FLAG-tagged dn-c-Jun was assessed by Western blot analysis with antibodies to FLAG.
Analysis for Cell Viability
PC12 cells and primary neurons, seeded in a PDL-coated 96-well plate (1 × 104 cells/well), were treated with/without Cd (10 and 20 μM) for 24 h following pre-incubation with/without 3-MA (4 mM) or zVAD-fmk (100 μM) for 1 h, with 5 replicates of each treatment. Subsequently, cell viability, after incubation with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) reagent (one solution reagent) (20 μl/well) for 3 h, was evaluated by measuring the optical density (OD) at 490 nm using a Victor X3 Light Plate Reader (Perkin-Elmer, Waltham, MA, USA).
DAPI and TUNEL Staining
PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-LacZ, respectively, were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well, Next day, cells were treated with/without Cd (10 and 20 μM) for 24 h, or with/without Cd (10 μM) for 24 h following pre-incubation with/without 3-MA (4 mM), zVAD-fmk (100 μM), SP600125 (20 μM), U0126 (5 μM), PD169136 (20 μM), EGTA (100 μM), BAPTA/AM (20 μM) or 2-APB (100 μM) for 1 h, with 5 replicates of each treatment. Subsequently, the cells with fragmented and condensed nuclei were stained by adding DAPI (4 μg/ml in deionized water) as described [32]. For the cells pretreated with/without 3-MA or zVAD-fmk for 1 h and then exposed to Cd for 24 h, after DAPI staining, the terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) staining was used according to the protocol given by the manufacturer’s In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). Finally, the images were visualized and taken under a fluorescence microscope (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. For quantitative analysis of the fluorescence intensity using TUNEL staining, the integral optical density (IOD) was determined by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
MDC-Labeled Autophagic Vacuoles
Autofluorescent compound MDC, a specific autophagolysosome marker, as described [37, 38], was used as a tracer for detecting and imaging intracellular autophagic status. In brief, PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-LacZ, respectively, were seeded in a 6-well plate (5 × 105 cells/well) containing a PDL-coated glass coverslip per well. Next day, cells were exposed to Cd with different concentrations (0–20 μM) for 12 h. In some cases, cells were pretreated with/without 3-MA (4 mM), zVAD-fmk (100 μM), SP600125 (20 μM), U0126 (5 μM), PD169136 (20 μM), EGTA (100 μM), BAPTA/AM (20 μM) or 2-APB (100 μM) for 1 h, followed by exposure to Cd (10 μM) for 12 h. After treatment, the cells were labeled with 50 μM MDC in PBS for 1 h at 37 °C, and then washed 3 times with PBS, followed by cell imaging under a fluorescence microscopy and measuring IOD value of fluorescence intensity using MDC-labeled vacuoles as described above.
GFP-LC3 Assay
PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-LacZ, respectively, were infected with Ad-GFP-LC3 and seeded at a density of 5 × 105 cells/well in a 6-well plate containing a glass coverslip per well. Next day, cells were exposed to Cd with different concentrations (0–20 μM) for 12 h, with 5 replicates of each treatment. Afterwards, the cells were fixed using 4% paraformalde-hyde solution in PBS for 30 min at 4 °C and washed 3 times with PBS, followed by imaging and counting the numbers of LC3 puncta (green) per cell to estimate autophagosome formation.
[Ca2+]i Detection
PC12 cells and primary neurons were seeded in a PDL-coated 6-well plate at a density of 5 × 105 cells/well and kept overnight at 37 °C humidified incubator with 5% CO2. Next day, cells were treated with/without Cd (10 μM) for 24 h following pre-incubation with/without EGTA (100 μM), BAPTA/AM (20 μM) or 2-APB (100 μM) for 1 h, with 5 replicates for each treatment. Subsequently, the cells were harvested and washed 3 times with PBS, followed by dilution to 2 × 106 cells/ml with PBS, and cell suspensions (100 μl) for [Ca2+]i analysis were loaded with 20 μl of 5 μM Fluo-3/AM for 40 min at 37 °C in the dark, and then washed 3 times with PBS to remove the extracellular Fluo-3/AM. PBS, replacing Fluo-3/AM, served as a negative control. Finally, the cells for each example were re-suspended with 1 ml PBS to detect status of [Ca2+]i by excitation at 488 nm and emission at 535 nm for adding the suspension into a 96-well plate (150 μl/well) using a Victor X3 Light Plate Reader.
Western Blot Analysis
After treatments, the indicated cells were briefly washed with cold PBS, and then on ice, lysed, followed by Western blotting, as described previously [32]. The following antibodies were used: phosphorylated JNK (p-JNK) (Thr183/Tyr185), JNK, p-c-Jun (Ser63), c-Jun, Erk2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-Erk1/2 (Thr202/Tyr204), p-p38 (Thr180/Tyr182), p38, cleaved-caspase-3 (Cell Signaling Technology, Beverly, MA, USA), LC3, Atg3, Atg4, Atg5-Atg12, Beclin 1, Atg7, Atg10, Atg12, Atg14, Atg16, Ambra 1, FLAG and β-tubulin (Sigma), horseradish peroxidase-coupled goat anti-rabbit IgG, goat anti-mouse IgG, or rabbit anti-goat IgG (Sciben Biotech Company).
Statistical Analysis
All data were expressed as mean values ± standard error (means ± SE). Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using one-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. The criterion for the statistical significance was P < 0.05.
Results
Cd Induces Expansion of Autophagosomes with a Concomitant Abnormal Expression of Atg Proteins in Neuronal Cells
A previous report shows that induction of autophagy by Cd may play a cytoprotective role in PC12 cells [39]. However, we demonstrate that Cd induction of autophagy is involved in cell death in PC12 cells and primary neurons [18]. Here, using autofluorescent compound MDC, a specific autophagolysosome marker [37, 38], we also exhibited that the accumulation of MDC was significantly induced by Cd dose-dependently in PC12 cells and primary neurons, as evidenced by the fluorescence intensity (in green) and quantification based on the incorporation of MDC (Fig. 1a, b). To corroborate the finding, we extended the studies by pinpointing autophagic vacuoles with GFP-LC3 localization. The results showed that when PC12 cells and primary neurons, infected with Ad-GFPLC3, were exposed to Cd (0–20 μM) for 12 h, the number of GFP-LC3 puncta of Cd-treated cells significantly increased compared to that of the vehicle-treated cells in a concentration-dependent fashion (Fig. 1c, d). The results underline that Cd induces expansion of autophagosomes in neuronal cells.
Fig. 1.
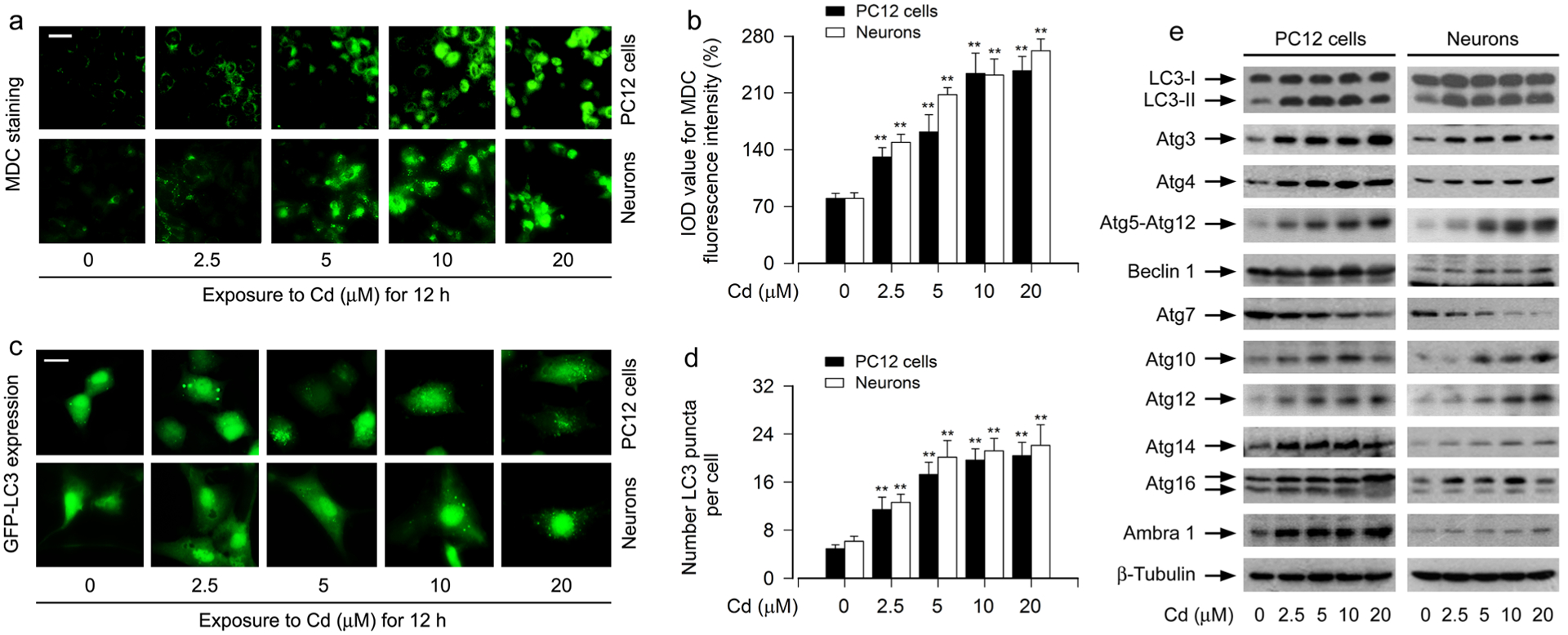
Cd induces expansion of autophagosomes with a concomitant abnormal expression of Atg proteins in neuronal cells. PC12 cells and primary neurons, or PC12 cells and primary neurons infected with Ad-GFP-LC3, respectively, were exposed to Cd (0–20 μM) for 12 h. a, b The cells were labeled using a specific autophagolysosome marker MDC staining and then the fluorescence intensity (in green) for MDC-labeled vacuoles was imaged (a) and quantified (b). Scale bar: 20 μm. c, d Shown were representative GFP-LC3 fluorescence images (in green) (c) and quantified number (d) for GFP-LC3 puncta in the cells. Scale bar: 2 μm. e Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. For b and d, all data were expressed as means ± SE. n = 5, *P < 0.05, **P < 0.01, difference with control group
Since autophagosome initiation and formation is related to a series of Atg proteins [11, 15], we next investigated the manifestation of Atg proteins’ expression, including Atg3, Atg4, Atg5-Atg12, Beclin 1/Atg6, Atg7, LC3/Atg8, Atg10, Atg12, Atg14, Atg16 and Ambra 1, in Cd-treated neuronal cells. Western blot analysis showed that Cd dose-dependently upregulated the expression of LC3-II protein (Fig. 1e), a marker for monitoring the number of autophagosomes [16, 17]. Furthermore, except the down-regulation of Atg7 expression, the upregulation of other Atg proteins, including Atg3, Atg4, Atg5-Atg12, Beclin 1, Atg10, Atg12, Atg14, Atg16 and Ambra 1, were seen in PC12 cells and primary neurons in response to Cd (Fig. 1e), consistent with the observation of Cd-triggered autophagosomes’ expansion (Fig. 1a–d). Collectively, our data indicate that Cd induces expansion of autophagosomes with a concomitant abnormal expression of Atg proteins in neuronal cells.
Cd Elicits Autophagosome Expansion-Dependent Apoptosis in Neuronal Cells
It is well known that autophagy is an important player in the life and death of cells [40]. To further determine the relationship between increased autophagosomes and apoptosis in neuronal cells in response to Cd, PC12 cells and primary neurons were treated with/without Cd (10 μM) for 12 h or 24 h following pretreatment with/without of 3-MA (4 mM), a pharmacological inhibitor for autophagosome formation via suppressing class III PI3K [41], for 1 h, or with/without zVAD-fmk (100 μM), a pan caspase inhibitor, for 1 h. MTS assay showed that pretreatment with 3-MA or zVAD-fmk significantly attenuated Cd-induced cell viability reduction in the cells, respectively (Fig. 2a). Consistently, using DAPI and concurrently TUNEL staining (Fig. 1b), we also observed that pretreatment with 3-MA or zVAD-fmk profoundly reduced the percentage of the cells with nuclear fragmentation and condensation (arrows) and the number of TUNEL-positive cells with fragmented DNA (in green) in PC12 cells and primary neurons triggered by Cd exposure (Fig. 2b–d). Of interest, 3-MA, but not zVAD-fmk, markedly attenuated Cd-induced accumulation of intracellular MDC and LC3-II expression (Fig. 2e, f) though both 3-MA or zVAD-fmk potently prevented Cd-evoked robust cleavage of caspase-3 in the cells (Fig. 2f), as determined by MDC staining and Western blotting. These findings support that Cd elicits an autophagosome expansion-dependent apoptotic cell death in neuronal cells.
Fig. 2.
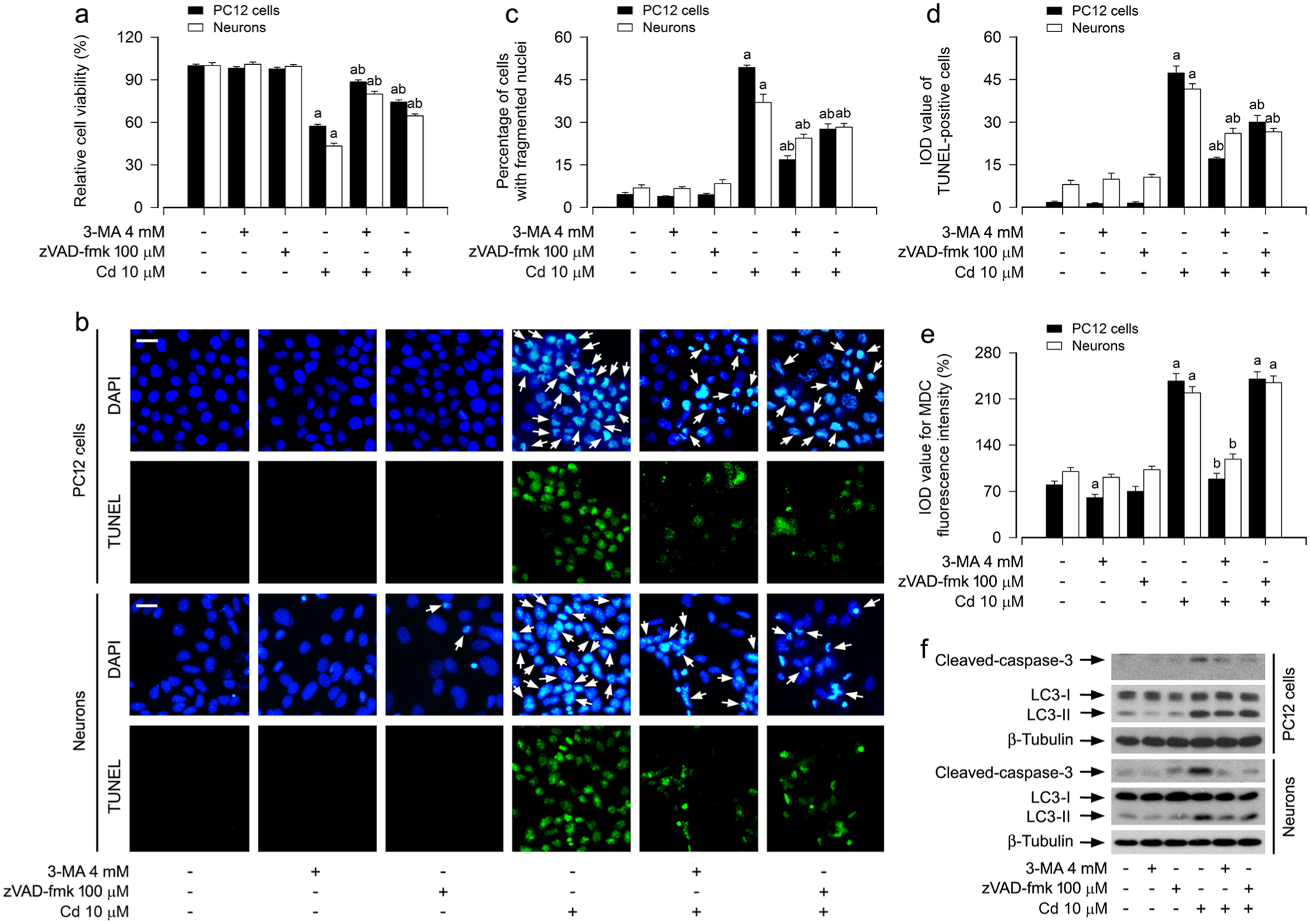
Cd elicits autophagosome expansion-dependent apoptosis in neuronal cells. PC12 cells and primary neurons were pretreated with/without 3-MA (4 mM) or zVAD-fmk (100 μM) for 1 h, followed by exposure to Cd (10 μM) for 12 h (for MDC staining, Western blotting) or 24 h (for cell viability analysis, DAPI and TUNEL staining). a The cell viability was determined by the MTS assay. b The apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining (upper panel) and concurrently by in situ detection of fragmented DNA (in green) using TUNEL staining (lower panel). Scale bar: 20 μm. c, d The percentages of cells with fragmented nuclei (c) and the number of TUNEL-positive cells (d) were quantified. e The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. f Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. For a, c, d and e, all data were expressed as means ± SE. n = 5, aP < 0.05, difference with control group; bP < 0.05, difference with 10 μM Cd group
Cd Induces Autophagosome Expansion-Dependent Neuronal Apoptosis by Activating the JNK Pathway
A number of studies have documented that the activity of MAPKs including JNK, Erk1/2 and/or p38 under various conditions plays a key role in autophagic cell survival and death [19–21]. Our group has demonstrated that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 participate in Cd-induced apoptosis in neuronal cells [31, 32]. This led us to hypothesize that Cd might induce autophagosome expansion-dependent neuronal apoptosis by activating MAPK pathway. To this end, PC12 cells and primary neurons were pretreated with/without SP600125 (20 μM, a specific JNK inhibitor), U0126 (5 μM, a specific inhibitor of MKK1/2, upstream kinases of Erk1/2) and PD169136 (20 μM, a specific p38 inhibitor) for 1 h, then exposed to Cd (10 and/or 20 μM) for 12 h or 24 h, respectively. We showed that SP600125 or U0126, but not PD169136, substantially attenuated Cd-induced apoptosis and cleaved-caspase-3 in the cells, respectively (Fig. 3a, b), in line with our previous findings [31, 42]. Interestingly, Cd-increased LC3-II was remarkably blocked only by SP600125 (Fig. 3b). Consistently, SP600125 robustly suppressed Cd-induced cell MDC accumulation as well (Fig. 3c). Furthermore, SP600125, but not U0126 or PD169136, obviously mitigated Cd-induced abnormal expression of Atg proteins in the cells (Fig. 3d). These findings imply that Cd-activated JNK pathway contributes to expansion of autophagosomes and abnormal expression of Atg proteins, leading to neuronal apoptosis.
Fig. 3.
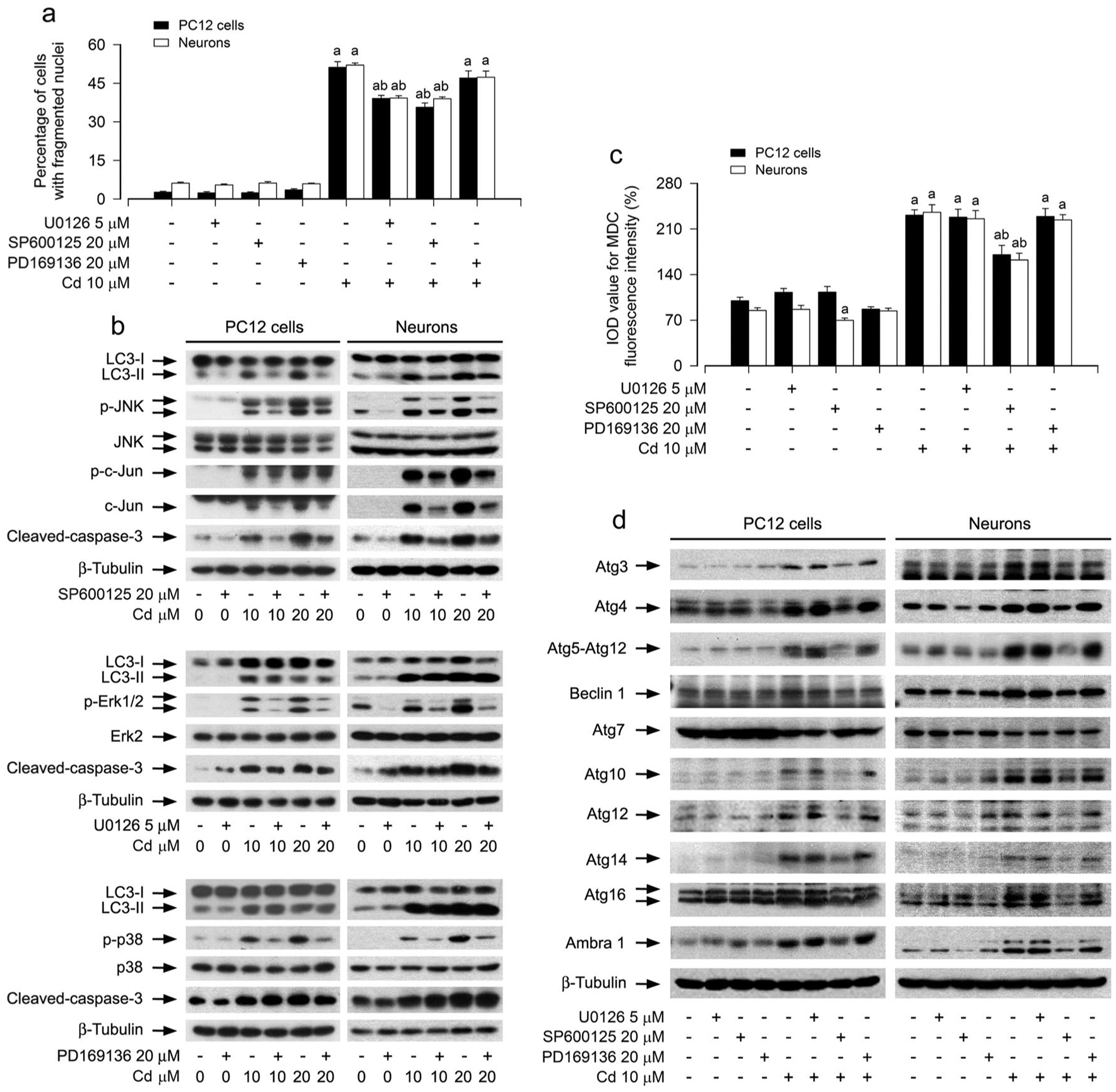
Pharmacological inhibition of JNK mitigates Cd-induced expansion of autophagosomes, abnormal expression of Atg proteins, and apoptosis in neuronal cells. PC12 cells and primary neurons were pretreated with/without U0126 (5 μM), SP600125 (20 μM) or PD169136 (20 μM) for 1 h, followed by exposure to Cd (10 μM) for 12 h (for Western blotting and MDC staining) or 24 h (for DAPI staining). a The apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. b, d Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. c The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. For a and c, all data were expressed as means ± SE. n = 5, aP < 0.05, difference with control group; bP < 0.05, difference with 10 μM Cd group
To validate the role of Cd-activated JNK pathway in contributing to abnormal Atg proteins and expansion of autophagosomes related to neuronal apoptosis, genetic approaches were taken. Ectopic expression of dn-c-Jun, but not LacZ, substantially blocked Cd-induced increases of total and phosphorylated c-Jun in PC12 cells (Fig. 4a). Consistently, Cd-induced increase in LC3-II and cleaved-caspase-3, abnormal expression of Atg proteins, as well as apoptosis were obviously attenuated by expression of dn-c-Jun in the cells (Fig. 4a–c). Using MDC-stained and GFP-LC3-labelled autophagosomes, we observed that expression of dn-c-Jun also reduced the number of Cd-elicited cell MDC accumulation (Fig. 4d) and the number of GFP-LC3 puncta in the cells (Fig. 4e). Collectively, our results clearly indicate that Cd induces autophagosome expansion-dependent apoptosis in neuronal cells, at least in part, through activating the JNK pathway.
Fig. 4.
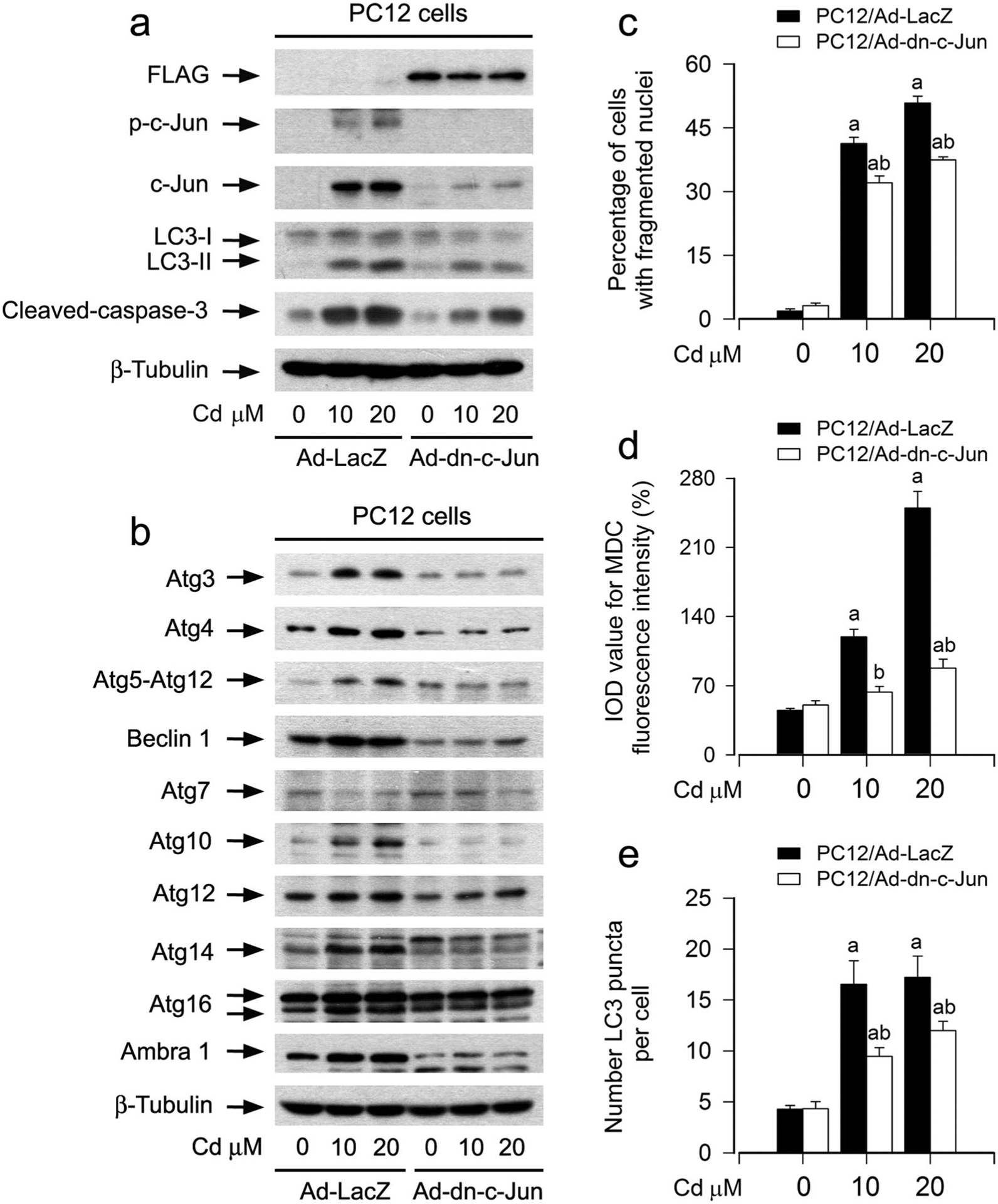
Ectopic expression of dominant negative c-Jun attenuates Cd-induced expansion of autophagosomes, abnormal expression of Atg proteins, and apoptosis in neuronal cells. PC12 cells, infected with Ad-dn-c-Jun or Ad-LacZ (as control) and/or infected with Ad-GFP-LC3, respectively, were exposed to Cd (10 and 20 μM) for 12 h (for Western blotting and MDC staining) or 24 h (for DAPI staining). a, b Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. c The apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. d The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. e Quantified number for GFP-LC3 puncta in the cells. For c, d and e, all data were expressed as means ± SE. n = 5, ap < 0.05, difference with control group; bp < 0.05, Ad-dn-c-Jun group versus Ad-LacZ group
Cd Activates JNK-Mediated Autophagosome Expansion in a Ca2+-Dependent Manner
Ca2+ is an important second messenger in the signal transduction pathways regulating numerous cellular events, such as proliferation, growth, survival and death including autophagy [24, 25]. Our previous studies have demonstrated that Cd elevates the [Ca2+]i level, which activates MAPK pathway leading to neuronal cell death [33]. Next, we determined whether Cd activates JNK pathway leading to autophagosome expansion and subsequent apoptosis in neuronal cells through a Ca2+-dependent mechanism. For this, PC12 cells and primary neurons were pretreated with/without EGTA (100 μM), an extracellular Ca2+ chelator, BAPTA/AM (20 μM), an intracellular Ca2+ chelator, or 2-APB (100 μM), an inhibitor for both inositol 1,4,5-trisphosphate (IP3) receptors and the Ca2+ release activated Ca2+ (CRAC) channels, for 1 h, followed by exposure to Cd (10 μM) for 12 h or 24 h. In line with our previous studies, pretreatment with EGTA, BAPTA/AM or 2-APB significantly attenuated Cd-induced [Ca2+]i elevation and apoptosis in the cells (Fig. 5a, b). Of importance, pretreatment with EGTA, BAPTA/AM or 2-APB remarkably repressed Cd-induced cell MDC increases (Fig. 5c), suggesting that Cd-elevated [Ca2+]i may promote accumulation of autophagosomes, resulting in neuronal apoptosis. Further studies showed that chelating [Ca2+]i with BAPTA/AM or preventing [Ca2+]i elevation using EGTA or 2-APB remarkably alleviated Cd-induced phosphorylation of JNK/c-Jun, increase of LC3-II and cleaved-caspase-3, and abnormal expression of Atg proteins in the cells (Fig. 5d, e). Collectively, the results underscore the concept that Cd activates JNK-mediated autophagosome expansion leading to neuronal apoptosis in a Ca2+-dependent manner.
Fig. 5.
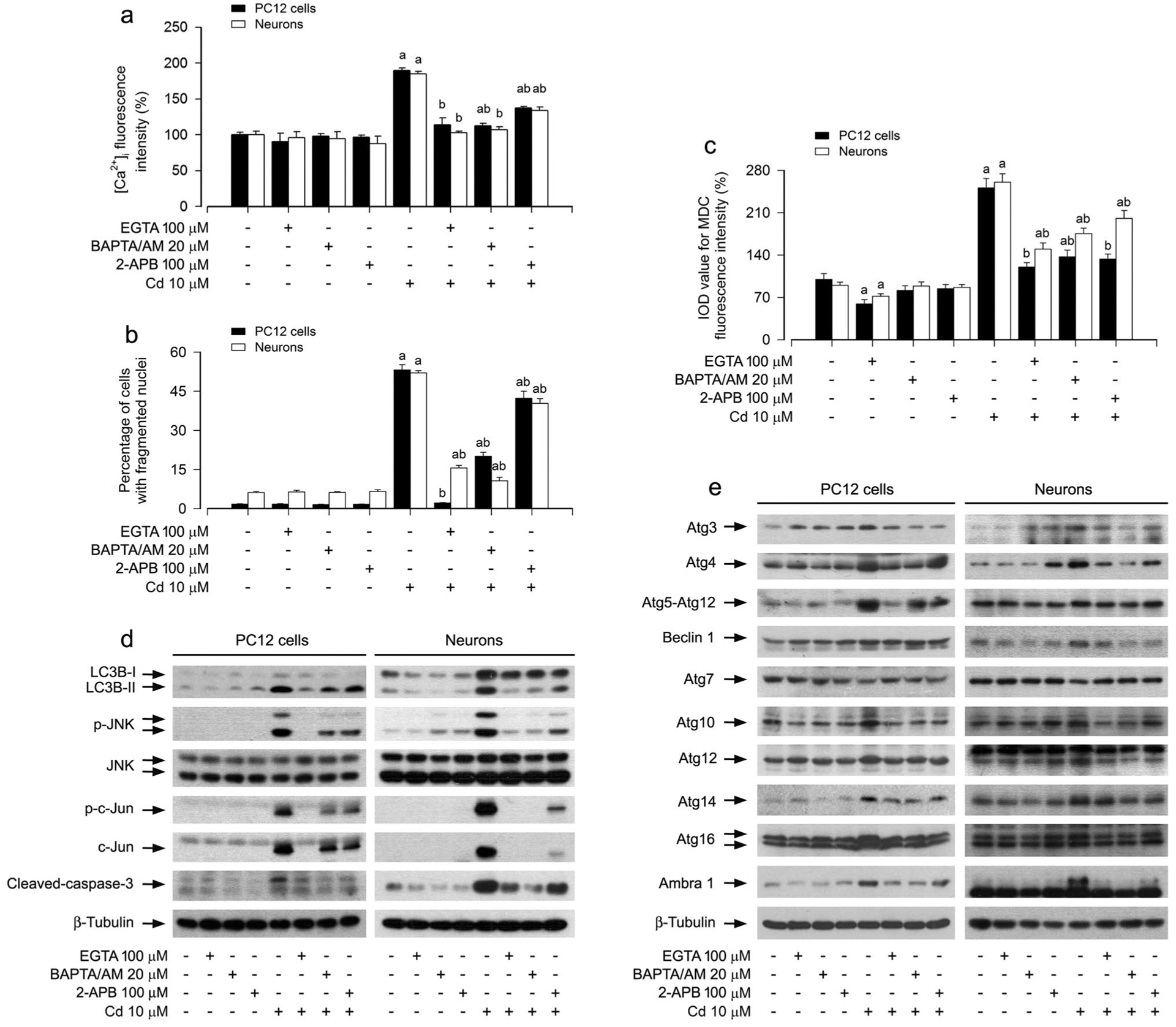
Cd activated JNK pathway contributing to expansion of autophagosomes, abnormal expression of Atg proteins, and neuronal apoptosis in a Ca2+-dependent manner. PC12 cells and primary neurons were pretreated with/without EGTA (100 μM), BAPTA/AM (20 μM) or 2-APB (100 μM) for 1 h, followed by exposure to Cd (10 μM) for 12 h (for Western blotting and MDC staining) or 24 h (for [Ca2+]i detection and DAPI staining). a [Ca2+]i manifestation was detected by a microplate reader using an intracellular Ca2+ indicator dye Fluo-3/AM. b The apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. c The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. d, e Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. For a, b and c, all data were expressed as means ± SE. n = 5, aP < 0.05, difference with control group; bP < 0.05, difference with 10 μM Cd group
Discussion
Autophagy plays a critical role in the neuronal homeostasis, and its dysfunction is closely associated with various neurodegenerative diseases, such as AD, PD, ALS and HD [10, 14, 43]. Cd, a well-known heavy metal pollutant, is neurotoxic [8–11]. How Cd causes autophagy dysfunction is poorly understood. Our previous work has shown that Cd causes neuronal apoptosis by elevating [Ca2+]i levels and subsequently activating MAPK pathway [33]. Here, we presented evidence that Cd induced Ca2+-dependent activation of JNK pathway, which contributed to expansion of autophagosomes and abnormal expression of Atg proteins, leading to neuronal apoptosis. Our findings suggest that Cd-impaired autophagy is an important mechanism in the neurotoxicity of Cd.
Several lines of evidence have described that the autophagy dysfunction may occur due to reduced autophagy induction, enhanced autophagy repression, altered cargo recognition, inefficient autophagosome/lysosome fusion, and/or inefficient degradation of the autophagic cargo in lysosomes [10, 11, 14, 43]. In the present study, we observed that Cd increased expansion of autophagosomes, as detected by MDC-stained autophagosomes and using GFP-LC3-labeling (Fig. 1a–d). As autophagosome formation is a key event in autophagy, it is regulated by a serious of Atg proteins. Current knowledge indicates that the Atg proteins in mammals are divided into several functional units: Unc51-like kinase 1 (ULK1) complex, class III PI3K complex, Atg12 conjugation system, LC3 conjugation system and others (such as Atg2A/B, Atg9L1, WIPI1/2/3/4, etc.) [15, 22, 23]. The ULK1 complex, including ULK1, Atg13, RB1CC1/FIP200 and Atg101, is negatively regulated by mTORC1, a classical autophagy initiation pathway [11, 15, 23]. However, the initiation of autophagy by inactivation of mTORC1 via the ULK1 complex is at odds with our previous findings that Cd evokes striking activation of mTORC1 pathway, leading to neuronal cell death [2, 31]. Therefore, we reasoned that Cd-induced expansion of autophagosomes may involve other systems instead of the ULK1 complex. In this study, we observed that Cd induced upregulation of LC3-II, a hallmark autophagosome-associating protein, in PC12 cells and primary neurons (Fig. 1e), not only supporting that Cd triggers expansion of autophagosomes, but also suggesting that Cd may alter the expression of other Atg proteins. Intriguingly, for the first time, we found that Cd did upregulate the expression of Atg3 and Atg4 (members of LC3 conjugation system), Atg5-Atg12, Atg10, Atg12 and Atg16 (members of Atg12 conjugation system) and Beclin 1, Atg14 and Ambra 1 (members of class III PI3K complex) in the neuronal cells (Fig. 1e). These data strongly imply that Cd might induce the formation of autophagosomes by upregulating the expression of those Atg proteins, thereby leading to expansion of autophagosomes in neuronal cells. However, it is worth mentioning that Atg7, an important E1-like enzyme shared by LC3 and Atg12 conjugation system, was downregulated by Cd (Fig. 1e). Although the mammalian autophagy occurs through at least Atg7-dependent and Atg7-independent pathways, both of which can lead to autophagosome formation, LC3-II generation has not been observed in Atg7-deficient cells [44–46]. Also, it is likely that Cd inhibits autophagic flux [18], especially blocks the degradation step in autolysosomes, resulting in the accumulation of LC3-II and autophagosomes. Therefore, the mechanism for Cd-induced expansion of autophagosomes in neuronal cells may be complex, involving a conventional pathway, an Atg7-independent alternative pathway, or the disorder of autophagy flux. Further studies are needed to uncover the relationship of Cd-induced abnormal expression of Atg proteins with the expansion of autophagosomes in the future.
As autophagy is essential in maintaining the normal physiological and biochemical functions of neuronal cells, and there exists a very low basal level autophagy under normal conditions [13], Cd-induced excessive accumulation of autophagosomes might affect neuronal homeostasis, leading to neurotoxicity. Since Cd causes neurotoxicity via induction of apoptosis [31], this prompted us to investigate the relationship between autophagy and apoptosis in neuronal cells in response to Cd. Our experiments showed that inhibition of autophagosome formation by 3-MA rescued cells from Cd-triggered apoptosis (Fig. 2a–d), while inhibition of caspase-dependent apoptosis by zVAD-fmk failed to suppress Cd-induced increases of autophagosomes and LC3-II (Fig. 2e, f). These results imply that Cd-induced expansion of autophagosomes occurs before the caspase cascade is activated. Possibly, a rapid autophagy response is induced at the beginning when the Cd-induced stress is not lethal, and apoptotic program is activated later when Cd-induced stress exceeds a critical duration or an intensity threshold. The fact that many signal transduction pathways, such as serine/threonine kinases that are elicited by cell-intrinsic stress, regulate both autophagy and apoptosis might explain the sequential activation of both processes [47].
It has been reported that MAPKs, including JNK, Erk1/2 and p38, play a key role in autophagic cell survival and death under various conditions [19–21]. MAPKs are involved in the neurotoxicity of Cd, showing that Cd activates Erk1/2, JNK and/or p38 MAPK contributing to neuronal apoptosis [1, 31, 32]. Some studies have pointed out the relationship between JNK and autophagy [15, 22, 23]. For example, IRE1-JNK activation is required for autophagosome formation after endoplasmic reticulum (ER) stress [15]; JNK-mediated Beclin 1 expression and p53 phosphorylation promotes the autophagic cell death in cancer cells [22]; JNK-dependent accumulation of p62 and AMP-activated protein kinase (AMPK) pathway participate in resveratrol-induced autophagic cell death [23]. In this study, we showed that inhibition of Cd-activated JNK and Erk1/2 pathways by SP600125 (a specific JNK inhibitor) and U0126 (a specific inhibitor of MKK1/2, upstream kinases of Erk1/2), respectively, obviously relieved Cd-induced neuronal apoptosis (Fig. 3a, b); but Cd-induced accumulation of autophagosomes and abnormal expression of Atg proteins were attenuated only by SP600125 (Fig. 3c, d). This implies a critical role of JNK pathway in controlling neuronal autophagy and apoptosis in response to Cd. To corroborate this finding, we extended our studies using genetic manipulation of c-Jun, showing that ectopic expression of dominant negative c-Jun (dn-c-Jun) potently alleviated Cd-induced expansion of autophagosomes and abnormal expression of Atg proteins, as well as apoptosis in PC12 cells (Fig. 4a–e). Taken together, our data support the notion that activation of JNK pathway is required for Cd-induced expansion of autophagosomes/abnormal expression of Atg proteins and apoptosis in neuronal cells.
In this study, via chelating [Ca2+]i with BAPTA/AM or preventing [Ca2+]i elevation using EGTA or 2-APB, we demonstrated that [Ca2+]i elevation plays a crucial role not only in apoptosis but also in expansion of autophagosomes/abnormal expression of Atg proteins in neuronal cells in response to Cd (Fig. 5). It has been shown that stimulating agents such as vitamin D, ionomycin, and thapsigargin induce autophagy via endoplasmic reticulum (ER) Ca2+ mobilization, and GFP-LC3 aggregates can be inhibited by BAPTA/AM, suggesting that there exists a Ca2+-dependent mechanism for autophagosome formation [48]. In addition, optimal Ca2+ transfer maintains efficient mitochondrial respiration and inhibits reactive oxygen species (ROS) formation and autophagy, and mitochondrial membrane potential imbalance can impair Ca2+ uptake leading to mitochondrial Ca2+ release, which occurs during mitophagy [25]. In mesangial cells, Cd induces both autophagy and apoptosis through elevation of [Ca2+]i-dependent Erk1/2 signaling pathway [28]. Taking these studies and our findings into consideration, we tentatively conclude that Cd-activated JNK pathway via evoking [Ca2+]i level, leading to expansion of autophagosomes/abnormal expression of Atg proteins and subsequent neuronal apoptosis, which may involve various factors, including ER stress induction, mitochondrial Ca2+ signaling distribution, and/or ROS production.
In summary, here we identify that Cd induces expansion of autophagosomes with a concomitant abnormal expression of Atg proteins, subsequently leading to neuronal apoptosis. Mechanistically, Cd impairs autophagy leading to apoptosis by Ca2+-dependent activation of JNK signaling pathway in neuronal cells. Our findings highlight that manipulation of intracellular Ca2+ level and/or JNK activity to ameliorate autophagy may be a promising intervention against Cd-induced neurotoxicity and neurodegeneration.
Acknowledgements
This work was supported in part by the grants from National Natural Science Foundation of China (No. 81873781; LC), NIH (CA115414; SH), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010; LC), Natural Science Foundation of Jiangsu Province (BK20180562; CX), and American Cancer Society (RSG-08-135-01-CNE; SH).
Abbreviations
- AD
Alzheimer disease
- ALS
Amyotrophic lateral sclerosis
- AMPK
AMP-activated protein kinase
- 2-APB
2-Aminoethoxydiphenyl borane
- Atg
Autophagy-related
- BAPTA/AM
1,2-Bis (o-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid tetra (acetoxymethyl) ester
- Ca2+
Calcium ion
- Cd
Cadmium
- CRAC
Ca2+-release activated Ca2+
- DAPI
4′, 6-Diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- EGTA
Ethylene glycol tetra-acetic acid
- ER
Endoplasmic reticulum
- Erk1/2
Extracellular signal-regulated kinase ½
- FBS
Fetal bovine serum
- GFP
Green fluorescent protein
- HD
Huntington’s disease
- JNK
C-Jun N-terminal kinase
- LacZ
β-Galacftosidase
- LC3
Microtuble-associated protein 1 light chain 3
- 3-MA
3-Methyladenine
- MAPK
Mitogen-activated protein kinase
- MDC
Monodansylcadaverine
- mTOR
Mammalian target of rapamycin
- mTORC1
MTOR complexes 1
- PBS
Phosphate buffered saline
- OD
Optical density
- PD
Parkinson disease
- PDL
Poly-D-lysine
- PI3K
Phosphatidylinositol 3’-kinase
- ROS
Reactive oxygen species
- TUNEL
The terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling
- ULK1
Unc51-like kinase 1
- zVAD-fmk
Z-Val-Ala-Asp-CH2F
Footnotes
Declarations
Conflict of interest The authors declared that there are no conflict of interest.
References
- 1.Wang B, Du Y (2013) Cadmium and its neurotoxic effects. Oxid Med Cell Longev 2013:898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu C, Liu C, Liu L, Zhang R, Zhang H, Chen S et al. (2015) Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology 97:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panayi AE, Spyrou NM, Iversen BS, White MA, Part P (2002) Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J Neurol Sci 195:1–10 [DOI] [PubMed] [Google Scholar]
- 4.Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP (2003) Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol 138:901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendez-Armenta M, Rios C (2007) Cadmium neurotoxicity. Environ Toxicol Pharmacol 23:350–358 [DOI] [PubMed] [Google Scholar]
- 6.Okuda B, Iwamoto Y, Tachibana H, Sugita M (1997) Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg 99:263–265 [DOI] [PubMed] [Google Scholar]
- 7.Goncalves JF, Fiorenza AM, Spanevello RM, Mazzanti CM, Bochi GV, Antes FG et al. (2010) N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chem Biol Interact 186:53–60 [DOI] [PubMed] [Google Scholar]
- 8.Feng Y, Yao Z, Klionsky DJ (2015) How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 25:354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M et al. (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49 [DOI] [PubMed] [Google Scholar]
- 10.Cerri S, Blandini F (2019) Role of autophagy in Parkinson’s disease. Curr Med Chem 26:3702–3718 [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, Klionsky DJ (2020) Autophagy and disease: unanswered questions. Cell Death Differ 27:858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Miah M, Culbreth M, Aschner M (2016) Autophagy in neurodegenerative diseases and metal neurotoxicity. Neurochem Res 41:409–422 [DOI] [PubMed] [Google Scholar]
- 14.Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997 [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132 [DOI] [PubMed] [Google Scholar]
- 16.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM et al. (2011) Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 7:188–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Dong X, Zhao R, Zhang R, Xu C, Wang X et al. (2019) Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal 55:26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q et al. (2014) p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 344:174–179 [DOI] [PubMed] [Google Scholar]
- 20.Xie CM, Chan WY, Yu S, Zhao J, Cheng CH (2011) Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med 51:1365–1375 [DOI] [PubMed] [Google Scholar]
- 21.Chen SY, Chiu LY, Maa MC, Wang JS, Chien CL, Lin WW (2011) zVAD-induced autophagic cell death requires c-Src-dependent ERK and JNK activation and reactive oxygen species generation. Autophagy 7:217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamacher-Brady A (2012) Autophagy regulation and integration with cell signaling. Antioxid Redox Signal 17:756–765 [DOI] [PubMed] [Google Scholar]
- 23.Hurley JH, Young LN (2017) Mechanisms of autophagy initiation. Annu Rev Biochem 86:225–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harr MW, Distelhorst CW (2010) Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol 2:a005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.East DA, Campanella M (2013) Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy 9:1710–1719 [DOI] [PubMed] [Google Scholar]
- 26.Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X (2010) Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci 113:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemarie A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, Vernhet L (2004) Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor. Free Radic Biol Med 36:1517–1531 [DOI] [PubMed] [Google Scholar]
- 28.Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65:3640–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang LY, Wu KH, Chiu WT, Wang SH, Shih CM (2009) The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy 5:571–572 [DOI] [PubMed] [Google Scholar]
- 30.Shen HM, Dong SY, Ong CN (2001) Critical role of calcium overloading in cadmium-induced apoptosis in mouse thymocytes. Toxicol Appl Pharmacol 171:12–19 [DOI] [PubMed] [Google Scholar]
- 31.Chen L, Liu L, Luo Y, Huang S (2008) MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem 105:251–261 [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Liu L, Huang S (2008) Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45:1035–1044 [DOI] [PubMed] [Google Scholar]
- 33.Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H et al. (2011) Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 6:e19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H et al. (2010) Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest 90:762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29:629–643 [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W et al. (2010) Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem 285:38362–38373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biederbick A, Kern HF, Elsasser HP (1995) Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 66:3–14 [PubMed] [Google Scholar]
- 38.Munafo DB, Colombo MI (2001) A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 114:3619–3629 [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Zhu J, Zhang K, Jiang C, Wang Y, Yuan Y et al. (2013) Induction of cytoprotective autophagy in PC-12 cells by cadmium. Biochem Biophys Res Commun 438:186–192 [DOI] [PubMed] [Google Scholar]
- 40.Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P (2012) Autophagy: for better or for worse. Cell Res 22:43–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS et al. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C, Zhang R, Sun C, Zhang H, Xu C, Liu W et al. (2015) Resveratrol prevents cadmium activation of Erk1/2 and JNK pathways from neuronal cell death via protein phosphatases 2A and 5. J Neurochem 135:466–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge KL, Chen WF, Xie JX, Wong MS (2010) Ginsenoside Rg1 protects against 6-OHDA-induced toxicity in MES23.5 cells via Akt and ERK signaling pathways. J Ethnopharmacol 127:118–123 [DOI] [PubMed] [Google Scholar]
- 44.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T et al. (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658 [DOI] [PubMed] [Google Scholar]
- 45.Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J et al. (2013) Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol 15:1067–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ra EA, Lee TA, Won Kim S, Park A, Choi HJ, Jang I et al. (2016) TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat Commun 7:11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swerdlow S, Distelhorst CW (2007) Bcl-2-regulated calcium signals as common mediators of both apoptosis and autophagy. Dev Cell 12:178–179 [DOI] [PubMed] [Google Scholar]
- 48.Hoyer-Hansen M, Jaattela M (2007) AMP-activated protein kinase: a universal regulator of autophagy? Autophagy 3:381–383 [DOI] [PubMed] [Google Scholar]