

Abstract
Herein we report a machine-assisted and scaled-up synthesis of propofol, a short-acting drug used in procedural sedation, which is extensively in demand during this COVID-19 pandemic. The continuous-flow protocol proved to be efficient, with great potential for industrial translation, reaching a production up to 71.6 g per day with process intensification (24 h-continuous experiments). We have successfully telescoped a continuous flow approach obtaining 5.74 g of propofol with productivity of 23.0 g/day (6 h-continuous experiment), proving the robustness of the method in both separated and telescoped modes. Substantial progress was also achieved for the in-line workup, which provides greater safety and less waste, also relevant for industrial application. Overall, the synthetic strategy is based on the Friedel-Crafts di-isopropylation of low-cost p-hydroxybenzoic acid, followed by a decarboxylation reaction, giving propofol in up to 84% overall yield and very low by-product formation.

The continuous flow synthesis of propofol 3 is presented as a two-step protocol. The isopropylated intermediate 2 was obtained from 4-hydroxybenzoic acid (1) in up 43.8 g, 85% yield and 30 min residence time. Propofol 3 was then obtained in 71.6 g, 87% yield, and 16 min residence time. A safe and cost-competitive machine-assisted protocol is described with a process intensification demonstration (24 h experiments) and a telescoped process intensification (6 h).
Keywords: Continuous flow synthesis, Telescoped, Active pharmaceutical ingredient, Propofol, Covid-19, Anesthetic
Introduction
The synthesis of active pharmaceutical ingredients (APIs) using enabling technologies has become an active field of research [1]. The use of continuous flow technologies for this purpose [2–6] is now prominent in both academia and industry, showing great advantages over the traditional batch processes. This is especially true in terms of sustainability, efficiency, safety, and fine reaction control, thus being a disruptive and modern frontier technology [7–9].
We are currently experiencing the COVID-19 pandemic, and this has brought to the forefront a worldwide problem, which is the low capacity to support demanding peaks for specific APIs. For example, during this current pandemic, the sudden and large increase of patients requiring ventilation caused a propofol shortage in several countries resulting in serious hospital treatment breakdowns, e.g. in Brazil [10]. Propofol is marketed, for example, as Diprivan® (AstraZeneca). It is a fast-acting intravenous anesthetic agent with worldwide demand, being applied for the induction and maintenance of anesthesia and sedation for medical procedures in adult and pediatric patients [11, 12]. Propofol has also been used to avoid cardiopulmonary complications in patients maintained under mechanical ventilation during long periods, as this drug minimizes the natural resistance of the body to this invasive procedure [13, 14]. The mechanisms of action involve several neurotransmitter receptors, especially the GABAA receptor, and is considered a distinct sedative drug with low toxicity and rapid onset and metabolization [15]. In addition, evidence indicates that propofol can potentialize the antitumor effect of chemotherapy drugs, and studies of its application as a synergistic drug are under investigation [16, 17]. Old uses and the recent needs justify the recognition of this drug as an essential medicine by the WHO since 2013 [18].
In general, the strategies for obtaining propofol involve direct syntheses from phenol [19–23], or by using the protected 4-position of phenol such as 4-hydroxybenzoic acid [24–26] or 4-chlorophenol [27, 28]. The Friedel-Crafts alkylation between phenol and propylene gas (Scheme 1 – Method A) [19–23] requires dramatic reaction conditions (high pressures), and several by-products are obtained, such as 2,4-diisopropylphenol, 2,5-diisopropylphenol and 2,4,6-triisopropylphenol. A resulting concern is how to purify propofol fulfilling the regulatory standards for medicinal application, as these by-products have similar physical properties. On the other hand, from the substituted phenols (Scheme 1 – Method B) [24, 25, 29], isopropanol in a strongly acidic medium is an option for the in-situ generation of propylene gas, which, followed by alkylation and removal of the para-substituent group of the phenol, also provides propofol. Attempts to improve the industrial isolation process of propofol have been reported with simplification of the extraction step, validation, and batch kilogram scale [24]. However, there is no doubt at all that continuous processing can introduce fast and improved control in propofol manufacturing, delivering a fine temperature control and minimizing by-products formation. Up to now, only the recent publication from the Poisson [30] group has reported a synthetic route to propofol using continuous flow conditions, with the solution of some problems, but still requiring optimizations of the H2SO4 equivalents, starting material concentration, continuous scale-up demonstration of the last synthetic step and other variables (Scheme 1 – Method C) [30].
Scheme 1.

Synthetic approaches and processes for propofol
In Big Pharma, synthetic routes for different intravenous anesthetics such as Cipepofol are currently being planned, but the necessity for several reaction steps, high temperatures and stoichiometric organometallics (n-butyl lithium or Grignard reagents) raise new problematic synthetic issues, especially related to the safety of a large-scale process and drug purification [31].
Considering the industrial process and the academic literature, several gaps remain unsolved for the synthesis of propofol, such as less waste generation and purifications, less dependence on acids, better productivity, identification of by-products, process intensification and modernization of the scale up process, among others.
Aiming at improving propofol production and following on our previous results in continuous flow chemistry [32–38], we now report a scaled-up and telescoped synthesis of propofol under continuous-flow conditions in 2 reaction steps, via Friedel-Crafts alkylation followed by a decarboxylation reaction. The synthesis of 4-hydroxy-3,5-diisopropylbenzoic acid and then 2,6-diisopropylphenol (propofol) was carried out individually, with a comprehensive screening of the reaction conditions in both batch and continuous flow, also demonstrating 24-hour experiments, additional advantages and the delivery of propofol in up to 71.6 g-scale. Then the telescoped continuous flow process intensification was developed as described below (Scheme 1 – Method D).
Results and discussion
Initially, the optimization of the synthesis of 4-hydroxy-3,5-diisopropylbenzoic acid (2) was performed in batch conditions to explore initial reaction conditions which are compatible with microflow reactors, which mean the highest possible concentration with no evidence of precipitation. Despite the methodology for the synthesis of 2 (Table 1) having been described previously in the literature by Pramanik et al. [24], these batch reaction conditions were not planned to be promptly adapted in flow. Additionally, the seminal flow methodology reported by the Poisson group [30] was not optimized, remaining the use of excessive H2SO4 (45% more), starting material 1 processed at 0.2 M, intermediate 2 processed in only a few milligrams, a less-efficient tube reactor, among others, as pointed out in Scheme 1 – Method C.
Table 1.
Optimization for the synthesis of 2 in batch.[a]
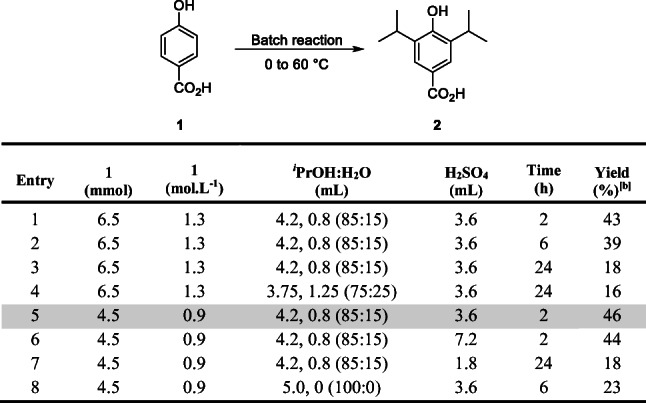
[a] 4-hydroxybenzoic acid 1, iPrOH:H2O and H2SO4. [b] Isolated by column chromatography.
Thus, isopropanol was carefully added to a cooled mixture of 4-hydroxybenzoic acid (1), water and concentrated sulfuric acid (from 0 °C to 60 °C) in different concentrations and solvent proportions (Table 1). We observed that with longer reaction times, the yield decreased with major by-products formation, possibly due to parallel and/or subsequent reactions from 2 (Table 1, entries 1–3). An increase in water concentration was not fruitful, as a lower solubility of 1 was observed with evident precipitation (Table 1, entry 4) whereas a lower concentration of 1 gave better solubility (Table 1, entry 5). The increase of the H2SO4 concentration did not improve the transformation (Table 1, entry 6). With less H2SO4 the reaction required more time to consume the starting material in batch, providing more by-products (evidenced by TLC) (Table 1, entry 7). In the absence of water (Table 1, entry 8), several by-products were also observed, showing that water plays a fundamental role, probably preventing the formation of the corresponding isopropyl ester of 2 and controlling the bis-alkylation. Therefore, our best reaction condition (Table 1, entry 5) in batch allowed us to obtain 2 in 46% yield. Importantly, this condition presented a lower by-product formation and no precipitation during the 2 h reaction time.
After the orientational studies in batch, it was possible to prospect the best initial conditions for the continuous flow experiments. A high-precision flow pump coupled to a continuous flow PFA tubing reactor was used (ASIA® modules from Syrris). Initially, the temperature of 80 °C was chosen to prevent clogging and allow us to increase the flow rate as much as possible. The optimization with longer residence times yielded 2 with low yields (Table 2, entries 1–4). At 80 °C, the best condition was achieved with 30 min residence time, delivering product 2 in 75% isolated yield, and up to 38.6 g/day (Table 2, entry 4). The temperature was lowered to 60 °C (Table 2, entries 5 and 6), and the best performance was obtained with 30 min residence time, an overall flow rate at 0.533 mL/min, giving 2 in 92% yield in up to 47.4 g/day.
Table 2.
Optimization for the synthesis of 2 under continuous-flow conditions
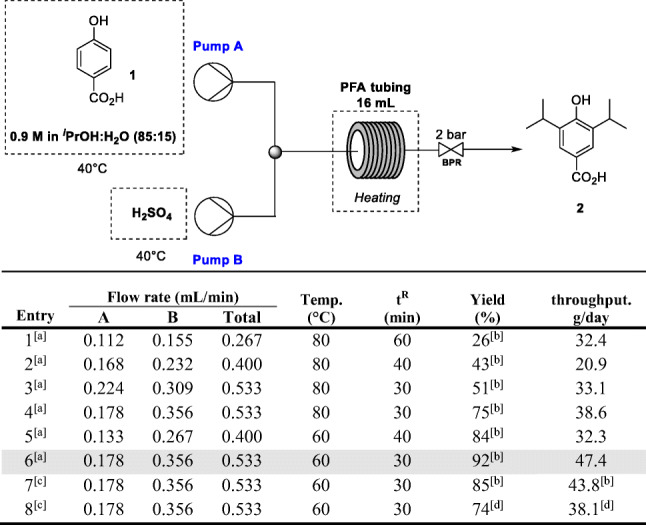
[a] Pump A: 4-hydroxybenzoic acid 1 (500 mg, 3.62 mmol) in iPrOH:H2O (85:15, ca 4.0 mL) to give a 0.9 M solution; Pump B: H2SO4 (8 mL). [b] Isolated by column chromatography. [c] Process intensification using a 0.9 M solution of 1 (32 g, 231.8 mmol) in iPrOH:H2O (85:15, ca 250 mL) (Pump A) and H2SO4 (Pump B); 24 h experiment. [d] Isolated after acid-base extraction (NaOH and then HCl) and crystallization from hexanes.
To confirm the efficiency of the present method, as well as the process intensification, a continuous flow reaction was performed for 24 h (Table 2, entry 7) with full virtual control (using the Syrris Software, webcam, and remote access), delivering 43.8 g (197.0 mmol) of highly pure intermediate 2. We emphasize that the experimental 24 h productivity is very similar to that estimated in entry 6 (Table 2) showing that our continuous setup and reaction condition are reliable for process intensification. Additionally, to minimize purification costs and avoid chromatography, an acid-base liquid-liquid extraction was applied after a new 24 h experiment delivering the highly pure intermediate 2 in 74% yield (38.1 g) after crystallization from hexanes (Table 2, entry 8). It is important to explain that in all our experiments in flow it is necessary to pre-heat the reagents/reaction mixture to 40 °C before pumping to avoid precipitations and thus clogging in the micromixer and entry of the flow reactor (See the supp. Information, S2 and S3).
A study of the decarboxylation of 2 in batch was then carried out to find again orientational conditions for the translation to flow, varying solvents, bases, temperature and reaction times (Table 3). When ethylene glycol was used as solvent with 3.0 equiv. of NaOH, a very viscous mixture was obtained, which would not be suitable for continuous flow conditions (Table 3, entry 1). Exchanging for DMF as solvent and NaOH as the base, the viscosity issue was solved, but the transformation was not satisfactory, and several by-products were observed (Table 3, entry 2). Other bases were evaluated, such as DBU, TEA and the very cheap nBuNH2 (Table 3, entries 3–8). 9.0 Equivalents of nBuNH2 at 150 °C showed the best performance, delivering propofol in 91% yield after 24 h (Table 3, entry 7). With a decrease in base concentration from 9.0 to 5.0 equivalents, a decrease in the yield was observed from 91 to 66% (Table 3, entry 8). Using toluene as solvent propofol (3) was obtained in 73% yield (Table 3, entry 9).
Table 3.
Optimization for the synthesis of 3 in batch.[a]
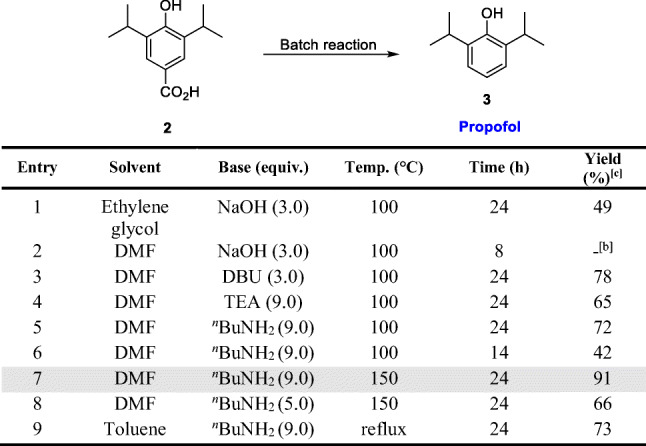
[a] 4-hydroxy-3,5-diisopropylbenzoic acid 2 (222 mg, 1.0 mmol) in solvent (3 mL) and base. [b] 3 not isolated, several by-products. [c] Isolated by filtration over silica gel.
The synthesis of propofol (3) was then adapted to flow using the best batch condition (entry 7, Table 3), and the flow optimizations were carried out using a stainless-steel continuous flow reactor (ASIA® modules from Syrris, Table 4). Considering our previous results in batch, it was determined that the best reaction mixture for the synthesis of 3 from 2 was to use n-butylamine as the base (9.0 equiv.) in DMF as solvent. The reagents were all mixed into a single solution (40 °C) for pumping, and the reactor temperature was initially set at 130 °C. As the flow was increased from 0.100 mL/min to 0.500 mL/min, a decrease in yield was observed, despite the increase in productivity per day (Table 4, entries 1–3). Concomitantly, increasing the flow to 0.600 mL/min and the temperature to 140 °C, propofol (3) was obtained in 93% yield (Table 4, entry 4). Increasing the temperature to 150 °C (Table 4, entries 5–9), we found the best results with up to 95% yield (62.6 g/day) (Table 4, entry 6). However, increasing the flow to 1.0 mL/min, despite the small decrease in the isolated yield (89%), optimal productivity of 73.3 g/day was achieved in only 16 min residence time (Table 4, entry 8).
Table 4.
Optimization for the synthesis of propofol (3) under continuous-flow conditions.

[a] Pump: 4-hydroxy-3,5-diisopropylbenzoic acid 2 (300 mg, 1.35 mmol, nBuNH2 (9.0 equiv., 1.19 mL) and DMF giving 2 at 0.32 M solution. [b] Isolated by extraction and filtration over silica gel. [c] Process intensification using 2 (102.85 g, 463.3 mmol), nBuNH2 (411.4 mL) and DMF (ca 1000 mL) to give a 0.32 M solution of 2; 24 h experiment, thus obtaining 71.6 g of 3.
To confirm the efficiency of the process intensification, a continuous flow reaction was performed during 24 h (Table 4, entry 9), affording 71.6 g (402.2 mmol) of propofol (3), after a simple filtration over silica gel and solvent evaporation. This result is in agreement with the predicted value (predicted 73.3 g/day vs 71.6 g/day obtained experimentally).
Telescoping
The optimal conditions obtained during the step-by-step flow experiments (Tables 2 and 4) were applied to a telescoped flow sequence (Table 5). Initially, the best condition for the extraction of 4-hydroxy-3,5-diisopropylbenzoic acid 2 was determined. Attempts for an in-line extraction with water and Et2O were performed, but with no success due to the strong heating of the reaction mixture while in contact with Et2O, H2SO4 and water. The in-line extraction with toluene was well-succeeded and the intermediate 2 recovered in compatible yields when compared to our previous experiments. For the in-line separation, the Biotage® hydrophobic membrane phase separator, and toluene were established as the best option for extraction and separation. Thus, the toluene extract containing 2 was then pumped simultaneously (Pump E, Table 5) together with a flow of pure nBuNH2 as the base, but we observed precipitation of salts and clogging. To solve this problem a solution of nBuNH2:DMF (6:4) and a preheating step at 100 °C was successfully used. With these changes in hand, the reaction conditions were established and the fully telescoped protocol performed (Table 5). With Pump E at 0.800 mL/min, Pump F at 0.100 mL/min and reactor 2 at 150 °C, the productivity achieved was 20.6 g/day in 67% overall yield for propofol (3) (Table 5, entry 1). By reducing the Pump F flow to 0.080 mL/min a slight improvement was observed, delivering 3 in 70% yield (Table 5, entry 2). Lower temperatures were ineffective while at 120 °C the yield was lower (49%, Table 5, entry 3). With the configuration set to Pump E = 0.700 mL/min, Pump F = 0.080 mL/min, and reactor 2 at 150 °C, propofol (3) was obtained in 82% overall yield and 22.2 g/day (Table 5, entry 4). All the experiments of this telescoped protocol were realized on a 3.62 mmol scale, and to confirm the efficiency of the process intensification, a 6 h-experiment was also performed (Table 5, entry 5), affording 5.74 g (32.2 mmol) 3 in 84% of overall yield (estimated 23.0 g/day), proving the robustness of our telescoped protocol.
Table 5.
Telescoped protocol for propofol synthesis under continuous-flow conditions.

[a] Pump A: 4-hydroxybenzoic acid 1 (500 mg, 3.62 mmol) in iPrOH:H2O (85:15) to give a 0.9 M solution pumped at 0.178 mL.min−1; Pump B: H2SO4 pumped at 0.356 mL.min−1; Pump C: Toluene (27 mL); Pump D: Water (27 mL); Pump F: nBuNH2:DMF (6:4). [b] Isolated by column chromatography. [c] Process intensification in 6.0 h experiment.
By-products identification
During the optimization of the 4-hydroxy-3,5-diisopropylbenzoic acid (2) synthesis (Table 1 and 2), some by-products were detected and we decided to investigate them taking into account future API manufacturing controls. Using entry 6 (Table 2) as a model, the reaction was quenched and directly analyzed by LC-MS (TOF/Q-TOF Mass Spectrometer), which revealed the formation of 3 minor by-products, (4, 5 and 6 – Fig. 1). Analyzing the LC-MS results for the synthesis of 2, only 5% of 4, 4% of 5 and 8% of 6 (relative peak areas) were observed. Their structures were proposed based upon the exact mass determinations. The main product 2 was quantified with an 83% area, very close to the isolated yield of 85% (Table 2, entry 7), also showing that our mass balance is compatible with the total starting material conversion and the formation of these impurities which were easily removed by crystallization from hexanes (Table 2, entry 8).
Fig. 1.

LC-MS chromatographic evaluation of products 2, and GC-MS of 3
In the analysis of eventual impurities during the synthesis of propofol (3), the reaction was quenched and directly analyzed by GC-MS (Fig. 1) using entry 6 conditions (Table 4) as the model. In this case, no unreacted 2 nor by-products were observed, presenting a very clean chromatogram even without previous purification (the crude reaction product was analyzed).
Conclusions
Considering the current need for more cost-competitive and safer synthetic processes, we have developed an enhanced continuous synthesis of propofol with greater efficiency, attenuating problems that remained unresolved in the literature, such as high-volume dependence of sulfuric acid, tedious and expensive column chromatography purification of intermediate 2, process intensification in the two steps, in-line work up and a telescoped continuous flow protocol. During the analyses carried out by LC-MS and GC-MS, it was possible to better understand the transformations, identify the minor by-products, as well as control them in the reaction medium. The first step is a Friedel–Crafts alkylation of 1, and provides the bis-alkylated product 2 in up to 43.8 g (real 24 h experiment), using a 16 mL PFA reactor. Another improvement was achieved by using purification via acid-base extraction without the need for tedious column chromatography purification. The second step is a decarboxylation reaction, and applying relatively mild conditions and short reaction time, propofol (3) was obtained in up to 71.6 g/day (real 24 h experiment) using a continuous flow 16 mL stainless steel reactor. Considering the optimal residence time of the first continuous step (30 min) and the residence time of the second (16 min) we can affirm that our protocol involves just 46 min of processing to obtain 3. The telescoped continuous flow protocol was performed in up to 6 h, thus affording 5.74 g (32.2 mmol) of propofol (3) with a productivity of 23.0 g/day, highlighting its innovation and attractiveness for industrial settings.
Experimental procedure
Continuous flow synthesis of 3,5-diisopropyl-4-hydroxybenzoic acid (2)
In pump A (Table 2) a solution of 4-hydroxybenzoic acid 1 (32.0 g, 231.8 mmol, 0.9 M) in iPrOH:H2O (85:15, ca 250 mL) was heated to 40 °C and pumped at 0.178 mL/min flow rate. Pump B was equipped with a flask containing conc. H2SO4 (heated to 40 °C) and pumped at 0.356 mL/min flow rate. An Asia Flow Syringe Pump and an Asia Tube Reactor (PFA, 16 mL) from Syrris were used. The pumps were connected by a T-mixer and flowed directly into the reactor at 60 °C. After completion of the reaction (24 h), the resulting mixture was quenched with cold water (1500 mL), and extracted with ethyl ether (3 × 800 mL). The organic phase was washed with brine (500 mL), dried over MgSO4, and concentrated under reduced pressure. Flash chromatography using silica gel and hexanes:EtOAc (95:5) as eluent provided the desired product 2 as a white solid (85%, 197 mmol, 43.8 g).
Acid-base liquid-liquid purification of 2
The purification of intermediate 2 was also performed by acid-base extraction. Thus, the crude reaction (same conditions from Table 2, entry 8) product was treated with NaOH (ca 2.6 L, 30%) up to pH 12, and extracted with ethyl ether (1 L). The aqueous phase was treated with HCl (ca 40 mL, 37%) up to pH 5, and 2 was extracted with ethyl ether (3 × 500 mL). The organic phase was washed with brine (500 mL), dried over MgSO4 and concentrated under reduced pressure, providing 2 as a white solid (38.1 g, 171.3 mmol, 74%) after crystallization from hexanes.
Continuous flow synthesis of 2,6-diisopropylphenol - Propofol (3)
The pump was equipped with a solution of 3,5-diisopropyl-4-hydroxybenzoic acid 2 (102.85 g, 463.3 mmol), nBuNH2 (9.0 equiv., 411.4 mL) and sufficient DMF to achieve a 0.32 M solution of 2 (ca 1000 mL of DMF). The mixture was homogenized at 40 °C and pumped at a flow rate of 1.0 mL/min. An Asia Flow Syringe Pump and an Asia Tube Reactor (Stainless - Steel, 16 mL) from Syrris were used. The pump was connected to the reactor at 150 °C. After completion of the reaction (24 h), the resulting mixture was diluted with ethyl ether (2000 mL) and extracted with water (3 × 2000 mL). The organic phase was washed with brine (500 mL), dried over MgSO4, and concentrated under reduced pressure. A simple filtration over silica gel using hexanes:EtOAc (98:2) provided propofol 3 as a yellowish oil (71.6 g, 403 mmol, 87%) after the solvent evaporation.
Telescoped continuous flow synthesis of 2,6-diisopropylphenol - Propofol (3)
In pump A (Table 5) a solution of 4-hydroxybenzoic acid 1 (32.0 g, 231.8 mmol, 0.9 M) in iPrOH:H2O (85:15, ca 250 mL) was heated to 40 °C and pumped at 0.178 mL/min flow rate. Pump B was equipped with a flask containing conc. H2SO4 (heated to 40 °C) and the acid was pumped at 0.356 mL/min flow rate. An Asia Flow Syringe Pump and an Asia Tube Reactor (PFA, 16 mL) from Syrris were used for processing this first step of the telescoped protocol. The pumps were connected by a T-mixer and flowed directly into the reactor at 60 °C. The reaction mixture was continuously extracted (in-line) using a dynamic mixer connected to two pumps (C and D). Pump C was equipped with a flask containing toluene, which was pumped at 0.900 mL/min flow rate. Likewise, pump D was equipped with a flask containing water which was pumped at 0.900 mL/min flow rate. For the in-line extraction, a dynamic mixer with a magnetic stirrer and a hydrophobic membrane phase separator (Biotage®) were coupled. The organic phase containing 2 and toluene was continuously collected and pumped using an Asia Flow Syringe Pump at 0.700 mL/min (Pump E), connected by a T-mixer also coupled to Pump F equipped with a solution containing nBuNH2:DMF (6:4) (pumped at 0.080 mL/min flow rate). The reaction mixture was continuously preheated to 100 °C and then to an Asia Tube Reactor (Stainless - Steel, 16 mL) from Syrris at 150 °C. After completing the reaction (6 hours process intensification), the resulting mixture was treated with HCl (ca 15 mL, 3.0 M) up to pH 5, and 3 was extracted with ethyl acetate (3 × 50 mL). The organic phase was washed with brine (100 mL), dried over MgSO4 and concentrated under reduced pressure. Flash chromatography using silica gel and hexanes:EtOAc (99:1) as eluent provided the desired propofol (3) as a yellowish oil (5.74 g, 32.2 mmol, 84%).
Acknowledgments
The authors would like to thank the São Paulo Research Foundation FAPESP (grant numbers: 2013/07276-1 (V.S.B), 2019/27176-8 (K.T.O.), 2020/06874-6 (K.T.O.) and 2021/01259-4 (G. M. M.)) as well as the Conselho Nacional de Pesquisa - CNPq (K.T.O. research fellowship 303890/2019-3), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Financial Code 001. We also thank the ACCERT Chemistryand Biotechnology Co. for sharing one of the Asia pumps and one reactor module for the telescope experiments (FAPESP grant 2015/11801-0, coordinatedby Dr. Leandro C. Alves).
Prof. Kleber T. de Oliveira
was awarded his Ph.D. in Organic Chemistry (Organic Synthesis) in 2006 from the University of São Paulo in Brazil, under the direction of Prof. Mauricio Constantino. He then carried out two post-doctorals, first with Prof. Cavaleiro at the University of Aveiro, and then with Prof. Iamamoto at the University of São Paulo, also spending 6 months at the Florida State University under the guidance of Prof D. Tyler McQuade in 2015. In 2010 he became a professor at the Federal University of São Carlos (Brazil), integrating the group of Bioorganic Chemistry, and in 2014 started the FlowChem team. His research group is totally focused on the design and synthesis of new photosensitizers, photo, and electrocatalysis as well as API synthesis under continuous flow conditions.

Declarations
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.de Souza AAN, Paez EBA, de Assis FF et al (2021). Topics in Med. Chem. 317–371. 10.1007/7355_2021_117
- 2.Baumann M, Baxendale IR. Beilstein J. Org. Chem. 2015;11:1194–1219. doi: 10.3762/bjoc.11.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gérardy R, Emmanuel N, Toupy T, et al. Eur. J. Org. Chem. 2018;2018:2301–2351. doi: 10.1002/ejoc.201800149. [DOI] [Google Scholar]
- 4.Baumann M, Moody TS, Smyth M, Wharry S. Org. Process. Res. Dev. 2020;24:1802–1813. doi: 10.1021/acs.oprd.9b00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloemendal VRLJ, Janssen MACH, van Hest JCM, Rutjes FPJT. React Chem Eng. 2020;5:1186–1197. doi: 10.1039/D0RE00087F. [DOI] [Google Scholar]
- 6.Di Filippo M, Baumann M. Molecules. 2021;26:6992. doi: 10.3390/molecules26226992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagandira CR, Nqeketo S, Mhlana K, et al. React Chem Eng. 2022;7:214–244. doi: 10.1039/D1RE00483B. [DOI] [Google Scholar]
- 8.Plutschack MB, Pieber B, Gilmore K, Seeberger PH. Chem. Rev. 2017;117:11796–11893. doi: 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- 9.Porta R, Benaglia M, Puglisi A. Org. Process. Res. Dev. 2016;20:2–25. doi: 10.1021/acs.oprd.5b00325. [DOI] [Google Scholar]
- 10.Seifert, J., Mueller, M., Williamson, I., Osterholm, M.T.: CIDRAP Viewp Part 6, Ensuring a Resilient US Prescription Drug. (2020)
- 11.Chidambaran V, Costandi A, D’Mello A. CNS Drugs. 2015;29:543–563. doi: 10.1007/s40263-015-0259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinbacher DM. Anesth. Prog. 2001;48:66–71. [PMC free article] [PubMed] [Google Scholar]
- 13.O’Gara B, Subramaniam B, Shaefi S, et al. Trials. 2019;20:312. doi: 10.1186/s13063-019-3400-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Lv B, West R, et al. BMC Anesthesiol. 2022;22:51. doi: 10.1186/s12871-022-01589-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yip GMS, Chen Z-W, Edge CJ, et al. Nat. Chem. Biol. 2013;9:715–720. doi: 10.1038/nchembio.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saha P, Das A, Chatterjee N, et al. Fundam. Clin. Pharmacol. 2022;36:49–71. doi: 10.1111/fcp.12732. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Pan S, Jiang W, et al. Cell Prolif. 2020;53:e12867. doi: 10.1111/cpr.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.World Health Organization Model List of Essential Medicines– 22nd List (2021). https://www.who.int/groups/expert-committee-on-selection-and-use-of-essential-medicines/essential-medicines-lists. Accessed 25 March 2022
- 19.Kealy TJ, Coffman DD. J. Organomet. Chem. 1961;26:987–992. doi: 10.1021/jo01063a001. [DOI] [Google Scholar]
- 20.Ecke GG, Kolka AJ (1966). United States patent, US3271314A
- 21.Napolitano, J.P.: United States patent, US3367981A. (1968)
- 22.Firth, B.E.: United States patent, US4275248A. (1981)
- 23.Nandiwale KY, Bokade VV. RSC Adv. 2014;4:32467–32474. doi: 10.1039/C4RA03004D. [DOI] [Google Scholar]
- 24.Pramanik C, Kotharkar S, Patil P, et al. Org. Process. Res. Dev. 2014;18:152–156. doi: 10.1021/op400300t. [DOI] [Google Scholar]
- 25.Baltalksne, A.E., Zitzmanis, A.H.: Patent, SU443019. (1974)
- 26.Sharma, A.K., Pandey, M., Giri, A., et al.: Patent, WO2021191832A1. (2021)
- 27.Tsutsumi S, Yoshizawa T, Koyama K. Nippon Kagaku Zasshi. 1956;77:737–738. doi: 10.1246/nikkashi1948.77.737. [DOI] [Google Scholar]
- 28.Keller, S., Jens, S. Patent, WO2012152665A1. (2012)
- 29.Kirti Prakash, J., Dhananjay Uddhavrao, E., Harpreet Singh, M., Gurpreet Singh, M. (2011)
- 30.Mougeot R, Jubault P, Legros J, Poisson T. Molecules. 2021;26:7183. doi: 10.3390/molecules26237183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Yu S, Liu Z, et al.: Org process res dev acs.Oprd.1c00306. (2022)
- 32.de Souza JM, Brocksom TJ, McQuade DT, de Oliveira KT. J. Organomet. Chem. 2018;83:7574–7585. doi: 10.1021/acs.joc.8b01307. [DOI] [PubMed] [Google Scholar]
- 33.Cao Y, Adriaenssens B, de Bartolomeu A, et al. J Flow Chem. 2020;10:191–197. doi: 10.1007/s41981-019-00070-9. [DOI] [Google Scholar]
- 34.Batista GMF, de Castro PP, Dos Santos HF, et al. Org. Lett. 2020;22:8598–8602. doi: 10.1021/acs.orglett.0c03187. [DOI] [PubMed] [Google Scholar]
- 35.Aguillón AR, Leão RAC, de Oliveira KT, et al. Org. Process. Res. Dev. 2020;24:2017–2024. doi: 10.1021/acs.oprd.0c00131. [DOI] [Google Scholar]
- 36.De Souza JM, Galaverna R, de SouzaA AAN, et al. An. Acad. Bras. Cienc. 2018;90:1131–1174. doi: 10.1590/0001-3765201820170778. [DOI] [PubMed] [Google Scholar]
- 37.de Souza AAN, Silva NS, Müller AV, et al. J. Organomet. Chem. 2018;83:15077–15086. doi: 10.1021/acs.joc.8b02355. [DOI] [Google Scholar]
- 38.Carmona-Vargas CC, de Alves LC, Brocksom TJ, de Oliveira KT. React Chem Eng. 2017;2:366–374. doi: 10.1039/C6RE00207B. [DOI] [Google Scholar]