

Abstract

Enantiomerically pure 1,2-amino alcohols are important compounds due to their biological activities and wide applications in chemical synthesis. In this work, we present two multienzyme pathways for the conversion of l-phenylalanine into either 2-phenylglycinol or phenylethanolamine in the enantiomerically pure form. Both pathways start with the two-pot sequential four-step conversion of l-phenylalanine into styrene via subsequent deamination, decarboxylation, enantioselective epoxidation, and enantioselective hydrolysis. For instance, after optimization, the multienzyme process could convert 507 mg of l-phenylalanine into (R)-1-phenyl-1,2-diol in an overall isolated yield of 75% and >99% ee. The opposite enantiomer, (S)-1-phenyl-1,2-diol, was also obtained in a 70% yield and 98–99% ee following the same approach. At this stage, two divergent routes were developed to convert the chiral diols into either 2-phenylglycinol or phenylethanolamine. The former route consisted of a one-pot concurrent interconnected two-step cascade in which the diol intermediate was oxidized to 2-hydroxy-acetophenone by an alcohol dehydrogenase and then aminated by a transaminase to give enantiomerically pure 2-phenylglycinol. Notably, the addition of an alanine dehydrogenase enabled the connection of the two steps and made the overall process redox-self-sufficient. Thus, (S)-phenylglycinol was isolated in an 81% yield and >99.4% ee starting from ca. 100 mg of the diol intermediate. The second route consisted of a one-pot concurrent two-step cascade in which the oxidative and reductive steps were not interconnected. In this case, the diol intermediate was oxidized to either (S)- or (R)-2-hydroxy-2-phenylacetaldehyde by an alcohol oxidase and then aminated by an amine dehydrogenase to give the enantiomerically pure phenylethanolamine. The addition of a formate dehydrogenase and sodium formate was required to provide the reducing equivalents for the reductive amination step. Thus, (R)-phenylethanolamine was isolated in a 92% yield and >99.9% ee starting from ca. 100 mg of the diol intermediate. In summary, l-phenylalanine was converted into enantiomerically pure 2-phenylglycinol and phenylethanolamine in overall yields of 61% and 69%, respectively. This work exemplifies how linear and divergent enzyme cascades can enable the synthesis of high-value chiral molecules such as amino alcohols from a renewable material such as l-phenylalanine with high atom economy and improved sustainability.
Keywords: biocatalysis; biocatalytic cascades; amine dehydrogenases; transaminases, alcohol dehydrogenases; alcohol oxidases; phenylethanolamine; 2-phenylglycinol
1. Introduction
Over the past two decades, biocatalysis has made a major contribution toward sustainable chemical synthesis, in particular for the highly selective syntheses of high-value chiral molecules.1−10 In this context, the utilization of biocatalysis to convert biomass-derived starting materials into chiral molecules as intermediates or final products for the manufacture of active pharmaceutical ingredients, flavors, fragrances, agrochemicals, and fine chemicals can make a decisive contribution to enabling a circular economy.11−18 Natural amino acids are abundant and inexpensive biobased feedstocks produced by fermentation that have been marginally used as starting materials for the chemical synthesis of chiral molecules.19−21 For instance, the fermentative production of l-phenylalanine with a titer above 70 g per liter of culture can be accomplished from glucose or glycerol using Escherichia coli strains in which the l-phenylalanine biosynthesis pathway (i.e., the shikimate pathway) has been engineered.22−26 In contrast, chiral 1,2-amino alcohol motifs are widespread in biologically active compounds and bioactive natural products such as antibiotics, neurotransmitters, β-adrenergic blockers, and antiviral drugs.27−29 They also find application in asymmetric organic synthesis as ligands, chiral auxiliaries, and even organocatalysts.30−32 As illustrated in Figure 1, chiral phenylethanolamines and 2-phenylglycinols are particularly important in this context for their chemical and biological properties.
Figure 1.
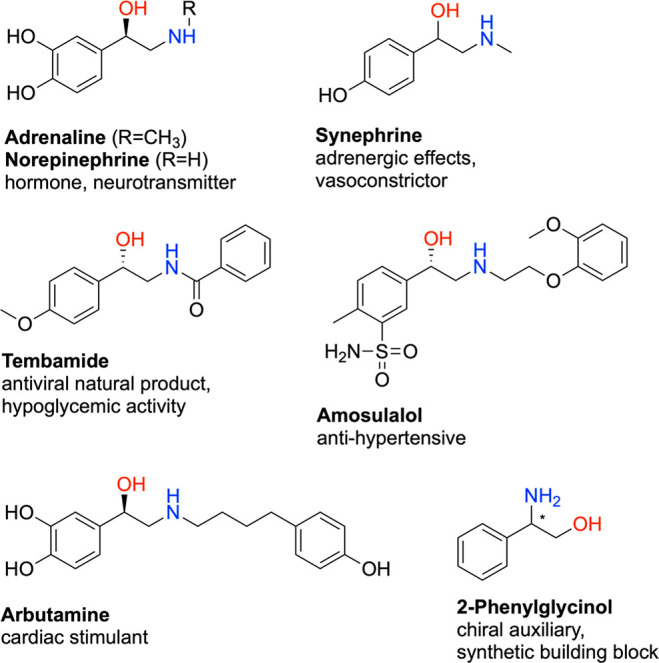
Examples of biologically active compounds and chiral auxiliaries bearing phenylethanolamine or 2-phenylglycinol moieties.
Highly selective asymmetric synthesis methods remain widely sought after to obtain these and many other valuable compounds and intermediates thereof in high chemical and optical purities. Synthetically applied methods comprise the Sharpless asymmetric aminohydroxylation of terminal olefins,33,34 the asymmetric hydrogenation of prochiral amino ketones,35 and the ring-opening of an epoxide with an amine as the nucleophile.36−39 Nevertheless, these methods have some drawbacks related to their selectivity and sustainability, as the phenylethanolamines are hardly ever obtained in their enantiomerically pure forms and toxic metals and reagents are often required in superstoichiometric amounts.40−42 Consequently, resolution techniques are still mainly used for the preparation of optically pure 1,2-amino alcohols.35 Notably, biocatalytic strategies for the synthesis of chiral 1,2-amino alcohols have also been developed;43−52 however, only a few methods are currently available for the specific synthesis of chiral phenylethanolamines and 2-phenylglycinols.44,49,53,54
A chemo-biocatalytic route toward phenylethanolamines entails the ring-opening of styrene oxide with ammonia under microwave irradiation; styrene oxide is obtained from the bioepoxidation of styrene with a styrene monooxygenase.55 In full biocatalytic approaches, linear cascades have been demonstrated to be viable options due to their often high selectivity and atom efficiency.56,57 Notable examples are a multienzymatic cascade for the asymmetric synthesis of (R)-2-phenylglycinol ((R)-7) from racemic styrene oxide (4);58 a one-pot three-step enzymatic process to convert a series of halo ketones to the corresponding amino alcohols, including the natural antiviral product (S)-tembamide;29 the conversion of styrene to (S)-phenylethanolamine ((S)-9) by modular cascade biocatalysis;59 and the engineered hemoprotein-catalyzed direct enantioselective aminohydroxylation of olefins.28
In this work, we exemplify the potential of biocatalysis for the synthesis of high-value aromatic 1,2-amino alcohols such as optically active phenylethanolamines and 2-phenylglycinols through enzymatic cascade reactions starting from l-phenylalanine as a renewable material. These cascades harness some of the engineered enzymes and reactions that our group has developed over the past five years.
2. Results and Discussion
2.1. Conversion of l-Phenylalanine into (R)-1-Phenylethane-1,2-diol
Scheme 1 depicts the first part of our synthetic strategy in which l-phenylalanine (l-1) is converted into (R)-1-phenylethane-1,2-diol ((R)-5) through two-pot four-step sequential biocatalytic cascades (or two-pot four-stage cascades, as all four steps are separated in time) with only one intermediate extraction step. l-1 (20 mM, 3.1 mmol, 507 mg) was first deaminated to cinnamic acid (2) with >99% conversion and >99% chemoselectivity (see Figure S2). The reaction was catalyzed by lyophilized E. coli cells (3 g, 20 mg mL–1) expressing a tyrosine ammonia lyase from Rhodobacter sphaeroides (TAL),60 which were suspended in a KPi buffer (pH 9, 50 mM, 150 mL) for 24 h at 30 °C. Following the removal of the cell pellets by centrifugation, the reaction mixture from step 1 was directly reacted in the subsequent step. The pH of the mixture was lowered to 6.5–7 via the addition of HCl, and lyophilized E. coli cells (3 g, 20 mg mL–1) expressing a ferulic acid decarboxylase from Saccharomyces cerevisiae (FDC1/tPAD1)61 were added to perform the decarboxylation of 2 to styrene (3) over 24 h at 30 °C. At the end of the reaction (conversion step 2 > 99%, see Figure S3), 3 was extracted with n-heptane and used directly in the next one-pot two-step (or one-pot two-stage) sequential cascade. The first step of the second pot (Scheme 1, pot B) was a biocatalytic epoxidation performed in an organic/aqueous biphasic system. The influence of the ratio of the two phases on the conversion was initially tested on an analytical scale (total volume of 1 mL), which showed that a 1:1 volume ratio was the optimal condition (see SI section 3.1). Therefore, the solution of n-heptane (150 mL) containing intermediate 3 was combined with a KPi buffer (pH 8, 50 mM, 150 mL) containing lyophilized E. coli cells (5 mg mL–1, related to the aqueous phase) expressing our previously reported chimeric styrene monooxygenase (Fus-SMO), where the reductive (StyB) and monooxygenase (StyA) enzyme units were fused together using a flexible linker.62 This biocatalytic epoxidation step required reducing equivalents that were provided by a catalytic amount of the reduced nicotinamide adenine dinucleotide coenzyme (NADH), which was generated in situ and recycled from NAD+ (1 mM) by a formate dehydrogenase (Cb-FDH) and sodium formate (100 mM, 5 equiv). Notably, Fus-SMO and Cb-FDH were produced together in the same E. coli cells through cloning and the balanced expression of both genes in a Duet-vector. This is an improvement on previous work in which the enzymes were coexpressed using different plasmids in the same host.62 After a 20 hr reaction time at 30 °C (>99% conversion and perfect chemoselectivity), the same reaction pot containing the (S)-styrene epoxide ((S)-4) product was subjected to the next step, where lyophilized E. coli cells (3 g, 20 mg mL–1) expressing an epoxide hydrolase from Solanum tuberosum St(R)-EH were simply added to the mixture.63−65 The regioselective biocatalytic hydrolysis step was run for an additional 24 h and proceeded with the full inversion of the stereochemical configuration of (S)-4 to yield (R)-1-phenylethane-1,2-diol ((R)-5) in a quantitative conversion and 94% chemoselectivity. The product was recovered by separation between the n-heptane phase and the aqueous phase, the latter of which was further extracted with methyl-tert-butyl ether. The organic phases were then dried with anhydrous MgSO4, and the solvent was removed. In summary, at the end of this two-pot four-step sequential biocatalytic process and workup (see the Experimental Part for details), (R)-5 was recovered from l-1 in a 75% overall isolated yield (320 mg) with high chemical (>99%) and optical (ee > 99%) purities. Notably, intermediate purification steps were not required, thereby minimizing waste generation and work time. (S)-Configured 1-phenylethane-1,2-diol ((S)-5) could also be generated (98–99% ee, 70% yield, 240 mg from (S)-4) in a similar manner under not-fully optimized conditions by changing the selectivity of the epoxide hydrolase in the last hydrolytic step (see SI section 3.2). We performed this reaction using an epoxide hydrolase from Sphingomonas sp. HXN200 (Sp(S)-EH).63−65
Scheme 1. Two-Pot Four-Step Sequential Biocatalytic Cascades for the Conversion of l-Phenylalanine (l-1) into (R)- or (S)-1-Phenylethane-1,2-diol ((R)- or (S)-5).

There is only one intermediate extraction work-up after step 2.
Products (R)-5 and (S)-5 were used as starting materials in two subsequent and distinct one-pot biocatalytic cascades for the synthesis of either optically pure 2-phenylglycinol (7) or phenylethanolamine (9), as described in the following sections.
2.2. Conversion of (R)-1-Phenylethane-1,2-diol into (S)- and (R)-2-Phenylglycinol
At this stage, we initially intended to convert (R)- or (S)-5, obtained as previously reported, into (R)- or (S)-2-phenylglycinol ((R)- or (S)-7) through the one-pot combination of a “secondary” NAD-dependent alcohol dehydrogenase (ADH) and an amine dehydrogenase (AmDH), thus following our previously developed strategy for the synthesis of optically pure phenylpropanolamines.63,66 However, all the tested AmDHs proved to be unsuitable for this process (data not shown). Therefore, we turned our attention to the alcohol amination by combining an ADH with an ω-transaminase (ωTA), thus following our alternative strategy for the synthesis of phenylpropanolamines (Scheme 2).64,67
Scheme 2. One-Pot Concurrent Interconnected Two-Step Biocatalytic Cascade for the Conversion of (R)-1-Phenylethane-1,2-diol ((R)-5)) into (R)- or (S)-Phenylglycinol ((R)- or (S)-7)).

In this alcohol amination cascade, Aa-ADH oxidizes (R)-5 to the hydroxyketone intermediate (6) and then the ωTA performs the transamination of the carbonyl moiety to yield either (R)-7 or (S)-7; additionally, the NAD+ coenzyme and alanine as the amine donor are internally recycled from NADH and pyruvate, respectively, by an alanine dehydrogenase from Bacillus sphaericus (Bs-AlaDH)68 at the expense of the ammonia and ammonium species that are provided by the reaction buffer.
The optimal biocatalysts for this transformation were initially tested for the separated reactions on an analytical scale. First, we investigated the oxidation of commercially available rac-5 (20 mM) to the hydroxyketone (6) in a Tris-HCl buffer (pH 7.5, 50 mM; final reaction volume of 1 mL) using 12 different alcohol dehydrogenases, namely Bs-BDHA,69,70 Pp-ADH,71,72 Sy-ADH,73 Rs-ADH,74 Ls-ADH,75 three variants of Te-ADH (v1, v2, and v3),76 Aa-ADH,77 Lbv-ADH,78 and Lb-ADH.79 For details on the abbreviations of enzyme names and related preparations, see SI section 1 and Table S1. The reaction with Ls-ADH was carried out in KPi buffer at pH 6.5 (100 mM) rather than pH 7.5 because previous tests performed in our laboratory demonstrated that this enzyme was more sensitive toward higher pH values. The tests were conducted in the presence of either NAD+ or NADP+ (1 mM) depending on the selectivity of the ADH. NAD+ and NADP+ were internally recycled with specific oxidoreductases (10 μM), which consume molecular oxygen as oxidant. NOx from Streptococcus mutans(80) and YcnD from Bacillus subtilis(81) were used for the reoxidation of NADH and NADPH, respectively. High conversions into 6 were observed when Aa-ADH, Lbv-ADH, Bs-BDHA, and Lb-ADH were used (considering both that the substrate 5 was used as racemate and that these ADHs have a preference toward one of the two enantiomers). Moderate conversions were also observed with Ls-ADH and Rs-ADH, while the remaining ADHs were not active toward the target substrate (SI section 4 and Table S3). Among this latter group, Sy-ADH, Pp-ADH, and Te-ADH-v3 are described as “non-stereoselective” ADHs. Since these enzymes were found not to be catalytically active toward 5, the utilization of rac-5 as a possible intermediate was ruled out at this stage. However, this is not a synthetic limitation since optically pure 5 can be efficiently obtained via the enzymatic strategy illustrated in this work. Therefore, we tested the best-performing ADH from the previous set of experiments with either enantiopure (R)-5 or enantiopure (S)-5 (10 mM, SI section 4 and Table S4). Among these four best ADHs, we excluded Lb-ADH because it was NADP-dependent. In fact, the use of a NAD-dependent ADH is more suitable for our intended final cascades, and NAD+ is also cheaper than NADP+. Lbv-ADH converted (S)-5 into 6 with >99% conversion, whereas Aa-ADH and Bs-BDHA converted (R)-5 into 6 with 84% and 69% conversion, respectively (SI section 4 and Table S4).
In the next step, we investigated the cascade from (R)- or (S)-5 to (R)- or (S)-7 using combinations of the three previously selected ADHs (i.e., Aa-ADH, LBv-ADH, and Bs-BDHA) and five stereocomplementary ω-transaminases, namely At-ωTA,82−84 Cv-ωTA,85 Bm-ωTA,86 Ac-ωTA,87,88 and Vf-ωTA.89,90 For details on the abbreviations of enzyme names, see SI section 1 and Table S1. The reactions were carried out at 30 °C for 48 h in a HCOONH4 buffer (pH 8.5, 1 M) supplemented with NAD+ (1 mM), d- or l-alanine (50 mM), ω-TA (varied concentrations of 35–60 μM), ADH (varied concentrations of 24–70 μM), Bs-AlaDH (20 μM), and substrate (10 mM). The experiments showed the inherently lower activity of Cv-ωTA toward the in situ generated intermediate 6 compared with those of At-ωTA and Bm-ωTA. Furthermore, selected experiments where Ac-ωTA and Vf-ωTA were used did not yield any conversion (see SI section 5 and Tables S5 and S6). Under the optimal reaction conditions (Table 1), both (S)-7 and (R)-7 were obtained from (R)-5 in high yields and excellent optical purities using Aa-ADH from Aromatoleum aromaticum combined with At-ωTA from Aspergillus terreus and Bm-ωTA from Bacillus megaterium.
Table 1. One-Pot Alcohol Amination of (R)-5 (10 mM) to Yield Either (S)-7 or (R)-7a.

| entry | enzymes | total conv. [%] | conv. into 7 [%] | ee of 7 [%]b |
|---|---|---|---|---|
| 1 | Aa-ADH/At-ωTA | >99 | 97 ± < 1 | >99 (S) |
| 2 | Aa-ADH/Bm-ωTA | 71 ± < 1 | 70 ± < 1 | >99 (R) |
The reaction was catalyzed by Aa-ADH from Aromatoleum aromaticum (70 μM), which was combined with either At-ωTA from Aspergillus terreus or Bm-ωTA from Bacillus megaterium (35 μM) in HCOONH4 buffer (pH 8.5, 1 M) at 30 °C for 48 h.
Determined by RP-HPLC (C18 HD column) following the derivatization of the amino group with GITC. Reactions were performed in duplicate, and results are reported as the average of the two samples.
To prove the synthetic applicability of the one-pot biocatalytic amination, the bioconversion of (R)-5 into (S)-7 was performed on a 101 mg scale. The product (S)-7 was obtained in an 81% isolated yield with 98% purity and >99.4% ee (see the Experimental Part for details).
2.3. Conversion of (S)- and (R)-1-Phenylethane-1,2-diol into (S)- and (R)-Phenylethanolamine
At this stage, we intended to convert (R)-5 or (S)-5, obtained as previously reported, into either (R)- or (S)-phenylethanolamine ((R)- or (S)-9) through the one-pot combination of a “primary” ADH and an amine dehydrogenase (AmDH). We initially tested the oxidation of intermediate 5 into 2-hydroxy-2-phenylacetaldehyde (8) using an alcohol dehydrogenase. The hT-ADH from Bacillus stearothermophilus,91 and the HL-ADH from Equus caballus (i.e., horse liver)92 were tested, as both have been known to oxidize the primary alcohol functionalities of molecules similar to 5. Ht-ADH was used in its purified form (50 μM), whereas HL-ADH was a commercially available enzyme and was used as the lyophilized cell lysate (2 mg mL–1 crude enzyme; activity of 0.52 U mg–1; 20% protein content). In both cases, the biocatalytic oxidation of rac-5 (20 mM) was performed in a Tris-HCl buffer (pH 7.5, 50 mM; final reaction volume of 1 mL) in the presence of a NAD+ recycling system (1 mM NAD+; 10 μM NOx). Contrary to our expectations, hT-ADH exhibited no activity toward substrate rac-5, while HL-ADH produced the hydroxyketone isomer 6 rather than the desired product 8, thus actually acting as a secondary ADH. Such activity of HL-ADH on secondary alcohol moieties with certain molecules was also reported in the literature.93−97 Therefore, we envisioned an alternative strategy for the amination of the primary alcohol moiety of 5 that combined a variant of the choline oxidase (AcCO6) originated from Arthrobacter chlorophenolicus(98) with an AmDH (Ch1-AmDH).99,100 Thus, this cascade for the bioamination of the diol 5 is comprised of two concurrent, albeit disconnected, steps that must confer a more favorable thermodynamic equilibrium (Scheme 3).
Scheme 3. One-Pot Concurrent Disconnected Two-Step Biocatalytic Cascade for the Conversion of (R)- or (S)-1-Phenylethane-1,2-diol ((R)-5 or (S)-5) into (R)- or (S)-Phenylethanolamine ((R)- or (S)-9).

Another advantage of AcCO6 is that it is an oxidase; therefore, its activity does not depend on the NADH/NAD+ coenzyme. However, a catalase must be added to prevent the possible deactivation of any enzyme in the reaction mixture due to the formation of H2O2 as a side product in the oxidation reaction. Furthermore, the addition of a catalytic amount of NAD+ and a formate dehydrogenase from Candida boidinii (Cb-FDH) is required in the second reductive amination step of the cascade for the in situ generation and recycling of NADH.76,100
The reactions were performed in a HCOONH4 buffer (pH 8.5, 1 M) that provided both the source of the amino group and the reducing hydride (i.e., for NADH regeneration) for the reductive amination. The initial set of experiments (SI section 6 and Table S7) were carried out using an equimolar ratio of AcCO6 and Ch1-AmDH (50 μM each) in a HCOONH4 buffer (pH 8.5, 1 M; 0.5 mL) supplemented with a catalytic amount of NAD+ (1 mM) and a catalase (0.1 mg mL–1). 5 was used as the substrate either as a racemate or as a single enantiomer in the R- or S-absolute configuration (10 or 20 mM). The quantitative conversion of rac-5 into the target amino alcohol 9 was detected at the 10 mM scale, and oxidation at the 20 mM scale afforded 90% conversion. Due to the racemic form of the substrate, the enantiomeric excess of the product was low in both cases (10% ee (S)). Therefore, although AcCO6 did not strongly discriminate between the two enantiomers of 5, we observed a small preference for the oxidation of (S)-5 over (R)-5. Next, the same reaction and conditions were investigated to convert optically pure 5 as substrates, obtained from l-phenylalanine (l-1) via the first cascades, into chiral products 9. (S)-5 was converted, resulting in a 98% (at 10 mM substrate concentration) or 88% (at 20 mM substrate concentration) yield of (S)-9. As expected, the enantiomeric excess of the starting material (S)-5 (98–99% ee) was the same as that in the final product (S)-9. (R)-5 was also converted into (R)-9, with 92% or 72% conversion at a substrate concentration of 10 or 20 mM, respectively, and showed the same enantiomeric excess of the starting material (>99% ee (R)). The slightly lower conversion for (R)-5 compared with that for (S)-5 again indicates the slightly higher preference of AcCO6 to oxidize the (S)-enantiomer of the substrate. Notably, the isomerization of the aldehyde intermediate 8 to the more stable hydroxyacetophenone (6) via keto–enol tautomerization was not detected (see Figure S10). However, we observed the formation of benzylamine (11) as a side product (from 2% to 8%) in nearly all tests; the only exception was the reaction of rac-5 at the 10 mM concentration for which quantitative conversion into 9 was detected. A possible explanation for this result is that the aldehyde intermediate 8 undergoes cleavage to benzaldehyde (10), which is then converted to benzylamine (11) by Ch1-AmDH (see Figure S10). To investigate that possibility, (S)-5 (20 mM) was incubated in a HCOONH4 buffer (pH 8.5, 1 M, 1 mL) in the presence of AcCO6 (50 μM) and a catalase (0.1 mg mL–1). A negative control experiment was performed by incubating the substrate in the same reaction mixture devoid of AcCO6 and the catalase (for details, see SI section 7). The reactions in the presence of AcCO6 and the catalase led to 27% and 32% benzaldehyde (10) formation within 6 and 48 h of incubation, respectively. In contrast, we did not observe any formation of 10 in the negative control experiment after 48 h (see Figure S11). Therefore, benzylamine (11) was indeed formed by the Ch1-AmDH-catalyzed reductive amination of side product 10.
Different enzyme loadings were tested to improve the conversions, especially in the case of (R)-5 (SI section 6 and Table S8). Three sets of experiments were carried out in which the molar ratio between AcCO6 and Ch1-AmDH was varied. The reaction of (S)-5 (20 mM) at a higher AcCO6 loading (70 μM) led to quantitative substrate conversion, although 11% of 11 was formed along with 89% of the desired product (S)-9. At lower AcCO6 loadings (10 or 24 μM), (S)-9 was obtained with 41–77% conversion along with traces of 11 (2–4%). Therefore, the use of equimolar amounts of AcCO6 and Ch1-AmDH (50 μM each) turned out to be the optimal condition for the amination of (S)-5, as reported in the initial experiments. In contrast, the optimal conditions for the amination of (R)-5 (10 mM) were found to be 70 μM AcCO6 and 35 μM Ch1-AmDH, which resulted in 98% conversion into product (R)-9 and traces of 11 (2%).
In summary, we could obtain our target products (S)-9 and (R)-9 with high conversions (98%) and enantiomeric excesses (ee up to >99%) by tuning the substrate and enzyme loadings to enhance the formation of the desired product while also limiting the side production of benzylamine (Table 2).
Table 2. One-Pot Concurrent Oxidation–Reduction Two-Step (Disconnected) Bioamination of (S)- or (R)-5 (10 mM) to Optically Active (S)- or (R)-9 Catalyzed by AcCO6 Combined with Ch1-AmDH.

| entry | substrate | total conv. [%]a | conv. into 9 [%] | ee of 9 [%]b |
|---|---|---|---|---|
| 1c | (S)-5 | >99 | 98 ± < 1 | >98 (S) |
| 2d | (R)-5 | >99 | 98 ± < 1 | >99 (R) |
Reactions were performed in duplicate, and results are reported as the average of the two samples; we detected the formation of 2 ± <1% benzylamine (11) in the reactions with each substrate.
Determined by RP-HPLC (C18 HD column) following the derivatization of the amino group with GITC.
AcCO6/Ch1-AmDH 50:50 μM.
AcCO6/Ch1-AmDH 70:35 μM.
To prove the synthetic applicability of the one-pot biocatalytic amination, the bioconversion of (R)-5 to (R)-9 was performed on a 104 mg scale. The product (R)-9 was obtained in a 92% isolated yield with 98% purity and >99.9% ee (see the Experimental Part for details).
3. Conclusion
In this work, we have presented the stereoselective synthesis of both enantiomers of 2-phenylglycinol (7) and those of phenylethanolamine (9) in highly optically pure forms (>99% ee) through consecutive and divergent biocatalytic routes starting from l-phenylalanine (l-1) as renewable material. In the first route, l-1 was converted into (R)-5 or (S)-5 at a ca. 500 mg scale with total isolated yields of 75% and 70%, respectively.
At this stage, two divergent routes were envisioned to lead to the formation of optically pure enantiomers of either 2-phenylglycinol (7) or phenylethanolamine (9). In the first route, (R)-5 was converted into (S)-7 or (R)-7 in a 97% or 70% yield, respectively, on an analytical scale. The ca. 100 mg scale conversion of (R)-5 under the same reaction conditions produced (S)-7 in an 81% isolated yield with >99.4% ee. In the second route, (R)- or (S)-5 was converted into (R)- or (S)-9 on an analytical scale with 98% conversion. The ca. 100 mg scale conversion of (R)-5 under the same reaction conditions produced (R)-9 in a 92% isolated yield with >99.9% ee.
In summary, this work exemplifies the potential impact of linear biocatalytic cascade reactions on the highly atom efficient and sustainable syntheses of high-value chiral molecules from available and inexpensive renewable material such as l-phenylalanine. For instance, this amino acid is produced by fermentation and is a suitable starting material for further biotransformations, both in vivo and in vitro. In fact, the fermentation product mixtures of l-phenylalanine normally contain low amounts of byproducts, namely acetate, lactate, and succinate. These compounds are not known to significantly interfere with or inhibit other enzymes.24 However, purified l-phenylalanine can be obtained by integrating a reactive extraction step into the fermentation process, as described elsewhere at the 300 L scale.101 In this work, l-phenylalanine could be converted into either (S)-phenylglycinol or (R)-phenylethanolamine in a total combined yield of 61% or 69%, respectively (see Figure 2).
Figure 2.
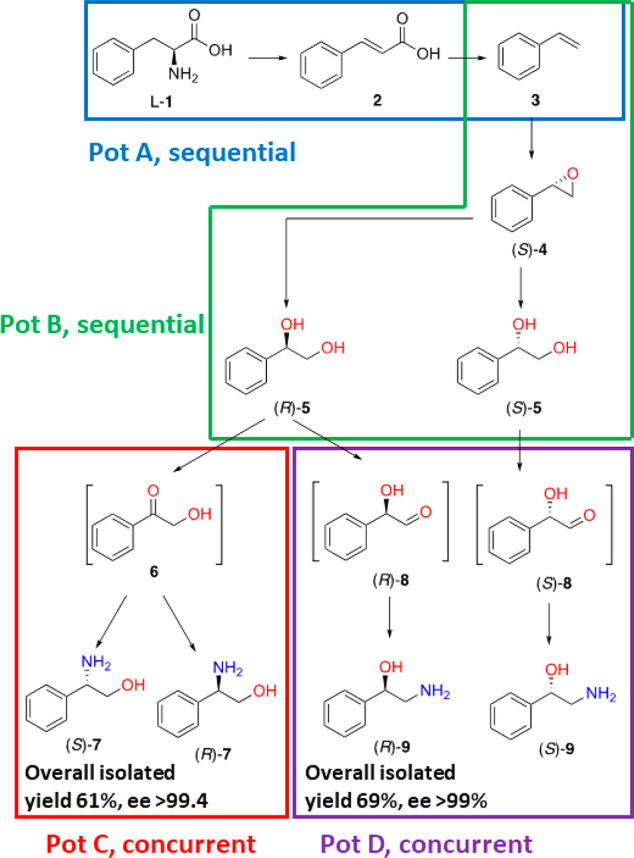
Summary of the biocatalytic pathways developed in this work and related synthetic strategies.
Notably, the process for converting l-phenylalanine into enantiomerically pure phenylethanolamine is comprised of a total of six steps performed in three pots. The reaction formally consumes only dioxygen as a simple and innocuous reagent and produces stoichiometric hydrogen carbonate as the sole byproduct, while water and ammonia molecules are formally exchanged along the process. These results also pave the way for the future metabolic engineering of E. coli whole-cell systems in which all the required enzymes for a certain cascade are coexpressed, thereby potentially improving the efficiency of the biochemical process.24,102
4. Experimental Part
4.1. Two-Pot Sequential Four-Step Cascades for the Conversion of l-Phenylalanine (l-1) into (R)-1-Phenylethane-1,2-diol ((R)-5) at a ca. 500 mg Scale
Step 1: A KPi buffer (pH 8.0, 50 mM, 150 mL) and l-1 (20 mM, 3.1 mmol, 507 mg) were added into a 250 mL Erlenmeyer flask. Then, the pH of the mixture was adjusted to 9.0–10.0 by adding KOH (10 M), and to the mixture were added lyophilized E. coli whole-cells carrying overexpressed tyrosine ammonia lyase (TAL, 3 g, 20 mg mL–1). The reaction mixture was incubated at 30 °C and 170 rpm for 24 h. Following the removal of the cell debris by centrifugation (10 min, 14000 rpm, 18800 × g), an aliquot of the reaction mixture (0.5 mL) was analyzed by RP-HPLC (method A) to determine the conversion into the desired cinnamic acid intermediate (2). In this work, we determined the conversion using the following ratio: (observed product formation)/(observed product formation + observed remaining substrate).
Step 2: Without any intermediate workup, the pH of the reaction mixture in the same pot from step 1 was lowered to 6.5–7.0 by adding concentrated HCl. Next, to the mixture were added lyophilized E. coli cells carrying overexpressed ferulic acid decarboxylase (FDC1/tPAD1, 3 g, 20 mg mL–1). The reaction mixture was incubated at 30 °C and 170 rpm for 24 h. Following removal of the cell debris by centrifugation (10 min, 14000 rpm, 18800 × g), an aliquot of the reaction mixture (0.5 mL) was analyzed by RP-HPLC (method A) to determine the conversion into the styrene (3) . Next, 3 was extracted with n-heptane (3 × 50 mL), and the obtained organic solution was used directly in the subsequent step.
Step 3: A KPi buffer (pH 8.0, 50 mM, 150 mL), lyophilized E. coli cells carrying coexpressed chimeric styrene monooxygenase and formate dehydrogenase (Fus-SMO/FDH, 5 mg mL–1), NAD+ (1 mM), FAD (50 μM), HCOONa (100 mM, 5 equiv), and a catalase (0.1 mg mL–1) were added in a 500 mL tribaffled flask. Then, to the mixture was added the solution of 3 in n-heptane (150 mL) obtained from step 2. The reaction mixture was incubated at 30 °C and 200 rpm for 20 h. The conversion was monitored by GC-FID (method A) using an aliquot of the reaction mixture. When the conversion was quantitative, the reaction proceeded to step 4.
Step 4: Lyophilized E. coli cells carrying overexpressed epoxide hydrolase (St(R)-EH, 3 g, 20 mg mL–1) were added to the same pot from step 3 without any intermediate workup. The reaction mixture was further incubated at 30 °C and 170 rpm for 24 h. The n-heptane phase was then separated from the aqueous phase. The latter phase was saturated with solid NaCl and extracted with MTBE (3 × 100 mL). The combined organic phase was dried over anhydrous MgSO4 and concentrated under reduced pressure to yield product (R)-5 (320 mg, 75% total isolated yield calculated from the l-phenylalanine (l-1) starting material; ee > 99% (R); 93% purity). The conversion and purity of the isolated product were analyzed by GC-FID (method A), and the enantiomeric excess was determined using chiral NP-HPLC (method B); see SI section 3.3 and Figures S4 and S5. 1H NMR (see Figure S6) spectra were recorded after column chromatography with petroleum ether and ethyl acetate (1:1, v v–1) as the eluent (Rf = 0.3), which afforded the quantitative yield of purification.
4.2. One-Pot Simultaneous Interconnected Two-Step Cascade for the Conversion of (R)-1-Phenylethane-1,2-diol ((R)-5) into (S)-2-Phenylglycinol ((S)-7) at a ca. 100 mg Scale
An ammonium formate buffer (2 M, pH 8.5, 37.5 mL), H2O (28 mL), NAD+ (1 mM, 49 mg), PLP (1 mM, 19 mg), d-alanine (50 mM, 322 mg), and substrate (R)-5 (10 mM, 101 mg) were added to a 250 mL Erlenmeyer flask. The pH was adjusted to 8.5 with ammonia. Then, to the mixture were added Bs-AlaDH (20 μM), Aa-ADH (70 μM), and At-ωTA (35 μM). The total reaction volume was 73 mL. The reaction mixture was incubated at 30 °C for 70 h. The aqueous reaction mixture was basified with KOH (10 M, 9 mL), saturated with solid NaCl, and extracted with EtOAc (2 × 40 mL). Following the drying of the combined organic phases over anhydrous MgSO4, the organic phase was concentrated under reduced pressure to yield product (S)-7 (orange oil, 81% isolated yield (81 mg), 98% purity, ee > 99.4%). The conversion and purity of the isolated product were measured by GC-FID (method A), 1H NMR, and 13C NMR (see SI section 8 and Figures S12, S15, and S16). The enantiomeric excess was determined by RP-HPLC following derivatization with GITC (method D); see Figure S13.
4.3. One-Pot Simultaneous Disconnected Two-Step Cascade for the Conversion of (R)-1-Phenylethane-1,2-diol ((R)-5) into (R)-Phenylethanolamine ((R)-9) at a ca. 100 mg Scale
An ammonium formate buffer (2 M, pH 8.5, 37.5 mL), H2O (16 mL), NAD+ (1 mM, 49 mg), a catalase (0.1 mg mL–1, 7.3 mg), and substrate (R)-5 (10 mM, 104 mg) were added to a 250 mL Erlenmeyer flask. The pH was adjusted to 8.5 with ammonia. Then, to the mixture were added Cb-FDH (10 μM), AcCO6 (70 μM), and Ch1-AmDH (35 μM). The total reaction volume was 73 mL. The reaction mixture was incubated at 30 °C for 70 h. The aqueous reaction mixture was basified with KOH (10 M, 9 mL), saturated with solid NaCl, and extracted with EtOAc (2 × 40 mL). Following the drying of the combined organic phases over anhydrous MgSO4, the organic phase was concentrated under reduced pressure to yield product (R)-9 (yellow oil, 92% isolated yield (95 mg), 98% purity, ee > 99.9%). The conversion and purity of the isolated product were measured by GC-FID (method A), 1H NMR, 13C NMR (see SI section 8 and Figures S12, S17, and 18). The enantiomeric excess was determined by RP-HPLC following derivatization with GITC (method D); see Figure S14.
Acknowledgments
This work was financed by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO), Grant “ECHO Chemistry in Relation to Technology and Sustainability 2013 CW”, project number 717.014.007. F.G.M. received funding from a European Research Council (ERC) starting grant (H2020, Grant 638271, BioSusAmin). Dutch funding from the NWO Sector Plan for Physics and Chemistry is also acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.oprd.1c00490.
Details on the enzymes used in this work; general procedures and references for enzyme expression and purification; experimental procedures for the cascade reactions and other experiments on an analytical scale; analytical procedures for the determination of conversions, enantiomeric excesses, and the derivatization of compounds; HPLC and GC chromatograms; and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sheldon R. A.; Brady D. Broadening the Scope of Biocatalysis in Sustainable Organic Synthesis. ChemSusChem 2019, 12, 2859–2881. 10.1002/cssc.201900351. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Woodley J. M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. 10.1021/acs.chemrev.7b00203. [DOI] [PubMed] [Google Scholar]
- Wu S.; Snajdrova R.; Moore J. C.; Baldenius K.; Bornscheuer U. T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem., Int. Ed. 2021, 60, 88–119. 10.1002/anie.202006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestl B. M.; Hammer S. C.; Nebel B. A.; Hauer B. New Generation of Biocatalysts for Organic Synthesis. Angew. Chem., Int. Ed. 2014, 53, 3070–3095. 10.1002/anie.201302195. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 2013, 135, 12480–12496. 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- Choi J. M.; Han S. S.; Kim H. S. Industrial Applications of Enzyme Biocatalysis: Current Status and Future Aspects. Biotechnol. Adv. 2015, 33, 1443–1454. 10.1016/j.biotechadv.2015.02.014. [DOI] [PubMed] [Google Scholar]
- Bell E. L.; Finnigan W.; France S. P.; Green A. P.; Hayes M. A.; Hepworth L. J.; Lovelock S. L.; Niikura H.; Osuna S.; Romero E.; Ryan K. S.; Turner N. J.; Flitsch S. L. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. 10.1038/s43586-021-00044-z. [DOI] [Google Scholar]
- Hauer B. Embracing Nature’s Catalysts: A Viewpoint on the Future of Biocatalysis. ACS Catal. 2020, 10, 8418–8427. 10.1021/acscatal.0c01708. [DOI] [Google Scholar]
- Yi D.; Bayer T.; Badenhorst C. P. S.; Wu S.; Doerr M.; Hohne M.; Bornscheuer U. T. Recent Trends in Biocatalysis. Chem. Soc. Rev. 2021, 50, 8003–8049. 10.1039/D0CS01575J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudroff F.; Mihovilovic M. D.; Gröger H.; Snajdrova R.; Iding H.; Bornscheuer U. T. Opportunities and Challenges for Combining Chemo- and Biocatalysis. Nat. Catal. 2018, 1, 12–22. 10.1038/s41929-017-0010-4. [DOI] [Google Scholar]
- Devine P. N.; Howard R. M.; Kumar R.; Thompson M. P.; Truppo M. D.; Turner N. J. Extending the Application of Biocatalysis to meet the Challenges of Drug Development. Nat. Rev. Chem. 2018, 2, 409–421. 10.1038/s41570-018-0055-1. [DOI] [Google Scholar]
- Truppo M. D. Biocatalysis in the Pharmaceutical Industry: The Need for Speed. ACS Med. Chem. Lett. 2017, 8, 476–480. 10.1021/acsmedchemlett.7b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M. P.; Peñafiel I.; Cosgrove S. C.; Turner N. J. Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals. Org. Process Res. Dev. 2019, 23, 9–18. 10.1021/acs.oprd.8b00305. [DOI] [Google Scholar]
- Patel R. N. Biocatalysis for Synthesis of Pharmaceuticals. Bioorg. Med. Chem. 2018, 26, 1252–1274. 10.1016/j.bmc.2017.05.023. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A. Biocatalysis and Biomass Conversion in Alternative Reaction Media. Chemistry 2016, 22, 12984–12999. 10.1002/chem.201601940. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Kosjek B. Recent Preparative Applications of Redox Enzymes. Curr. Opin. Chem. Biol. 2019, 49, 105–112. 10.1016/j.cbpa.2018.11.011. [DOI] [PubMed] [Google Scholar]
- Li G.; Wang J. B.; Reetz M. T. Biocatalysts for the Pharmaceutical Industry created by Structure-guided Directed Evolution of Stereoselective Enzymes. Bioorg. Med. Chem. 2018, 26, 1241–1251. 10.1016/j.bmc.2017.05.021. [DOI] [PubMed] [Google Scholar]
- Bezborodov A. M.; Zagustina N. A. Enzymatic Biocatalysis in Chemical Synthesis of Pharmaceuticals. Appl. Biochem. Microbiol. 2016, 52, 237–249. 10.1134/S0003683816030030. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Wu S.; Li Z. Cascade Biocatalysis for Sustainable Asymmetric Synthesis: From Biobased L-Phenylalanine to High-Value Chiral Chemicals. Angew. Chem., Int. Ed. 2016, 55, 11647–11650. 10.1002/anie.201606235. [DOI] [PubMed] [Google Scholar]
- Rodriguez A.; Martinez J. A.; Flores N.; Escalante A.; Gosset G.; Bolivar F. Engineering Escherichia coli to overproduce Aromatic Amino Acids and Derived Compounds. Microb. Cell Fact. 2014, 13, 126. 10.1186/PREACCEPT-2032244031131285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchtenberger W.; Huthmacher K.; Drauz K. Biotechnological Production of Amino Acids and Derivatives: Current Status and Prospects. Appl. Microbiol. Biotechnol. 2005, 69, 1–8. 10.1007/s00253-005-0155-y. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Xu Y.; Ding D.; Wen J.; Zhu B.; Zhang D. Genetic Engineering of Escherichia coli to improve l-Phenylalanine Production. BMC Biotechnol. 2018, 18, 5. 10.1186/s12896-018-0418-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-P.; Xiao M.-R.; Zhang L.; Xu J.; Ding Z.-Y.; Gu Z.-H.; Shi G.-Y. Production of L-Phenylalanine from Glucose by Metabolic Engineering of Wild Type Escherichia coli W3110. Process Biochem. 2013, 48, 413–419. 10.1016/j.procbio.2013.02.016. [DOI] [Google Scholar]
- Sekar B. S.; Lukito B. R.; Li Z. Production of Natural 2-Phenylethanol from Glucose or Glycerol with Coupled Escherichia coli Strains Expressing l-Phenylalanine Biosynthesis Pathway and Artificial Biocascades. ACS Sustain. Chem. Eng. 2019, 7, 12231–12239. 10.1021/acssuschemeng.9b01569. [DOI] [Google Scholar]
- Weiner M.; Albermann C.; Gottlieb K.; Sprenger G. A.; Weuster-Botz D. Fed-batch Production of L-Phenylalanine from Glycerol and Ammonia with Recombinant Escherichia coli. Biochem. Eng. J. 2014, 83, 62–69. 10.1016/j.bej.2013.12.001. [DOI] [Google Scholar]
- Khamduang M.; Packdibamrung K.; Chutmanop J.; Chisti Y.; Srinophakun P. Production of L-phenylalanine from Glycerol by a Recombinant Escherichia coli. J. Ind. Microbiol. Biotechnol. 2009, 36, 1267–1274. 10.1007/s10295-009-0606-z. [DOI] [PubMed] [Google Scholar]
- Metro T. X.; Appenzeller J.; Pardo D. G.; Cossy J. Highly Enantioselective Synthesis of β-Amino Alcohols. Org. Lett. 2006, 8, 3509–3512. 10.1021/ol061133d. [DOI] [PubMed] [Google Scholar]
- Cho I.; Prier C. K.; Jia Z. J.; Zhang R. K.; Gorbe T.; Arnold F. H. Enantioselective Aminohydroxylation of Styrenyl Olefins Catalyzed by an Engineered Hemoprotein. Angew. Chem., Int. Ed. 2019, 58, 3138–3142. 10.1002/anie.201812968. [DOI] [PubMed] [Google Scholar]
- Schrittwieser J. H.; Coccia F.; Kara S.; Grischek B.; Kroutil W.; d’Alessandro N.; Hollmann F. One-pot Combination of Enzyme and Pd Nanoparticle Catalysis for the Synthesis of Enantiomerically Pure 1,2-Amino Alcohols. Green Chem. 2013, 15, 3318–3331. 10.1039/c3gc41666f. [DOI] [Google Scholar]
- Ager D. J.; Prakash I.; Schaad D. R. 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96, 835–876. 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]
- Vicario J.; Badia D.; Carrillo L.; Reyes E.; Etxebarria J. Amino Acids, Amino Alcohols and Related Compounds as Chiral Auxiliaries, Ligands and Catalysts in the Asymmetric Aldol Reaction. Curr. Org. Chem. 2005, 9, 219–235. 10.2174/1385272053369105. [DOI] [Google Scholar]
- Everaere K.; Mortreux A.; Carpentier J.-F. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Carbonyl Compounds with 2-Propanol and Ephedrine-Type Ligands. Adv. Synth. Catal. 2003, 345, 67–77. 10.1002/adsc.200390030. [DOI] [Google Scholar]
- Bodkin J. A.; McLeod M. D. The Sharpless Asymmetric Aminohydroxylation. J. Chem. Soc., Perkin Trans. 1 2002, 2733–2746. 10.1039/b111276g. [DOI] [Google Scholar]
- Nilov D.; Reiser O. The Sharpless Asymmetric Aminohydroxylation – Scope and Limitation. Adv. Synth. Catal. 2002, 344, 1169–1173. . [DOI] [Google Scholar]
- Klingler F. D. Asymmetric Hydrogenation of Prochiral Amino Ketones to Amino Alcohols for Pharmaceutical use. Acc. Chem. Res. 2007, 40, 1367–1376. 10.1021/ar700100e. [DOI] [PubMed] [Google Scholar]
- Lohse O.; Spöndlin C. Efficient Preparation of (R)- and (S)-2-Amino-1-Phenylethanol. Org. Process Res. Dev. 1997, 1, 247–249. 10.1021/op9600264. [DOI] [Google Scholar]
- Azizi N.; Saidi M. R. Highly Chemoselective Addition of Amines to Epoxides in Water. Org. Lett. 2005, 7, 3649–3651. 10.1021/ol051220q. [DOI] [PubMed] [Google Scholar]
- Pastó M.; Rodríguez B.; Riera A.; Pericàs M. A. Synthesis of Enantiopure Amino Alcohols by Ring-opening of Epoxyalcohols and Epoxyethers with Ammonia. Tetrahedron Lett. 2003, 44, 8369–8372. 10.1016/j.tetlet.2003.09.124. [DOI] [Google Scholar]
- Murugan A.; Kadambar V. K.; Bachu S.; Rajashekher Reddy M.; Torlikonda V.; Manjunatha S. G.; Ramasubramanian S.; Nambiar S.; Howell G. P.; Withnall J. Regio-selective Synthesis of 1,2-Aminoalcohols from Epoxides and Chlorohydrins. Tetrahedron Lett. 2012, 53, 5739–5741. 10.1016/j.tetlet.2012.08.013. [DOI] [Google Scholar]
- Chang H.-T.; Sharpless K. B. A Practical Route to Enantiopure 1,2-aminoalcohols. Tetrahedron Lett. 1996, 37, 3219–3222. 10.1016/0040-4039(96)00534-5. [DOI] [Google Scholar]
- Buesking A. W.; Ellman J. A. Convergent, Asymmetric Synthesis of Vicinal Amino Alcohols via Rh-Catalyzed Addition of α-amido trifluoroborates to Carbonyls. Chem. Sci. 2014, 5, 1983–1987. 10.1039/C4SC00084F. [DOI] [Google Scholar]
- Reiser O. The Sharpless Asymmetric Aminohydroxylation of Olefins. Angew. Chem., Int. Ed. 1996, 35, 1308–1309. 10.1002/anie.199613081. [DOI] [Google Scholar]
- Lihammar R.; Millet R.; Bäckvall J.-E. An Efficient Dynamic Kinetic Resolution of N-Heterocyclic 1,2-Amino Alcohols. Adv. Synth. Catal. 2011, 353, 2321–2327. 10.1002/adsc.201100466. [DOI] [Google Scholar]
- Gupta P.; Mahajan N. Biocatalytic Approaches Towards the Stereoselective Synthesis of Vicinal Amino Alcohols. New J. Chem. 2018, 42, 12296–12327. 10.1039/C8NJ00485D. [DOI] [Google Scholar]
- Rouf A.; Gupta P.; Aga M. A.; Kumar B.; Parshad R.; Taneja S. C. Cyclic trans-β-Amino Alcohols: Preparation and Enzymatic Kinetic Resolution. Tetrahedron: Asymmetry 2011, 22, 2134–2143. 10.1016/j.tetasy.2011.11.019. [DOI] [Google Scholar]
- Chaubey A.; Parshad R.; Gupta P.; Taneja S. C.; Qazi G. N.; Rajan C. R.; Ponrathnam S. Arthrobacter sp. Lipase Immobilization for Preparation of Enantiopure Masked Beta-Amino Alcohols. Bioorg. Med. Chem. 2009, 17, 29–34. 10.1016/j.bmc.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Rehdorf J.; Mihovilovic M. D.; Bornscheuer U. T. Exploiting the Regioselectivity of Baeyer-Villiger Monooxygenases for the Formation of Beta-amino acids and Beta-amino alcohols. Angew. Chem., Int. Ed. 2010, 49, 4506–4508. 10.1002/anie.201000511. [DOI] [PubMed] [Google Scholar]
- Rehdorf J.; Mihovilovic M. D.; Fraaije M. W.; Bornscheuer U. T. Enzymatic Synthesis of Enantiomerically Pure Beta-Amino Ketones, Beta-Amino Esters, and Beta-Amino Alcohols with Baeyer-Villiger Monooxygenases. Chemistry 2010, 16, 9525–9535. 10.1002/chem.201001480. [DOI] [PubMed] [Google Scholar]
- Baer K.; Dückers N.; Hummel W.; Gröger H. Expanding the Application Range of Aldolases: Novel Asymmetric Syntheses of α-Methylated β-Hydroxy α-Amino Acids and β-Amino Alcohols. ChemCatChem. 2010, 2, 939–942. 10.1002/cctc.201000007. [DOI] [Google Scholar]
- Sehl T.; Hailes H. C.; Ward J. M.; Wardenga R.; von Lieres E.; Offermann H.; Westphal R.; Pohl M.; Rother D. Two Steps in One Pot: Enzyme Cascade For The Synthesis of Nor(pseudo)ephedrine from Inexpensive Starting Materials. Angew. Chem., Int. Ed. 2013, 52, 6772–6775. 10.1002/anie.201300718. [DOI] [PubMed] [Google Scholar]
- Matosevic S.; Lye G. J.; Baganz F. Immobilised Enzyme Microreactor for Screening of Multi-step Bioconversions: Characterisation of a De novo Transketolase-Omega-Transaminase Pathway to synthesise Chiral Amino Alcohols. J. Biotechnol. 2011, 155, 320–329. 10.1016/j.jbiotec.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Ingram C. U.; Bommer M.; Smith M. E.; Dalby P. A.; Ward J. M.; Hailes H. C.; Lye G. J. One-pot Synthesis of Amino-alcohols using a De-novo Transketolase and Beta-Alanine: Pyruvate Transaminase Pathway in Escherichia coli. Biotechnol. Bioeng. 2007, 96, 559–569. 10.1002/bit.21125. [DOI] [PubMed] [Google Scholar]
- Steinreiber J.; Schürmann M.; van Assema F.; Wolberg M.; Fesko K.; Reisinger C.; Mink D.; Griengl H. Synthesis of Aromatic 1,2-Amino Alcohols Utilizing a Bienzymatic Dynamic Kinetic Asymmetric Transformation. Adv. Synth. Catal. 2007, 349, 1379–1386. 10.1002/adsc.200700051. [DOI] [Google Scholar]
- Steinreiber J.; Schurmann M.; Wolberg M.; van Assema F.; Reisinger C.; Fesko K.; Mink D.; Griengl H. Overcoming Thermodynamic and Kinetic Limitations of Aldolase-catalyzed Reactions by Applying Multienzymatic Dynamic Kinetic Asymmetric Transformations. Angew. Chem., Int. Ed. 2007, 46, 1624–1626. 10.1002/anie.200604142. [DOI] [PubMed] [Google Scholar]
- Sello G.; Orsini F.; Bernasconi S.; Gennaro P. D. Synthesis of Enantiopure 2-Amino-1-phenyl and 2-Amino-2-phenyl Ethanols using Enantioselective Enzymatic Epoxidation and Regio- and Diastereoselective Chemical Aminolysis. Tetrahedron: Asymmetry 2006, 17, 372–376. 10.1016/j.tetasy.2006.01.009. [DOI] [Google Scholar]
- Schrittwieser J. H.; Velikogne S.; Hall M.; Kroutil W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. 10.1021/acs.chemrev.7b00033. [DOI] [PubMed] [Google Scholar]
- France S. P.; Hepworth L. J.; Turner N. J.; Flitsch S. L. Constructing Biocatalytic Cascades: In Vitro and in Vivo Approaches to de Novo Multi-Enzyme Pathways. ACS Catal. 2017, 7, 710–724. 10.1021/acscatal.6b02979. [DOI] [Google Scholar]
- Sun Z. B.; Zhang Z. J.; Li F. L.; Nie Y.; Yu H. L.; Xu J. H. One Pot Asymmetric Synthesis of (R)-Phenylglycinol from Racemic Styrene Oxide via Cascade Biocatalysis. ChemCatChem. 2019, 11, 3802–3807. 10.1002/cctc.201900492. [DOI] [Google Scholar]
- Wu S.; Zhou Y.; Wang T.; Too H. P.; Wang D. I.; Li Z. Highly regio- and Enantioselective Multiple Oxy- and Amino-Functionalizations of Alkenes by Modular Cascade Biocatalysis. Nat. Commun. 2016, 7, 11917. 10.1038/ncomms11917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilbak S.; Farkas O.; Poppe L. Mechanism of the Tyrosine Ammonia Lyase Reaction-tandem Nucleophilic and Electrophilic Enhancement by a Proton Transfer. Chemistry 2012, 18, 7793–7802. 10.1002/chem.201103662. [DOI] [PubMed] [Google Scholar]
- Mertens M. A. S.; Sauer D. F.; Markel U.; Schiffels J.; Okuda J.; Schwaneberg U. Chemoenzymatic Cascade for Stilbene Production from Cinnamic Acid Catalyzed by Ferulic Acid Decarboxylase and an Artificial Metathease. Catal. Sci. Technol. 2019, 9, 5572–5576. 10.1039/C9CY01412H. [DOI] [Google Scholar]
- Corrado M. L.; Knaus T.; Mutti F. G. A Chimeric Styrene Monooxygenase with Increased Efficiency in Asymmetric Biocatalytic Epoxidation. ChemBioChem. 2018, 19, 679–686. 10.1002/cbic.201700653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M. L.; Knaus T.; Mutti F. G. Regio- and Stereoselective Multi-enzymatic Aminohydroxylation of β-Methylstyrene using Dioxygen, Ammonia and Formate. Green Chem. 2019, 21, 6246–6251. 10.1039/C9GC03161H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M. L.; Knaus T.; Mutti F. G. High Regio- and Stereoselective Multi-enzymatic Synthesis of All Phenylpropanolamine Stereoisomers from Beta-Methylstyrene. ChemBioChem. 2021, 22, 2345–2350. 10.1002/cbic.202100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Chen Y.; Xu Y.; Li A.; Xu Q.; Glieder A.; Li Z. Enantioselective trans-Dihydroxylation of Aryl Olefins by Cascade Biocatalysis with Recombinant Escherichia coli Coexpressing Monooxygenase and Epoxide Hydrolase. ACS Catal. 2014, 4, 409–420. 10.1021/cs400992z. [DOI] [Google Scholar]
- Mutti F. G.; Knaus T.; Scrutton N. S.; Breuer M.; Turner N. J. Conversion of Alcohols to Enantiopure Amines through Dual-enzyme Hydrogen-borrowing Cascades. Science 2015, 349, 1525–1529. 10.1126/science.aac9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler J. H.; Fuchs M.; Tauber K.; Mutti F. G.; Faber K.; Pfeffer J.; Haas T.; Kroutil W. Redox Self-Sufficient Biocatalyst Network for the Amination of Primary Alcohols. Angew. Chem., Int. Ed. 2012, 51, 9156–9159. 10.1002/anie.201204683. [DOI] [PubMed] [Google Scholar]
- Ohashima T.; Soda K. Purification and Properties of Alanine Dehydrogenase from Bacillus sphaericus. Eur. J. Biochem. 1979, 100, 29–30. 10.1111/j.1432-1033.1979.tb02030.x. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wu S.; Wu J.; Li Z. Enantioselective Cascade Biocatalysis via Epoxide Hydrolysis and Alcohol Oxidation: One-Pot Synthesis of (R)-α-Hydroxy Ketones from Meso- or Racemic Epoxides. ACS Catal. 2015, 5, 51–58. 10.1021/cs5016113. [DOI] [Google Scholar]
- Zhang J.; Xu T.; Li Z. Enantioselective Biooxidation of Racemic trans-Cyclic Vicinal Diols: One-Pot Synthesis of Both Enantiopure (S,S)-Cyclic Vicinal Diols and (R)-α-Hydroxy Ketones. Adv. Synth. Catal. 2013, 355, 3147–3153. 10.1002/adsc.201300301. [DOI] [Google Scholar]
- Lavandera I.; Holler B.; Kern A.; Ellmer U.; Glieder A.; de Wildeman S.; Kroutil W. Asymmetric Anti-Prelog Reduction of Ketones Catalysed by Paracoccus pantotrophus and Comamonas sp Cells via Hydrogen Transfer. Tetrahedron: Asymmetry 2008, 19, 1954–1958. 10.1016/j.tetasy.2008.08.005. [DOI] [Google Scholar]
- Lavandera I.; Kern A.; Schaffenberger M.; Gross J.; Glieder A.; de Wildeman S.; Kroutil W. An Exceptionally DMSO-tolerant Alcohol Dehydrogenase for the Stereoselective Reduction of Ketones. ChemSusChem 2008, 1, 431–436. 10.1002/cssc.200800032. [DOI] [PubMed] [Google Scholar]
- Lavandera I.; Kern A.; Resch V.; Ferreira-Silva B.; Glieder A.; Fabian W. M.; de Wildeman S.; Kroutil W. One-way Biohydrogen Transfer for Oxidation of sec-Alcohols. Org. Lett. 2008, 10, 2155–2158. 10.1021/ol800549f. [DOI] [PubMed] [Google Scholar]
- Lavandera I.; Kern A.; Ferreira-Silva B.; Glieder A.; de Wildeman S.; Kroutil W. Stereoselective Bioreduction of Bulky-bulky Ketones by a Novel ADH from Ralstonia sp. J. Org. Chem. 2008, 73, 6003–6005. 10.1021/jo800849d. [DOI] [PubMed] [Google Scholar]
- Inoue K.; Makino Y.; Itoh N. Purification and Characterization of a Novel Alcohol Dehydrogenase from Leifsonia sp. strain S749: a Promising Biocatalyst for an Asymmetric Hydrogen Transfer Bioreduction. Appl. Environ. Microbiol. 2005, 71, 3633–3641. 10.1128/AEM.71.7.3633-3641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus T.; Cariati L.; Masman M. F.; Mutti F. G. In vitro Biocatalytic Pathway Design: Orthogonal Network for the Quantitative and Stereospecific Amination of Alcohols. Org. Biomol Chem. 2017, 15, 8313–8325. 10.1039/C7OB01927K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffken H. W.; Duong M.; Friedrich T.; Breuer M.; Hauer B.; Reinhardt R.; Rabus R.; Heider J. Crystal Structure and Enzyme Kinetics of the (S)-Specific 1-Phenylethanol Dehydrogenase of the Denitrifying bacterium strain EbN1. Biochemistry 2006, 45, 82–93. 10.1021/bi051596b. [DOI] [PubMed] [Google Scholar]
- Schlieben N. H.; Niefind K.; Muller J.; Riebel B.; Hummel W.; Schomburg D. Atomic Resolution Structures of R-Specific Alcohol Dehydrogenase from Lactobacillus brevis provide the Structural Bases of its Substrate and Cosubstrate Specificity. J. Mol. Biol. 2005, 349, 801–813. 10.1016/j.jmb.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Niefind K.; Muller J.; Riebel B.; Hummel W.; Schomburg D. The crystal Structure of R-Specific Alcohol Dehydrogenase from Lactobacillus brevis suggests the Structural Basis of its Metal Dependency. J. Mol. Biol. 2003, 327, 317–828. 10.1016/S0022-2836(03)00081-0. [DOI] [PubMed] [Google Scholar]
- Matsumoto J.; Higuchi M.; Shimada M.; Yamamoto Y.; Kamio Y. Molecular Cloning and Sequence Analysis of the Gene Encoding the H2O-forming NADH Oxidase from Streptococcus mutans. Biosci Biotechnol Biochem 1996, 60, 39–43. 10.1271/bbb.60.39. [DOI] [PubMed] [Google Scholar]
- Morokutti A.; Lyskowski A.; Sollner S.; Pointner E.; Fitzpatrick T. B.; Kratky C.; Gruber K.; Macheroux P. Structure and Function of YcnD from Bacillus subtilis, a Flavin-containing Oxidoreductase. Biochemistry 2005, 44, 13724–13733. 10.1021/bi0510835. [DOI] [PubMed] [Google Scholar]
- Hohne M.; Schatzle S.; Jochens H.; Robins K.; Bornscheuer U. T. Rational Assignment of Key Motifs for Function Guides in Silico Enzyme Identification. Nat. Chem. Biol. 2010, 6, 807–813. 10.1038/nchembio.447. [DOI] [PubMed] [Google Scholar]
- Mutti F. G.; Fuchs C. S.; Pressnitz D.; Sattler J. H.; Kroutil W. Stereoselectivity of Four (R)-Selective Transaminases for the Asymmetric Amination of Ketones. Adv. Synth. Catal. 2011, 353, 3227–3233. 10.1002/adsc.201100558. [DOI] [Google Scholar]
- Schätzle S.; Steffen-Munsberg F.; Thontowi A.; Höhne M.; Robins K.; Bornscheuer U. T. Enzymatic Asymmetric Synthesis of Enantiomerically Pure Aliphatic, Aromatic and Arylaliphatic Amines with (R)-Selective Amine Transaminases. Adv. Synth. Catal. 2011, 353, 2439–2445. 10.1002/adsc.201100435. [DOI] [Google Scholar]
- Kaulmann U.; Smithies K.; Smith M. E. B.; Hailes H. C.; Ward J. M. Substrate Spectrum of ω-Transaminase from Chromobacterium violaceum DSM30191 and its Potential for Biocatalysis. Enzyme Microb. Technol. 2007, 41, 628–637. 10.1016/j.enzmictec.2007.05.011. [DOI] [Google Scholar]
- Hanson R. L.; Davis B. L.; Chen Y.; Goldberg S. L.; Parker W. L.; Tully T. P.; Montana M. A.; Patel R. N. Preparation of (R)-Amines from Racemic Amines with an (S)-Amine Transaminase from Bacillus megaterium. Adv. Synth. Catal. 2008, 350, 1367–1375. 10.1002/adsc.200800084. [DOI] [Google Scholar]
- van Oosterwijk N.; Willies S.; Hekelaar J.; Terwisscha van Scheltinga A. C.; Turner N. J.; Dijkstra B. W. Structural Basis of the Substrate Range and Enantioselectivity of Two (S)-Selective omega-Transaminases. Biochemistry 2016, 55, 4422–4431. 10.1021/acs.biochem.6b00370. [DOI] [PubMed] [Google Scholar]
- Pressnitz D.; Fuchs C. S.; Sattler J. H.; Knaus T.; Macheroux P.; Mutti F. G.; Kroutil W. Asymmetric Amination of Tetralone and Chromanone Derivatives Employing ω-Transaminases. ACS Catal. 2013, 3, 555–559. 10.1021/cs400002d. [DOI] [Google Scholar]
- Shin J. S.; Yun H.; Jang J. W.; Park I.; Kim B. G. Purification, Characterization, and Molecular Cloning of a Novel Amine:Pyruvate Transaminase from Vibrio fluvialis JS17. Appl. Microbiol. Biotechnol. 2003, 61, 463–471. 10.1007/s00253-003-1250-6. [DOI] [PubMed] [Google Scholar]
- Mutti F. G.; Fuchs C. S.; Pressnitz D.; Turrini N. G.; Sattler J. H.; Lerchner A.; Skerra A.; Kroutil W. Amination of Ketones by Employing Two New (S)-Selective ω-Transaminases and the His-Tagged ω-TA from Vibrio fluvialis. Eur. J. Org. Chem. 2012, 2012, 1003–1007. 10.1002/ejoc.201101476. [DOI] [Google Scholar]
- Cannio R.; Rossi M.; Bartolucci S. A Few Amino Acid Substitutions are Responsible for the Higher Thermostability of a Novel NAD(+)-dependent Bacillar Alcohol Dehydrogenase. Eur. J. Biochem. 1994, 222, 345–352. 10.1111/j.1432-1033.1994.tb18873.x. [DOI] [PubMed] [Google Scholar]
- Quaglia D.; Pori M.; Galletti P.; Emer E.; Paradisi F.; Giacomini D. His-tagged Horse Liver Alcohol Dehydrogenase: Immobilization and Application in the Bio-based Enantioselective Synthesis of (S)-Arylpropanols. Process Biochem. 2013, 48, 810–818. 10.1016/j.procbio.2013.03.016. [DOI] [Google Scholar]
- Davies J.; Jones J. B. Enzymes in organic synthesis. 16. Heterocyclic Ketones as Substrates of Horse Liver Alcohol Dehydrogenase. Stereospecific Reductions of 2-Substituted Tetrahydrothiopyran-4-ones. J. Am. Chem. Soc. 1979, 101, 5405–5410. 10.1021/ja00512a048. [DOI] [Google Scholar]
- van Osselaer T. A.; Lemière G. L.; Merckx E. M.; Lepoivre J. A.; Alderweireldt F. C. Enzymatic “in vitro” Reduction of Ketones. IV. (1) Preparative Scale Reductions of Cyclohexanone Derivatives in an Ethanol-NAD+-HLAD System. Bull. Soc. Chim. Belg. 1978, 87, 799–800. 10.1002/bscb.19780871009. [DOI] [Google Scholar]
- Jones J. B.; Schwartz H. M. Enzymes in Organic Syntheses. 19. Evaluation of the Stereoselectivities of Horse Liver Alcohol Dehydrogenase; Catalyzed Oxidoreductions of Hydroxy- and Ketothiolanes, -thianes, and -thiepanes. Can. J. Chem. 1981, 59, 1574–1579. 10.1139/v81-232. [DOI] [Google Scholar]
- Jones J. B.; Takemura T. Enzymes in organic synthesis. 30. Reaction Conditions – Control of Enantiomeric Purities. Horse Liver Alcohol Dehydrogenase-catalyzed Reductions of 2-Alkylcyclohexanones and their Thiopyran Analogs. Can. J. Chem. 1984, 62, 77–80. 10.1139/v84-015. [DOI] [Google Scholar]
- Lam L. K. P.; Gair I. A.; Jones J. B. Enzymes in Organic Synthesis. 42. Stereoselective Horse Liver Alcohol Dehydrogenase Catalyzed Reductions of Heterocyclic Bicyclic Ketones. J. Org. Chem. 1988, 53, 1611–1615. 10.1021/jo00243a004. [DOI] [Google Scholar]
- Heath R. S.; Birmingham W. R.; Thompson M. P.; Taglieber A.; Daviet L.; Turner N. J. An Engineered Alcohol Oxidase for the Oxidation of Primary Alcohols. ChemBioChem. 2019, 20, 276–281. 10.1002/cbic.201800556. [DOI] [PubMed] [Google Scholar]
- Bommarius B. R.; Schurmann M.; Bommarius A. S. A Novel Chimeric Amine Dehydrogenase shows Altered Substrate Specificity Compared to its Parent Enzymes. Chem. Commun. 2014, 50, 14953–14955. 10.1039/C4CC06527A. [DOI] [PubMed] [Google Scholar]
- Knaus T.; Bohmer W.; Mutti F. G. Amine Dehydrogenases: Efficient Biocatalysts for the Reductive Amination of Carbonyl Compounds. Green Chem. 2017, 19, 453–463. 10.1039/C6GC01987K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerigk M. R.; Maass D.; Kreutzer A.; Sprenger G.; Bongaerts J.; Wubbolts M.; Takors R. Enhanced Pilot-scale Fed-batch l-Phenylalanine Production with Recombinant Escherichia coli by Fully Integrated Reactive Extraction. Bioprocess Biosyst. Eng. 2002, 25, 43–52. 10.1007/s00449-002-0280-2. [DOI] [PubMed] [Google Scholar]
- Sekar B. S.; Li X.; Li Z. Bioproduction of Natural Phenethyl Acetate, Phenylacetic Acid, Ethyl Phenylacetate, and Phenethyl Phenylacetate from Renewable Feedstock. ChemSusChem 2022, e202102645. 10.1002/cssc.202102645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.