

Abstract
Recombinant DNA is a fundamental tool in biotechnology and medicine. These DNA sequences are often built, replicated, and delivered in the form of plasmids. Validation of these plasmid sequences is a critical and time-consuming step, which has been dominated for the last 35 years by Sanger sequencing. As plasmid sequences grow more complex with new DNA synthesis and cloning techniques, we need new approaches that address the corresponding validation challenges at scale. Here we prototype a high-throughput plasmid sequencing approach using DNA transposition and Oxford Nanopore sequencing. Our method, Circuit-seq, creates robust, full-length, and accurate plasmid assemblies without prior knowledge of the underlying sequence. We demonstrate the power of Circuit-seq across a wide range of plasmid sizes and complexities, generating full-length, contiguous plasmid maps. We then leverage our long-read data to characterize epigenetic marks and estimate plasmid contamination levels. Circuit-seq scales to large numbers of samples at a lower per-sample cost than commercial Sanger sequencing, accelerating a key step in synthetic biology, while low equipment costs make it practical for individual laboratories.
Keywords: plasmid, sequencing, long-reads, assembly
Introduction
Plasmids are the core building block of recombinant DNA. Researchers can combine and synthesize novel DNA sequences to manipulate cellular biology, from emerging therapeutics like CAR-T cells to synthetic biology circuits that perturb cellular functions.1,2 These constructs are often sensitive to imperfections and require careful sequence validation. Plasmid sequences are typically verified using chain-termination sequencing (also known as Sanger sequencing), which was first described in 1977.3 Sanger sequencing requires a complementary oligonucleotide to bind upstream of the sequence of interest, from which random-terminated fragments are used to create a consensus sequence.4 However, plasmid sequences now routinely contain more than 10 kilobases (kb), requiring a large number of custom primers, and repetitive regions or imbalanced nucleotide content can be especially challenging for Sanger sequencing.5 To circumvent these limitations, the field has developed a number of secondary techniques to validate the entirety of the sequence, including restriction enzyme mapping and successive primer walking. The collective reagents and time required to validate individual sequences can halt progress or limit the scope of scientific questions. As we incorporate advances in DNA construction and decreased DNA synthesis costs, we expect these challenges of scale and accuracy only to grow more acute.6,7
Our lab’s need to validate complex plasmid assemblies at scale led us to assess the available methods for high-throughput sequencing and assembly. Recent work has demonstrated the utility of high-throughput plasmid verification using Illumina sequencing technologies.8,9 The results have been impressive: Gallegos et al. were able to fully assemble a 96-well plate of 2.5 to 3.3 kb plasmids. Unfortunately, these short-read approaches often struggle to achieve high contiguity for more complex plasmids: the same pipeline in Gallegos et al. was able to assemble only 25% of a more complex plasmid pool containing longer repeat elements.9 Sequencing cost is also an issue, as large numbers of plasmids must be pooled to overcome reagent costs, and upfront machine costs are often prohibitive for laboratories that do not have direct access to an Illumina machine.9
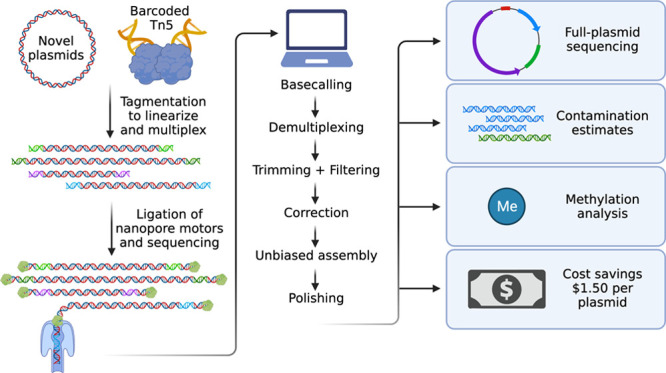
Inspired by a recent publication of a PCR-based plasmid sequencing method10 and the low-cost Nanopore Flongle flow cell, we turned our attention to the Oxford Nanopore long-read sequencing platform. We reasoned that long-read sequences could overcome challenging repeat regions, especially if we were able to preserve the contiguity of the underlying sequence through library generation. This key advantage, coupled with recent advances in sequencing output and computational improvements, could allow us to accurately assemble even the most complex plasmids. Here we detail our approach, circular reconstruction of cut in vitro transposed plasmids (Circuit-seq), which generates complete maps of synthetic DNA constructs within 24 h at a cost that is competitive with single chain-terminating sequencing reactions. These assemblies can then be annotated with epigenetic base modifications, and our long-read data can be used to estimate input contamination levels, providing a comprehensive sequence characterization for a wide variety of downstream applications. We have packed the full computational process into a publicly available Nextflow pipeline, which is available at https://github.com/mckennalab/Circuitseq.11
Results
Tagmentation Leads to Robust Multiplexing of a Diverse Set of 96 Plasmids
Multiplexing approaches for sequencing have increased throughput and decreased per-sample costs.23 Thus, we set out to develop an approach that multiplexed the capture and assembly of full-length plasmid sequences on a long-read sequencing platform. We initially considered two library generation approaches: (1) restriction digestion of plasmids followed by barcode ligation (Figure S1) and (2) barcode tagmentation with the Tn5 transposase (Figure 1A).24 While both approaches proved to be successful, we decided to employ the sequence-agnostic Tn5 transposition technique.
Figure 1.
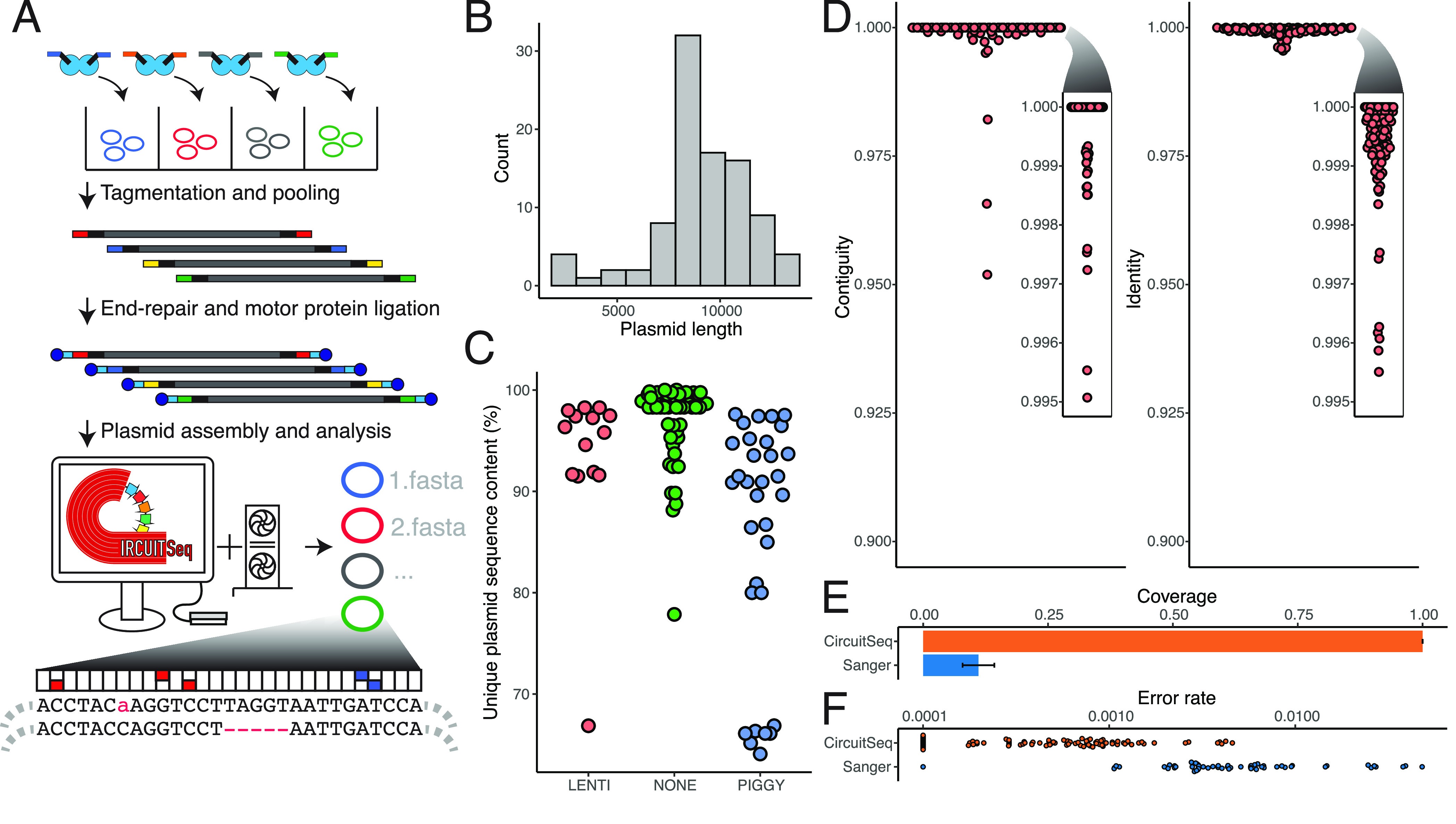
(A) Schematic representation of Circuit-seq. Plasmids are arranged in a 96-well plate and are tagmented with well-specific barcodes. Samples are then pooled, end-repaired, adapter-ligated, and processed on an Oxford Nanopore Flongle flow cell. The generated data are then run through a custom NextFlow pipeline, producing the final assembled sequences. (B) distribution of plasmid sizes used in our experiment. (C) Unique (nonrepetitive) DNA fraction for our input plasmid pool. Lentiviral (LENTI) and PiggyBac (PIGGY) payload plasmids have more repetitive sequence in their backbones, a key criterion to challenge our assembly pipeline. (D) Contiguity and identity scores for the polished assemblies were calculated by comparison to the known reference. (E) Proportions of the full plasmid covered by Circuit-seq assemblies and Sanger sequencing. (F) Error rates for Circuit-seq assemblies and Sanger sequencing calculated by comparison with the known reference. Sanger sequences were prefiltered to remove sequences with >10% error to exclude technical errors from the analysis.
The Tn5 enzyme is commonly used for transposase-mediated adapter insertion and subsequent sequencing.25 Tn5 is loaded in vitro with an oligonucleotide (oligo) containing the Tn5 mosaic sequence.24−26 To uniquely tag individual plasmids, we extended these 19 bp mosaic sequences with a set of 96 error-resistant 17 bp barcodes from Hawkins et al. (Figure 1A and Table S1).12 Our goal was a limited tagmentation of each plasmid to preserve contiguity and to lower the per-reaction cost. We used limiting quantities of oligonucleotides in an excess of Tn5 to restrict tagmentation, which we tested over a range of concentrations near the lowest levels in Picelli et al.(27) Tn5 tagmented libraries were then pooled and prepared using the Oxford Nanopore ligation sequencing kit and loaded onto an individual Flongle flow cell. The results of our optimization (Figure S2A) show that our approach is robust over the tested conditions, generating a sizable proportion of full-length reads. Increasing the amount of Tn5 or the duration of tagmentation increased the number of tagmentations per plasmid, though we recovered sufficient fragments from all conditions to generate correct assemblies (Figure S2B).
We next tagmented a diverse set of 96 plasmids using the optimized conditions above. Each input plasmid had a known sequence map, ranging in size from 2433 to 13 286 bp (Figure 1B). The input plasmids had a wide range of repetitive sequence content (ranging from 0 to 36% of the known map) incorporating both lentiviral and PiggyBac integration vectors (Figure 1C). We also used an improved set of Tn5 sample barcodes (v2) with increased hamming distance to improve barcode recovery (Table S2). We then demultiplexed and processed individual plasmids with a custom NextFlow pipeline.11 Reads assigned to plasmid barcode IDs accounted for 81% of reads, resulting in 150–4619× (median 1118) coverage of our plasmid set with little correlation to input plasmid length (r = −0.16; Figure S3A). As expected, we observed peaks in fragment sizes at known plasmid lengths, with increasing proportions of smaller fragments that are presumably from DNA fragmentation in library preparation and sequencer length bias (Figure S3B).28 Consistent with this hypothesis, we observed more full-length fragments in smaller plasmids (Figure S3C). We then filtered and corrected each sample’s reads to generate error-corrected consensus sequences where the distribution more closely matches single and double tagmentation events (Figure S3D).18
De Novo Plasmid Assembly and Validation
We then assembled plasmids de novo from the corrected reads, avoiding any biases from reference-based assembly approaches.29 We used a two-pronged assembly approach, leveraging both Flye and Miniasm long-read assemblers to generate competing initial consensuses from which the longest single-contig assembly was taken forward19,20 (https://github.com/lh3/miniasm). This was followed by subsequent rounds of refinement with Medaka and custom postprocessing steps to eliminate whole-genome duplications and other common assembly artifacts (https://github.com/nanoporetech/medaka). Our pipeline was able to sequence and assemble 95 of 96 plasmids to a 100.00% median contiguity and 99.95% identity (Figure 1D), an improvement over existing Illumina sequencing and assembly approaches.9 No reads for the missing assembly were recovered from the raw data, suggesting that it was due to technical error during sample preparation.
We next wanted to determine the accuracy of our approach compared with conventional validation techniques. We collected 64 Sanger sequencing reactions from 36 different plasmids within our Nanopore assembly data set. We converted the resulting trace files and filtered them by quality score using Mott’s algorithm to clip low-quality regions.30 We further removed seven Sanger sequencing results where the error rate was above 10% to avoid biasing our results from potential user error (Figure S3E,F). The 57 remaining Sanger sequencing results covered a median of 10.3% of their respective plasmids with an average aligned length of 1023 bases (Figure 1E). The Sanger median per-base error rate was 0.4%, approximately a 10-fold higher rate than our Nanopore assembly approach (Figure 1F). In aggregate, our pipeline successfully assembled maps with high contiguity and identity even when faced with highly repetitive sequences.
Plasmid Epigenetics
Given the success of our assembly pipeline, we wondered whether we could use our rich sequencing data to provide validation that is inaccessible from chain-terminating sequencing results. Bacterial epigenetic patterns are carried over to plasmid sequences; both dcm and dam methylation can be transferred to matching recognition sites in plasmids propagated by Escherichia coli.31 This methylation has a number of implications for synthetic biology, including methylation-sensitive restriction enzymes, the stability of repeat sequences, and downstream sensitivity issues in the target organism.32,33 Given Oxford Nanopore’s ability to directly capture DNA modifications, we were interested to see whether we could profile the epigenetic status of our assemblies. We then extended our pipeline to call methylation modifications present in each plasmid assembly. As an initial validation of this approach, we created consensus DNA sequence logos from the 6-methyladenine (6mA) and 5-methylcytosine (5mC) patterns detected in our 96-plasmid run, recovering known sequence motifs of dam (GATC) and dcm (CCYGG) methylation (Figure 2A).
Figure 2.
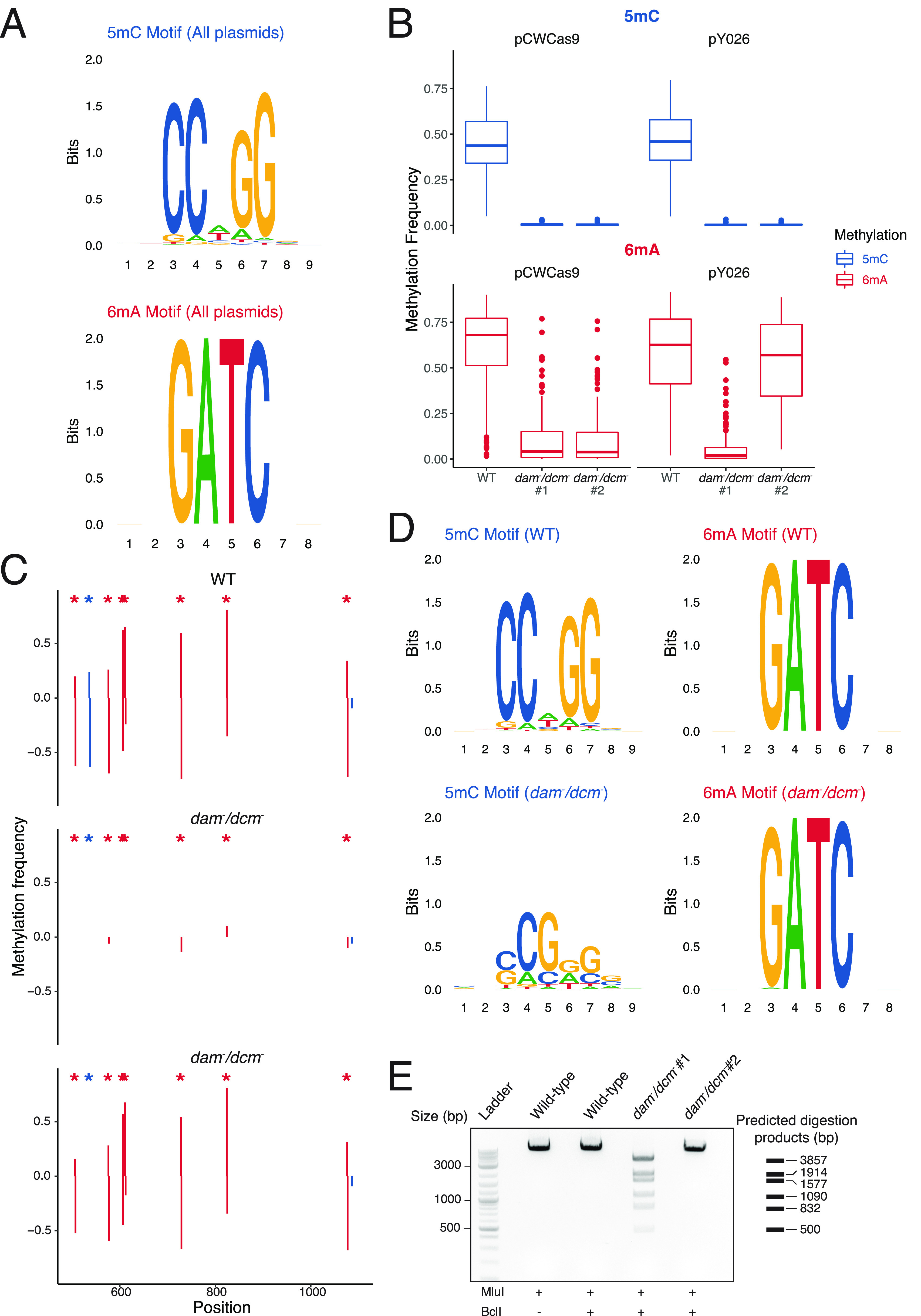
(A) DNA sequence logos generated from methylated 5-methylcytosine (5mC) and 6-methyladenine (6mA) plasmid sequences, capturing known dcm and dam consensus sequences, respectively. (B) Global 5mC and 6mA rates from plasmids grown in conventional dcm+/dam+E. coli (WT) and dcm–/dam–E. coli. (C) Visualization of methylation frequencies across a region of pY026 plasmid shows signal localization to consensus motifs (asterisks) in pairs; positive and negative frequencies correlate to the top and bottom strands, respectively. The first dcm–/dam– samples exhibits complete loss of 5mC and 6mA methylation, while the second dcm–/dam– sample shows complete loss of 5mC but partial recovery of 6mA due to conversion to dam+. (D) DNA sequence logo plots from methylation sites of plasmids grown in WT and dcm–/dam–E. coli showing the loss of 5mC but recovery of 6mA. (E) Validation of computational methylation calls using 6mA-sensitive BclI digestion of MluI linearized pY026 plasmid. BclI digestion failed to cleave the methylated plasmid as well as the dam+ recovered plasmid but achived nearly complete digestion of the dam– plasmid. Predicted digestion products are annotated at the right.
To further validate the accuracy of our approach, we then transformed plasmids into both NEB Stable Competent cells (WT) and dam–/dcm– competent cells that lack both methyltransferases. One WT plasmid and two separate colonies of dam–/dcm– plasmids were processed according to our normal Circuit-seq protocol. In the dam–/dcm– plasmids, methylation calls at motif sequences showed a drastic drop of 6mA and nearly complete loss of 5mC (Figure 2B). The low, but not abolished, levels of 6mA can be explained by spontaneous reactivation of dam through loss of the knockout transposon sequence.34 This is evident in the second replicate of pY026 dam–/dcm– which has nearly complete reactivation of dam but complete absence of dcm activity. Highlighting a stretch of this pY026 assembly, we see a mirroring of methylation patterns on each of the positive and negative strands, confirming that the regained dam activity is consistent with true methylation patterns (Figure 2C). As can also be seen in the DNA sequence motifs from the various conditions, there was no clear sequence motif enrichment in the 5mC motif in the dam–/dcm– samples, while 6mA successfully recreated the dam motif (Figure 2D). Finally, to experimentally validate the methylation levels, we digested plasmids with a dam-sensitive restriction enzyme, BclI. The resulting gel showed nearly complete digestion of the first dam–/dcm– plasmid but no visible fragments in the second colony, validating the computational characterization (Figure 2E).
Contamination Detection
Plasmid preparations can be contaminated by bacterial DNA or other passenger plasmids that confound downstream experiments. Given the number of sequencing reads produced for each plasmid, we wondered whether we could leverage this read depth to estimate potential contamination levels. Given the shared components of many plasmids, i.e., the origin of replication or lentivirus components, we reasoned that previously developed SNV-based contamination estimation methods would be ill-suited for the task.22,35 Instead, we used a simple Bayesian model to leverage the proportion of unaligned bases (“clipped” bases) as well as unmapped reads during sequence alignment, taking into account the shared nature of plasmid components (Figure 3A).
Figure 3.

(A) Contamination rates calculated from the proportion of unaligned bases and the global shared sequence proportions between plasmids, estimated over a range of contamination hypotheses. (B) In silico contamination estimates of assembled Addgene plasmids using simulated reads over a range of plasmid lengths (upper panel; XS = extra small to XL = extra large maps). Our estimates track the programmed rates well (r = 0.97, RSME = 9.1), though at higher contamination levels our assemblies had issues that resulted in poor contamination estimates, which can be seen in the assembly completeness dropoff for small plasmids at >70% contamination and at >90% for larger plasmids (lower panel). (C) Correlation of an in vitro mixing experiment of two known plasmids prior to sequencing and assembly. Contamination estimates were well-correlated to the mixing proportion (r = 0.997, RSME = 4.66), though we failed to assemble the plasmids at the two highest contamination levels (85% and 95% contaminated). (D) Estimated contamination rates significantly dropped with our improved Tn5 barcoding design (p = 0.00119).
We first validated our approach using computationally generated reads from commonly used plasmids available from the Addgene repository. Five plasmids spanning a range of sizes were chosen and subsequently contaminated by random reads from 91 other constructs. The simulated reads were then used as input to our existing pipeline, and our computational tool was used to predict contamination levels (Figure 3B, top). The contamination levels were relatively well-predicted by our simple method (r = 0.97, RSME = 9.1) but were less accurate at high contamination levels because of underlying assembly failures (Figure 3B, bottom and Figure S4A). When we estimated the contamination using known plasmid maps instead of our assemblies, we saw similar results, with more consistent estimates at higher contamination levels (r = 0.99, RSME = 7.2; Figure S4B).
To further validate our contamination pipeline, we experimentally mixed one plasmid preparation into another over a range of contamination rates. We then sequenced these mixtures in individual wells in an additional Circuit-seq run. Our two most contaminated plasmids, 85% and 95% contaminated, failed to assemble, but computational estimates in the remaining samples tracked the known experimental rates (r = 0.997, RSME = 4.66; Figure 3C). We then profiled overall contamination rates in both our v1 and v2 barcoding runs (Figure 3D). The contamination levels were correlated with plasmid assembly incompleteness (Pearson r = 0.32), suggesting an avenue to further improve the assembly results.
Another potential source of contamination is the bacterial genome. We analyzed our most representative sample, the r9.4.1 Flongle with the v2 barcodes, and found 339 reads out of 350 262 (0.1%) mapping to the E. coli genome. Individual plasmid samples had between 0 and 40 E. coli reads, proportional to the total number of reads per sample. Although it is tempting to use these values to estimate genomic contamination, we think that these values are heavily biased by technical aspects of the sequencing platform: shorter reads are more likely to be sequenced and longer reads are more likely to be lost during cleanups. Consistent with this hypothesis, the average length of these captured E. coli reads was only 2694 bp. One caveat of this analysis is that we preselected high-quality plasmid samples for sequencing. It is possible that a poorly purified sample would have more genomic contamination that could be flagged by downstream analysis.
Discussion
Here we detail our new high-throughput technique, Circuit-seq, an end-to-end plasmid validation pipeline that leads to near-perfect assemblies. This technique, complementing other second- and third-generation sequencing approaches, provides a comprehensive map of both simple and complex plasmids. In contrast to existing Illumina-based techniques, our long-read approach can assemble through large repetitive regions and characterize epigenetic marks, leveraging the unique advantages of the Nanopore platform. We are then able to couple this with computational tools to provide end users a comprehensive view of the resulting plasmid with errors rates that are an order of magnitude lower than those for Sanger sequencing (Figure 1F).
Our assemblies are also cost-effective. The reagents required per 96-well run cost approximately $140 (or less than $1.50 per plasmid), which is significantly cheaper than commercial Sanger sequencing (Table S3). Additionally, the low startup cost of the Oxford Nanopore platform allows researchers with even moderate validation needs to consider this approach. Higher-density barcoding (e.g., 384-well plates in parallel) would further reduce costs. Additionally, Circuit-seq can be run with the larger Minion flow cells that produce data at 5–10 times the speed, reducing the amount of time required to obtain assemblies. The Circuit-seq protocol is relatively straightforward once the reagents are obtained, and new users in our lab have successfully generated data in their initial experiment. To further enable the adoption of this technique, we have made all of the barcode sequences and computational tools publicly available.
In the future, we hope to adopt new computational and experimental improvements on the Oxford Nanopore platform into Circuit-seq, including higher-accuracy Nanopore kits and flow cells. It would be straightforward to include these improvements as they become available for the lower-output Flongle, further decreasing error rates and potentially solving our limited remaining contiguity challenges. We have also had preliminary success with direct sequencing of plasmids from bacterial colonies, replacing traditional colony PCR, although optimization is still needed to increase the consistency and yield.
High-throughput biological studies have resulted in increased quantity and sequence complexity of plasmids, but sequence validation efforts have lagged. Our lab often deals with highly repetitive or complex plasmid sequences, where Sanger sequencing results are hard to interpret or outright fail. We have found Circuit-seq to be of immense practical value. Even in routine experiments, the effort of Circuit-seq is rewarded when we discover a plasmid backbone mutation or mislabeled tube that would have gone undiagnosed until much later in the project. Because of this practical value combined with the competitive cost, we are excited by the prospect of Circuit-seq or other modern techniques supplanting Sanger sequencing, providing a single-pass, comprehensive validation of DNA constructs.
Materials and Methods
Plasmid Purification
The plasmids utilized in this work were purified with either the QIAprep Spin Miniprep Kit or the ZymoPURE II Plasmid Midiprep Kit. In unpublished results generated in the course of our other projects, we have found that any plasmid purification approach yields sufficiently high quality plasmid.
Restriction Digest Approach
We created six test ligation adapters with unique 20 bp barcode identifiers (Table S4), containing XhoI, EcoRI, XbaI, BmtI, BclI, and SacI restriction sites. Restriction sites were chosen by scanning a listing of all Addgene plasmid sequences, removing sequences shorter than 2000 bases, and generating a cover set of enzymes that would maximize the number of sequences with at least one single- or double-cut enzyme location (Figure S1). A common forward adapter and unique reverse adapters were annealed in an equimolar ratio and extended with a single cycle of Kapa HiFi polymerase using the manufacturer-recommended conditions and an extension time of 5 min. Adapters were then cut with the target restriction enzyme and purified with the Zymo Research Clean & Concentrate Kit. Target plasmids were processed similarly in parallel. Adapters were then ligated to their corresponding plasmids at 3-fold molar excess using New England Biolab’s Quick Ligation Kit for 10 min at room temperature, followed by a 0.5× Ampure cleanup to remove excess adapters and standard Oxford Nanopore ONT LSK-109 or ONT LSK-110 ligation protocols, and loaded onto an Oxford Nanopore Flongle flow cell.
Tn5 Barcode Design
Our v1 17 nucleotide (nt) barcodes were obtained from Hawkins et al. using a set with a minimum of two error corrections12 (Table S1). For our version 2 (v2) barcodes, we used 27 nt barcode sequences generated by combining Finkelstein’s 13 nt and 14 nt sequences, each with two error corrections. We sampled 10 000 combinations with the R package DNABarcodes and selected an optimized set with a median hamming distance of 21 and a guaranteed error correction of 4 bp.13 The resulting primers were then screened with an Integrated DNA Technologies (IDT) OligoAnalyzer to select 96 primers with optimal ΔG > −4 kcal/mol (Table S2). Barcodes were annealed with a common phosphorylated oligo (/5Phos/CTGTCTCTTATACACATCT) by heating to 95 °C for 5 min and slow cooling to room temperature.
Tn5 Purification and Storage
Tn5 was purified by the University of California Berkeley Quantitative Bioscience MacroLab Core Facility as per previously described protocols.14 A long-term storage stock solution of Tn5 at 4 mg/mL was kept at −80 °C in storage buffer (50 mM Tris-HCl, pH 7.5, 800 mM NaCl, 0.2 mM EDTA, 2 mM DTT, 10% glycerol). A stock solution of 40 μg/mL Tn5 was kept at −20 °C in the working buffer (50 mM Tris-HCl, pH 7.5, 800 mM NaCl, 0.2 mM EDTA, 2 mM DTT, 50% glycerol).
Tagmentation Reactions
For 96 plasmids on an r9.4 Flongle, 50 ng of plasmid was combined with 2.5 pmol of annealed barcode oligo and 40 ng of Tn5 in reaction buffer (50 mM Tris-acetate, pH 7.5, 150 mM potassium acetate, 10 mM magnesium acetate, 4 mM spermidine, 1 mM DTT) in a 5 μL reaction. For runs with fewer than 48 plasmids, all quantities can be doubled. Using a thermocycler, samples were incubated at 23 °C for 10 min, 37 °C for 10 min, and then 55 °C for 5 min. The reaction was stopped with 1 volume of 0.2% SDS for 5 min at 23 °C. Samples were pooled and cleaned up with 0.5 volume of Ampure beads according to established protocols and eluted in 25 μL of water. For the r10.3 minion run, all of the volumes were doubled, but the rest of the protocol remained the same.
Oxford Nanopore (ONT) Library Preparation
Plasmid sequences are available for our v1 and v2 efforts on our Github site (https://github.com/mckennalab/Circuitseq). Samples were prepared in accordance with the ONT LSK-110 manual for Flongle libraries. In brief, the purified tagmented libraries were repaired using a combined NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End Repair/dA-Tailing Module by incubation at 20 °C for 7.5 min and 65 °C for 7.5 min. Samples were purified with 0.5 volume of Ampure beads and eluted into 30 μL of water. Ligation was performed with NEB Quick Ligase, ONT ligation buffer, and Oxford Nanopore’s adapter mix (AMX) for 10 min at room temperature. Samples were purified with 0.5× Ampure beads and then washed with ONT long-fragment wash buffer instead of 70% ethanol and eluted into 7 μL of ONT elution buffer. A 5 μL sample of the resulting library was loaded into a Flongle as per ONT specifications.
Demethylated Plasmid Preparation
Plasmids were transformed into NEB dam–/dcm– competent E. coli (C2925I) as per the vendor’s protocols and plated onto LB agar with ampicillin. The following day we picked two colonies per plasmid and grew them overnight in LB broth with ampicillin for plasmid extraction with the Qiagen Miniprep Kit.
Data Generation and Assessment Workflow
We established a NextFlow pipeline using a prepackaged Docker container to facilitate adoption and reproducibility.11 This pipeline performs all of the computational steps from basecalling to assembly polishing with the ability to create assembly statistics if reference sequences are provided. The pipeline and related documentation are available on our GitHub site.
Basecalling was performed with ONT guppy software version 5.0.16+b9fcd7b5b using the r941_min_sup_g507 mode. When necessary, the pipeline parameter file can be modified to work with different basecalling models or flow cells. Fastq files are then binned using Oxford Nanopore’s Guppy demultiplexing function with a custom barcode configuration file. Porechop15,16 (https://github.com/rrwick/Porechop) is then used to trim the adapter sequences and discard chimeric reads resulting from aberrant ligation products. Sequences shorter than 500 bp are filtered out with nanofilt17 (https://github.com/wdecoster/nanofilt). Reads that pass filtering are corrected with Canu18 (https://github.com/marbl/canu) and assembled with miniasm (https://github.com/lh3/miniasm) and Flye assemblers.19 The assembly is then polished with one round of Medaka (https://github.com/nanoporetech/medaka) to reduce error, which increases the fidelity of duplication removal using DupScoop (https://github.com/mckennalab/DupScoop), a tool we developed to resolve a common error in Flye assemblies where the assembly is perfectly duplicated. The assemblies are then rotated 50% of their length. We found that the edges of the circular assemblies do not get polished as effectively, and rotating the assemblies allows these sequences that are now in the center of the assembly to be polished efficiently during the following three rounds of Medaka.
If a reference is provided, the assemblies undergo assessment of identity and contiguity,20 generating the values used within the paper. We then define each assembly’s “completeness”, in the range of [0, 1], as the value
This is a somewhat conservative calculation since the contiguity and identity values are not fully independent.
Methylation Calling and Analysis
The raw fast5 reads are demultiplexed using the Guppy demultiplexed fastq files and the fast5_subset from ont_fast5_api (https://github.com/nanoporetech/ont_fast5_api). The fast5 reads are then basecalled using Guppy version 4.4.2+9623c16 with the methylation-trained model dna_r9.4.1_450bps_modbases_dam-dcm-cpg_hac.cfg. The methylated sites are then detected and annotated using modPhred15 (https://github.com/novoalab/modPhred). For the purpose of creating unbiased DNA sequence logos, modPhred was run to detect minimum modification frequencies of 0%, to include all sites that were modified across all replicates. To compare methylation at frequencies between plasmids grown in WT versusdam–/dcm– competent E. coli, we restricted analysis to sites with consensus methylation sequences.
Contamination: Computational Simulation of Known Plasmids
We simulated reads from established plasmid maps downloaded from Addgene. Our goal was to sample from commonly used plasmids, so we first took sequences from the Addgene “top 15” list (https://blog.addgene.org/15-years-of-addgene-the-top-15-plasmids) and then randomly sampled 84 additional plasmids by traversing the Addgene “Blue Flame Award” list alphabetically and selecting a single Blue Flame plasmid from each lab, attempting to avoid any related plasmids. The full list of the simulated plasmids is given in Table S5. The resulting plasmid lengths ranged from 2830 to 16973 bases, with a mean of 8080 bases. Plasmid sequences were downloaded, and reads were simulated using a version of BadRead customized to empirically draw read lengths from our established fragment lengths over a circular reference, using standard BadRead parameters for “bad” reads (junk reads and chimeric reads) totaling 3%21 (https://github.com/aaronmck/Badread). Contamination was simulated for the five Addgene plasmids chosen as our controls (Addgene IDs 31815, 16337, 12251, 49792, and 52961) in five replicates each, representing relatively small to relatively large plasmids. These control plasmids were artificially contaminated with reads randomly drawn from all other plasmids at the following contamination rates: 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 95, 98, and 99%.
Contamination is estimated by first aligning Nanopore reads to a duplicated reference map of the plasmid (to capture reads spanning the circular breakpoint) using Minimap2 (https://github.com/lh3/minimap2). Both aligned and unaligned reads are then assessed using a custom Python script. Our Bayesian formulation assumes a flat prior across equally spaced, discrete contamination level hypotheses, much like our approach in ref (22):
where p(C) is our flat prior and the denominator p(B) is the same over all contamination (C) levels. We then need to evaluate the likelihood function. Assuming reads are independent and that our randomly sampled bases from the read-alignment pair are independent, controlling for read length, we can define the likelihood as a product of probabilities:
where i = 1, ..., N represent the individual sequencing reads. We can define the following piecewise function:
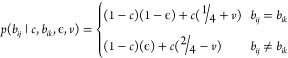 |
where bij and bik represent the corresponding aligned reference base j and sequencing base k when comparing read i, c is the contamination rate, and ϵ is the Phred-scaled error rate. The parameter v represents a constant shared sequence proportion between plasmids, estimated by comparing k-mer proportions between all sampled Addgene plasmid sequences (here set to 0.1984). Code to estimate this parameter is included in the GitHub repository. Contamination likelihoods are then normalized to 1 over the range [0, 1], and the mode of this distribution is found, representing the maximum a posteriori (MAP) score. For simplicity, in the work described in this paper we chose to sample over 100 bins from [0, 1], but increasing this resolution is trivial at the cost of computational time.
Acknowledgments
We thank the members of the McKenna lab for experimental help and advice and for providing the stream of complex plasmids needed to validate this approach. We also thank the members of the Dartmouth community who also contributed plasmids in the early development phase. We especially thank Rachel Saxe and Maryam Fathi for testing both the protocol and computational pipelines. The graphical abstract was made with Biorender.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.2c00126.
Supporting figures showing a schematic of the restriction digest approach, optimization of tagmentation, the read length distribution, and contamination analysis; supporting tables providing a cost analysis, sequences for the restriction digest approach, Tn5 oligo sequences, and the list of addgene plasmid IDs used to select restriction sites (PDF)
Author Contributions
F.E. and A.M. conceived and designed the study, gathered and analyzed the data, and wrote the manuscript. I.H. assisted in data analysis and participated in editing the manuscript. All of the authors had access to the study data and approved the decision to submit the manuscript.
This work was supported by funding from the Neukom Institute at Dartmouth College and The Norris Cotton Cancer Center (NCI 5P30CA023108-37). A.M. was supported by NIH/NHGRI (R00 HG010152-04), a Pew Biomedical Scholars Fellowship, and the V Foundation. F.E.E. was supported by a Tenney Fellowship, and I.H. was supported by the Neukom Institute and a Sophomore Research Scholarship at Dartmouth College.
The authors declare no competing financial interest.
Notes
The sequencing data used to generate the figures are available at ENA (ERP135565), and our analysis pipeline can be downloaded from GitHub (https://github.com/mckennalab/Circuitseq).
Supplementary Material
References
- Antebi Y. E.; Linton J. M.; Klumpe H.; Bintu B.; Gong M.; Su C.; McCardell R.; Elowitz M. B. Combinatorial Signal Perception in the BMP Pathway. Cell 2017, 170, 1184–1196. 10.1016/j.cell.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez R. A.; Yu W.; Cabral K. A.; Creasey O. A.; Lopez Pazmino M. D. P.; Tonai Y.; De Guzman A.; Mäkelä A.; Saksela K.; Gartner Z. J.; et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 2021, 371, 1166–1171. 10.1126/science.abc1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F.; Nicklen S.; Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 5463–5467. 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heather J. M.; Chain B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. 10.1016/j.ygeno.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieleczawa J.; Dunn J. J.; Studier F. W. DNA sequencing by primer walking with strings of contiguous hexamers. Science 1992, 258, 1787–1791. 10.1126/science.1465615. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R.-Y.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Hughes R. A.; Ellington A. D. Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harbor Perspect. Biol. 2017, 9, a023812 10.1101/cshperspect.a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapland E. B.; Holmes V.; Reeves C. D.; Sorokin E.; Durot M.; Platt D.; Allen C.; Dean J.; Serber Z.; Newman J.; et al. Low-Cost, High-Throughput Sequencing of DNA Assemblies Using a Highly Multiplexed Nextera Process. ACS Synth. Biol. 2015, 4, 860–866. 10.1021/sb500362n. [DOI] [PubMed] [Google Scholar]
- Gallegos J. E.; Rogers M. F.; Cialek C. A.; Peccoud J. Rapid, robust plasmid verification by de novo assembly of short sequencing reads. Nucleic Acids Res. 2020, 48, e106 10.1093/nar/gkaa727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin A.; Swainston N.; Dunstan M. S.; Jervis A. J.; Mulherin P.; Robinson C. J.; Taylor S.; Carbonell P.; Hollywood K. A.; Yan C.; et al. Highly multiplexed, fast and accurate nanopore sequencing for verification of synthetic DNA constructs and sequence libraries. Synth. Biol. 2019, 4, ysz025. 10.1093/synbio/ysz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P.; Chatzou M.; Floden E. W.; Barja P. P.; Palumbo E.; Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- Hawkins J. A.; Jones S. K. Jr.; Finkelstein I. J.; Press W. H. Indel-correcting DNA barcodes for high-throughput sequencing. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E6217–E6226. 10.1073/pnas.1802640115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann T.; Bystrykh L. V. Levenshtein error-correcting barcodes for multiplexed DNA sequencing. BMC Bioinf. 2013, 14, 272 10.1186/1471-2105-14-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig B. P.; Velten L.; Racke I.; Tu C. S.; Thoms M.; Rybin V.; Besir H.; Remans K.; Steinmetz L. M. Large-Scale Low-Cost NGS Library Preparation Using a Robust Tn5 Purification and Tagmentation Protocol. G3: Genes, Genomes, Genet. 2018, 8, 79–89. 10.1534/g3.117.300257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz L. P.; Novoa E. M. ModPhred: an integrative toolkit for the analysis and storage of nanopore sequencing DNA and RNA modification data. Bioinformatics 2021, 38, 257–260. 10.1093/bioinformatics/btab539. [DOI] [PubMed] [Google Scholar]
- Wick R. R.; Judd L. M.; Gorrie C. L.; Holt K. E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genomics 2017, 3, e000132 10.1099/mgen.0.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W.; D’Hert S.; Schultz D. T.; Cruts M.; Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S.; Walenz B. P.; Berlin K.; Miller J. R.; Bergman N. H.; Phillippy A. M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M.; Yuan J.; Lin Y.; Pevzner P. A. Assembly of Long Error-Prone Reads Using Repeat Graphs.. Nat. Biotechnol. 2019, 37, 540–546. 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Wick R. R.; Holt K. E. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research 2019, 8, 2138. 10.12688/f1000research.21782.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick R. Badread: simulation of error-prone long reads. J. Open Source Softw. 2019, 4, 1316. 10.21105/joss.01316. [DOI] [Google Scholar]
- Cibulskis K.; McKenna A.; Fennell T.; Banks E.; DePristo M.; Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 2011, 27, 2601–2602. 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J.; Balasubramanian S.; Church G. M.; Gilbert W.; Rogers J.; Schloss J. A.; Waterston R. H. DNA sequencing at 40: past, present and future. Nature 2017, 550, 345–353. 10.1038/nature24286. [DOI] [PubMed] [Google Scholar]
- Reznikoff W. S. Transposon Tn5. Annu. Rev. Genet. 2008, 42, 269–286. 10.1146/annurev.genet.42.110807.091656. [DOI] [PubMed] [Google Scholar]
- Adey A.; Morrison H. G.; Asan; Xun X.; Kitzman J. O.; Turner E. H.; Stackhouse B.; MacKenzie A. P.; Caruccio N. C.; Zhang X.; Shendure J. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010, 11, R119. 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A. C. Tagmentation-based single-cell genomics. Genome Res. 2021, 31, 1693–1705. 10.1101/gr.275223.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S.; Björklund A. K.; Reinius B.; Sagasser S.; Winberg G.; Sandberg R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014, 24, 2033–2040. 10.1101/gr.177881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarasinghe S. L.; Su S.; Dong X.; Zappia L.; Ritchie M. E.; Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G.; Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B.; Hillier L.; Wendl M. C.; Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Marinus M. G.; Løbner-Olesen A. DNA Methylation. EcoSal Plus 2014, 10.1128/ecosalplus.ESP-0003-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolek J.; Sedlar K.; Provaznik I.; Patakova P. Dam and Dcm methylations prevent gene transfer into Clostridium pasteurianum NRRL B-598: development of methods for electrotransformation, conjugation, and sonoporation. Biotechnol. Biofuels 2016, 9, 14 10.1186/s13068-016-0436-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol K.; Pearson C. E. CpG methylation modifies the genetic stability of cloned repeat sequences. Genome Res. 2002, 12, 1246–1256. 10.1101/gr.74502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New England Biolabs. Making Unmethylated (Dam- Dcm-) DNA. https://www.neb.com/tools-and-resources/usage-guidelines/making-unmethylated-dam-dcm-dna (accessed 2022-03-08).
- Jun G.; Flickinger M.; Hetrick K. N.; Romm J. M.; Doheny K. F.; Abecasis G. R.; Boehnke M.; Kang H. M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012, 91, 839–848. 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.