

Abstract
Purpose:
Comprehensive genomic profiling (CGP) is of increasing value for patients with metastatic castration-resistant prostate cancer (mCRPC). mCRPC tends to metastasize to bone, making tissue biopsies challenging to obtain. We hypothesized CGP of cell-free circulating tumor DNA (ctDNA) could offer a minimally invasive alternative to detect targetable genomic alterations that inform clinical care.
Experimental Design:
Using plasma from 3,334 patients with mCRPC (including 1,674 screening samples from TRITON2/3), we evaluated the landscape of genomic alterations detected in ctDNA and assessed concordance with tissue based CGP.
Results:
3,129 patients (94%) had detectable ctDNA with a median ctDNA fraction of 7.5%; BRCA1/2 were mutated in 295 (8.8%). In concordance analysis, 72/837 patients had BRCA1/2 mutations detected in tissue, 67 (93%) of which were also identified using ctDNA, including 100% of predicted germline variants. ctDNA harbored some BRCA1/2 alterations not identified by tissue testing, and ctDNA was enriched in therapy resistance alterations, as well as possible clonal hematopoiesis mutations (for example in ATM and CHEK2). Potential AR resistance alterations were detected in 940/2,213 patients (42%), including amplifications, polyclonal and compound mutations, rearrangements, and novel deletions in exon 8.
Conclusions:
Genomic analysis of ctDNA from patients with mCRPC recapitulates the genomic landscape detected in tissue biopsies, with a high level of agreement in detection of BRCA1/2 mutations but more acquired resistance alterations detected in ctDNA. CGP of ctDNA is a compelling clinical complement to tissue CGP, with reflex to tissue CGP if negative for actionable variants.
Keywords: circulating tumor DNA, prostate cancer, metastatic castration resistant prostate cancer, BRCA, androgen receptor
Introduction
Prostate cancer is the second most common cancer in men, accounting for 7% of all cancer-related deaths in this population (1). This cancer is driven by androgen receptor (AR) signaling, and androgen deprivation therapy is the first line treatment for metastatic prostate cancer. Duration of response varies, with a median of 1–2 years before the disease progresses to castration-resistant prostate cancer (mCRPC) (2). AR signaling inhibitors (ARSi) for treatment of mCRPC include abiraterone and enzalutamide, but primary and acquired resistance to these agents remain a challenge (3). Taxane-based chemotherapy has demonstrated efficacy prior and post-progression on abiraterone and enzalutamide (4,5), yet the 3-year overall survival rate for mCRPC remains under 50% (6).
The landscapes of genomic alterations (GAs) of primary prostate cancer (7) and mCRPC (8,9) have been previously characterized using tissue biopsies and used to identify mechanisms of resistance to ARSi (10–13). Some GAs enriched in mCRPC are emerging as potential therapeutic targets. Genomic profiling of recent specimens that captures somatic alterations may thus be more valuable than sequencing archival, pre-systemic treatment primary tumor tissue.
The most recent advances in mCRPC therapy target DNA repair defects in mCRPC using poly(ADP-ribose) polymerase inhibitors (PARPi) (14–16). Rucaparib (17) and olaparib have recently been FDA-approved for treatment of mCRPC with germline or somatic BRCA1/2 alterations. Germline alterations in homologous recombination repair (HRR) genes BRCA1/2 are a hereditary risk factor for prostate cancer (18) and mCRPC samples show enrichment in BRCA2 alterations compared to primary tumors, suggesting loss of HRR is a therapeutically-relevant driver of aggressive disease(19). Loss of function alterations in other DNA damage repair (DDR) genes are also enriched in mCRPC and may be targetable with PARPi but require further investigation (14–16).
While mutational status of DDR genes can be assessed with smaller sequencing panels, wider panels can detect other alterations targetable in mCRPC, such as PI3K pathway perturbations which have been targeted with ipatasertib (20), as well as genomic signatures such as tumor mutational burden (TMB) and microsatellite instability (MSI) that predict response to immunotherapy and are FDA-approved biomarkers for pembrolizumab in all solid tumors (21,22).
Despite the inherent advantages of profiling the latest available sample from a patient with advanced disease, mCRPC presents a technical challenge for collection of a tissue specimen, with metastases often confined to bone (23,24). Bone biopsies are invasive, technically difficult to collect, and have high failure rates of obtaining enough quality DNA for sequencing (10,25,26). Blood-based liquid biopsy and genomic profiling of cell-free circulating tumor DNA (ctDNA) from plasma provides a minimally invasive alternate method to profile mCRPC, with the added capability of detecting variants from multiple metastatic lesions that may have undergone clonal evolution. Here we build off previous studies of ctDNA in mCRPC (27–31) by analyzing a larger cohort of patients, characterizing the genomic landscape leveraging clinically available approaches, and evaluating concordance with tissue-based CGP.
Methods
A total of 3,334 liquid biopsy samples and 2,621 tissue samples were assayed with hybrid-capture based comprehensive genomic profiling (CGP). CGP was performed in a Clinical Laboratory Improvement Amendments-certified (CLIA), College of American Pathologists-accredited (CAP), New York State‐regulated reference laboratory (Foundation Medicine, Inc., Cambridge, MA, USA). Patients who submitted screening samples for TRITON2 or TRITON3 provided written informed consent before participation. Approval for the study of the FMI dataset, including a waiver of informed consent and Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (protocol 20152817). Studies were conducted in accordance with the Declaration of Helsinki.
Liquid biopsy specimens were obtained from three cohorts: screening samples from the TRITON2 (NCT02952534) and TRITON3 (NCT02975934) trials of rucaparib (collected Nov 2016-March 2019), and samples submitted to Foundation Medicine (FMI) for routine clinical testing (December 2013-March 2019). 20–100 ng of cell-free DNA (cfDNA) was extracted to create adapted sequencing libraries before hybrid capture and sample-multiplexed sequencing (FoundationACT™, FoundationOne® Liquid) as described previously (32). Two versions of the plasma assay were used, with 62 (FoundationACT™) or 70 genes (FoundationOne® Liquid). Genomic regions baited in the two different liquid biopsy assays are depicted in Table S1. Genomic alterations detected by both assays included base substitutions, insertions and deletions (short variants), rearrangements, and copy number changes. This study did not evaluate gene deletions. Table S2 depicts frequencies of all GAs assessed by liquid biopsy in the three different datasets.
Tissue specimens from metastatic sites submitted for routine clinical testing (December 2013-March 2019) were used for global comparisons of liquid biopsy to metastatic tissue (N = 2,006). Additional tissue specimens used only in the concordance analysis were screening samples collected from the TRITON2 (N = 337) and TRITON3 (N = 277). At least 50 ng of DNA was isolated from PCaA formalin-fixed, paraffin-embedded tumor specimens and sequenced to high, uniform ≥500X coverage, with larger gene panels inclusive of all 70 genes in liquid assays.
Microsatellite instability (MSI) status was determined in samples screened with the 70 gene panel, as described previously (33). For tumor specimens, zygosity and somatic/germline status for mutations was computationally predicted without matched normal tissue as previously described (34); in validation testing of 480 tumor-only predictions against matched normal specimens, accuracy was 95% for somatic and 99% for germline predictions (35).
Quantification of the ctDNA fraction was measured using two complementary methods: the proprietary tumor fraction estimator (TFE) and the maximum somatic allele frequency (MSAF) method. TFE is based on a measure of tumor aneuploidy that incorporates observed deviations in coverage across the genome for a given sample. Calculated values for this metric are calibrated against a training set based on samples with well-defined tumor fractions to generate an estimate of the tumor fraction. When lack of tumor aneuploidy limits the TFE’s ability to return an informative estimate, MSAF is used. MSAF calculates the allele fraction of all known somatic, likely somatic, and variant of unknown significance (VUS) substitution alterations detected at >2000X median unique coverage by non-PCR duplicate read pairs, excluding germline variants, and variants associated with clonal hematopoiesis.
Results
Patient characteristics and ctDNA shed
Liquid biopsy CGP results from a total of 3,334 patients with prostate cancer were included. Patients screened for TRITON3 (n = 818) had progressed on one prior ARSi therapy while patients screened for TRITON2 (n = 856) progressed on 1–2 lines of ARSi, followed by a taxane-based chemotherapy in the castration-resistant setting. 1,660 liquid biopsies were sourced from routine clinical testing at FMI of patients with advanced prostate cancer. The median age of patients was 72 years (range 38–97, IQR: 66–78) and was similar across all three datasets (Table 1, Fig S1).
Table 1.
Patient characteristics in the 3 liquid biopsy datasets.
| Liquid biopsy datasets and metastatic tissue biopsy dataset used as comparison (unmatched) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Liquid biopsies | Tissue biopsies | ||||||||||
| FMI routine clinical cohort | TRITON2 screening cohort | TRITON3 screening cohort | Total | Metastatic tissue biopsies | |||||||
| Cohort characteristics | Prostate cancer plasma samples submitted for CGP during routine clinical care | mCRPC, progressed on 1–2 next-generation ARSi and taxane-based chemotherapy | mCRPC, progressed on 1 next-generation ARSi | - | Prostate cancer tissue samples submitted for CGP during routine clinical care | ||||||
| Patients, N | 1,660 | 856 | 818 | 3,334 | 2,006 | ||||||
| Profiled with FACT, n | Profiled with F1L, n | 943 | 717 | 106 | 750 | 72 | 746 | 1,121 | 2,213 | - | |
| Median patient age, years (range) a | 71 (38–≥89b) |
72 (46–95) |
73 (43–97) |
72 (38–97) |
69 (39–≥89a) |
||||||
| Median ctDNA fraction, % (IQR) | 7.7 (0.6–35.7) |
18.1 (1.5–38.1) |
3.4 (0.8–23.3) |
7.5 (0.8–33.7) |
- | ||||||
| Percentage of samples with no ctDNA detected in sample | 8.6% (142/1,660) |
2.7% (23/856) |
4.9% (40/818) |
6.1% (205/3,334) |
- | ||||||
| Percentage of samples with ctDNA fraction of ≥20% | 39% (648/1,660) |
47% (401/856) |
28% (233/818) |
38% (1,282/3,334) |
- | ||||||
| 1 or more oncogenic/ deleterious GAs detected in sample c | 85.6% (614/717) |
90.3% (677/750) |
80.0% (597/746) |
85.3% (1,888/2,213) |
99.2%d (1,991/2,006) 89.6%e (1,797/2,006) |
||||||
| MSI-H detected c | 1.12% (8/717) |
2.00% (15/750) |
1.07% (8/746) |
1.40% (31/2,213) |
2.74% (55/2,006) |
||||||
| Site of metastatic biopsy | N/A | N/A | N/A | N/A | |||||||
| Other visceral sites | 29.0% | ||||||||||
| Subset of patients with available matched tissue | |||||||||||
| Patients with matched tissue, N | 223 | 337 | 277 | 837 | |||||||
| Profiled with FACT, n | Profiled with F1L, n | 155 | 68 | 52 | 285 | 27 | 250 | 234 | 603 | ||
| Patients with matched tissue collected within 30 days of liquid biopsy, N | 36 | 53 | 28 | 117 | |||||||
| Profiled with FACT, n | Profiled with F1L, n | 26 | 10 | 10 | 43 | 2 | 26 | 38 | 79 | ||
| Median Δ between liquid and tissue collection, months (IQR) | 11 (0.1–44) |
31 (5–74) |
30 (12–70) |
25 (3–61) |
|||||||
28 samples had no available age information: 1 from routine clinical cohort, 15 TRITON2, 12 TRITON3 screening samples
FMI routine clinical datasets do not include specific patient age above 89.
Only samples analyzed with F1L (70 gene panel) used for this comparison.
Oncogenic/deleterious GAs in all genes baited in tissue biopsy panel considered.
Oncogenic/deleterious GAs only in genes baited in 70 gene liquid panel considered.
Screening samples from the clinical trials contained detectable ctDNA in >95% of samples (as determined by the comprehensive tumor fraction estimation method, see Appendix for full method description). The median ctDNA fraction of these datasets was 7.5% (IQR: 0.8% - 34%) (Table 1, Fig 1A) and was higher in patients who had progressed on taxane (median 18.1% in TRITON2 versus 3.4% in TRITON3), consistent with higher ctDNA shed after more lines of therapy (29). The number of samples with no ctDNA detected (that is, no detection of aneuploidy or somatic alleles of known or unknown functional significance) was 40/746 (5.4%) in the TRITON3 screening cohort and only 23/856 (2.7%) in the more heavily treated TRITON2 screening cohort. A substantial 401/856 (47%) of the TRITON2 screening cohort had a ctDNA fraction of 20% or above, which allows for >90% sensitivity of detection for all three variant types reported by the liquid biopsy assay: substitutions/indels, rearrangements, and amplifications. Liquid biopsy identified at least one GA predicted to have deleterious/oncogenic effects on protein function in 2,651/3,334 (79.5%) of all patients and 1,888/2,213 (85.3%) of patients profiled using the 70 gene panel (Table 1).
Figure 1. Genomic landscape of prostate cancer in liquid biopsies.

A) Distribution of estimated tumor fraction within each liquid biopsy dataset B) Frequency and cooccurrence of alterations in genes associated with mCRPC across 2,213 liquid biopsy samples assayed with 70 gene panel. Variant type indicated by color legend at the top of the oncoprint. MSI-H status indicated in last row. Estimated tumor fraction indicated by bar below the oncoprint. Copy number deletions not reported by the liquid biopsy assay versions used in this study. C) Frequency of short variants detected in metastatic tissue samples versus frequency of short variants detected in liquid samples. Genes with short variants with significantly different frequencies in tissue and liquid are color coded to reflect p-value. *TP53 off-scale (45.4% in liquid vs 40.8% in tissue, p = 0.0096).
Genomic landscape of prostate cancer ctDNA
The most frequently altered genes were TP53 (46%) and AR (42%) (Fig 1B, Fig S2A), consistent with patient cohorts with prior ARSi exposure; both genes are commonly altered in tissue-based profiling of mCRPC and associated with resistance to ARSi (36,37). At least one DNA repair gene was altered in 30% of all patients, including BRCA2 (7.5%) and BRCA1 (1.4%). Genes in the PI3K/AKT/mTOR pathway were altered in 14% of samples, including activating mutations in PIK3CA and AKT1 (Fig S3A). WNT/β-catenin pathway genes were altered in 17% of patients (Fig S3B). Alterations in RAS/RAF/MEK pathway components were detected in 5% of all patients (Fig S2A, Fig S3C). Amplifications of FGFR1 were detected in 3% of patients, and 11 patients harbored rearrangements of FGFR1/2/3 with breakpoints in intron 17, which preserve the kinase domain and are predicted to be oncogenic (38) (Fig S3D). MSI-H status was found in 31/2,213 (1.4%) of patients (Table 1), comparable to the 2% of primary site and 3% of metastatic tissue biopsies (10,12).
The landscape of short variants (substitutions and indels) detected by liquid biopsy closely resembled the landscape detected in tissue biopsies from metastatic sites of origin (Fig 1C, Fig S4A) and previous reports (9,11,12). Rearrangements in most genes were detected at similar frequencies in liquid biopsies relative to metastatic tissue (Fig S4B), except AR, as discussed further on. Copy number amplifications were detected less frequently in liquid than tissue (Fig S4C), likely owing to decreased sensitivity of detection in samples with low levels of ctDNA (Fig S5). We examined the subset of samples with ≥20% ctDNA fraction (a threshold where there is >95% sensitivity of detection for amplifications (32)). This subset included 1,282/3,334 (38%) of all samples (Table 1). Amplifications were detected with significantly higher frequency in this subset: AR (344/781; 41%), FGFR1 (91/1,282; 7.1%), and MYC (67/1,282; 5.2%). It is important to note that the two liquid biopsy platforms used in this study did not report copy number losses, and common driver TMPRSS2-ERG fusion was not reported due to lack of baiting in these genes.
Frequencies of short variants in liquid biopsies were compared to those in 2,006 metastatic tissue biopsies (Fig 1C). While most genes were altered at similar rates, variants were detected significantly more often in liquid in 9 of 70 genes. Alterations in AR were enriched in ctDNA, likely representing resistance mechanisms acquired on therapy. Low level enrichment for JAK2, GNAS, and IDH2 (genes not often altered in mCRPC) likely represent signal from clonal hematopoiesis (CH) (39) rather than the tumor; the same applies to mutations in NF1 and the TERT promoter. Alterations in CHEK2, ATM, and TP53 occur with some frequency in mCRPC tissue biopsies but were also more prevalent among liquid biopsies; it is uncertain whether this is related to mCRPC biology or also to CH, as these have been detected in some studies of CH (31). In this study, no strong associations with age were observed for likely CH variants in JAK2, GNAS, and IDH2 (Fig S6), thus age association could not be used to distinguish CH variants from liquid-prevalent resistance mutations such as AR, sourced from the tumor. The lack of age association for CH variants may be the result of an older cohort in which 88% of patients were older than 60.
Overall concordance between tissue and liquid biopsies
Patient-matched tissue samples were available for 837 of the 3,334 liquid biopsies (Table 1). Tissue specimens were collected a median 758 days before plasma collection (range: 19.9 years before - 1.8 years after liquid biopsy) (Table 1, Fig S7A). 117 pairs were collected within 30 days of each other and were considered “contemporaneous pairs” in this concordance analysis.
Detection of short variants in genes included in the liquid assay showed 75.3% positive percent agreement to tissue as reference (PPA) (Table S3). PPA was 70.3% for rearrangements and 27.5% for amplifications. Among contemporaneous pairs, PPA increased to 87.2%, 91.7%, and 38.8% for short variants, rearrangements, and amplifications, respectively. ctDNA fraction was a major factor in concordance of liquid to tissue for amplifications since 20% ctDNA fraction is needed to detect amplifications with >95% sensitivity (32); above this threshold, PPA for amplifications was 50.8%. Alterations exclusively detected in tissue tended to be in sample pairs with a low ctDNA fraction in the liquid biopsy (Fig S7B).
BRCA1/2 alterations
BRCA1/2 was altered in 8.9% of all patients’ plasma samples (7.5% BRCA2, 1.6% BRCA1, 0.18% both genes). Curiously, BRCA2 was altered significantly more frequently in the TRITON2 versus TRITON3 screening cohorts (Table S4). BRCA2 inactivation is a predictor of poor response to docetaxel(40,41), and patients screened for TRITON2 had progressed on a taxane-based chemotherapy, which could account for a larger proportion of these patients in this cohort. BRCA1 alterations were also more frequent in the group that had received prior taxane. The combined BRCA1/2 alteration frequency within the TRITON2 screening cohort was 12.3%-- nearly twice the frequency in the TRITON3 screening cohort (6.4%) (Fig 2A).
Figure 2. BRCA1/2 alterations in liquid biopsy.
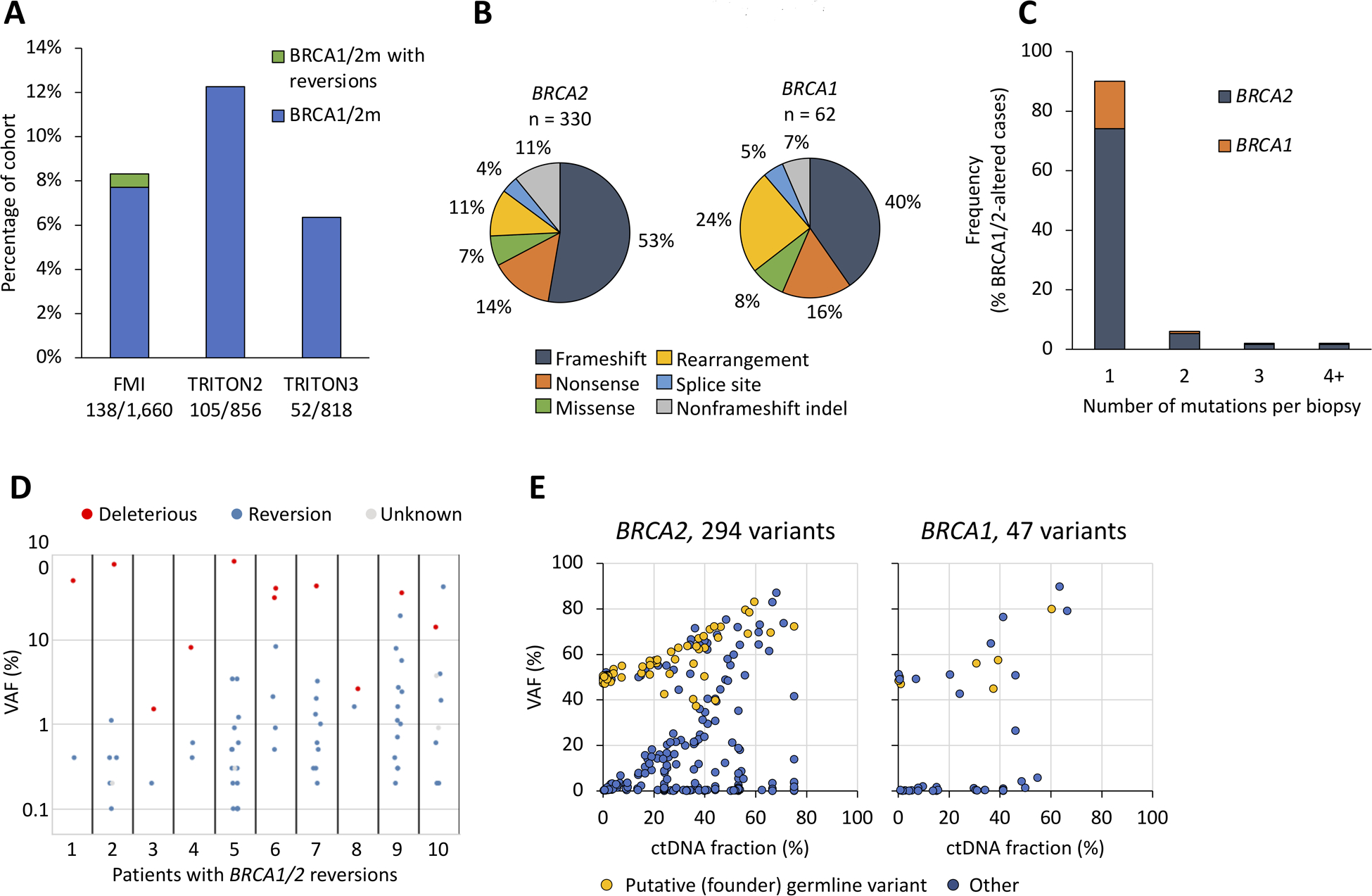
A) Prevalence of BRCA1/2 alterations in the three cohorts B) Types of detected BRCA1/2 alterations. 174 frameshifts among 157 patients for BRCA2, 25 frameshifts among 25 patients for BRCA1, 58 BRCA1/2 nonsense point mutations among 57 cases, 51 patients with rearrangements, 28 missense mutations among 22 patients, and 16 splice site alterations among 14 patients, 49 non-frameshift deletions among 7 patients (these indels were all reversion mutations). C) Numbers of BRCA1/2 alterations per patient D) Variant allele frequencies of BRCA1/2 short variants in ten patients with detected reversion mutations. Variants with unknown functional status are splice site mutations. See table S2 for details. E) Variant allele frequencies of short variants in BRCA1 and BRCA2 compared to the ctDNA fraction of the liquid biopsy. Germline variants were predicted using heuristic scoring of observed instances across all FMI datasets.
Frameshifts were the most common alteration in both BRCA2 and BRCA1, rearrangements were more common in BRCA1, and the frequencies of nonsense and missense mutations was similar between the two genes (Fig 2B). 90% of BRCA1/2-altered patients had a single mutation detected (Fig 2C). In 10 of the 30 patients with 2 or more mutations, the additional variants were reversions. All ten patients with BRCA1/2 reversions were from the routine clinical CGP dataset, not the TRITON2/3 screening cohorts (Fig 2A) and may have had exposure to platinum-based chemotherapy or PARPi (42–44). Reversions were defined as in-frame deletions spanning a frameshift or nonsense mutation, or missense mutations in the same position as the nonsense mutation (Table S5). These alterations were mostly subclonal in relation to the deleterious mutation, with up to 13 found in one sample (Fig 2D).
Studying the variant allele fractions (VAFs) of BRCA1 and BRCA2 short variants as compared to ctDNA fraction (Fig 2E), two distributions were seen, consistent with somatic and germline mutations. Variants with a VAF ≥40% detected at low ctDNA fraction were enriched for putative germline variants (i.e. founder mutations). When ctDNA fraction was high, somatic and germline variants were not clearly distinguishable. Some somatic variants tracked closely with ctDNA fractions, while others appear subclonal (variants close to the horizontal axis in Fig 2D). The VAF distribution of variants in additional DDR genes ATM, CHEK2, PALB2, and CDK12 is provided in Fig S8.
BRCA1/2 variant detection by liquid and tissue biopsy
Among 837 patients in the concordance analysis, 92 (11.0%) had a BRCA1/2 alteration detected by one or both assays. 67 (8.0%) had BRCA1/2 alterations detected concordantly in tissue and liquid, 5 (0.6%) exclusively in tissue biopsy, and 20 (2.4%) exclusively in the liquid biopsy (Fig 3A, 3B). The percent positive agreement (PPA) between the tissue and liquid assays was 93.1% on the patient-level (Fig 3B). The variant level concordance between the tissue and liquid assays for BRCA1/2 was high: the PPA was 95.2% and 85.0% in the TRITON2 and TRITON3 screening cohorts, respectively (Table S3).
Figure 3. Concordance of BRCA1/2 detection in liquid and tissue biopsy.

A) 92 tissue/liquid pairs where BRCA1/2 variants were detected among the tissue sample alone (n=5), the liquid sample alone (n=20), or both (n=67). Samples in each group arranged in ascending ctDNA fraction (gray bar). Variant allele frequency is indicated for each short variant. Rearrangements, for which VAF was not reported, indicated at the top of the chart. Tissue only variants, with no associated liquid VAF, also indicated at the top of the chart. For simplicity and clarity, all analyses presented in this figure omit 9 BRCA reversion mutations detected in one sample. All alterations presented are predicted deleterious to BRCA1/2 function. B) Patient-level BRCA1/2 mutant status was assigned in the presence of at least one deleterious alteration in BRCA1 or BRCA2 in a sample. No patient in this study had multiple discordant BRCA1/2 variants assigned in tissue and liquid tests. Positive percentage agreement (PPA): the number of patients assigned BRCA1/2-mutant status by both liquid and tissue biopsies divided by the total number of BRCA1/2-mutant patients identified by tissue biopsy. Negative percent agreement (NPA), was also calculated with tissue biopsy taken as standard. OPA calculated as patients assigned similarly by both tests divided by total patients in paired comparison. C) SGZ algorithm predictions of variants germline/somatic status using the tissue biopsy, and the proportions of these variants also detected in the matching liquid biopsy. D) Comparison of VAF of short variants in liquid versus matched tissue biopsy. Variants were classified as detected in liquid only, or detected in tissue.
Lack of detection of BRCA1/2 variants in liquid was mostly attributable to low ctDNA fraction: of 5 patients with tissue-only detection of a BRCA1/2 alteration, 4 had ctDNA fractions of ≤1% (Fig 3A). Some BRCA1/2 alterations identified exclusively in the liquid biopsy (colored red in Fig 3A) potentially represent alterations acquired after tissue specimens collection (median time difference in sample collection was 38 months, IQR: 15–112, Fig S7C). These variants, especially in BRCA1, were more likely to be subclonal—have VAFs significantly smaller than the ctDNA fraction of the sample. However, in 5 of the 20 samples where only liquid biopsy identified BRCA1/2 alterations, BRCA2 short variant VAF exceeded 50% of the ctDNA fraction.
BRCA1/2 alterations detected in tissue were predicted to be germline or somatic using a previously established and validated computational method(34). All 40 predicted germline mutations were detected in the corresponding liquid biopsies at >40% VAF (Fig 3C,D). 16/18 predicted somatic and 8/10 unknown status were detected in the corresponding liquid biopsy (Fig 3C), usually at lower VAF (Fig 3D).
Overall, liquid biopsy was able to detect 100% of predicted germline BRCA1/2 alterations and reliably detect somatic alterations identified in tissue when ctDNA fraction was over 1%. In samples where BRCA1/2 VAF was lower than ctDNA fraction, it is possible that the variant represents a subclone, a monoallelic alteration, or even CH. It is worth noting the assays used in this study detected inactivating short variants and rearrangements but did not report BRCA1/2 copy loss, thus copy number deletions were not considered in concordance analysis.
Putative and novel resistance AR alterations
Liquid biopsy detected 1,090 AR alterations among 42% of evaluated patients (940/2,213). Among AR-altered patients, 45% had missense mutations, 33% had amplifications, 8% had rearrangements, and 14% had multiple types of alterations (Fig 4A). AR amplifications were identified in 419 patients (43% of AR-altered patients, 13% overall), with technically limited detection in samples with low ctDNA fractions (Fig S9). Among the subset of patients with ≥20% ctDNA fraction in their biopsy, AR amplifications were detected in 41% (344/781).
Figure 4. AR alterations in liquid biopsy.

A) Oncoprints of 940 AR-altered samples, divided as separate cohorts and the aggregate B) Polyclonality of AR activating mutations: numbers of AR short variants and rearrangements per sample. SV= short variant, RE= rearrangement C) Distribution of oncogenic missense mutations in AR. Letters indicate amino acid change when there is >1 missense mutation at the position D) Rare AR alterations identified near the C-terminus of the ligand binding domain: Compound missense mutations in cis (gold) and in-frame deletions (grey) spanning important androgen-binding residues H875, F877, and T878, all resulting in S885 moving into the 878 position (red). One sample contained an isoleucine insertion (light blue). F877L/T878A double mutants are predicted to have enhanced resistance to enzalutamide (bold). 16 compound mutations were found in 12 patients, and 11 in-frame deletions among as many patients. All patients were confirmed to have progressed on at least one of abiraterone, enzalutamide or apalutamide, except 2 patients with compound mutations for whom treatment information was not available. E) Map of AR rearrangements that describe breakpoints for translocations and deleted, duplicated, or inverted regions (22 translocations, 60 deletions, 53 duplications, and 25 inversions). X-axis is a schematic representations of the 8 exons in the AR gene (not to scale). Among the 160 patients with AR rearrangements, 138 were confirmed to have progressed on at least one of abiraterone, enzalutamide or apalutamide, and 22 had no available treatment information. F) Patient-matched sample pairs collected within 30 days of each other with ≥1 AR short variant detected, in ascending order of ctDNA fraction. Bar represents estimated ctDNA fraction of the liquid biopsy. Tumor fraction of tissue biopsy listed on left. Table to the right lists short variants identified exclusively in tissue (orange), in both tissue and liquid (blue), or exclusively in liquid (green). Ratio of VAF/ctDNA fraction listed in parentheses after each variant detected in liquid biopsy. * = VAF can exceed ctDNA fraction if mutation is an amplified copy of AR. G) Tissue-liquid pairs in which an AR amplification was detected in tissue. Correlation of copy number, ctDNA fraction and detection in the matched liquid biopsy.
Among patients with a mutation or rearrangement in AR, ~40% harbored ≥2 variants (up to 6) (Fig 4B, Fig S2B). Many mutations were subclonal, with variant allele fractions (VAFs) smaller than ctDNA fractions; some VAFs exceeded ctDNA fraction, which may result from mutations in amplified copies of AR (Fig S10). Hotspots concentrated in the ligand binding domain and are known to confer resistance to ARSi (45) (Fig 4C). The most frequent mutations included W742L/C (bicalutamide resistance), H875Y, F877L, and T878A (bicalutamide/enzalutamide/apalutamide resistance and promiscuous activation by progesterone) and L702H (resistance to abiraterone/enzalutamide as well as the AR proteolysis-targeting chimera ARV-110, and activation by corticosteroids) (45,46). Less common AR resistance mutations were found in V716M (15 patients), S889G (15 patients), and M896I/V/L (10 patients) (Fig 4C); these mutations have been detected in ctDNA from patients with mCRPC who progressed on bicalutamide or abiraterone (45,47,48).
Rare AR variants were detected in this large dataset, some of which have not been previously described and warrant further characterization. Double mutant F877L/T878A appeared in 11 patients (Fig 4D). This compound mutant has been shown to confer synergistic resistance to enzalutamide in preclinical studies (45,49). In-frame deletions spanning residues H875 to T878 were detected in 11 patients, each shifting S885 into the T878 position (Fig 4D). While it is not known whether these mutants mimic T878S, their appearance in multiple patients in this study raises the possibility of their being a mechanism of ARSi resistance. One patient harbored indel S647–648>F, impacting critical serine residues within a binding motif for the ubiquitin ligase SPOP, and predicted to stabilize the AR receptor by reducing proteasomal degradation (50) (Fig S11A).
AR rearrangements that truncate the reading frame just after exon 3 yield a receptor with an intact DNA binding domain (DBD) and without a ligand binding domain (LBD) to suppress its activity(51,52). These rearrangements were detected in 160 patients (17% of AR-altered patients, 7% overall), more commonly than the 2.2% frequency detected among metastatic tissue samples (12) (Fig 4E). The number of intra-AR rearrangements is consistent with ctDNA from patients with mCRPC who have undergone ARSi therapy (52). There were 7 patients with truncating mutations in AR that disrupt the LBD and retain the DBD; these may yield active ARSi-resistant receptors according to similar principles (53) (Fig S11A).
AR alterations were significantly more common among TRITON2 than TRITON3 screening samples (52% vs 37%, p=3.0E-8) (Table S4), consistent with progression on more lines of therapy and higher ctDNA fractions in the TRITON2 cohort. It is also possible that higher ctDNA fractions in the TRITON2 cohort allowed for more sensitive detection of certain variants, such as amplifications.
AR variants in liquid and tissue biopsies
Among patient-matched tissue/liquid sample pairs, liquid biopsy detected far more AR short variant mutations than tissue (18 shared, 10 tissue-only, 173 liquid-only). To exclude pairs where tissue was archival and collected prior to exposure to ARSi, pairs collected within thirty days of each other were compared. Among these, 10 AR mutations were detected concordantly, 3 mutations exclusively in tissue, and 13 exclusively by liquid. In 7/17 patients, liquid biopsy provided the only evidence of AR mutations, and in 2/17 patient additional mutations were detected by liquid biopsy (Fig 4F). This analysis highlights the ability of liquid biopsy to detect subclonal resistance variants that may not exist in every metastatic lesion—and thus may not be detected in a tissue biopsy—but have relevance when choosing a course of treatment.
AR amplifications were detected in 72 of the tissue samples, with a median copy number (CN) of 20 (IQR: 13–35). Liquid biopsy detected 53% of these amplifications (38/72), and 91% of amplifications when ctDNA fraction was ≥20% (32/35). Amplifications with greater CNs could be detected at lower ctDNA fractions: liquid biopsy detected 87% of amplifications of CN>16 in samples with a ctDNA fraction as low as 5% (Fig 4G). AR amplifications were detected exclusively in liquid biopsy in 96 samples. Among the 79 contemporaneously collected pairs where AR was profiled (Table 1), 20 amplifications were detected concordantly, 16 exclusively in tissue, and 5 exclusively in liquid. Lack of detection in liquid biopsy was largely attributable to ctDNA fractions below 20% (Fig S11B). In contemporaneous pairs, one AR rearrangement was detected concordantly and 5 exclusively in liquid (Fig S11C).
Discussion
In the largest study of mCRPC liquid biopsy samples conducted to date, CGP of liquid biopsies from 3,334 patients with advanced prostate cancer recapitulated the genomic landscape detected in tissue biopsies, with a high level of agreement in detection of BRCA1/2 mutations. ctDNA also identified more acquired resistance alterations than tissue, including novel AR activating alterations and subclonal BRCA1/2 secondary mutations and reversions.
Analysis of cfDNA extracted from plasma was feasible in these cohorts, with 94% of patients having detectable ctDNA and with higher ctDNA fraction among patients with more advanced disease. The rich genomic signal within ctDNA, combined with its ease of use as compared to biopsy of a metastatic site, suggests that liquid biopsy could be a compelling option for identifying targetable GAs in patients with mCRPC.
94% of BRCA1/2 mutations and 90% of all BRCA1/2 variants detected by tissue CGP were detected in ctDNA, including all predicted germline variants. Liquid biopsy also demonstrated the ability to identify patients with somatic BRCA2 mutations not detected in tissue, some with a high VAF, suggesting HR deficiency acquired later in tumor evolution. Given the high percentage of patients with detectable ctDNA and the agreement with tissue-defined genomic landscape of mCRPC observed in this study, CGP of a minimally invasive plasma biopsy is a viable option to detect BRCA1/2 mutations. Of note, this analysis did not test for BRCA1/2 homozygous deletion; this variant is more challenging to detect in ctDNA but is measured on the latest version of the assay, which has recently been approved by the FDA (54). An estimated additional ~3% of patients with mCRPC might have been identified as having BRCA1/2 mutations with a platform that reports deletions (12). These patients are worth identifying because they may receive particularly sustained benefit from PARPi, having a BRCA defect incapable of reversion (55,56).
The most striking area of discordance between liquid and tissue variant detection was in the detection of a range of resistance mutations, in agreement with previous reports documenting the sensitivity of liquid biopsies in detecting AR-directed resistance mutations (27,47,48,57–60). AR activating alterations and subclonal somatic BRCA1/2 secondary mutations and reversions were far more prevalent in liquid biopsies, likely due to a combination of (a) differences in the patient populations, since patients submitting a liquid biopsy likely were exposed to more lines of therapy (Fig S7A) (b) the increased sensitivity of detection and lower reported VAFs in liquid biopsies over tissue testing, and (c) the ability of liquid biopsy to integrate acquired resistance signals from multiple metastatic sites (Fig 4F).
The sensitive detection of resistance mechanisms in liquid biopsies can potentially be used as a marker of therapy resistance and could provide additional ability detect patients who might benefit from a non-ARSi drug. In this study, liquid biopsy outperformed tissue in detecting AR mutations that have relevance to clinical decisions about choices of ARSi, including W742L (bicalutamide resistance), H875Y and T878A (bicalutamide/enzalutamide/apalutamide resistance), and L702H (resistance to the AR proteolysis-targeting chimera ARV-110) (45,46). Liquid biopsy’s sensitive detection of BRCA1/2 reversions offers potential to be used for monitoring of emerging resistance.
One established limitation of liquid biopsy genotyping is the identification of mutations derived from white blood cells (e.g. clonal hematopoiesis) (29,31,61), an age-related phenomenon which may be particularly relevant in patients with prostate cancer who tend to be older at metastatic diagnosis. Intuitively, alterations which confer a fitness advantage in hematopoietic progenitors (JAK2 V617F, IDH2 R140Q) are often suspected to originate from CH. Our study found an increased prevalence of mutations in ATM and CHEK2 in ctDNA compared to tissue, two potentially targetable HRR genes which can be mutated in cancer cells, can harbor germline mutations, and have been described as recurrently mutated in CH (31,62). Our study also identified enrichment for NF1 and TERT promoter mutations in ctDNA. NF1 inactivation has been detected in tumor infiltrating lymphocytes and white blood cells in previous studies (31,63). TERT promoter mutations have not been implicated as CH variants but have been linked to myeloid malignancies. These findings highlight that genomic discovery based on ctDNA genomics may benefit from paired-depth blood cell sequencing to clarify tumor-derived versus CH-derived signal. While such mutations are suspicious for CH, even classic CH genes may impact prostate cancer biology, with a recent study noting that TET2 and IDH1 are commonly altered in a novel subtype of prostate cancer (64). In clinical care, such paired-depth blood cell sequencing is not widely available, therefore clinicians must understand, even when using FDA approved assays, that CH mutations are common and mutations in ATM and CHEK2 may not always be tumor derived.
The key limitation of ctDNA analysis is the variable shed of ctDNA into plasma. In this study, 60% of samples had a ctDNA fraction below 20%, which reduced the ability to detect amplifications. Both liquid biopsy platforms used in this study were not designed to detect deletions, leading to marked under-detection of PI3K signaling perturbation (PTEN homozygous deletions account for more than half of alterations in that gene, ~30% to the 9% detected in this study). Other genes in the panel where deletions were likely missed were RB1, BRCA1/2, APC, and TP53. Deletions are captured with FMI’s next-generation assay (54). Finally, the routine clinical CGP prostate cancer samples available for this study do not uniformly represent mCRPC, and there is limited information on treatments the patients received prior to specimen collection. Nevertheless, this dataset closely resembles the defined mCRPC TRITON2/3 cohorts and demonstrates the utility of liquid to identify patients who may benefit from targeted therapies.
This study highlights the ability of the liquid platform to detect targetable alterations in patients with mCRPC, including somatic mutations acquired during or after the transition to castration-resistant, metastatic disease. It also exposes some of the weaknesses of ctDNA analysis, such as reduced detection of copy number changes in biopsies with low ctDNA. This reduced sensitivity with lower ctDNA content highlights that liquid biopsy cannot replace tissue CGP but may complement it. If tissue profiling fails due to specimen inadequacy, the high ctDNA shed in prostate cancer means liquid biopsy could serve as a back-up. Alternatively, if tissue is unavailable for profiling, liquid biopsy could be used first with tissue CGP as a reflex option when ctDNA analysis is negative – indeed clinicians ordering a liquid biopsy might in parallel request archival tissue for CGP so this can be analyzed if ctDNA is uninformative. Together these two diagnostic tools offer an opportunity to increase access to precision therapeutics in advanced prostate cancer.
Supplementary Material
Translational Relevance.
Comprehensive genomic profiling (CGP) in metastatic castration-resistant prostate cancer (mCRPC) is of increasing value given the diversity of emerging treatment options. While CGP by tissue testing remains the gold standard, bone metastases are challenging to sample and analyze. Genomic profiling of plasma cell-free circulating tumor DNA (ctDNA) offers a compelling, minimally invasive complement to tissue testing. Advanced prostate cancer has a high shed rate (ctDNA was detectable in 94% of patients). Using the largest cohort of patients with mCRPC to date, we demonstrate high concordance between alterations identified by liquid and tissue biopsy. ctDNA detected additional alterations, including a broad spectrum of AR resistance alterations and somatic BRCA1/2 mutations and reversions. ctDNA profiling can overcome the technical difficulties and high failure rates associated with bone metastasis biopsy and help to guide precision therapy in advanced prostate cancer.
Acknowledgments
The TRITON2 and TRITON3 trials are funded by Clovis Oncology, Inc. We thank and acknowledge all patients and their families and caregivers who are participating in the trials, along with the investigators. We thank Brady Forcier and Jeff Lee for help with data analysis. We thank Neeru Bhardwaj for critical reading and helpful discussions of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest:
COI: HT, RWM, JHC, EAS, LD, BJF, SM, LZ, RPG, JSR, BMA, JMV, and GRO are employees of Foundation Medicine, a wholly owned subsidiary of Roche, and have equity interest in Roche. TG, SPW, AS, and AL are employees of Clovis Oncology. WA has served as consultant or advisor to or received travel, accommodations or expenses from Clovis Oncology, Daiichi Sankyo, Glaxo-Kline Smith, Janssen, MORE Health and ORIC Pharmaceuticals. CJR has received honoraria or served as consultant or advisor to Advanced Accelerator Applications, Bayer, Bristol Myers Squibb, Dendreon, Janssen Oncology, Myovant Sciences, and Roivant. KF has received honoraria and travel, accommodations or expenses from, or served as consultant or advisor to, Astellas Pharma, Bayer, Curevac, Janssen Oncology, Orion Pharma GmbH, and Sanofi. SC has received honoraria from, served as consultant or advisor to, or is part of the Speaker’ Bureau for Astellas Pharma, Bayer, BeiGene, Clovis Oncology, Janssen-Cilag, Novartis, and Pfizer.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Kongseang C, Attawettayanon W, Kanchanawanichkul W, Pripatnanont C. Predictive factor of androgen deprivation therapy for patients with advanced stage prostate cancer. Prostate International. 2017; 5: 35–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nuhn P, de Bono JS, Fizazi K, Freedland SJ, Grilli M, Kantoff PW, et al. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology [Internet]. European Urology. 2019. page 88–99. [DOI] [PubMed] [Google Scholar]
- 4.de Wit R, de Bono J, Sternberg CN, Fizazi K, Tombal B, Wülfing C, et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. New England Journal of Medicine. 2019; 381: 2506–18. [DOI] [PubMed] [Google Scholar]
- 5.Oh WK, Cheng WY, Miao R, Vekeman F, Gauthier-Loiselle M, Duh MS, et al. Real-world outcomes in patients with metastatic castration-resistant prostate cancer receiving second-line chemotherapy versus an alternative androgen receptor-targeted agent (ARTA) following early progression on a first-line ARTA in a US community oncology setting. Urologic Oncology: Seminars and Original Investigations. 2018; 36: 500.e1–500.e9. [DOI] [PubMed] [Google Scholar]
- 6.Francini E, Gray KP, Shaw GK, Evan CP, Hamid AA, Perry CE, et al. Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer and Prostatic Diseases. 2019; 22: 420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015; 163: 1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. European Urology. 2013; 63: 920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson D, van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161: 1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precision Oncology. 2017; 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nature Genetics. 2018; 50: 645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ, et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precision Oncology. 2019; 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mateo J, Seed G, Bertan C, Rescigno P, Dolling D, Figueiredo I, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. Journal of Clinical Investigation. 2020; 130: 1743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. New England Journal of Medicine. 2015; 373: 1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. The Lancet Oncology. 2020; 21: 162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: analysis from the phase 2 TRITON2 study. Clinical Cancer Research. 2020; clincanres.0394.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abida W, Campbell D, Patnaik A, Sautois B, Shapiro JD, Vogelzang NJ, et al. Genomic characteristics associated with clinical activity of rucaparib in patients (pts) with BRCA1 or BRCA2 (BRCA) - mutated metastatic castration-resistant prostate cancer (mCRPC). . Journal of Clinical Oncology. 2020; 38: 178–178. [Google Scholar]
- 18.Pilarski R The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. American Society of Clinical Oncology Educational Book. 2019; 79–86. [DOI] [PubMed] [Google Scholar]
- 19.Pritchard CC, Mateo J, Walsh MF, de Sarkar N, Abida W, Beltran H, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. New England Journal of Medicine. 2016; 375: 443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bono JS, de Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating AKT blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clinical Cancer Research. 2019; 25: 928–36. [DOI] [PubMed] [Google Scholar]
- 21.Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA, et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncology. 2019; 5: 471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu YM, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell. 2018; 173: 1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong SK, Mohamad NV, Giaze TR, Chin KY, Mohamed N, Ima-Nirwana S. Prostate cancer and bone metastases: The underlying mechanisms. International Journal of Molecular Sciences. 2019; 20: 2587–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandaglia G, Abdollah F, Schiffmann J, Trudeau V, Shariat SF, Kim SP, et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. The Prostate. 2014; 74: 210–6. [DOI] [PubMed] [Google Scholar]
- 25.Sailer V, Schiffman MH, Kossai M, Cyrta J, Beg S, Sullivan B, et al. Bone biopsy protocol for advanced prostate cancer in the era of precision medicine. Cancer. 2018; 124: 1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorente D, Omlin A, Zafeiriou Z, Nava-Rodrigues D, Pérez-López R, Pezaro C, et al. Castration-Resistant Prostate Cancer Tissue Acquisition From Bone Metastases for Molecular Analyses. Clinical Genitourinary Cancer. 2016; 14: 485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyatt AW, Azad AA, Volik S v., Annala M, Beja K, McConeghy B, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncology. 2016; 2: 1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J Natl Cancer Inst. 2017; 109: djx118–djx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayrhofer M, de Laere B, Whitington T, van Oyen P, Ghysel C, Ampe J, et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Medicine. 2018; 10: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonpavde G, Agarwal N, Pond GR, Nagy RJ, Nussenzveig RH, Hahn AW, et al. Circulating tumor DNA alterations in patients with metastatic castration-resistant prostate cancer. Cancer. 2019; 125: 1459–69. [DOI] [PubMed] [Google Scholar]
- 31.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nature Medicine. 2019; 25: 1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical Validation of a Hybrid Capture–Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. Journal of Molecular Diagnostics. 2018; 20: 686–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gowen K, Clark TA, Gregg JP, Greene MZ, Murphy A, White J, et al. MSI-H testing via hybrid capture based NGS sequencing of liquid biopsy samples. Journal of Clinical Oncology. 2019; 37: 504–504.30615549 [Google Scholar]
- 34.Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLOS Computational Biology. 2018; 14: e1005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sokol ES, Pavlick D, Khiabanian H, Frampton GM, Ross JS, Gregg JP, et al. Pan-Cancer Analysis of BRCA1 and BRCA2 Genomic Alterations and Their Association With Genomic Instability as Measured by Genome-Wide Loss of Heterozygosity. JCO Precision Oncology. 2020; 442–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discovery. 2018; 8: 444–57. [DOI] [PubMed] [Google Scholar]
- 37.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2019; 166: 11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine and Growth Factor Reviews. 2015. page 425–49. [DOI] [PubMed] [Google Scholar]
- 39.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine. 2014; 20: 1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nientiedt C, Heller M, Endris V, Volckmar AL, Zschäbitz S, Tapia-Laliena MA, et al. Mutations in BRCA2 and taxane resistance in prostate cancer. Scientific Reports. 2017; 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castro E, Romero-Laorden N, del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. Journal of Clinical Oncology. 2019; 37: 490–503. [DOI] [PubMed] [Google Scholar]
- 42.Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DnA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discovery. 2017; 7: 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng HH, Salipante SJ, Nelson PS, Montgomery B, Pritchard CC. Polyclonal BRCA2 Reversion Mutations Detected in Circulating Tumor DNA After Platinum Chemotherapy in a Patient With Metastatic Prostate Cancer. JCO Precision Oncology. 2018; 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simmons AD, Nguyen M, Pintus E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: A case report. BMC Cancer. 2020; 20: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lallous N, Volik S v., Awrey S, Leblanc E, Tse R, Murillo J, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biology. 2016; 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Proof-of-Concept with PROTACs in Prostate Cancer. Cancer Discovery. 2020; 10: 1084–1084. [DOI] [PubMed] [Google Scholar]
- 47.Sumiyoshi T, Mizuno K, Yamasaki T, Miyazaki Y, Makino Y, Okasho K, et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as a biomarker for treatment of castration-resistant prostate cancer. Scientific Reports. 2019; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Azad AA, Volik S v., Wyatt AW, Haegert A, le Bihan S, Bell RH, et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clinical Cancer Research. 2015; 21: 2315–24. [DOI] [PubMed] [Google Scholar]
- 49.Prekovic S, van Royen ME, Voet ARD, Geverts B, Houtman R, Melchers D, et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol Cancer Ther. 2016; 15: 1702–12. [DOI] [PubMed] [Google Scholar]
- 50.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of Full-Length Androgen Receptor by Wild-Type SPOP, but Not Prostate-Cancer-Associated Mutants. Cell Reports. 2014; 6: 657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KAT, et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 17492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nature Communications. 2016; 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han D, Gao S, Valencia K, Owiredu J, Han W, Waal E de, et al. A novel nonsense mutation in androgen receptor confers resistance to CYP17 inhibitor treatment in prostate cancer. Oncotarget. 2017; 8: 6796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodhouse R, Dennis L, Li M, Burns C, Ma P, Meng W, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-gene blood-based comprehensive genomic profiling assay. Journal of Clinical Oncology. 2020; 38: e13685–e13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Antonarakis ES, Madison R, Snider J, Snow T, Sokol E, Chung J, et al. Association of BRCA alteration (alt) type with real-world (RW) outcomes to PARP inhibitors (PARPi) in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2020; 38: 5527–5527. [Google Scholar]
- 56.Abida W, Campbell D, Patnaik A, Sautois B, Shapiro JD, Vogelzang NJ, et al. Genomic characteristics associated with clinical activity of rucaparib in patients (pts) with BRCA1 or BRCA2 (BRCA) - mutated metastatic castration-resistant prostate cancer (mCRPC). . Journal of Clinical Oncology. 2020; 38: 178–178. [Google Scholar]
- 57.Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Annals of Oncology. 2017; 28: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conteduca V, Jayaram A, Romero-Laorden N, Wetterskog D, Salvi S, Gurioli G, et al. Plasma Androgen Receptor and Docetaxel for Metastatic Castration-resistant Prostate Cancer. European Urology. 2019; 75: 368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Torquato S, Pallavajjala A, Goldstein A, Valda Toro P, Silberstein JL, Lee J, et al. Genetic Alterations Detected in Cell-Free DNA Are Associated With Enzalutamide and Abiraterone Resistance in Castration-Resistant Prostate Cancer. JCO Precision Oncology. 2019; 3: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kohli M, Tan W, Zheng T, Wang A, Montesinos C, Wong C, et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine. 2020; 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clinical Cancer Research. 2018; 24: 4437–43. [DOI] [PubMed] [Google Scholar]
- 62.Jensen K, Konnick EQ, Schweizer MT, Sokolova AO, Grivas P, Cheng HH, et al. Association of Clonal Hematopoiesis in DNA Repair Genes With Prostate Cancer Plasma Cell-free DNA Testing Interference. JAMA Oncology. 2021; 7: 107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ptashkin RN, Mandelker DL, Coombs CC, Bolton K, Yelskaya Z, Hyman DM, et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncology. 2018. page 1589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjamp M, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet. 2020; 52: 778–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.