

Abstract
Purpose
Neurolysin (Nln) is a peptidase that functions to preserve the brain following ischemic stroke by hydrolyzing various neuropeptides. Nln activation has emerged as an attractive drug discovery target for treatment of ischemic stroke. Among first-in-class peptidomimetic Nln activators, we selected three lead compounds (9d, 10c, 11a) for quantitative pharmacokinetic analysis to provide valuable information for subsequent preclinical development.
Methods
Pharmacokinetic profile of these compounds was studied in healthy and ischemic stroke-induced mice after bolus intravenous administration. Brain concentration and brain uptake clearance (Kin) was calculated from single time point analysis. The inter-relationship between LogP with in-vitro and in-vivo permeability was studied to determine CNS penetration. Brain slice uptake method was used to study tissue binding, whereas P-gp-mediated transport was evaluated to understand the potential brain efflux of these compounds.
Results
According to calculated parameters, all three compounds showed a detectable amount in the brain after intravenous administration at 4 mg/kg; however, 11a had the highest brain concentration and brain uptake clearance. A strong correlation was documented between in-vitro and in-vivo permeability data. The efflux ratio of 10c was ~6-fold higher compared to 11a and correlated well with its lower Kin value. In experimental stroke animals, the Kin of 11a was significantly higher in ischemic vs. contralateral and intact hemispheres, though it remained below its A50 value required to activate Nln.
Conclusions
Collectively, these preclinical pharmacokinetic studies reveal promising BBB permeability of 11a and indicate that it can serve as an excellent lead for developing improved drug-like Nln activators.
Keywords: enzyme activators, ischemic stroke, Neuropeptidase, pharmacokinetic study, small molecules
Introduction
Focal brain ischemia results from a thrombus or an embolus in cranial blood vessels and can cause a complex series of pathophysiological events resulting in localized brain tissue injury. Reperfusion strategies, using mechanical devices and thrombolytic agents, have been shown to be effective for recanalizing large occluded vessels in a subset of patients within a window of 0–24 h of symptom onset (1). A neuroprotective approach, on the other hand, proposes that the delivery of neuroprotective agent(s) could prevent or lessen the injurious biochemical and molecular events of the ischemic cascade and lessen the size of the infarction. However, this approach for acute ischemic stroke has been challenged to translate successfully in randomized clinical trials (2, 3). An opportunity also exists for combining reperfusion therapy with neuroprotection which could maximize ischemic tissue salvage. However, the class of neuroprotective drug needs to be carefully selected with regard to the timeline of this treatment regimen. Considering the complexity of the ischemic cascade, it has been proposed that drugs with multifaceted mechanisms of action are theoretically most efficacious (4).
Neurolysin is a zinc metallo endopeptidase that belongs to the M3 family of metallopeptidases and is involved in the processing of bioactive peptides that are expressed by neuronal and glial cells (5). It is widely distributed in the CNS and found in cytosolic, mitochondrial and membrane-bound forms (6, 7). Neurotensin, substance P and bradykinin are the most studied extracellular substrates of Nln in the setting of ischemic stroke, and have various functions during acute and chronic phases of the disease (8, 9). More specifically, in the acute post-ischemic brain these neuropeptides promote hyperpermeability of the blood brain barrier (BBB), neuroinflammation, excitotoxicity, and oxidative stress, and their inactivation by Nln facilitates cerebroprotection (10, 11). Three other bioactive peptides, angiotensin-(1–7), Leu- and Met-enkephalins, which are generated from their precursors as a result of Nln hydrolysis, function to enhanced ischemic tolerance, reduce inflammation, improve cerebral blood flow and afford neuroprotection after stroke. Based on this putative mechanism, to simultaneously inactivate several deleterious and generate cerebroprotective peptides, Nln is viewed as a potential therapeutic target which can modulate function of several, independent peptidergic systems to protect the brain from ischemic injury. Importantly, this notion has been confirmed in an experimental stroke model, demonstrating that inhibition of Nln after ischemia exacerbates brain infarction and edema, BBB dysfunction, neuroinflammation and neurological impairment, whereas brain overexpression of the peptidase before stroke leads to substantially improved disease outcomes (11). Remarkably, the hypothesized cerebroprotective function appears to be the reason for the observed compensatory upregulation of Nln one day after ischemic insult in primary cortical neurons (7, 12) and stroke-affected mouse brain (13). Therefore, there is growing interest in the pharmacological activation of Nln to harness its multi-mechanism cerebroprotective function in the setting of acute brain injury and substantial effort has been made to develop small, ‘drug-like’ molecules that can selectively enhance the catalytic activity of the peptidase (14).
In recent in vitro drug development studies in our lab, we introduced peptidomimetic activators of Nln that showed improved metabolic stability and in vitro BBB permeability compared to dipeptide hit compounds (14, 15). Based on these findings, the main goal of this study was to determine the in vivo pharmacokinetic profile and brain penetration of the leading peptidomimetic Nln activators 9d, 10c and 11a (15), with the objective to develop high-potency, ‘drug-like’ Nln activators with optimal pharmacokinetic properties, which can reach the brain at pharmacological concentrations upon peripheral administration and enhance activity of the peptidase. These activator molecules will be used for future structure activity studies to eventually evaluate the effect of Nln activation on stroke outcome.
Drug metabolism and pharmacokinetics (DMPK) play a crucial role in compound selection and progression in modern drug discovery. However, drug development for the central nervous system remains challenging because of the increased protection and regulation afforded to the brain compared to other organs (16). Knowledge about the physicochemical properties that determine CNS penetration, the blood-brain barrier (BBB)’s impact on CNS uptake, and the application of rational measures of CNS penetration related to drug efficacy could result in successful drug development. Within drug discovery programs, determination of brain drug level is regularly performed in experimental animals. Ultimately, by utilizing these methods we wanted to find a relationship between in vivo and in vitro potency, recognizing that application of total drug amount in the brain (Abrain) for subsequent evaluation of efficacy has some limitations. It is well-known that unbound drug concentration is responsible for exerting the effect at the target. However, a large amount of drug in brain tissue does not necessarily mean that high concentrations are available for the receptor because drug molecules might bind to tissue components. The measurement of drug amount can often be misleading. Therefore, initial assessment of the unbound drug concentration in brain interstitial fluid (ISF) is essential in directing early drug discovery research (17). Due to the active efflux and influx transporters at the BBB, the unbound drug concentration in the brain is not equal to the unbound drug concentration in plasma. Drug concentration in cerebrospinal fluid (CSF) is more closely related to the unbound drug concentration in brain interstitial fluid (Cu,brainISF) because of the presence of the blood-CSF barrier. However, CSF has a different turnover compared to brain ISF (18, 19).
In the present investigation, we performed a thorough pharmacokinetic study to measure the in vivo BBB permeability of candidate, peptidomimetic Nln activators and obtained their plasma profiles. We also completed ex-vivo brain uptake studies using a brain slice technique to obtain the Vu, brain (unbound volume of distribution) for these compounds. Further, we used Vu, brain to calculate various parameters (Kp,uu,cell, Kp,uu,brain, etc.) to estimate their CNS distribution. Moreover, using P-gp overexpressing cells, we performed an efflux transporter assay in vitro. Lastly, one Nln activator was selected, based on its high brain concentration and brain uptake clearance, for further studies in experimental stroke animals to compare its pharmacokinetic profile and brain uptake in healthy and disease conditions.
Materials and Methods
Chemicals and Reagents
Unless stated otherwise, chemicals were purchased from Fisher Scientific. We purchased heparin solution from APP Pharmaceuticals (Schaumburg, IL, USA). AssayReady MDR1-MDCK 12-well for efflux transport assay was ordered from MB Biosciences LLC. Cyclosporine A (CsA) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). All other chemicals were analytical grade and purchased from commercial sources.
Animals
Adult male C57BL/6 J mice aged 2–3 months were purchased from Charles River (Wilmington, MA). The mice were kept in ventilated cages in a temperature and humidity-controlled environment along with 12-h dark-light cycles and free access to food and water. All animal procedures were approved by the Institutional Animal Care and Use Committee at Texas Tech University Health Sciences Center and complied with the National Research Council guidelines for care and use of animals.
In Vivo Brain Uptake Study of Peptidomimetic Nln Activators
For the pharmacokinetic study, the adult male mice (n = 4–6 animals per time point) with 23–27 g body weight were injected with 4 mg/kg of compounds 11a, 9d, or 10c (dissolved in saline) through IV bolus injection. A silicone face mask was used to apply isoflurane (4% for induction, 1.5–2% v/v for maintenance) in 70% nitrous oxide/30% oxygen at a flow rate of 1 L/min. The jugular veins were exposed at the neck by skin incision for IV injections and blood sampling. Blood samples (40 μL) were collected at 1, 5, 10, 20, 30, and 60 min in microtubes containing heparin. The samples were collected to generate plasma concentration-time curves in each animal. At the last time point, trans cardiac perfusion was performed for vascular space correction. Briefly, the thorax was opened immediately, and 40 mL phosphate-buffered saline (pH 7.4) at room temperature was used to perform the vascular perfusion via the heart’s left ventricle (flow rate of 5 mL/min) using a Harvard syringe pump. Collected blood samples were centrifuged for 10 min at 6000 g, and supernatant plasma was separated. The brain was collected after euthanasia by cervical dislocation. Meninges were removed from the collected brains, and the forebrains were weighted without olfactory bulbs, cerebellum, or brain stem. Then, the brain samples were homogenized, and plasma samples were diluted according to the sample preparation steps described previously (15). The homogenized brain and diluted plasma were stored at −80°C until the LC-MS/MS system measurement.
Pharmacokinetic Analysis
The apparent terminal elimination rate constants (k) were determined by linear least-squares regression through all samples’ last three plasma-concentration time points. The apparent elimination half-life (t1/2) was calculated as 0.693/k. The area under the plasma concentration-time curves (AUC0–T) was determined using the logarithmic trapezoidal method from zero to the last sampling time.
Brain Uptake Clearance of Nln Activators Using Single Time Point Analysis
The initial brain uptake clearance (Kin) was calculated using the below equation:
| (1) |
AUC0-T denotes the area under the plasma concentration-time curve from time point 0 to the terminal sampling time. Cbr is the brain concentration of Nln activators reported as percent injected dose (%ID/mL). It should be noted that Kin values in this study are calculated based on total plasma concentration.
In Vitro-In Vivo Correlation
For a comparison between in vitro and in vivo permeability of Nln activators, the Kin values or permeability surface area products (PS) were converted to permeability coefficients, taking 120 cm2/g of the brain as the surface area of the BBB in vivo (20). In vitro permeability coefficient for each compound was obtained from in vitro studies of Nln activators as described previously using co-culture model of bEnd3 cells and primary astrocytes (15).
P-Glycoprotein (Efflux Transporter) Substrate Assay
MDR1-MDCK Cells (P-Gp-Overexpressing Cells) and Culture Condition
MDR1-MDCK cells that had been transfected with the human MDR1 gene to overexpress P-gp were purchased from MB Biosciences LLC (Natick, MA). The cells were maintained in DMEM supplemented with 10% FBS, non-essential amino acid, penicillin/streptomycin, L-glutamine, and colchicine as recommended by the supplier. Monolayer cultures were grown at 37°C in a 5% CO2/95% air atmosphere on a microporous polycarbonate membrane in 12-well Costar Transwell inserts. After the shipping medium was changed to a fresh MDR1-MDCK cell culture medium, the plate was kept in the incubator for 24 h before performing the experiment.
P-Gp Substrate Assay Using P-Gp-Overexpressing Cell Monolayers
The transport of Nln activators from apical to basolateral side (A-to-B) of Transwell inserts was assessed in the presence or absence of 10 μM cyclosporine A (CsA) which completely inhibits P-gp function at this concentration (21). Approximately 1 h before the initiation of the transport experiment, the medium in both compartments was replaced with transport medium, Hanks’ balanced salt solution, with or without the inhibitor. The transport experiment was started by replacing the apical chamber medium with HBSS containing 10 μg/mL of peptidomimetic Nln activators. The medium on the basal compartment was replaced with fresh HBSS with or without inhibitor. The inhibitor was present on both sides of the membrane during the preincubation and transport period. After 2 h of incubation at 37°C, samples were collected from the basal chamber. To confirm the integrity of the cell monolayer in each well, sodium fluorescein was added with the test compounds at the beginning of the experiment to the apical chamber. The concentration of test compounds in the transport buffer was measured by LC-MS/MS. The apparent permeability coefficient (Papp; cm/s) was calculated using the literature method (22). Papp in the absence (Papp,A-to-B(−CsA)) or presence (Papp,A-to-B(+CsA)) of the P-gp inhibitor CsA was used to calculate unidirectional flux ratio (UFR), which is defined by the following equation:
| (2) |
In a parallel study, the test compounds were added to the basolateral compartment, and samples were collected from the apical chamber at the end of 2-h incubation to evaluate the basolateral to apical (B-to-A) permeability of Nln activators. From this method, the efflux ratio (ER) was determined as shown in eq. 3, which is defined as the fold difference between the basolateral-to-apical (B-to-A) transfer rate of the compound across a cell monolayer relative to its apical-to-basolateral (A-to-B) transfer rate.
| (3) |
Brain Slice Method for Studying Drug Distribution in the CNS
Brain slices were prepared according to the method described in the literature (23, 24). Briefly, drug-naïve mice are anesthetized using 5% isoflurane inhalation. After reaching deep anesthesia, the animal is decapitated, the brain is isolated, and immediately placed in blank ice-cold artificial extracellular fluid (aECF) bubbled with oxygen. Then using the McIlwain tissue chopper, 8–10 brain slices with the thickness of 300 μm are cut on a coronal plane. Slices with intact edges are transferred to a beaker filled with cold oxygenated blank aECF.
The incubation process starts by transferring brain slices from the storage beaker into the flat-bottom glass beaker containing 10 mL of the aECF with 1 μM of Nln activators. The beaker is then covered and placed on a shaker at 37°C for 5 h. The rotation speed of the beaker is 45 rpm, and continuous oxygen flow is maintained at 70–80 mL/min.
200 μL sample of aECF is collected directly from the incubation beaker at the beginning and the end of the incubation period in duplicate without taking any brain debris. The aECF samples are then transferred to Eppendorf tubes containing 200 μL 5D blank brain homogenate prepared beforehand with blank aECF. The brain homogenate is included to match the matrix of the slice homogenates as required for LC-MS/MS analysis. Then brain slices are collected individually, dried with filter paper, weighed, and homogenized with 9 volumes of blank aECF using a tissue homogenizer. The samples are then stored at −20°C for further analysis. Bioanalytically determined compound concentrations in brain slices and aECF are used to determine Vu, brain, Vu,cell, Kp,uu,cell, and Kp,uu,brain (25, 26). LDH assay was also performed to confirm the viability of brain slices after each experiment using the cytotoxicity detection kit (Sigma Aldrich, IN, USA). Data from experiments with 85–90% viability range was considered for analysis (26).
Brain Uptake Profile and Uptake Clearance of 11a Using Multiple Time-Point Analysis
The Kin values for 11a were also estimated using multiple time point analysis. For this purpose, after IV injection of 4 mg/kg 11a to animals, brains were collected at 5, 10, 20, 30, and 60 min after performing trans cardiac perfusion. Plasma and brain samples were analyzed using LC-MS/MS.
In-Vivo Model: Middle Cerebral Artery Occlusion (MCAO) in Mice
MCAO surgery was performed in male C57BL/6 mice (25–28 g) as previously reported (27). The surgery was performed using a Zeiss OPMI pico I surgical microscope (Carl Zeiss GmbH, Jena, Germany) on an S100 suspension system with a 3 CCD Toshiba digital camera. Using a facemask and a SurgiVet Vaporizer (Smiths Medical North America, Waukesha, WI, USA), mice were anesthetized with 4% isoflurane and maintained at 1.5% isoflurane in N2O/O2 mixture (70/30). Continuous blood flow was measured using a laser Doppler probe (Moor Instruments, Wilmington, DE, USA). After exposing the skull with a small incision, the laser probe was placed directly above the left middle cerebral artery (MCA) with the support of a 2 cm long tube. Body temperature was maintained at 37°C throughout the procedure and was monitored by a thermostatic blanket (TC-1000 Temperature Controller, CWE, USA). The animal was placed in the supine position, and a 1.5 cm long incision was made on the midline of the neck. The left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were carefully from surrounding tissue. Then the common carotid artery was occluded, ECA was ligated, and a micro clip was placed on the ICA. Using a small incision on the ECA, a 6–0 nylon microfilament with a round tip (0.20–0.25 mm) was introduced to block the middle cerebral artery bifurcation. 80% decrease of blood flow from baseline detected by laser doppler was considered a successful occlusion. After 60 min of occlusion, the nylon filament was removed to restore blood flow and achieve reperfusion. An increase of 70% or more occlusion blood flow was considered successful reperfusion; otherwise, the animal was excluded from the experimental group.
To confirm the incidence of ischemic stroke and to observe the infarct area, TTC (2,3,5-triphenyl tetrazolium chloride) staining was used after 3 h of reperfusion in some animals (28).
Pharmacokinetic and Brain Uptake of 11a in MCAO-Induced Mice
To compare the PK profile of 11a in healthy and stroke animals, 4 mg/kg of 11a was injected to mice exposed to 1-h occlusion and 3 h reperfusion through the jugular vein as described in section 2.3. Plasma samples were collected at 1, 5, 10, 20, and 30 min. The brain was extracted after trans cardiac perfusion and plasma, and brain samples were processed for LC-MS/MS analysis as described previously. Plasma concentration-time profile, brain concentration, AUC, and Kin were determined in all animals and compared with the control group.
Statistical Analysis
Data are presented as mean ± SD. Statistical analysis was performed in Prism 9 (GraphPad Software, La Jolla, CA). Data with more than two groups were analyzed by 1-way ANOVA followed by Tukey’s multiple comparison test. In all cases, p < 0.05 was considered significant.
Results
Single Time Point Analysis
To investigate the PK profile of Nln activators, 4 mg/kg of 11a, 9d, or 10c was injected into mice through IV bolus injection, and plasma samples were collected at different time points until 60 min then the brain was extracted. As depicted in fig. 1.A, the plasma profiles of the three compounds were similar, and they showed a biexponential decline in plasma with a rapid distribution phase and a slower elimination phase. AUC was calculated using the logarithmic trapezoidal rule from time zero to the last sampling time. As shown in Table 1, the AUC values for these activators are significantly different, and 9d showed the highest AUC compared to 10c and 11a. The apparent elimination half-life was calculated, and 10c and 11a showed a similar plasma half-life of almost 30 min; however, the elimination half-life of 9d is significantly shorter (9.17 ± 1.8 min, p < 0.0001) compared to the other two compounds (Table 1). Comparing the brain concentrations of these activators indicated a significant difference (5-fold) in the 11a group compared to 9d and 10c, which showed similar brain concentrations. Brain concentration (%ID/mL) for 11a was 0.04 ± 0.005 whereas it was 0.007 ± 0.0008 and 0.004 ± 0.001 for 10c and 9d respectively (p < 0.0001). Similarly, the Kin value for 11a was significantly higher than that of the other compounds (5- and 2-fold higher than 9d and 10c, respectively). Although the brain concentration of 10c and 9d was similar, the brain uptake clearance of 10c was significantly more than that of 9d (p < 0.001) (Fig. 1.B). All compounds had a higher brain concentration and Kin value compared to [14C] sucrose (data not shown).
Fig. 1. Pharmacokinetic profile of peptidomimetic Nln activators up to 60 min after IV bolus injection.
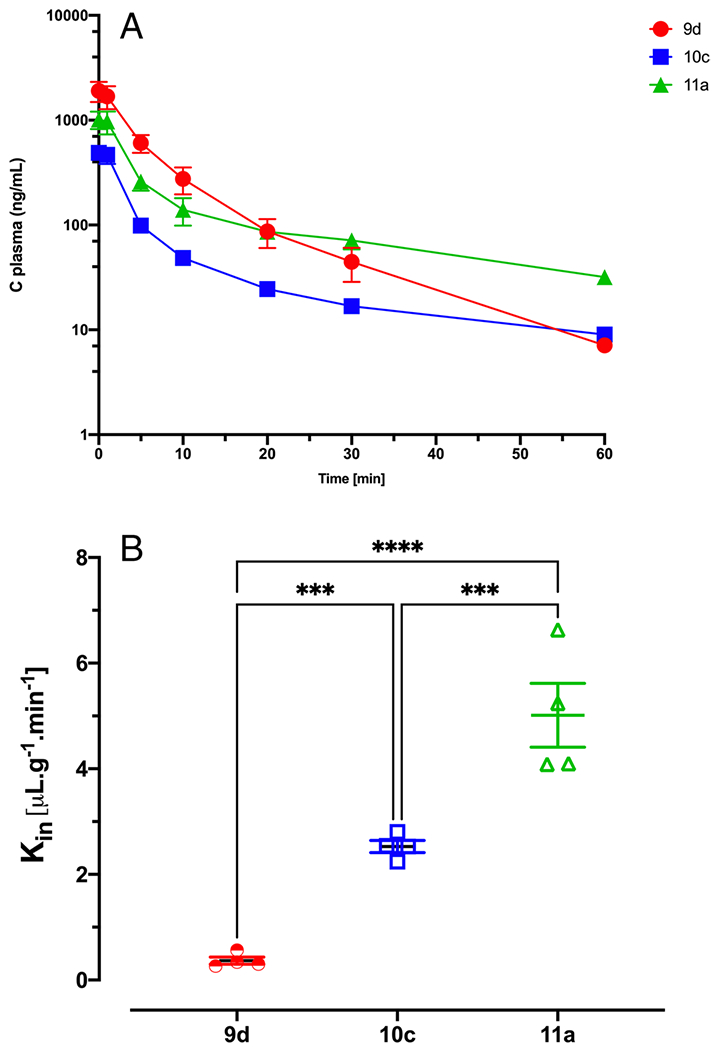
(A) Plasma concentration-time profile of Nln activators showed biexponential decline. (B) Brain uptake clearance (Kin) of Nln activators using single time point analysis. 11a showed the highest brain concentration and brain uptake clearance compared to other compounds. Data are presented as mean ± SD of 4 animals per compound. In all cases, p < 0.05 was considered significant.
Table 1.
Nln activators’ PK parameters
| Structure | AUC (%ID/mL−1.min) | t1/2 (min) | |
|---|---|---|---|
| 11a |
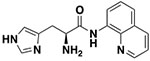
|
8 ± 1.1 | 27.94 ± 2.76 |
| 9d |
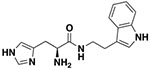
|
12.6 ± 0.53 | 9.2 ± 1.8 |
| 10c |
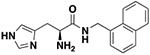
|
3 ± 0.46 | 29.41 ± 4.9 |
Data are presented as mean ± SD of 4 animals per compound
In Vitro-In Vivo Correlation of Nln Activators Based on Permeability Coefficient
The inter-relationship between log octanol/water partition coefficients with in-vitro and in-vivo permeability were studied to determine the extent of central nervous system penetration. For comparing in-vitro and in-vivo permeability data, Kin values were converted to permeability coefficient using the surface area of the BBB in-vivo (120 cm2/g brain). There is a strong correlation between in-vitro and in-vivo permeability data, and compound 9d has the lowest permeability coefficient in both platforms, reflecting its lower LogP value (Fig. 2).
Fig. 2.
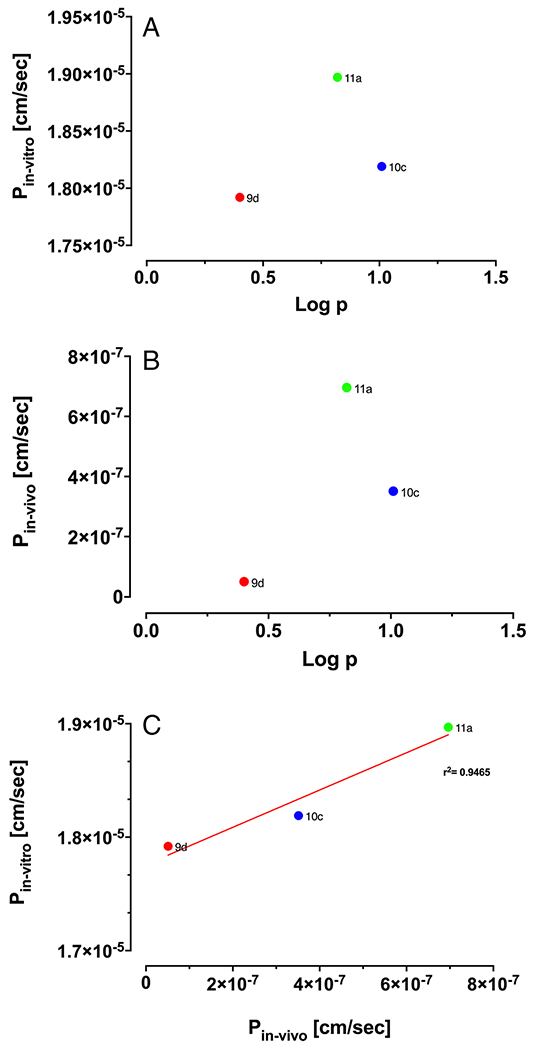
(A) Correlation of in vitro permeability coefficient (n = 4) and LogP (n = 3). (B) Correlation of in vivo permeability coefficient (n = 4) and LogP (n = 3) (C) In vitro and in vivo correlation of Nln activators based on permeability coefficient.
P-Gp Substrate Assay
The permeability of the reference compounds was assessed in the apical to basolateral (A-to-B) direction in Transwell inserts in the presence or absence of the P-gp inhibitor CsA (10 μM), using P-gp-overexpressing cells that had been stably transfected with the human MDR1 gene. This experiment was used to investigate if the compounds are substrate of P-gp efflux transporter and to predict the level of P-gp transport potential (21). The results for the unidirectional membrane transport of these reference compounds across the P-gp-overexpressing cell monolayers, along with the UFRs and ERs, are summarized in Fig. 3 and Table 2. In the presence of a p-gp inhibitor, the permeability of 10c and 11a increased significantly; however, the addition of inhibitor did not change the permeability of 9d in the A-to-B direction (Fig. 3A). Moreover, the permeability coefficient of 11a and 10c in the basal to apical (B-to-A) direction was significantly more than their permeability coefficients in the A-to-B direction which results in higher efflux ratio. The permeability direction did not affect the permeability of 9d significantly.
Fig. 3. Permeability coefficient of Nln activators in MDR1-MDCA P-gp overexpressed cell monolayer.
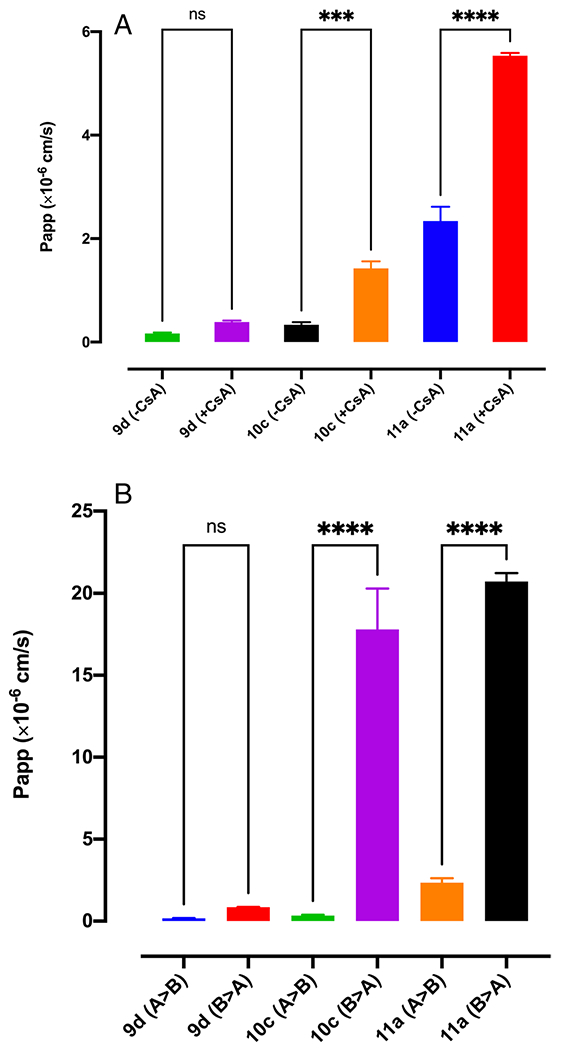
(A) Permeability of Nln activators in the presence and absence of P-gp inhibitor (CsA) (n = 4). (B) Permeability of Nln activators in apical to basolateral (A > B) and basolateral to apical (B > A) direction (n = 3).
Table 2.
UFR and ER of Nln Activators Obtained from Permeability Assay with MDR1-MDCK P-gp Overexpressed Cells
| UFR (+CsA/−CsA) | Efflux Ratio | |
|---|---|---|
| 11a | 2.36 ± 0.04 | 8.85 ± 0.3 |
| 9d | 2.31 ± 0.29 | 5.1 ± 0.1 |
| 10c | 4.24 ± 0.7 | 52.98 ± 5.25 |
Nln Activators’ Distribution in the CNS Using Brain Slice Uptake
Using the brain slice uptake method, first, we obtained the volume of distribution of the unbound drug in the brain (Vu,brain). As shown in Table 3, all three compounds showed very high Vu,brain, which can be due to their slightly basic structure that results in trapping these molecules in cellular compartments such as lysosomes. Then using the brain homogenate binding experiment and taking Vcell into account in the dilution factor, the affinity of compounds for physical binding inside the cells (Vu, cell) was obtained. It was observed that Nln activators showed extensive tissue binding and distribution to the intracellular spaces. Kp,uu,cell, the ratio of intracellular to extracellular unbound drug concentrations was calculated for each compound using Vu,brain from the slice method, and Vu,cell from the homogenate method. All compounds had a Kp,uu,cell greater than 1 (Table 3). The basic nature and extensive tissue binding and distribution to the intracellular space of the compounds result in higher intracellular exposure. The active transport system may also play a role in high intracellular exposure of Nln activators. The last application of Vu,brain, was the determination of unbound brain-to-plasma concentration ratio for Nln activators. The brain-to-plasma AUC ratio (Kp,brain) of Nln activators obtained from in vivo brain uptake was corrected with fu,plasma, and Vu,brain to estimate the BBB net flux (Kp,uu,brain) or percent unbound drug passing from plasma to brain (Table 3). The brain-to-blood concentration ratios of the unbound drug (Kp,uu,brain) were found to be lower than 1 for all three activators, suggesting an active efflux phenomenon at the BBB. However, it was found that ~5% of unbound 11a is entering the brain from plasma, and this is around 1% and 3% for 10c and 9d, respectively.
Table 3.
Nln Activators’ Distribution in the CNS using Brain Slice Uptake
| Vu, brain (mL/g) | Vu,cell (mL/g) | Kp,uu,cell | %Unbound passing from plasma to brain | |
|---|---|---|---|---|
| 9d | 47.23 ± 8.1 | 8.29 ± 0.35 | 7.88 ± 1.16 | 3.5 ± 0.75 |
| 10c | 101.26 ± 18.27 | 23.06 ± 0.54 | 6.28 ± 0.56 | 1.37 ± 0.51 |
| 11a | 71.03 ± 9.16 | 20.65 ± 0.44 | 5.1 ± 0.45 | 4.8 ± 0.75 |
Data represents the mean ± SD of 3 independent experiments (6–8 brain slices/compound/experiment)
Brain Uptake Profile and Uptake Clearance of 11a
The plasma and brain concentration-time profile of 11a is shown in fig. 4.A. Brain concentration (%ID/mL) of 11a is similar at different time points following IV injection of 4 mg/kg 11a with a slight increase until 30 min and then reduction at 60 min (Fig. 4.B). Kin values were calculated for each time point and are shown in fig. 4.C. According to the data, the Kin value of 11a declines by 40–60% at higher sampling time points which is due to the predominance of efflux to influx at later time points. The average Kin value of 11a at 5 min was 12.31 ± 1.86 μL.min−1.g−1 while at 30 and 60 min it reduced to 5.03 ± 1.4 and 5.01 ± 1.2 μL.min−1.g−1.
Fig. 4. Brain uptake profile and uptake clearance of 11a using multiple time point analysis.
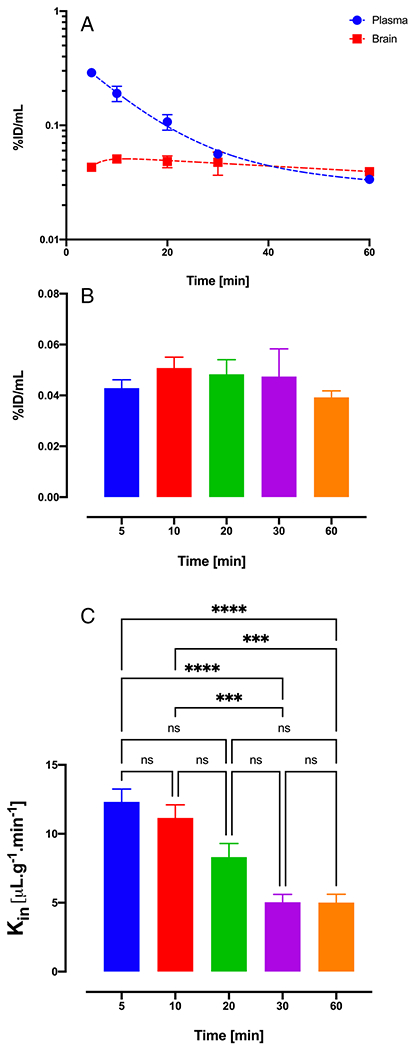
(A) Plasma and brain pharmacokinetic profile reported as percent injected dose of 11a. (B) Brain concentrations of 11a at different time points until 60 min. (C) Brain uptake clearance of 11a using multiple time points. Data are represented as mean±SD. (n=4 animals for each time point).
Pharmacokinetic Profile and Brain Uptake Clearance of 11a in Experimental Stroke Mouse Model (MCAO)
To investigate the PK profile and brain uptake clearance of 11a in stroke condition, MCAO surgery was performed on C57BL/6 mice by 1 h of occlusion followed by 3 h of reperfusion. One-hour occlusion was selected based on previous publications about the upregulation of Nln in the ischemic brain following stroke (13). Animals with an 80% decrease in blood flow during occlusion followed by an increase of 70% or more occlusion blood flow were included in the PK study. Figure 5. is representative TTC staining of the ischemic brain following 1-h occlusion and 3 h reperfusion and shows infarct area within the ischemic hemisphere. The plasma concentration-time profile of 11a in MCAO-induced animals is shown in fig. 6. The plasma profile of 11a was similar for control and stroke animals when blood samples were collected until 30 min after IV injection. Overall, the AUC of plasma concentration-time was higher for the MCAO group. Also, the total concentration of 11a was more elevated in the ischemic brain compared to the control brain. Interestingly, the brain concentration of 11a was significantly higher in the ischemic hemisphere compared to the control brain, and there was a 2.5-fold increase in brain concentration. However, the concentration of 11a in the ischemic hemisphere was not significantly higher than the non-ischemic one. Regarding the brain uptake clearance of 11a, the Kin value for the ischemic hemisphere was considerably higher than the non-ischemic hemisphere and control brain. Kin of 11a for the ischemic hemisphere was 9.4 ± 0.8, whereas it was 7 ± 1.2 and 5 ± 1.4 μL.min−1.g−1 for the non-ischemic hemisphere and control brain (Fig. 7).
Fig. 5. Middle cerebral artery occlusion.
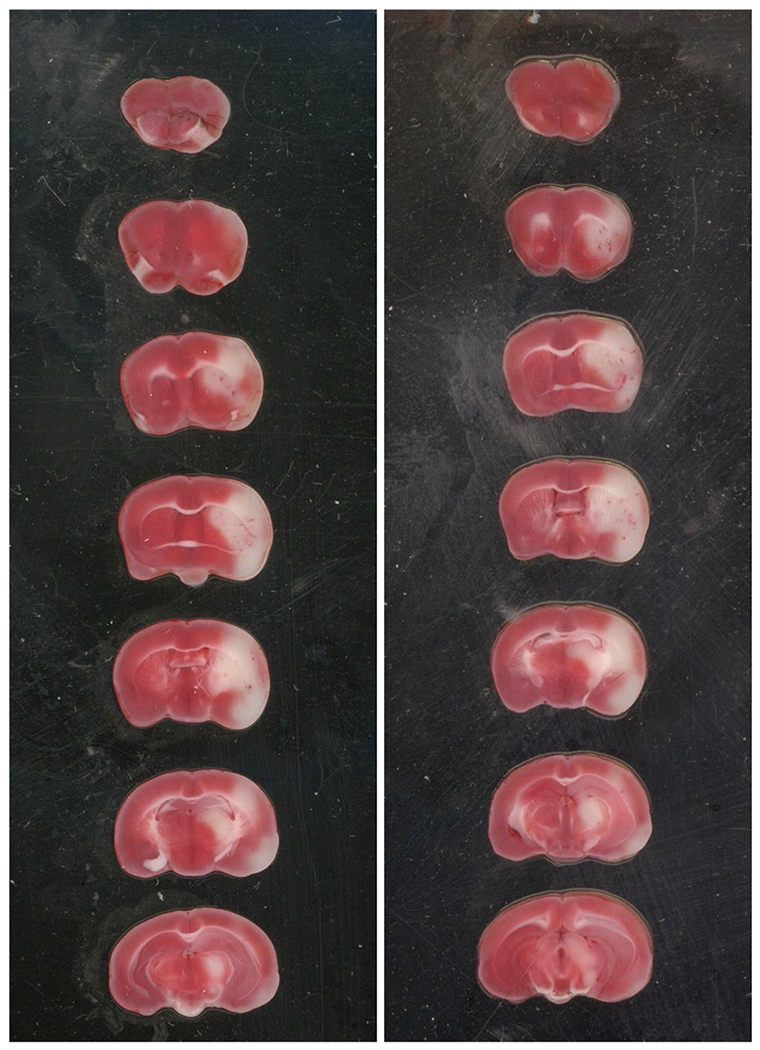
Representative TTC staining of brain slices after 1 h occlusion and 3 h reperfusion.
Fig. 6. Pharmacokinetic profile of 11a in experimental stroke mouse model (MCAO).
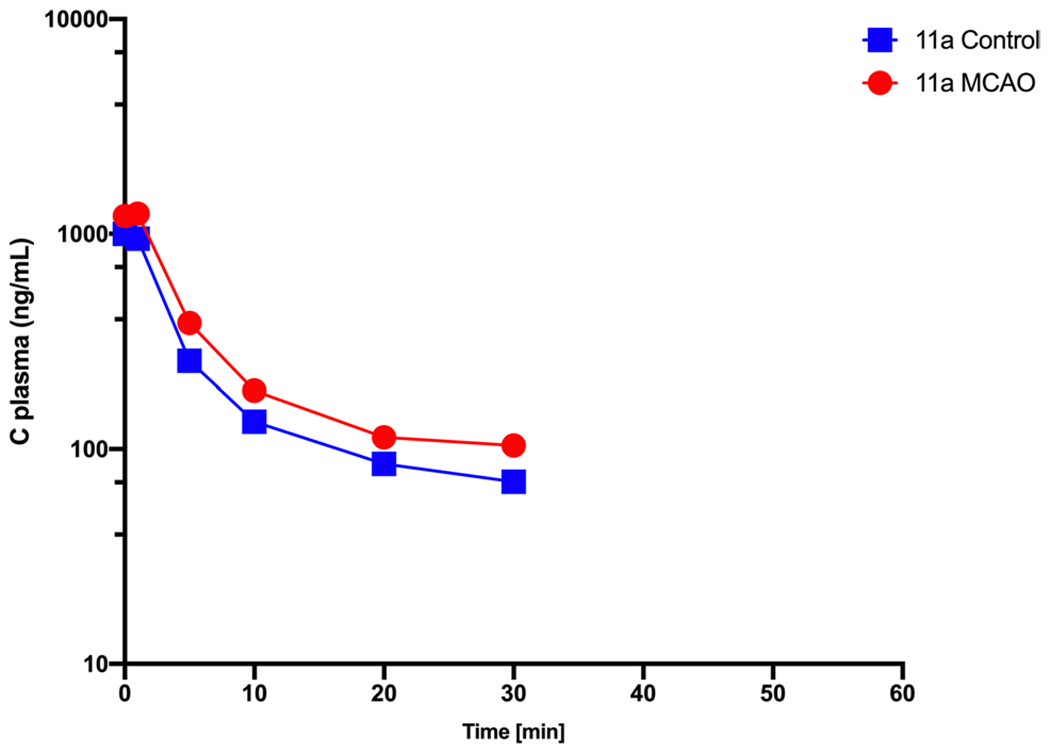
Plasma concentration-time profile of 11a in control and MCAO group. Plasma profile of stroke animals is like control group and follows biexponential decline. Data are represented as mean ± SD (n = 4 animals).
Fig. 7. Brain profile of 11a in MCAO animals.
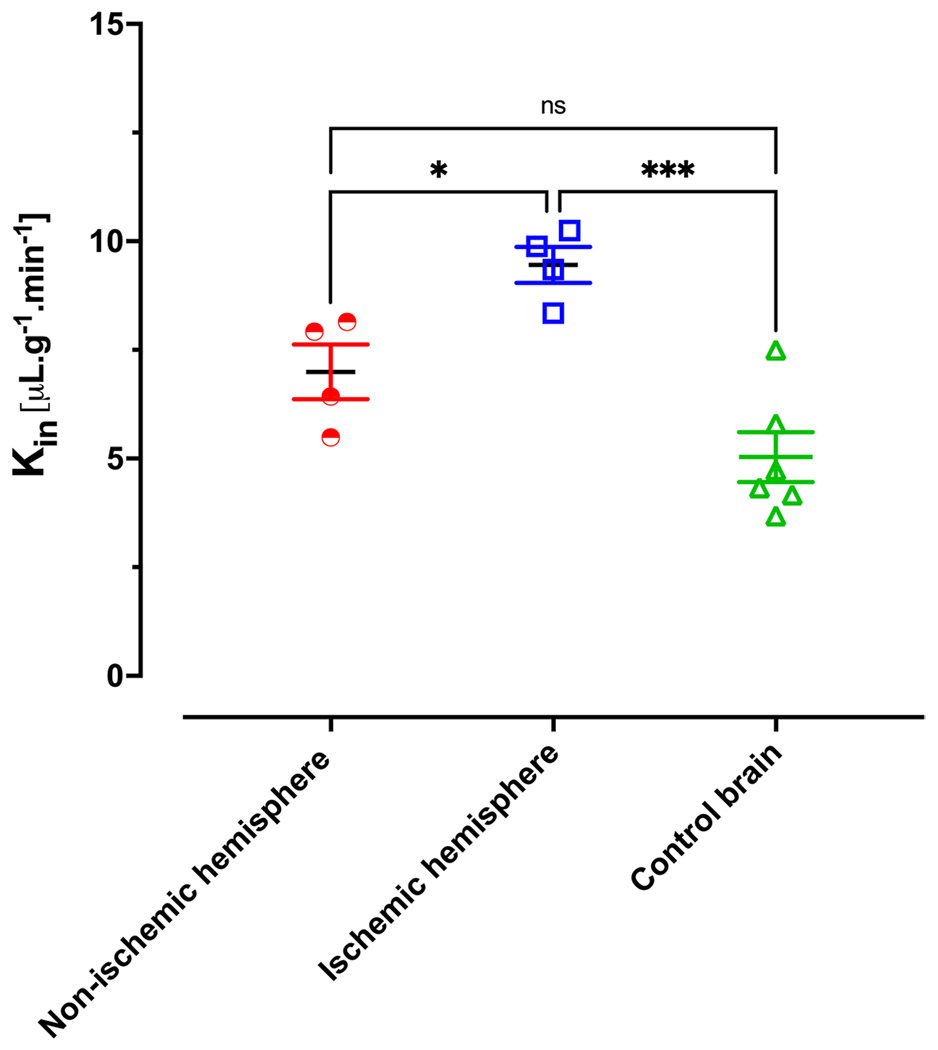
Brain uptake clearance of 11a. Kin values for ischemic hemisphere is significantly more than non-ischemic hemisphere and control brain however, non-ischemic hemisphere has similar brain uptake with control brain. Data are represented as mean ± SD (n = 4–6 animals).
Discussion
The present study demonstrated that peptidomimetic Nln activators show a biexponential decline in plasma, and they have a rapid distribution phase followed by an elimination phase from plasma. Among these activators, 11a showed the highest brain concentration and brain uptake clearance compared to other compounds. Physicochemical properties and a delicate balance between active uptake and efflux transporters are significant determinants of a drug’s CNS penetration. As lipophilicity of a drug increases, the unbound fraction in the brain decreases, so there might be a reduction in available unbound drug for the target despite enough CNS penetration (29). Therefore, there should be a balance between sufficient CNS penetration and adequate free drug. Using polarized cell monolayers, the bidirectional transport of drug molecules is assessed to study the interplay between efflux and uptake transporters. In most cell lines, uptake transporters are confined in the basolateral membranes whereas the efflux transporters are localized in the apical membrane (30). Transwell assays are used to measure drug movement through cell monolayers, and a comparison of the ER and UFR determined in MDR1-MDCK cells is a commonly used method to assess whether a compound has limitations in accessing the brain. The UFR values increase depending on the potential of the P-gp substrate, and a compound is a P-gp substrate when the efflux ratio is >1.99 (21, 31). Although 10c showed more lipophilicity than 11a, it had lower permeability both in vitro and in vivo, so either 10c is an efflux transporter substrate, or active influx plays a significant role in the uptake of 11a. Following efflux assay, we observed that in the presence of CsA, the permeability of 10c and 11a increased significantly. Moreover, these two compounds’ permeability coefficient was substantially higher when the permeability experiment was performed in the B-to-A direction. Following calculation of UFR and ER, it was observed that 10c is a stronger efflux transporter substrate, and it has almost 6-fold higher ER than 11a which could be due to the aromatic group in the structure of 10c that is more hydrophobic compared to that of 11a (32, 33). Therefore, this compound’s in vitro and in vivo permeability is significantly lower than 11a despite their similar LogP values. Based on the reported data, it seems that these factors are interrelated, and compounds with low permeability that are also P-gp substrates may still enter the brain if tissue binding provides enough driving force to overcome the influence of the efflux transporters.
Determination of Vu,brain values using fresh brain slices are more relevant to the in vivo situation and is associated with a more accurate assessment of Cu,brainISF (i.e., Cu,buffer), which represents the unbound drug concentration that is responsible for occupying extracellular receptors and exerting therapeutic effects. Wang et al. introduced the unbound volume of distribution in the brain (Vu,brain) to relate Cu,brainISF to Abrain (34). Thus, Vu,brain describes the distribution of drugs inside the brain regardless of brain-to-plasma distribution. A low value for Vu,brain, close to the interstitial space volume (0.2 mL. gram−1 brain−1), would thus describe predominantly extracellular distribution. In contrast, a high value would indicate that the drug enters brain cells and binds to tissue components (35). All three peptidomimetic activators of Nln showed high ranges of Vu,brain; therefore, they have extensive tissue binding and are binding to tissue components.
The Vu,brain reflects the intracellular protein binding or membrane partitioning and the concentration gradient of the unbound drug across the cell membranes (Kp,uu,cell). Kp,uu,cell is related to the ion class of the drug molecule. Kp,uu,cell is higher for the basic compounds than for the neutral or acidic compounds. The intracellular exposure of Nln activators was also higher due to their basic nature. Influx or efflux transporters at the cell membrane of the brain cells could also influence Kp,uu’cell. There are no good or optimal values for Vu’brain, and it would, accordingly, be rational to determine Vu’brain only for those compounds that have available in vivo data for the amount of drug in the brain, i.e., the total brain concentration (26, 36). In drug molecules with Kp,uu’brain > 1, influx transport across the BBB is more efficient than efflux or bulk flow, and for Nln activators, this value was less than 1. However, by comparing this value with the in-vivo brain to plasma concentration ratio for each activator, we found that ~5% of unbound 11a is entering the brain from the plasma, which confirms our finding of higher concentration and brain uptake clearance of this compound in mice compared to 10c and 9d. One potential caveat is that our brain uptake studies are done in anesthetized mice, and it is possible that isoflurane could alter brain uptake of these experimental agents.
Since 11a showed better brain penetration than 10c and 9d, we selected this compound for further PK studies. First, we sought to find the optimum time of brain uptake clearance for 11a through an investigation of the brain profile of this compound at different time points. Initial brain uptake value (Kin) is higher at earlier time points since the transport of compounds from circulation to the brain predominates over the action of efflux transporters and this value tends to decrease over time since efflux becomes more substantial. We observed that within the studied time course, brain concentration of 11a increases until 30 min in the brain, although not statistically significant, and then decreases at 60 min. We also obtained brain uptake clearance of this compound at each time point, and after 30 min, the Kin value decreases 40–60% compared to initial time points due to an increase in the efflux component; therefore, based on the brain concentration and Kin, we selected 30 min as the optimum time of brain collection after 11a injection.
We also investigated the PK profile of 11a in mice after ischemic stroke and compared it with control animals. Pathophysiologic PK analysis, using a model of ischemic stroke, more precisely determines the brain disposition of an agent into brain regions that are damaged (ischemic hemisphere) versus contralateral (non-ischemic hemisphere). In early phases of acute ischemic stroke, ischemic core is formed. In the case of delayed reperfusion, the ischemic core is highly susceptible to become infarcted. The ischemic core is surrounded by a salvageable and reversibly injured brain tissue which is called penumbra. The survival of neurons is especially important in the penumbra and the apoptotic cascade can be blocked to save these injured neurons therefore, penumbra is the pharmacological target of acute stroke therapy (37–39).
Brain and plasma samples were collected thirty minutes after injection of 11a (4 mg/kg) in stroke-induced animals exposed to 1 h of occlusion and 3 h of reperfusion. It has been shown that 3 to 48 h after MCAO, reperfusion results in reperfusion-induced BBB damage hence brain edema and additional brain injuries (40). The plasma concentration-time profile was similar between the two groups; however, the brain concentration of 11a was higher in the injured brain compared to the control brain. As expected, the ischemic hemisphere contained significantly more 11a compared to the control brain. Moreover, brain uptake clearance of 11a was markedly higher in the ischemic hemisphere than in both the non-ischemic hemisphere and healthy brains. A higher amount of this compound on the ischemic side could be due to the opening of the BBB at the ischemic core and penumbra, upregulation and involvement of different uptake transporters or dysfunction of efflux transporters at the BBB level.
Overall, the reported data in this study support the hypothesis that BBB permeability, transporters, and the free fraction of drug in blood and brain all play crucial roles in determining the level of CNS penetration of a drug in vivo. Partitioning into brain tissue may act as a driving force to overcome some of the effects of efflux transporters at the BBB to promote CNS penetration. Therefore, it cannot be assumed that because a compound is a P-gp substrate, it cannot access the brain. In Table 4 we have summarized various pharmacologic parameters of compound 11a obtained from in vivo and in vitro drug development studies of this compound.
Table 4.
In Vitro and in Vivo Drug Development Parameters of Compound 11a
| Structure | LogP (octanol-water) | fu Brain and plasma | Pe in-vitro (cm/sec) | Kin 30 min(μL.g−1.min−1) | A50 (μ) | Brain concentration 30 min (μM) |
|---|---|---|---|---|---|---|
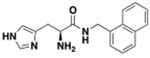
|
1.14 ± 0.1 | 0.062 ± 0.011 & 0.41 ± 0.081 | 18.97 ± 0.9 × 10−6 | 5.03 ± 1.01 | 7 ± 1.76 | 0.26 ± 0.07 |
Data represents the mean ± SD (n = 4–6)
Conclusion and Future Studies
Overall, although compound 11a showed higher brain concentration and brain uptake clearance compared to the other two lead compounds, it is not the ideal candidate for performing in vivo efficacy studies in an experimental stroke model since the brain concentration is still several fold lower than the A50 value (~7 μM) for this compound, shown in Table 4. However, compound 11a is now considered an excellent lead molecule for developing further compounds with higher potency for the activation of Nln and possible cerebroprotective effects after stroke injury. Further work is required to understand the role of other drug transporters (both influx and efflux) in CNS disposition and the applicability of estimating fu(brain) to understand drug efficacy at the level of the brain, where local free concentrations may differ from that in the bulk milieu.
Funding
This project was supported by NIH grant R01NS106879 to P.C.T., V.T.K. and T.J.A.
Footnotes
Acknowledgments and Disclosures Authors declare no conflict of interest.
References
- 1.Savitz SI, Baron J-C, Fisher M, Consortium SX. Stroke treatment academic industry roundtable X: brain cytoprotection therapies in the reperfusion era. Stroke. 2019;50(4):1026–31. [DOI] [PubMed] [Google Scholar]
- 2.Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2007;61(5):396–402. [DOI] [PubMed] [Google Scholar]
- 3.Tymianski M Combining neuroprotection with endovascular treatment of acute stroke: is there hope? Stroke. 2017;48(6):1700–5. [DOI] [PubMed] [Google Scholar]
- 4.Fisher M, Ratan R. New perspectives on developing acute stroke therapy. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2003;53(1):10–20. [DOI] [PubMed] [Google Scholar]
- 5.Checler F, Ferro ES. Neurolysin: from initial detection to latest advances. Neurochem Res. 2018;43(11):2017–24. [DOI] [PubMed] [Google Scholar]
- 6.Shrimpton CN, Smith AI, Lew RA. Soluble metalloendopeptidases and neuroendocrine signaling. Endocr Rev. 2002;23(5):647–64. [DOI] [PubMed] [Google Scholar]
- 7.Wangler NJ, Santos KL, Schadock I, Hagen FK, Escher E, Bader M, et al. Identification of membrane-bound variant of metalloendopeptidase neurolysin (EC 3.4. 24.16) as the non-angiotensin type 1 (non-AT1), non-AT2 angiotensin binding site. J Biol Chem. 2012;287(1):114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karamyan VT. The role of peptidase neurolysin in neuroprotection and neural repair after stroke. Neural Regen Res. 2021;16(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Ahmad AJ, Pervaiz I, Karamyan VT. Neurolysin substrates bradykinin, neurotensin and substance P enhance brain microvascular permeability in a human in vitro model. J Neuroendocrinol. 2021;33(2):e12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karamyan VT. Peptidase neurolysin is an endogenous cerebroprotective mechanism in acute neurodegenerative disorders. Med Hypotheses. 2019;131:109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jayaraman S, Al Shoyaib A, Kocot J, Villalba H, Alamri FF, Rashid M, et al. Peptidase neurolysin functions to preserve the brain after ischemic stroke in male mice. J Neurochem. 2020;153(1):120–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rashid M, Arumugam TV, Karamyan VT. Association of the novel non-ATI, non-AT2 angiotensin binding site with neuronal cell death. J Pharmacol Exp Ther. 2010;335(3):754–61. [DOI] [PubMed] [Google Scholar]
- 13.Rashid M, Wangler NJ, Yang L, Shah K, Arumugam TV, Abbruscato TJ, et al. Functional up-regulation of endopeptidase neurolysin during post-acute and early recovery phases of experimental stroke in mouse brain. J Neurochem. 2014;129(1):179–89. [DOI] [PubMed] [Google Scholar]
- 14.Jayaraman S, Kocot J, Esfahani SH, Wangler NJ, Uyar A, Mechref Y, et al. Identification and characterization of two structurally related dipeptides that enhance catalytic efficiency of neurolysin. J Pharmacol Exp Ther. 2021;379(2):191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahman MS, Kumari S, Esfahani SH, Nozohouri S, Jayaraman S, Kinarivala N, et al. Discovery of first-in-class Peptidomimetic Neurolysin activators possessing enhanced brain penetration and stability. J Med Chem. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nozohouri S, Sifat AE, Vaidya B, Abbruscato TJ. Novel approaches for the delivery of therapeutics in ischemic stroke. Drug Discov Today. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25(8):1737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45(4):545–52. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, et al. Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos. 2006;34(9):1443–7. [DOI] [PubMed] [Google Scholar]
- 20.Pardridge WM. The isolated brain microvessel: a versatile experimental model of the blood-brain barrier. Front Physiol. 2020;11:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohashi R, Watanabe R, Esaki T, Taniguchi T, Torimoto-Katori N, Watanabe T, et al. Development of simplified in vitro P-glycoprotein substrate assay and in silico prediction models to evaluate transport potential of P-glycoprotein. Mol Pharm. 2019;16(5):1851–63. [DOI] [PubMed] [Google Scholar]
- 22.Nozohouri S, Noorani B, Al-Ahmad A, Abbruscato TJ. Estimating Brain Permeability Using In Vitro Blood-Brain Barrier Models. 2020. [DOI] [PubMed] [Google Scholar]
- 23.Friden M, Ducrozet F, Middleton B, Antonsson M, Bredberg U, Hammarlund-Udenaes M. Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab Dispos. 2009;37(6):1226–33. [DOI] [PubMed] [Google Scholar]
- 24.Rice ME. Use of ascorbate in the preparation and maintenance of brain slices. Methods. 1999;18(2):144–9. [DOI] [PubMed] [Google Scholar]
- 25.Harashima H, Sugiyama Y, Sawada Y, Iga T, Hanano M. Comparison between in-vivo and in-vitro tissue-to-plasma unbound concentration ratios (Kp, f) of quinidine in rats. J Pharm Pharmacol. 1984;36(5):340–2. [DOI] [PubMed] [Google Scholar]
- 26.Loryan I, Fridén M, Hammarlund-Udenaes M. The brain slice method for studying drug distribution in the CNS. Fluids and Barriers of the CNS. 2013;10(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Wang H, Shah K, Karamyan VT, Abbruscato TJ. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011;1383:307–16. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Islam MR, Karamyan VT, Abbruscato TJ. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res. 2015;1609:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith QR. A review of blood-brain barrier transport techniques. The Blood-Brain Barrier. 2003:193–208. [DOI] [PubMed] [Google Scholar]
- 30.Brouwer KL, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, et al. In vitro methods to support transporter evaluation in drug discovery and development. Clinical Pharmacology & Therapeutics. 2013;94(1):95–112. [DOI] [PubMed] [Google Scholar]
- 31.Summerfield SG, Stevens AJ, Cutler L, del Carmen OM, Hammond B, Tang S-P, et al. Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J Pharmacol Exp Ther. 2006;316(3):1282–90. [DOI] [PubMed] [Google Scholar]
- 32.Demeule M, Régina A, Jodoin J, Laplante A, Dagenais C, Berthelet F, et al. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood–brain barrier. Vasc Pharmacol. 2002;38(6):339–48. [DOI] [PubMed] [Google Scholar]
- 33.Seelig A, Landwojtowicz E. Structure–activity relationship of P-glycoprotein substrates and modifiers. Eur J Pharm Sci. 2000;12(1):31–40. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Welty DR. The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res. 1996;13(3):398–403. [DOI] [PubMed] [Google Scholar]
- 35.Gupta A, Chatelain P, Massingham R, Jonsson EN, Hammarlund-Udenaes M. Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: Kp, Kp, u, and Kp, uu. Drug Metab Dispos. 2006;34(2):318–23. [DOI] [PubMed] [Google Scholar]
- 36.Friden M, Gupta A, Antonsson M, Bredberg U, Hammarlund-Udenaes M. In vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids. Drug Metab Dispos. 2007;35(9):1711–9. [DOI] [PubMed] [Google Scholar]
- 37.Liu S, Levine SR, Winn HR. Targeting ischemic penumbra: part I-from pathophysiology to therapeutic strategy. Journal of experimental stroke & translational medicine. 2010;3(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goyal M, Ospel JM, Menon B, Almekhlafi M, Jayaraman M, Fiehler J, et al. Challenging the ischemic core concept in acute ischemic stroke imaging. Stroke. 2020;51(10):3147–55. [DOI] [PubMed] [Google Scholar]
- 39.De Saint-Hubert M, Prinsen K, Mortelmans L, Verbruggen A, Mottaghy FM. Molecular imaging of cell death. Methods. 2009;48(2):178–87. [DOI] [PubMed] [Google Scholar]
- 40.Liu S, Levine SR, Winn HR. Targeting ischemic penumbra part II: selective drug delivery using liposome technologies. Journal of experimental stroke & translational medicine. 2011;4(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]