

Abstract
Background and Aims
Patient‐derived human‐induced pluripotent stem cells (hiPSCs) differentiated into hepatocytes (hiPSC‐Heps) have facilitated the study of rare genetic liver diseases. Here, we aimed to establish an in vitro liver disease model of the urea cycle disorder ornithine transcarbamylase deficiency (OTCD) using patient‐derived hiPSC‐Heps.
Approach and Results
Before modeling OTCD, we addressed the question of why hiPSC‐Heps generally secrete less urea than adult primary human hepatocytes (PHHs). Because hiPSC‐Heps are not completely differentiated and maintain some characteristics of fetal PHHs, we compared gene‐expression levels in human fetal and adult liver tissue to identify genes responsible for reduced urea secretion in hiPSC‐Heps. We found lack of aquaporin 9 (AQP9) expression in fetal liver tissue as well as in hiPSC‐Heps, and showed that forced expression of AQP9 in hiPSC‐Heps restores urea secretion and normalizes the response to ammonia challenge by increasing ureagenesis. Furthermore, we proved functional ureagenesis by challenging AQP9‐expressing hiPSC‐Heps with ammonium chloride labeled with the stable isotope [15N] (15NH4Cl) and by assessing enrichment of [15N]‐labeled urea. Finally, using hiPSC‐Heps derived from patients with OTCD, we generated a liver disease model that recapitulates the hepatic manifestation of the human disease. Restoring OTC expression—together with AQP9—was effective in fully correcting OTC activity and normalizing ureagenesis as assessed by 15NH4Cl stable‐isotope challenge.
Conclusion
Our results identify a critical role for AQP9 in functional urea metabolism and establish the feasibility of in vitro modeling of OTCD with hiPSC‐Heps. By facilitating studies of OTCD genotype/phenotype correlation and drug screens, our model has potential for improving the therapy of OTCD.
Abbreviations
- ALB
albumin
- AQP9
aquaporin 9
- ARG1
arginase 1
- ASL
argininosuccinate lyase
- ASS
argininosuccinate synthetase
- CPS1
carbamoylphosphate synthetase 1
- GFP
green fluorescent protein
- gDNA
genomic DNA
- hiPSC
human‐induced pluripotent stem cell
- hiPSC‐Hep
hiPSC‐derived hepatocyte
- OTC
ornithine transcarbamylase
- OTCD
OTC deficiency
- PHH
primary human hepatocyte
- UCD
urea cycle disorder
- UCE
urea cycle enzyme
INTRODUCTION
Human‐induced pluripotent stem cell (hiPSC)–derived hepatocytes (hiPSC‐Heps) generated from patients with genetically encoded liver diseases have been used successfully to model liver diseases in vitro, leading to a better understanding of disease mechanisms and the identification of new therapeutic agents.[ 1 ]
Urea cycle disorders (UCDs) are a group of eight inborn errors of metabolism that can be life‐threatening and for which treatment options are limited.[ 2 , 3 ] Urea is the waste product arising from ammonia (NH4 +) detoxification in the urea cycle, which consists of five urea cycle enzymes (UCEs) located in mitochondria (carbamoylphosphate synthetase 1 [CPS1] and ornithine transcarbamylase [OTC]) or cytoplasm (argininosuccinate synthetase [ASS], argininosuccinate lyase [ASL], and arginase 1 [ARG1]) of hepatocytes (Figure 1A). An additional UCD is caused by deficiency of N‐acetylglutamate synthase (NAGS), which is required for CPS1 activation. The two remaining UCDs are caused by defects in mitochondrial citrin and ornithine transporters, leading to citrin deficiency and hyperornithinemia‐hyperammonemia‐homocitrullinuria syndrome, respectively.
FIGURE 1.
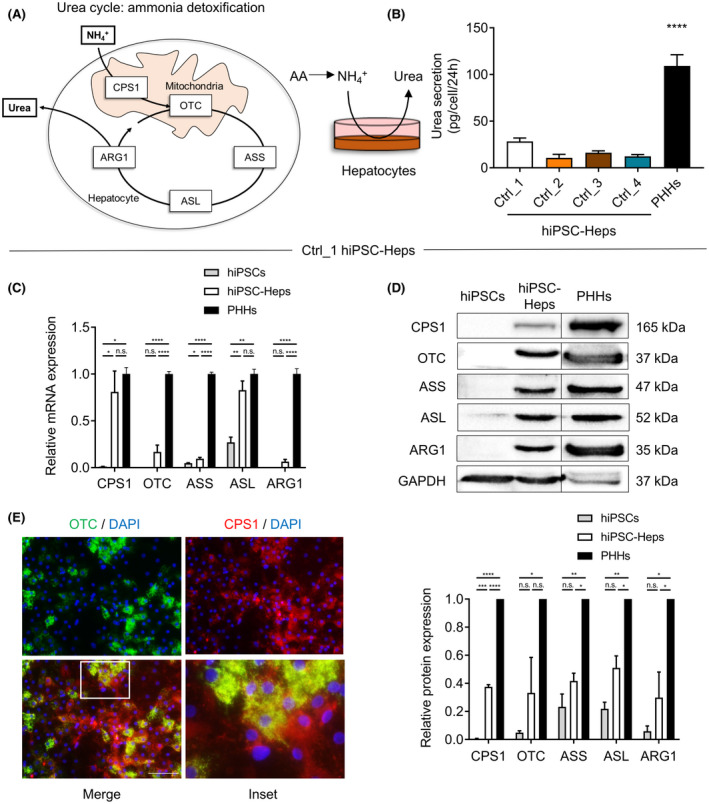
Urea metabolism is impaired in patient‐derived human‐induced pluripotent stem cells (hiPSCs) differentiated into hepatocytes (hiPSC‐Heps). (A) Scheme showing the urea cycle detoxifying ammonia (NH4 +) into nontoxic urea in five consecutive urea cycle enzyme (UCE)–mediated reactions in mitochondria in hepatocytes. (B) Urea secretion in four control hiPSC‐Hep lines (Ctrl_1‐4) as compared with PHHs. Data represent the average of 4 to 12 independent biological samples. (C,D) Relative mRNA and protein expression of UCEs analyzed by real‐time quantitative PCR (C) and western blot (D) in Ctrl_1 hiPSCs and hiPSC‐Heps compared with primary human hepatocytes (PHHs). All samples shown were loaded on the same gel. The line indicates that the image was cropped. Data represent the average of two to four independent biological samples. One‐way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Error bars represent SEM. (E) Immunofluorescent images showing abundant mitochondrial expression and co‐localization of carbamoyl phosphate synthetase 1 (CPS1) and ornithine transcarbamylase (OTC) in Ctrl_1 hiPSC‐Heps. Nuclei are labeled with DAPI. Scale bar = 100 µM. Inset in merged image shown as bottom‐right image. ARG1, arginase 1; ASL, argininosuccinate lyase; ASS, argininosuccinate synthetase; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; n.s., not significant
To date, only ARG1 deficiency, ASS deficiency, and citrin deficiency have been modeled using hiPSC‐Heps.[ 4 , 5 , 6 ] A hiPSC‐Hep‐based model of OTC deficiency (OTCD) has yet to be reported. Although OTCD is the most common UCD,[ 7 ] resulting in substantial clinical experience, its prognosis is unpredictable because disease onset and severity are affected by a large number of different mutations[ 8 ] and variable effects of X‐chromosome inactivation in women.[ 9 ] The current treatment, consisting of low‐protein diet and supplementation of essential amino acid mixtures,[ 10 ] is challenging and often fails to prevent adverse outcomes in patients,[ 11 ] leading to a high mortality rate.[ 12 ] A faithful disease model would help to better understand the relationship between gene defect and disease manifestation, allowing prediction of the disease course in individual patients, and could be used to develop novel therapeutics.
A potential reason for the lack of a hiPSC‐Hep model of OTCD is that ureagenesis is generally low in hiPSC‐Heps, both in hiPSC‐Heps derived from normal controls and in hiPSC‐Heps, in which the genetic defect causing the UCD was corrected.[ 4 , 5 , 6 , 13 ] This functional deficiency is consistent with the general notion that hiPSC‐Heps generated with current protocols are not as differentiated as adult primary human hepatocytes (PHHs) but retain some fetal characteristics.[ 14 ]
We overcame this roadblock by identifying lack of expression of aquaporin 9 (AQP9), a membrane channel protein that mediates passage of water, glycerol, and urea,[ 15 , 16 ] as the reason for low ureagenesis in hiPSC‐Heps. Taking advantage of this insight to establish functional urea metabolism in hiPSC‐Heps, we developed an in vitro model of genotype‐specific manifestation of OTCD.
MATERIALS AND METHODS
Patients
Written informed consent was obtained from all study subjects. These studies were approved by the local ethics committee in Bern, Switzerland (project ID: 2020‐02979).
Reprogramming of fibroblasts into hiPSCs and directed differentiation of hiPSCs into hiPSC‐Heps
hiPSCs were generated and cultured as previously described.[ 17 ] Hepatocyte differentiation of hiPSCs recapitulating critical stages of development was performed as we previously described for Ctrl_1 line[ 18 ] (see Supporting Methods).
Cell culture and origin of PHHs
PHHs were purchased from BioIVT (catalog number M00995‐P; lot numbers BVI, FLO, and JFC). Biological replicates were generated using all three PHH lots. Cells were plated on rat tail collagen type I–coated plates at a density of 6.0 104 cells/cm2 in Dulbecco’s minimum essential medium supplemented with 10% fetal bovine serum, glutaMax, 50 U/ml penicillin, 50 μg/ml streptomycin, and 1 μmol/l dexamethasone and insulin. After overnight culture, the medium was replaced by serum‐free Williams E medium containing the same concentrations of penicillin/streptomycin, dexamethasone, and insulin.
Urea assay and albumin ELISA
To assess cellular urea and/or albumin (ALB) secretion, we collected cell culture supernatants at the indicated time points (usually after 24 h), centrifuged them at 700 g for 5 min at 4°C and either directly measured or stored samples at 80°C and later determined urea and/or ALB concentration with the Quantichrom Urea Assay Kit (Bioassay Systems) and/or ALB ELISA (Bethyl Laboratories). The absolute amount of secreted urea and/or ALB was expressed in pg/cell/24 h.
Quantitative PCR
For RNA isolation we used the RNAeasy Mini Kit (Qiagen). All samples were treated with DNAse. Reverse transcription was performed using qScript cDNA Supermix (Quanta Biosciences). Quantitative PCR was performed using a SYBR Green Supermix (Affymetrix) on an Applied Biosciences ViiA7 Real‐Time PCR System (Thermo Fisher Scientific). Reactions were performed in triplicate, and expression was normalized to glyceraldehyde 3‐phosphate dehydrogenase or 18S gene expression and quantified using the ΔΔCt method. Primers are listed in the Supporting Methods.
Western blot
Western blot was performed using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis as described previously.[ 19 ] Further details and antibodies are given in the Supporting Methods.
Immunofluorescence
Immunofluorescence imaging was performed as described previously.[ 20 ] Further details and antibodies are given in the Supporting Methods.
Relative mRNA expression in liver tissue and in hiPSC‐Heps
A microarray data set previously published by Bonder et al.,[ 21 ] deposited in the National Center for Biotechnology Information gene expression omnibus (GEO) database under accession number GSE61279, was analyzed for relative mRNA expression of AQP9 and other enzymes and transporters involved in ammonia detoxification and urea metabolism in fetal and adult human liver tissue.
In addition, expression of these genes in hiPSC‐Heps and PHHs was assessed in four additional data sets deposited in the GEO database.[ 22 , 23 , 24 , 25 ] More information about these data sets is given in the Supporting Methods.
Lentiviral transduction
Lentiviral vectors were custom‐designed at and purchased from VectorBuilder. All lentiviral vectors had viral titers of at least 5 108 TU/ml and expressed AQP9 (vector ID: VB180418‐1015mqd), OTC (vector ID: VB191009‐1069yvq), or green fluorescent protein (GFP) (vector ID: VB180418‐1016dym). The genes were under the control of the ubiquitously active EF1A promoter. Vector details are available at https://en.vectorbuilder.com. Cells were transduced on day 18 of the hiPSC‐Heps differentiation protocol using a multiplicity of infection of 20 in hepatocyte culture medium (HCM) (Lonza; for details see Supporting Methods) supplemented with 10 µg/ml polybrene. After overnight transduction, cells were washed with PBS and HCM was added.
15NH4Cl challenge and isotopic [15N]Urea enrichment
One‐molar aqueous stock solutions of NH4Cl and 15NH4Cl (Sigma Aldrich) were prepared and filter‐sterilized before use in cell cultures at working concentrations of 0, 1, 2, and 10 mM. Samples of the cell culture supernatants were taken at 2 h and 24 h. Isotopic [15N]urea enrichment and semi‐quantitative amino acid concentrations were measured by adjustment for cell culture supernatants using a previously described method.[ 26 ]
Ornithine transcarbamylase activity assay
An adapted version of a previously established OTC activity assay[ 27 ] was performed to determine OTC activity in cell culture lysates. Briefly, 25‐50 µg of whole cell lysates were used to assess OTC activity. The substrates ornithine and carbamoylphosphate were added in excess to the lysates. Citrulline, which is produced in the OTC enzyme reaction, was measured spectrophotometrically after a color reaction with diacetylmonoxim/antipyrin/Fe. The intensity of the absorption is proportional to the amount of produced citrulline, which is proportional to the OTC enzyme activity.
Statistical analysis
All experiments were repeated at least twice with a minimum of two biological replicates. Student t test or ordinary one‐way ANOVA was used to compare groups, with significance set at p < 0.05. Data are expressed as the mean, and error bars represent SEM.
RESULTS
Urea metabolism is impaired in hiPSC‐Heps
Although hiPSC‐Heps express many functions of mature adult PHHs. They also exhibit characteristics of immature fetal PHHs, including reduced capacity to secrete urea.[ 14 ] Therefore, as the first step toward using patient‐derived hiPSC‐Heps to create an in vitro model of OTCD, we investigated ammonia detoxification and urea metabolism in hiPSC‐Heps derived from normal controls. We generated four hiPSC lines from normal controls and differentiated them into hiPSC‐Heps (Ctrl_1‐4 hiPSC‐Heps) using our step‐wise hepatocyte differentiation protocol (Figure S1A; for details see Supporting Methods and Lee‐Montiel et al.[ 18 ]). Ctrl_1 hiPSC‐Heps showed characteristic hepatocyte morphology (Figure S1A) and well‐developed hepatocyte functions, including ALB secretion (Figure S1B), CYP3A4‐mediated drug metabolism (Figure S1C), LDL receptor–mediated LDL uptake (Figure S1D), and ALB and hepatocyte nuclear factor 4 mRNA expression (Figure S1E). However, Ctrl_1‐4 hiPSC‐Heps showed significantly lower urea secretion than PHHs (Figure 1B). Analysis of mRNA and protein expression in Ctrl_1 hiPSC‐Heps showed that the five UCEs were expressed, some at similar levels and some at lower levels than in PHHs (Figure 1C,D). As expected, UCE expression in hiPSCs was very low or absent. Immunofluorescent staining of the two most abundant UCEs CPS1 and OTC showed mitochondrial localization as expected (Figure 1E and Figure S1F).
These results confirmed that hiPSC‐Heps secrete lower amounts of urea than PHHs. The overall reduction in UCE expression likely contributed to the decreased urea secretion found in hiPSC‐Heps, but the extent to which urea secretion was impaired in hiPSC‐Heps suggested the contribution of factors beyond the UCEs.
Urea metabolism is impaired in hiPSC‐Heps due to lack of AQP9
Considering that hiPSC‐Heps maintain many characteristics of fetal hepatocytes, such as alpha‐fetoprotein (AFP) mRNA expression (Figure S1E), we reasoned that comparing mRNA expression in human fetal and adult liver tissue will reveal genes responsible for low urea secretion in hiPSC‐Heps. For this, we analyzed published mRNA expression profiling of 14 fetal and 92 adult human liver tissue samples (GSE62179)[ 21 ] (Figure 2A). mRNA expression of the five UCEs, NAGS, and two mitochondrial transporters (encoded by SLC25A13 and SLC25A15) involved in ureagenesis showed little variation between fetal and adult human liver tissue samples (Figure 2A). However, expanding the analysis revealed AQP9 as one of the most differentially expressed genes in GSE62179 (p < 1.0−07), exhibiting higher abundance in adult (≥ 70‐fold higher expression) than in fetal liver (Figure 2A).
FIGURE 2.
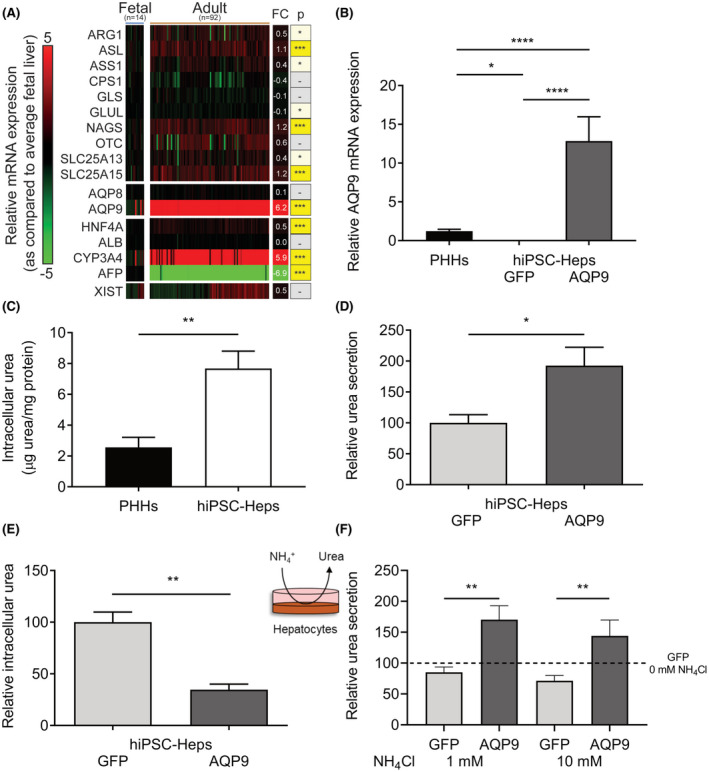
Urea metabolism is impaired in hiPSC‐Heps due to lack of aquaporin 9 (AQP9). (A) Comparison of log2 fold change (FC) in relative mRNA expression in 14 fetal and 92 adult human liver‐tissue samples as compared with average fetal human liver expression. AQP9 is one of the most differentially expressed genes between adult and fetal human liver tissue. (B) Relative mRNA expression of AQP9 in hiPSC‐Heps transduced with lentiviruses expressing AQP9 or green fluorescent protein (GFP; negative control) and PHHs (positive control). Data represent the average of three to seven independent biological samples. (C) Intracellular urea levels are significantly higher in hiPSC‐Heps than in PHHs. Data represent the average of three (hiPSC‐Heps) and six (PHHs) independent biological samples. (D,E) hiPSC‐Heps were transduced with lentiviruses expressing AQP9 or GFP, and relative urea secretion (D) and intracellular urea concentration (E) were determined. Data represent the average of two independent experiments. (F) Seven days after transduction with lentiviruses expressing AQP9 or GFP, hiPSC‐Heps were challenged with 1 or 10 mM NH4Cl for 24 h. Relative urea secretion was determined in the supernatant and compared with the nonchallenged (0 mM NH4Cl) GFP‐transduced negative control. Data represent the average of two independent experiments. One‐way ANOVA (A,B) or Student’s t test (C‐F); *p < 0.05, **p < 0.01, and ****p < 0.0001. Error bars represent SEM. AFP, alpha fetoprotein; ALB, albumin; AQP8, aquaporin 8; CYP3A4, cytochrome P450 family 3 subfamily A member 4; GLS, glutaminase; GLUL, glutamate‐ammonia ligase; HNF4A, hepatocyte nuclear factor 4 alpha; NAGS, N‐acetylglutamate synthase; SLC25A13, solute carrier family 25 member 13; SLC25A15, solute carrier family 25 member 15; XIST, X inactive specific transcript
AQP9 is located in the hepatocyte plasma membrane and is required for urea transport across this membrane.[ 28 , 29 ] As in human fetal liver, comparison to PHHs showed that AQP9 expression was lacking in hiPSC‐Heps, as evidenced not only by analysis of hiPSC‐Heps generated with our current protocol (Figure 2B) but also hiPSC‐Heps generated with a previous version of our protocol (GSE52309)[ 25 ] or three additional gene‐expression profiles of hiPSC‐Heps generated in other laboratories[ 22 , 23 , 24 ] (Figure S2A and Supporting Methods).
If lack of AQP9 expression was contributing to low ureagenesis in hiPSC‐Heps, urea should be accumulating intracellularly. Indeed, intracellular urea levels were higher in hiPSC‐Heps than in PHHs (Figure 2C). Moreover, lentiviral expression of AQP9 in hiPSC‐Heps (Figure S2B) restored urea secretion (Figure 2B,D) and decreased intracellular urea levels (Figure 2E). To further study ammonia detoxification and urea metabolism in hiPSC‐Heps, we challenged the cells with ammonium chloride (NH4Cl). In contrast to PHHs, hiPSC‐Heps failed to increase urea secretion in response to NH4Cl (Figure S2C). However, lentiviral AQP9 expression in hiPSC‐Heps normalized the response to NH4Cl challenge (i.e., increased urea secretion) (Figure 2F). Taken together, our findings show that hiPSC‐Heps generated with current protocols lack AQP9 expression, and its restoration normalizes basal and ammonia‐induced urea secretion.
AQP9 expression normalizes response of hiPSC‐Heps to ammonia challenge
To further characterize ammonium detoxification and urea metabolism and to demonstrate functional ureagenesis in AQP9‐expressing hiPSC‐Heps, we challenged the cells with NH4Cl labeled with the stable isotope [15N] (15NH4Cl) to measure the enrichment of secreted [15N]‐labeled urea (Figure 3A,B). If ureagenesis is functional, three different isotopes of urea are detectable after such a challenge: nonlabeled urea, mono‐labeled urea, and double‐labeled urea. Mono‐labeled urea receives the labeled [15N]‐atom from NH4 + in carbamoylphosphate or aspartate. Double‐labeled urea receives one [15N]‐atom from carbamoylphosphate and one [15N]‐atom from aspartate (Figure 3A). Challenging the AQP9‐expressing hiPSC‐Heps with 1, 2, or 10 mM of 15NH4Cl led to an increase of [15N]‐mono‐labeled and double‐labeled urea when compared with untreated (0 mM) cells, which demonstrated functional ureagenesis (Figure 3B).
FIGURE 3.
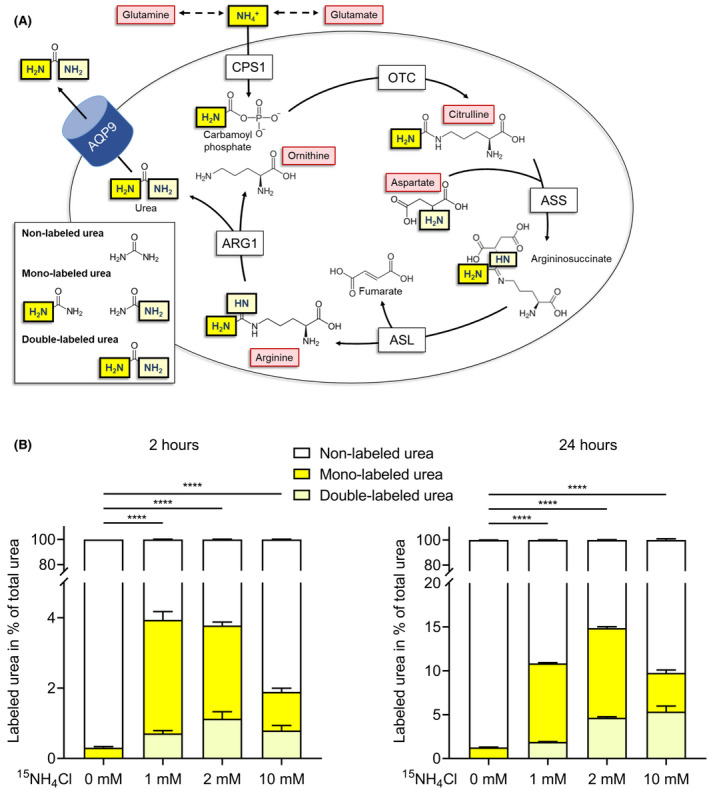
Demonstration of fully functional ureagenesis in AQP9‐expressing hiPSC‐Heps. (A) Schematic depicting the enzyme reactions in the urea cycle. 15NH4 + (yellow) enters the urea cycle and is converted to carbamoylphosphate by CPS1. Next, OTC converts carbamoylphosphate and ornithine to citrulline, which further reacts with aspartate, the donor of the second amino group found in urea, in the ASS enzyme reaction to produce argininosuccinate. ASL cleaves off fumarate from argininosuccinate to build up arginine. In the last step of the urea cycle, ARG1 cleaves arginine to urea and ornithine, which reenters the urea cycle. After challenge of AQP9‐expressing hiPSC‐Heps with 15NH4Cl, three different isotopes of urea are found: nonlabeled urea, mono‐labeled urea, and double‐labeled urea. (B) Mono‐labeled and double‐labeled urea was detected in supernatants of AQP9‐expressing hiPSC‐Heps following challenge with 15NH4Cl (0, 1, 2 and 10 mM) after 2 h (left panel) or 24 h (right panel). Ureagenesis is most pronounced when cells are challenged with 1 and 2 mM 15NH4Cl for 24 h, resulting in a [15N]‐enrichment of approximately 11% and 15%, respectively (expressed as percentage of 15‐N‐labeled urea over total urea). Data represent the average of three independent biological samples. One‐way ANOVA; ****p < 0.0001. Error bars represent SEM
Further analysis of amino acid concentrations in the cell culture supernatants revealed glutamine and alanine as the two most abundant amino acids (levels between approximately 700 µM and 2200 µM) (Figure S3). After hiPSC‐Heps were challenged with 1 mM of NH4Cl for 24 h, glutamine concentrations increased by a factor of approximately 1.6 (from 700 µM to 1200 µM) (Figure S3), consistent with glutamine acting as an ammonia‐scavenging back‐up system as observed in vivo.[ 30 ]
Taken together, these results show that expression of AQP9 in hiPSC‐Heps normalized the reaction to ammonia challenge (i.e., ammonia conversion into urea and its secretion out of the cells into the culture medium). In addition to demonstrating functional ureagenesis, these results show physiological ammonia flux into urea and amino acid changes in AQP9‐expressing hiPSC‐Heps.
Generation of hiPSC‐Heps from patients with ornithine transcarbamylase deficiency
We generated hiPSC lines from fibroblasts of 2 patients who died from OTCD.[ 31 ] Analysis of OCT3/4 and NANOG mRNA (Figure S4A) and OCT3/4 and SSEA4 protein expression (Figure S4B) confirmed the cells’ pluripotency. One patient, designated OTCD_1, was male and died in the neonatal period. Mutational analysis of the patient`s fibroblasts revealed a previously described hemizygous mutation in the OTC gene, which is located on the X chromosome (exon 6; c.548A>G [p.Tyr183Cys]).[ 32 ] We confirmed the presence of the mutation in hiPSC‐Heps generated from the patient’s fibroblasts (Figure 4A). Due to the X‐chromosomal inheritance of OTCD, male patients often suffer from a fatal disease course; in female patients, the disease course is affected by X inactivation and therefore more variable. The second patient (OTCD_2) was female and developed fatal acute liver failure at the age of 6 years, suggesting skewed X inactivation strongly favoring the mutated allele. She had a previously described heterozygous stop mutation in the OTC gene (exon 3; c.274C>T [p.Arg92*]),[ 33 ] which we confirmed to be present heterozygously in genomic DNA (gDNA) in hiPSC‐Heps (Figure 4A). Analysis of OTC mRNA in hiPSC‐Heps revealed complete inactivation of the normal allele of the OTC gene, leading to only the mutated allele being expressed, which is in line with the skewed X inactivation found in the patient (Figure 4A,B).
FIGURE 4.
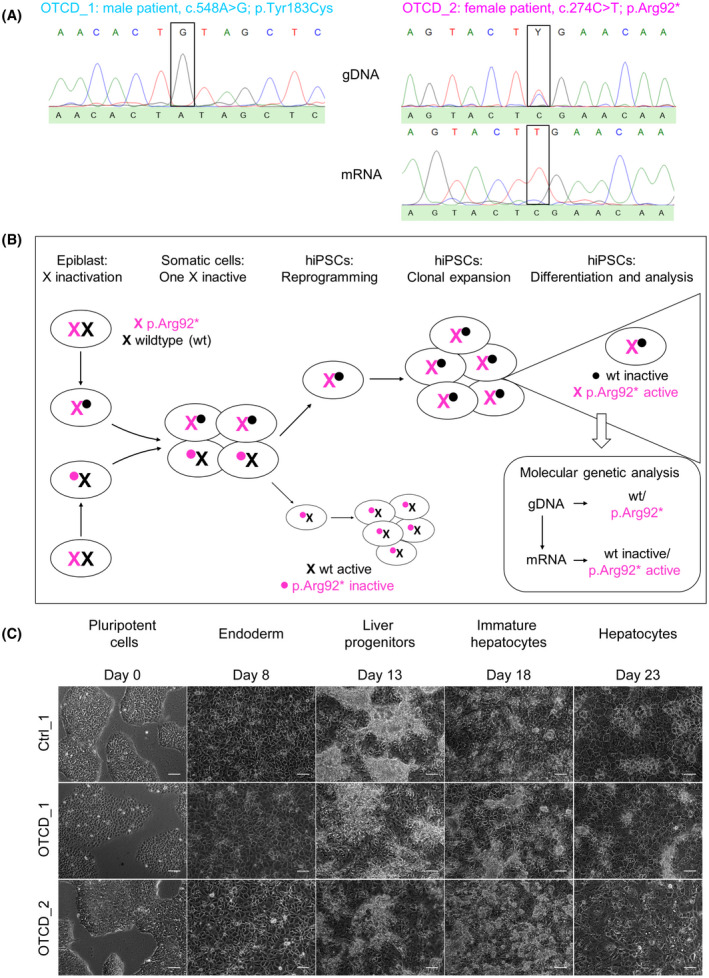
Generation of OTC‐deficient patient‐derived hiPSC‐Heps. (A) Mutational analysis confirmed presence of OTC mutations in genomic DNA (gDNA) and mRNA of hiPSC‐Heps. OTCD_1 is a male patient, and his mutation was confirmed at the gDNA level. OTCD_2 is a female patient with one normal allele (c.274C) and one mutated allele (c.274T) at the gDNA level. The patient’s sequence on top shows a Y for the two pyrimidine bases C and T. At the mRNA level only, the mutated allele was expressed due to X‐chromosome inactivation–mediated silencing of the normal allele. (B) Scheme of X chromosome inactivation in epiblast and hiPSCs in female OTCD_2 patient. (C) Brightfield microscopy showing representative images of the differentiation process (for details see Supporting Methods) from hiPSCs to hiPSC‐Heps for Ctrl_1 and OTCD_1 and OTCD_2. Scale bars = 100 µM
Differentiation of OTCD hiPSCs using our standard protocol (Figure S1A) produced hiPSC‐Heps with hepatocyte‐specific morphology (Figure 4C), albumin secretion (Figure S5A) and mRNA (Figure S5B) and protein (Figure S5C) expression.
Modeling ornithine transcarbamylase deficiency with patient‐derived hiPSC‐Heps
OTCD_1 and OTCD_2 hiPSC‐Heps transduced with lentiviruses expressing AQP9 showed significantly reduced urea secretion compared with AQP9‐expressing Ctrl_1 hiPSC‐Heps (Figure 5A). Analysis of mRNA (Figure 5B) and protein expression (Figure 5C) of the five UCEs showed complete absence of OTC protein in both OTCD_1 and OTCD_2 hiPSC‐Heps (Figure 5C). The other UCEs were expressed at normal levels in OTCD_1 hiPSC‐Heps; in OTCD_2 hiPSC‐Heps, the UCE protein levels were generally lower than in Ctrl_1 and OTCD_1 hiPSC‐Heps (Figure 5C). Relative OTC activity was significantly decreased in OTCD_1 and nearly absent in OTCD_2 hiPSC‐Heps, respectively (Figure 5D). In agreement, OTC protein expression was abundant in Ctrl_1 hiPSC‐Heps but undetectable by immunofluorescence in OTCD_1 and OTCD_2 hiPSC‐Heps (Figure 5E). Together, these results show that OTCD patient–derived hiPSC‐Heps replicate the disease as evidenced by reduced/absent OTC expression and activity, leading to impaired urea secretion.
FIGURE 5.
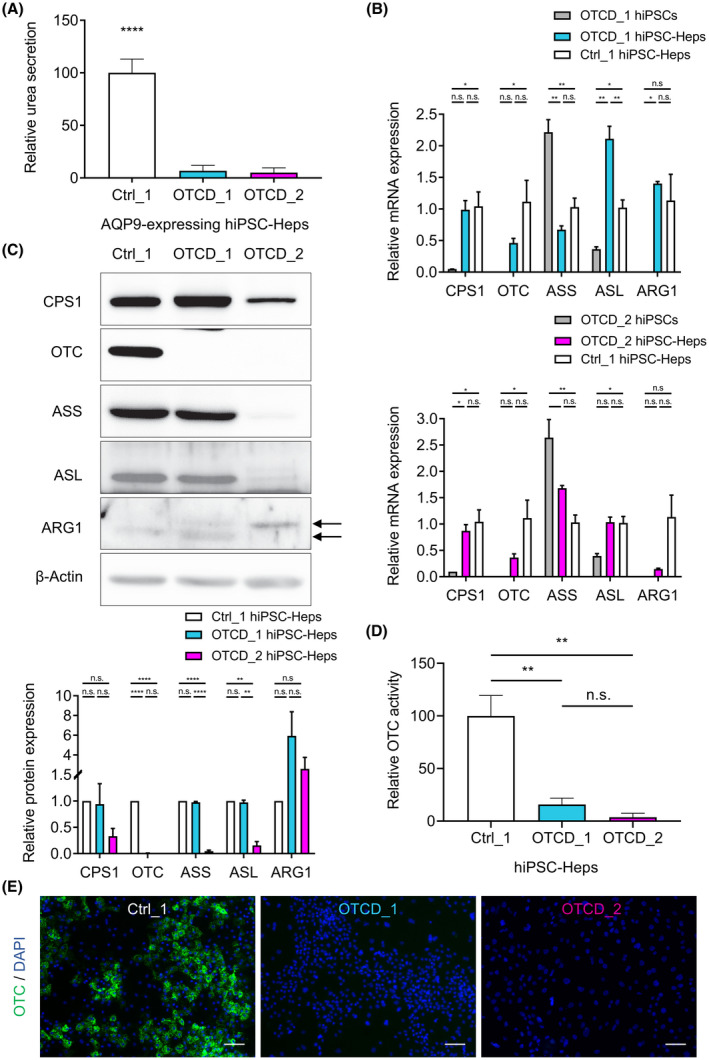
Modeling ornithine transcarbamylase deficiency with patient‐derived hiPSC‐Heps. (A) Relative urea secretion is significantly impaired in AQP9‐expressing OTCD_1 and OTCD_2 hiPSC‐Heps compared with AQP9‐expressing Ctrl_1 hiPSC‐Heps. Data represent the average of 7 to 11 independent biological samples. (B) Relative mRNA expression of UCEs by real‐time quantitative PCR in OTCD_1 and OTCD_2 hiPSCs and hiPSC‐Heps compared with Ctrl_1 hiPSC‐Heps. Data represent the average of two to four independent biological samples. (C) Western blot of UCE in Ctrl_1, OTCD_1 and OTCD_2 hiPSC‐Heps and quantification of relative protein expression of two independent biological samples each. Double arrows indicate the two bands detected for ARG1. (D) Relative OTC enzyme activity in OTCD_1 and OTCD_2 hiPSC‐Heps compared with Ctrl_1 hiPSC‐Heps. Data represent the average of three to five independent biological samples. (E) Representative immunofluorescent images showing abundant OTC expression in Ctrl_1 and absent expression in OTCD_1 and OTCD_2 hiPSC‐Heps. Nuclei are labeled with DAPI. Scale bars = 100 µM. One‐way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Error bars represent SEM
OTC and AQP9 co‐expression restores ureagenesis in OTC‐deficient patient‐derived hiPSC‐Heps
To correct the OTCD phenotype, we co‐transduced patient‐derived hiPSC‐Heps with lentiviruses expressing OTC and AQP9. OTC expression was assessed by western blot (Figure 6A) and analysis of OTC activity (Figure 6B), and both significantly increased upon OTC transduction. We also measured citrulline—the product of the OTC‐mediated transcarbamylation of ornithine—in cell culture supernatant. Its abundance significantly increased following OTC expression in Ctrl_1, OTCD_1 and OTCD_2 hiPSC‐Heps (Figure 6C). In accord, ureagenesis in OTCD_1 and OTCD_2 hiPSC‐Heps increased significantly following OTC transduction (Figure 6D). Whereas in OTCD_1 hiPSC‐Heps ureagenesis was normalized and reached levels comparable to Ctrl_1 hiPSC‐Heps, restoration was less pronounced in OTCD_2 hiPSC‐Heps (Figure 6D). This deficiency was likely due to the low levels of distal UCE expression (ASS and ASL) in OTCD_2 hiPSC‐Heps (Figure 5C). Thus, co‐transduction of AQP9 and OTC resulted in correction of OTC expression and activity as well as in significant increases of citrulline as the direct product of the OTC reaction and ureagenesis in hiPSC‐Heps from both patients with OTCD.
FIGURE 6.
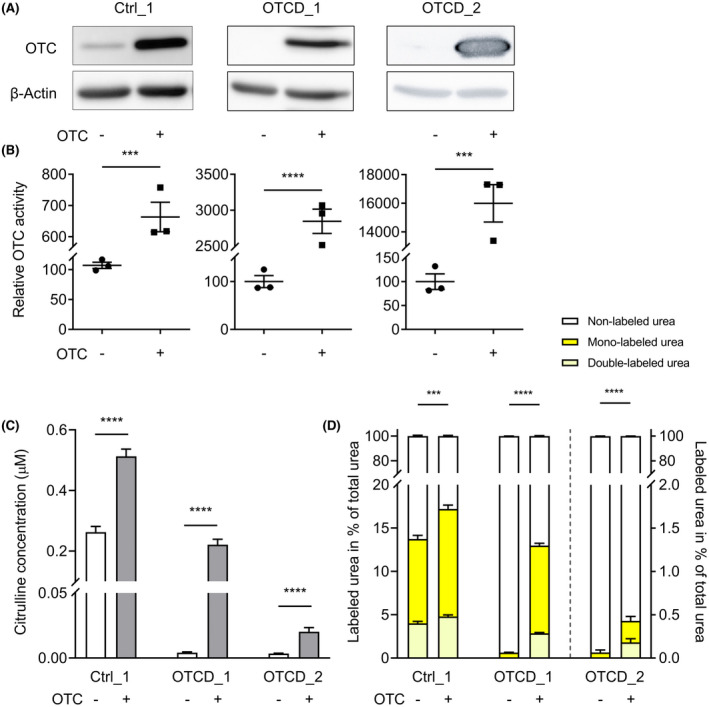
OTC and AQP9 co‐expression corrects the disease phenotype in OTC‐deficient patient‐derived hiPSC‐Heps and restores ureagenesis. (A,B) Rescue of OTC deficiency by lentiviral OTC gene correction in AQP9‐expressing Ctrl_1, OTCD_1 and OTCD_2 hiPSC‐Heps as assessed by western blot (A) and OTC enzyme activity assay (B). (C) Semi‐quantitative determination of extracellular citrulline concentrations after cell treatment as in (A) and (B). (D) Determination of mono‐labeled and double‐labeled urea in cell culture supernatants following a challenge with 1 mM of 15NH4Cl for 24 h in cells treated as in (A)‐(C). Data in (B)‐(D) represent the average of each three independent biological samples. Student’s t test; ***p < 0.001 and ****p < 0.0001. Error bars represent SEM
Finally, we explored induction of endogenous AQP9 expression as a physiological alternative to lentiviral overexpression. Previous findings of a glucocorticoid‐responsive element motif in the AQP9 promoter suggest that AQP9 is regulated by the catabolic hormones glucagon and glucocorticoids.[ 16 ] In agreement, we found that glucagon and the potent glucocorticoid dexamethasone induce AQP9 expression and urea secretion in hiPSC‐Heps (Figure S6A‐D).
DISCUSSION
UCDs are rare inborn errors of metabolism caused by mutations in one of the UCEs.[ 34 ] Affected patients suffer from recurrent and life‐threatening hyperammonemic decompensations and from severe neurological sequelae.[ 35 ] In addition to the neurological phenotype found in all UCDs, involvement of the liver in the form of acute organ failure or chronic hepatic disease is particularly common in OTCD.[ 31 , 36 ] It has been suggested that high ammonia levels contribute to acute liver failure observed in more than 50% of patients with OTCD.[ 31 ] In fact, acute liver failure accounts for a substantial number of deaths among patients with OTCD, as seen in our OTCD_2 patient.[ 31 ] Current treatment strategies including dietary protein restriction, nitrogen scavengers, and avoidance of catabolism are unfortunately not sufficient to fully suppress recurrent metabolic crises and the resulting morbidity.[ 34 ] Although another therapeutic option, liver transplantation, is curative, this approach is not only invasive but has several important limitations such as liver donor shortage and requirement of life‐long immunosuppressive therapy.[ 37 ] Thus, alternative strategies for the treatment of UCDs are needed, especially for patients with severe disease and early life‐threatening hyperammonemic crises. One such approach could be gene addition or gene editing, and indeed adeno‐associated viruses were shown to correct OTCD in preclinical animal models[ 38 , 39 , 40 ] and are currently being investigated in clinical trials in adult human subjects suffering from mild OTCD (NCT02991144).
In the preclinical phase of treatment development, cellular or animal models of the target disease are essential, which prompted us to develop a model of OTCD based on patient‐derived hiPSC‐Heps. By accurately replicating the disease at the genetic and functional level, such a model would facilitate detailed analyses of patient‐specific pathophysiology that could inform individual risk assessment and disease management, including genotype‐phenotype correlation. In addition, a faithful in vitro model of OTCD could be used to identify (patient‐specific) drugs by high‐throughput screening.
The gold standard for determining differences in urea cycle activity in OTCD and other UCDs is to assess enrichment of secreted [15N]‐labeled urea after challenging cells with NH4Cl labeled with the stable isotope [15N].[ 26 , 41 ] However, to be able to apply this sensitive assay to OTCD patient‐derived hiPSC‐Heps, we first had to address the question of why hiPSC‐Heps from normal controls secrete less urea than PHHs.
Human iPSC‐Heps resemble fetal rather than mature adult hepatocytes.[ 14 ] Therefore, we analyzed published gene‐expression profiles of fetal and adult human liver tissues[ 21 ] to identify differentially expressed genes that could explain the difference in urea secretion between hiPSC‐Heps and PHHs. Although some UCEs were expressed at lower levels in hiPSC‐Heps than in PHHs, their activity appeared sufficient for ammonia detoxification. In contrast, we found that AQP9, which is required for hepatocyte urea secretion[ 15 , 16 , 29 , 42 , 43 , 44 ] and virtually absent in human fetal liver tissue, is insufficiently expressed in hiPSC‐Heps.
Our results demonstrate that lack of AQP9 in hiPSC‐Heps causes reduced urea secretion and intracellular accumulation of urea. Overexpression of AQP9 facilitates urea secretion and normalizes the cellular response to ammonia challenge as confirmed by [15N]‐ureagenesis assay. Thus, normalizing AQP9 expression facilitates faithful modeling of OTCD and other UCDs in hiPSC‐Heps in vitro. This finding has implications for other research fields, as urea secretion is commonly used as a marker of hepatocyte function of hiPSC‐Heps.[ 1 ] To search for a potential role of AQP9 on ammonia metabolism in vivo, we investigated 10 human liver‐tissue samples from patients suffering from hyperammonemia of unknown origin. In contrast to hiPSC‐Heps, AQP9 was highly expressed in all of these liver tissue samples. Moreover, no possibly pathogenic AQP9 variants were detected in these patients or in the HGMD Professional database (QIAGEN) (Figure S7). Thus, impaired AQP9 expression is a technical limitation of hiPSC‐Heps in vitro but not an obvious disease‐causing factor in patients.
After overcoming the obstacle of reduced urea secretion in hiPSC‐Heps caused by lack of AQP9 expression, we focused on developing a hiPSC‐Hep‐based model of OTCD. Using hiPSCs from 2 patients with OTCD, we found that the characteristic hepatic disease phenotype of OTCD, including reduced OTC expression and activity and diminished ureagenesis, could be recapitulated in hiPSC‐Heps. We also observed differences in phenotypical severity between OTCD patient–derived hiPSC‐Heps, potentially reflecting the complex genetics of OTCD, including the impact of X inactivation in female patients.[ 12 , 34 ] In addition to low expression levels of several UCEs in hiPSC‐Heps, hiPSCs from the female patient with OTCD appeared more fragile during hepatocyte differentiation and contained more vacuoles, suggesting cell toxicity (data not shown). We confirmed skewed X inactivation, leading to exclusive expression of the mutated OTC gene,[ 45 ] in hiPSC‐Heps generated from two different hiPSC clones (data not shown). Thus, the severity of the OTCD phenotype in this patient—leading to death from acute liver failure at the age of 6 years—was recapitulated in our hiPSC‐Hep‐based disease model.
In OTCD, over 400 mutations have been identified that explain the broad spectrum of disease severity.[ 8 ] Many of the identified mutations are point mutations causing reduced enzyme activity. Mutated OTC protein could be stabilized by small molecule compounds functioning as pharmacological chaperones. We recently performed a high‐throughput screen in a noncellular system for compounds interacting and stabilizing human OTC protein (unpublished data). Our OTCD model using patient‐derived hiPSC‐Heps could be used to assess the therapeutic efficacy of the top hits from this screen.
In summary, our study identified AQP9 expression as a missing factor in urea metabolism in hiPSC‐Heps. By addressing this roadblock, we established the efficacy for modeling of OTCD of hiPSCs generated from patient‐derived fibroblasts, which are routinely banked in the clinical setting. Our results pave the way for studies aiming to tailor disease management to individual patients with OTCD and develop much‐needed new drugs.
CONFLICT OF INTEREST
Dr. Willenbring advises and owns stock in Ambys.
AUTHOR CONTRIBUTIONS
A.L. designed and performed the experiments, made the figures and wrote the manuscript. M.P. and J.H. developed the ureagenesis assay (enrichment of [15N]‐labeled urea) and analyzed cell culture supernatants. B.H. and M.B. performed differentiation experiments and provided critical review of the manuscript. V.R. performed the OTC and AQP9 sequencing and provided Figure 4A and Supporting Figure 7. J.R. performed the analysis of and provided Figure 2A. M.S. performed the analysis of and provided Supporting Figure 1F. J.M.N. provided critical review of the manuscript. J.H. and H.W. contributed to the conception of the study and interpretation of data, provided critical review of the figures and the manuscript and approved the final version for publication.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
The authors thank Ljubica Caldovic (Children’s National Medical Center) for sharing the rabbit anti‐human OTC antibody. They also thank Renata Gallagher, Simone Kurial, and Feng Chen (University of California San Francisco) and Matthias Gautschi (University of Bern) for their valuable input when discussing this project.
Laemmle A, Poms M, Hsu B, Borsuk M, Rüfenacht V, Robinson J, et al. Aquaporin 9 induction in human iPSC‐derived hepatocytes facilitates modeling of ornithine transcarbamylase deficiency. Hepatology. 2022;76:646–659. 10.1002/hep.32247
Johannes Häberle and Holger Willenbring contributed equally to this work.
Funding information
“Novartis Stiftung für Medizinisch‐Biologische Forschung” (Research fellowship No. 15B086 to A.L.); “Fondazione Ettore and Valeria Rossi” (Research fellowship to A.L.); and the “Batzenbär‐Stiftung des Inselspitals” (to A.L.); Swiss National Science Foundation (320030_176088 to J.H.); National Institutes of Health (UH3 TR000487 and P30 DK26743 to H.W.); and the California Institute for Regenerative Medicine (DISC2‐10088 to H.W.)
REFERENCES
- 1. Corbett JL, Duncan SA. iPSC‐derived hepatocytes as a platform for disease modeling and drug discovery. Front Med. 2019;6:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matsumoto S, Haberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K. Urea cycle disorders‐update. J Hum Genet. 2019;64:833–47. [DOI] [PubMed] [Google Scholar]
- 3. Soria LR, Ah Mew N, Brunetti‐Pierri N. Progress and challenges in development of new therapies for urea cycle disorders. Hum Mol Genet. 2019;28:R42–8. [DOI] [PubMed] [Google Scholar]
- 4. Lee PC, Truong B, Vega‐Crespo A, Gilmore WB, Hermann K, Angarita SAK, et al. Restoring ureagenesis in hepatocytes by CRISPR/Cas9‐mediated genomic addition to arginase‐deficient induced pluripotent stem cells. Mol Ther Nucleic Acids. 2016;5:e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshitoshi‐Uebayashi EY, Toyoda T, Yasuda K, Kotaka M, Nomoto K, Okita K, et al. Modelling urea‐cycle disorder citrullinemia type 1 with disease‐specific iPSCs. Biochem Biophys Res Commun. 2017;486:613–9. [DOI] [PubMed] [Google Scholar]
- 6. Kim Y, Choi JY, Lee SH, Lee BH, Yoo HW, Han YM. Malfunction in mitochondrial beta‐oxidation contributes to lipid accumulation in hepatocyte‐like cells derived from citrin deficiency‐induced pluripotent stem cells. Stem Cells Dev. 2016;25:636–47. [DOI] [PubMed] [Google Scholar]
- 7. Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype‐phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J Genet Genomics. 2015;42:181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maestri NE, Lord C, Glynn M, Bale A, Brusilow SW. The phenotype of ostensibly healthy women who are carriers for ornithine transcarbamylase deficiency. Medicine. 1998;77:389–97. [PubMed] [Google Scholar]
- 10. Adam S, Almeida MF, Assoun M, Baruteau J, Bernabei SM, Bigot S, et al. Dietary management of urea cycle disorders: European practice. Mol Genet Metab. 2013;110:439–45. [DOI] [PubMed] [Google Scholar]
- 11. Burgard P, Kolker S, Haege G, Lindner M, Hoffmann GF. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders—review and meta‐analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016;39:219–29. [DOI] [PubMed] [Google Scholar]
- 12. Batshaw ML, Tuchman M, Summar M, Seminara J, Members of the urea cycle disorders C . A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113:127–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, et al. Rescue of ATP7B function in hepatocyte‐like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum Mol Genet. 2011;20:3176–87. [DOI] [PubMed] [Google Scholar]
- 14. Baxter M, Withey S, Harrison S, Segeritz C‐P, Zhang F, Atkinson‐Dell R, et al. Phenotypic and functional analyses show stem cell‐derived hepatocyte‐like cells better mimic fetal rather than adult hepatocytes. J Hepatol. 2015;62:581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishibashi K, Kuwahara M, Gu Y, Tanaka Y, Marumo F, Sasaki S. Cloning and functional expression of a new aquaporin (AQP9) abundantly expressed in the peripheral leukocytes permeable to water and urea, but not to glycerol. Biochem Biophys Res Commun. 1998;244:268–74. [DOI] [PubMed] [Google Scholar]
- 16. Tsukaguchi H, Shayakul C, Berger UV, Mackenzie B, Devidas S, Guggino WB, et al. Molecular characterization of a broad selectivity neutral solute channel. J Biol Chem. 1998;273:24737–43. [DOI] [PubMed] [Google Scholar]
- 17. Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration‐free human iPS cells. Nat Methods. 2011;8:409–12. [DOI] [PubMed] [Google Scholar]
- 18. Lee‐Montiel FT, Laemmle A, Charwat V, Dumont L, Lee CS, Huebsch N, et al. Integrated isogenic human induced pluripotent stem cell‐based liver and heart microphysiological systems predict unsafe drug‐drug interaction. Front Pharmacol. 2021;12:667010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laemmle A, Hahn D, Hu L, Rüfenacht V, Gautschi M, Leibundgut K, et al. Fatal hyperammonemia and carbamoyl phosphate synthetase 1 (CPS1) deficiency following high‐dose chemotherapy and autologous hematopoietic stem cell transplantation. Mol Genet Metab. 2015;114:438–44. [DOI] [PubMed] [Google Scholar]
- 20. Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, et al. De novo formation of the biliary system by TGFbeta‐mediated hepatocyte transdifferentiation. Nature. 2018;557:247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonder MJ, Kasela S, Kals M, Tamm R, Lokk K, Barragan I, et al. Genetic and epigenetic regulation of gene expression in fetal and adult human livers. BMC Genom. 2014;15:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berger DR, Ware BR, Davidson MD, Allsup SR, Khetani SR. Enhancing the functional maturity of induced pluripotent stem cell‐derived human hepatocytes by controlled presentation of cell‐cell interactions in vitro. Hepatology. 2015;61:1370–81. [DOI] [PubMed] [Google Scholar]
- 23. Matz P, Wruck W, Fauler B, Herebian D, Mielke T, Adjaye J. Footprint‐free human fetal foreskin derived iPSCs: a tool for modeling hepatogenesis associated gene regulatory networks. Sci Rep. 2017;7:6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wruck W, Adjaye J. Human pluripotent stem cell derived HLC transcriptome data enables molecular dissection of hepatogenesis. Sci Data. 2018;5:180035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu S, Rezvani M, Harbell J, Mattis AN, Wolfe AR, Benet LZ, et al. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature. 2014;508:93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Allegri G, Deplazes S, Grisch‐Chan HM, Mathis D, Fingerhut R, Haberle J, et al. A simple dried blood spot‐method for in vivo measurement of ureagenesis by gas chromatography‐mass spectrometry using stable isotopes. Clin Chim Acta. 2017;464:236–43. [DOI] [PubMed] [Google Scholar]
- 27. Brown GW Jr, Cohen PP. Comparative biochemistry of urea synthesis. I. Methods for the quantitative assay of urea cycle enzymes in liver. J Biol Chem. 1959;234:1769–74. [PubMed] [Google Scholar]
- 28. Maeda N, Funahashi T, Shimomura I. Metabolic impact of adipose and hepatic glycerol channels aquaporin 7 and aquaporin 9. Nat Clin Pract Endocrinol Metab. 2008;4:627–34. [DOI] [PubMed] [Google Scholar]
- 29. Tsukaguchi H, Weremowicz S, Morton CC, Hediger MA. Functional and molecular characterization of the human neutral solute channel aquaporin‐9. Am J Physiol. 1999;277:F685–96. [DOI] [PubMed] [Google Scholar]
- 30. Hakvoort TBM, He Y, Kulik W, Vermeulen JLM, Duijst S, Ruijter JM, et al. Pivotal role of glutamine synthetase in ammonia detoxification. Hepatology. 2017;65:281–93. [DOI] [PubMed] [Google Scholar]
- 31. Laemmle A, Gallagher RC, Keogh A, Stricker T, Gautschi M, Nuoffer J‐M, et al. Frequency and pathophysiology of acute liver failure in ornithine transcarbamylase deficiency (OTCD). PLoS One. 2016;11:e0153358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum Mutat. 1993;2:174–78. [DOI] [PubMed] [Google Scholar]
- 33. Grompe M, Caskey CT, Fenwick RG. Improved molecular diagnostics for ornithine transcarbamylase deficiency. Am J Hum Genet. 1991;48:212–22. [PMC free article] [PubMed] [Google Scholar]
- 34. Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42:1192–230. [DOI] [PubMed] [Google Scholar]
- 35. Enns GM. Neurologic damage and neurocognitive dysfunction in urea cycle disorders. Semin Pediatr Neurol. 2008;15:132–9. [DOI] [PubMed] [Google Scholar]
- 36. Gallagher RC, Lam C, Wong D, Cederbaum S, Sokol RJ. Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J Pediatr. 2014;164:720–25 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu L, Rayhill SC, Hsu EK, Landis CS. Liver transplantation for urea cycle disorders: analysis of the united network for organ sharing database. Transplant Proc. 2015;47:2413–8. [DOI] [PubMed] [Google Scholar]
- 38. Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, et al. A dual AAV system enables the Cas9‐mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang L, Bell P, Morizono H, He Z, Pumbo E, Yu H, et al. AAV gene therapy corrects OTC deficiency and prevents liver fibrosis in aged OTC‐knock out heterozygous mice. Mol Genet Metab. 2017;120:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunningham SC, Siew SM, Hallwirth CV, Bolitho C, Sasaki N, Garg G, et al. Modeling correction of severe urea cycle defects in the growing murine liver using a hybrid recombinant adeno‐associated virus/piggyBac transposase gene delivery system. Hepatology. 2015;62:417–28. [DOI] [PubMed] [Google Scholar]
- 41. Yudkoff M, Daikhin Y, Nissim I, Jawad A, Wilson J, Batshaw M. In vivo nitrogen metabolism in ornithine transcarbamylase deficiency. J Clin Invest. 1996;98:2167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li C, Wang W. Molecular biology of aquaporins. Adv Exp Med Biol. 2017;969:1–34. [DOI] [PubMed] [Google Scholar]
- 43. Lindskog C, Asplund A, Catrina A, Nielsen S, Rutzler M. A systematic characterization of aquaporin‐9 expression in human normal and pathological tissues. J Histochem Cytochem. 2016;64:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Litman T, Sogaard R, Zeuthen T. Ammonia and urea permeability of mammalian aquaporins. Handb Exp Pharmacol. 2009;327–58. [DOI] [PubMed] [Google Scholar]
- 45. Lyu C, Shen J, Zhang J, Xue F, Liu X, Liu W, et al. The state of skewed X chromosome inactivation is retained in the induced pluripotent stem cells from a female with hemophilia B. Stem Cells Dev. 2017;26:1003–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material