

Abstract
Kaposi’s sarcoma herpesvirus or human herpesvirus-8 (KSHV/HHV-8) is the etiological agent of Kaposi’s sarcoma (KS), an AIDS-defining angioproliferative neoplasm that continues to be a major global health problem and, of primary effusion lymphoma (PEL), a rare incurable B-cell lymphoma. This review describes the research from our laboratory and its collaborators to uncover molecular mechanisms of viral oncogenesis in order to develop new pathogenesis-based therapies to the KSHV-induced AIDS malignancies KS and PEL. They include the discovery of the viral angiogenic oncogene G protein-coupled receptor (vGPCR), the development of mouse models of KSHV and oxidative stress-induced KS, the identification of the role of Rac1-induced ROS in viral oncogenesis of KS and the development of novel therapeutic approaches able to target both latent and lytic oncogenic KSHV infection.
Keywords: AIDS-associated malignancies, Targeted therapies, Antivirals, Human herpesvirus-8, Angiogenesis, Animal models of cancer
Kaposi’s sarcoma-associated herpesvirus, its constitutively active G protein-coupled receptor and the paracrine oncogenesis hypothesis
Our laboratory in the Department of Microbiology and Immunology of the University of Miami Miller School of Medicine focuses on studies of Kaposi’s sarcoma herpesvirus (KSHV/HHV-8) using cell and animal models. KSHV is the etiological agent of Kaposi’s sarcoma (KS) [1], an AIDS-defining angioproliferative neoplasm that in spite of the implementation of HAART continues to be a major global health problem [2, 3], of multicentric Castlemans disease [4] and of primary effusion lymphoma (PEL), a rare incurable AIDS-associated B-cell lymphoma [5, 6]. In 1994; DNA fragments of a novel human herpesvirus highly homologous to two oncogenic gammaherpesviruses, Epstein–Barr virus (EBV) and herpesvirus saimiri (HVS) were found in strict association with Kaposi’s sarcoma lesions, and thus named Kaposi’s sarcoma-associated herpesvirus DNA (KSHV-DNA) [7]. Our laboratory, which at what time was in Weill Cornell Medical College, in collaboration with Ethel Cesarman, Shahin Rafii, and Adam Asch was the first in isolating infectious particles from PEL cell lines and transmitting it to B cells [8, 9]. We showed that supernatants from PEL/body cavity B-cell lymphoma contained herpesviral-like particles contained encapsidated KSHV-DNA-containing viral genomes [9]. We found that the particles were infectious and able to virally transmit KSHV-DNA to CD19? B cells, since UV light and foscarnet, a herpesviral polymerase inhibitor blocked the transmission [9]. KSHV turned out to fulfill all necessary Koch-like postulates that would define them as KS etiologic agent. It was strictly associated with all clinical forms of KS, it infected KS spindle cells, KSHV infection preceded the onset of KS and KSHV seroprevalence was higher in areas of higher incidence of KS. Finally, the genome of KSHV was found to encode many genes with potential for oncogenesis [1, 2, 10, 11]. Therefore, we next focused on identifying genes of the virus that could explain its oncogenic capacity; in particular, we sought for genes able to confer the characteristic KS angiogenic phenotype. We studied the KSHV-encoded G protein-coupled receptor (vGPCR), characterized by our collaborators Ethel Cesarman and Marvin Gershenghorn [12]. Interestingly, vGPCR was most homologous to two human receptors, CXCR-1 and CXCR-2, which are receptors for the angiogenic chemokines IL-8 and Gro-a/MGSA and also found to be able to signal both in a constitutive manner [12]. Thus, we speculated that vGPCR could have potential in transformation and angiogenesis. We found that vGPCR was able to constitutively activate the MAPKs, JNK and p38, and was able to transform NIH 3T3 cells as shown by focus formation and tumorigenesis assays [13]. More importantly, we showed that vGPCR expression on NIH3T3 cells was able to induce an angiogenic phenotype, which was characterized by the ability of NIH3T3-vGPCR supernatants to induce HUVEC proliferation and microtube formation, and was mediated by vascular endothelial growth factor (VEGF) expression and secretion [13]. This finding clearly indicated that KSHV possessed the molecular armamentarium to cause the KS angioproliferative lesion. Our findings indicated that through its vGPCR, KSHV could exploit host cell signaling pathways to induce transformation and angiogenesis in KSHV-mediated oncogenesis. Through collaborative studies, we showed that vGPCR was able to activate all the MAPKs and to regulate the cytoskeleton via RAFTK/Pyk2 activation [14]. We also found that through these signaling cascades vGPCR activated HIF-1alpha-mediated VEGF transcription [15], further indicating the molecular potential of vGPCR as an angiogenic oncogene. However, to causally link vGPCR and KSHV oncogenesis of KS, there were constrains determined by herspesviral biology and KS lesion histopathology that needed to be considered. As all herpesviruses, KSHV has two replicative programs: (1) a latent restricted program in which the virus coexists as an episome and divides with the host cells and (2) a full lytic program of viral replication whereby all the viral components including DNA and structural proteins are produced, leading to infectious virion production and cytopathic killing of the host cell (hence the denomination lytic) [2]. Since vGPCR belonged to the lytic cycle of the virus, the finding of such a powerful angiogenic oncogene that would only be expressed in lytically infected cells that are doomed for virus-induced killing posed a paradox [13]. A second problem came from direct observation of KS tumors, which showed that the majority of the infected cells were latently infected, with only a slim 1–5 % being lytically infected. Thus, our laboratory became a major proponent of new oncogenesis hypotheses: vGPCR could participate in oncogenesis when transiently expressed in cells that do not complete the lytic cycle—a mechanism also referred as abortive lytic replication. We also proposed that a minority of lytically infected cells expressing the angiogenic oncogene vGPCR could participate in oncogenesis in a paracrine manner (paracrine oncogenesis) [13, 16]. This oncogenesis modality was characteristic of some lymphomas such as Hodgkin’s disease, in which a few transformed Reed–Sternberg cells drive proliferation of non-transformed lymphoid cells [17]. Lytically infected cells expressing vGPCR would secrete VEGF and other factors that could drive in a paracrine manner latently infected cell proliferation and angioproliferation (see Fig. 1). Our second study showing the oncogenicity of vGPCR in KSHV human target endothelial cells further supported these hypotheses. We showed that retroviral transduction of vGPCR to human umbilical vein endothelial cells (HUVEC) led to immortalization by autocrine and paracrine loops mediated by VEGF–VEGF receptor-2/KDR axis activation of the AKT and ERK1/s signaling cascades [18]. This indicated that; in human cells, vGPCR was able to induce an immortalizing oncogenic hit through autocrine stimulation of VEGF-R2/KDR. Using the vGPCR reverse agonist IP10 in combination with anti-VEGFR-2 antibodies and tyrosine kinase inhibitors, we were able to demonstrate that: although VEGF autocrine stimulation was essential for immortalized HUVEC cell survival mediated by AKT activation; suppression of signaling by vGPCR could be compensated by exogenous VEGF [18]. This oncogenic mechanism was compatible with both the transient and the paracrine scenario. In both scenarios, lytically or abortive–lytically infected cells expressing vGPCR and producing VEGF would be able to induce angiogenesis, and they would also be able to paracrinally support the growth of latent cells transformed by transient expression of vGPCR that no longer express vGPCR [18] (see Fig. 1). Important evidences in support of the ability of vGPCR to drive paracrine oncogenesis were provided by the S. Lira group, which showed that transgenic expression of vGPCR was able to induce tumors that were composed of a minority of vGPCR-expressing cells producing paracrine factors such as VEGF and PDGF that were necessary for tumor growth [19, 20]. Another important experimental support to this hypothesis was provided by the elegant work of the Silvio Gutkind and Silvia Montaner groups. Employing endothelial-specific retrovirus transduction (RCAS system), they showed that vGPCR was the only KSHV gene able to induce angioproliferative malignancies, showing that vGPCR was expressed in a minority of tumor cells [21]. They also employed this model to identify Rac1 as the major GTPase mediating vGPCR activation of NFjB-mediated angiogenesis [22], and to show that vGPCR-expressing cells were able to support in a paracrine manner the tumorigenicity of endothelial cells expressing KSHV latent genes [23]. These groups made also a key contribution to the identification of the PI3K–AKT–mTOR pathway as a major target in vGPCR oncogenesis [24]. All these basic and preclinical studies pointing to a role of KSHV vGPCR in KSHV oncogenesis, were further supported by clinical studies showing the efficacy of the mTOR inhibitor sirolimus/rapamycin in renal transplant KS [25], further reinforcing the concept that vGPCR was a bona fide KS therapeutic target [26].Yet, in order to validate vGPCR as target, its essential role in angiogenesis and viral oncogenesis had to be demonstrated in the context of a model of tumorigenicity induced by the whole KSHV virus. We therefore focused on developing such a model, since we thought that would also be an important tool for mechanistic and preclinical studies. A key step for achieving a murine model was the creation of an infectious bacterial artificial chromosome containing the whole KSHV genome by the Shou-Jiang Gao Laboratory in UTHSC San Antonio [27]. This tool opened the possibility of using cell transfection to target mouse cells, which until that time showed to be refractory to KSHV infection. Based on accumulating evidence and on pioneering work by the late Phil Browning showing the existence of circulating spindle cell progenitors in AIDS–KS, we decided to target a population enriched in endothelial progenitor cells (EPC) [28], with the help and expertise of the Shahin Rafii Laboratory. We succeeded in transfecting a bacterial chromosome encoding the full KSHV (KSHVBac36) to freshly isolated mouse bone marrow endothelial-lineage cells (mEC) to create a tumorigenic cell type, mECK36, able to induce highly vascularized spindle cell Kaposi’s sarcoma-like tumors in nude mice when injected s.c., and multifocal KS lung tumors in irradiated SCID mice when it was injected i.v. [29]. The identity of these Kaposi’s sarcoma-like murine tumors was validated using genomic microarrays for the host as well as by the use of PCR microarrays for the full viral genome carried out by our collaborator Dirk Dittmer at UNC Chappell Hill [29]. These analyses complemented with complete histological and immunohistochemical and cytofluorometry analysis, showed that mECK36 tumors displayed transcriptomes that were highly representative of both the viral and host KS transcriptome and displayed most of the phenotypic markers of KS lesions. The tumor cells expressed the typical KS histological markers including VEGFR-2,3 and the KS and endothelial lymphatic marker podoplanin [29]. Most importantly, we showed that these tumors were, as KS, composed of cells lytically and latently infected with KSHV. All the cells were episomally infected since they displayed the typical LANA punctuated nuclear pattern. Additionally, a noticeable percentage also expressed the lytic gene K8.1, indicating the presence of both latent and lytic cells. In order to use our model to study viral pathogenesis, we needed to prove that tumorigenicity was strictly KSHV dependent. We demonstrated that mECK36 cells which were forced to loose the episome in vitro by lack of antibiotic selection were unable to form tumors, indicating that KSHV viral gene expression provided an in vivo survival advantage to the infected cells. A related interesting finding was that in the tumors, most KSHV lytic genes were upregulated, in particular several viral host homologs that are potentially oncogenic including the lytic angiogenic oncogene vGPCR. This upregulation correlated with upregulation of host angiogenic growth factors and their receptors, suggesting that the KSHV lytic upregulation and angiogenic upregulation were causally linked [29]. An important evidence in support for this mechanism and also of the paracrine hypothesis was obtained by shRNA-mediated vGPCR silencing in the mECK36 tumors. We found that although mECK36 cells expressing vGPCRshRNA or control shRNA were indistinguishable by comparing their growth in vitro, only cells expressing vGPCR displayed high levels of VEGF expression and were tumorigenic and angiogenic in vivo. This result showed that although vGPCR was expressed only in lytically infected cells of the tumor, it played a non-redundant necessary role in inducing tumor angiogenesis and in driving KS tumorigenesis [29]. Main conclusions of these experiments were the following: (1) vGPCR and the vGPCR-induced paracrine mediators of oncogenesis were valid therapeutic targets and (2) our mECK36 tumor model was a powerful tool to study viral oncogenesis and for testing new anti-KSHV therapeutic approaches. Hence, the next focus was to use the mECK36 and other new animal models to identify critical host mediators in KSHV viral paracrine oncogenesis in order to develop new therapeutic interventions.
Fig. 1.
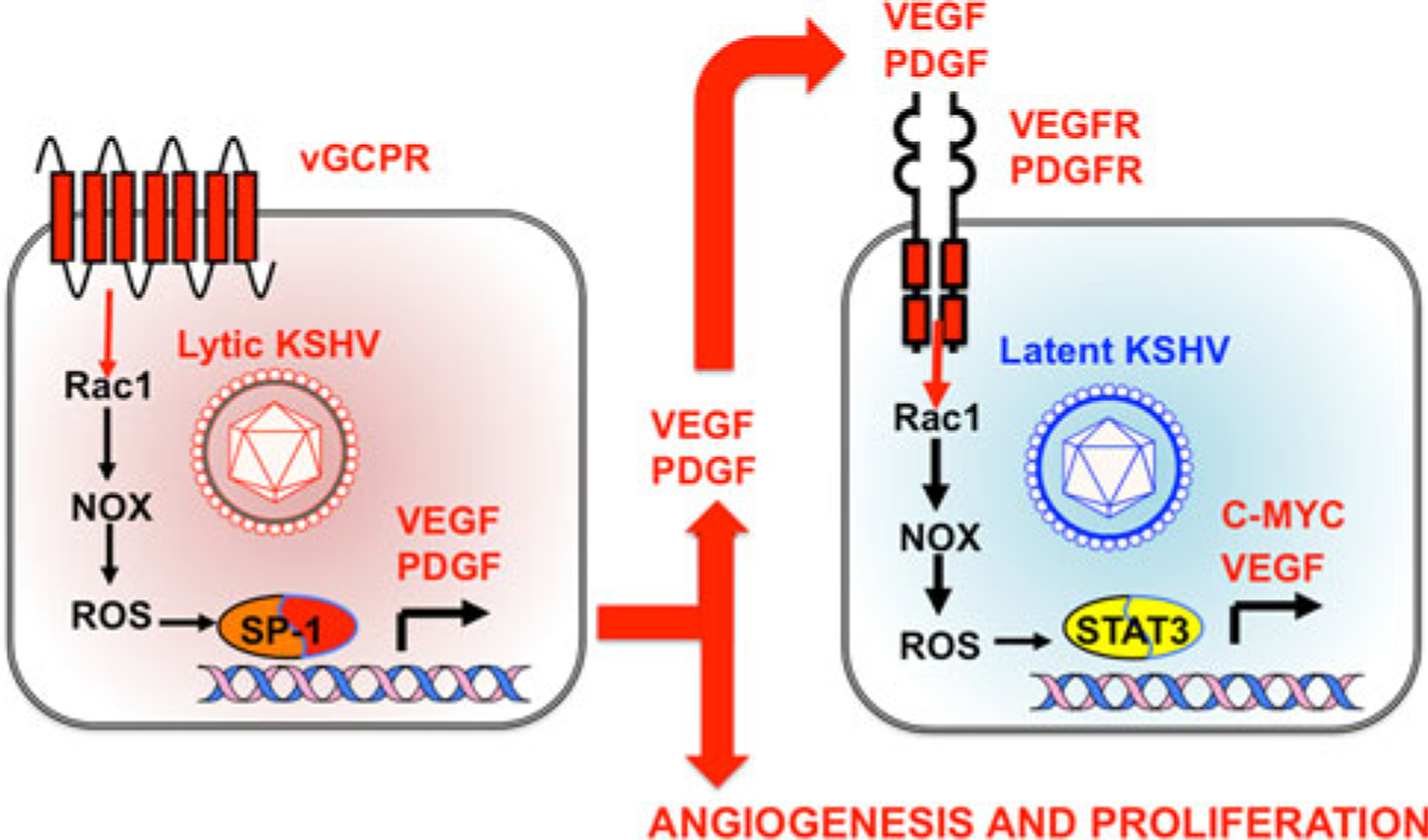
Role of Rac1 and ROS in mechanisms of KSHV-induced paracrine oncogenesis. In lytic or abortive lytic infected cells, expression of KSHV early lytic genes such as viral G protein-coupled receptor (vGPCR) activates the Rac1–NOX–ROS pathway, leading to the expression and secretion of angiogenic and proliferative factors including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) [9, 13, 32]. VEGF and PDGF stimulate their receptors in latently infected cells through a paracrine mechanism, complementing the oncogenic activities of KSHV latent genes and leading Rac1- and ROS-mediated STAT3 activation [32]. STAT3 activates transcription of c-myc and VEGF, increasing cell proliferation and angiogenesis [9, 32]. ROS inhibition by NAC could therefore block oncogenic mechanisms in lytic and latent cells, leading to inhibition of KSHV-induced angiogenesis and tumorigenesis [32]
Role of Rac1-induced oxidative stress in KSHV oncogenesis
In the recent years, key discoveries have substantiated the role of oxidative stress mediators in oncogenesis. ROS, first thought to be a spurious product of inflammation, have now a recognized broad function in oncogenic signaling. This is due in part to the discovery of non-phagocytic NADPH oxidases (NOX), a ROS-producing enzyme which is regulated by the inflammatory signaling mediator Rac. ROS production plays a role in cell cycle regulation, DNA damage and angiogenesis. Yet the specific molecular events linking ROS and these cancer hallmarks are still elusive. The Goldschmidt lab pioneered the field of oxidative stress in oncogenesis by showing that ROS plays a key role in Ras-mediated transformation. They demonstrated that a constitutively activated form of Ras induces a significant increase in intracellular ROS in NIH3T3 cells. They inhibited transformation not only with the ROS scavenger NAC, but also with the expression of a dominant negative mutant of Rac1 GTPase or with diphenylene iodonium (DPI), an inhibitor of NOX [30]. In the recent years, we teamed up with the Goldchmidt laboratory that developed a transgenic mouse model based on the expression of a constitutively activated mutant of Rac1 (RacV12), an activator of NOX-mediated ROS production which is also a major host mediator of KSHV vGPCR angiogenesis, under the control of the α-smooth muscle actin promoter (α-SMA). We found that; remarkably, these transgenic mice developed KS-like tumors in a manner that phenocopied vGPCR transgenic mice [31]. IHC analysis showed that tumors expressed KS and endothelial markers, α-SMA, and Rac1; while gene arrays from RacCA tumors showed a significant overlap with mouse-KSHV-induced and human AIDS–KS [31]. Most interestingly, we were able to show that Rac1 KS-like tumorigenesis was completely prevented by administration of the FDA-approved antioxidant N-acetyl l-cysteine (NAC) [31]. We also found that Rac1 was highly expressed in AIDS–KS lesions, making both Rac1 and ROS candidate targets in KSHV oncogenesis. The fact that Rac1 was expressed in all the KSHV-infected cells of the KS lesion and mouse KSHV tumors and not only in vGPCR cells was also supportive of the paracrine oncogenesis hypothesis since Rac1 is activated downstream of receptors that are activated through vGPCR-induced cytokines such as VEGF, PDGF and IL-8. We therefore proposed that vGPCR via Rac1 and paracrine mediators of oxidative stress played a major role in KS and that their targeting could have therapeutic utility. We next showed that, as predicted, ROS were induced by vGPCR via Rac1-NOX pathways, and that the antioxidant NAC was able to both prevent and block KSHV-induced tumorigenesis through anti-angiogenic and anti-proliferative mechanisms. These results pointed to ROS as attractive KS prevention and therapeutic targets [32] and NAC as an viable FDA-approved drug, particularly for resource-poor settings such as Africa, where KS continues to be the most prevalent cancer among men and children [3, 32]. Analysis of treated tumors revealed that NAC anti-tumor effect was mediated by a strong anti-angiogenic and anti-proliferative activity mediated by inhibition of the oncogenic transcription factor STAT3. We found that STAT3 transcription mediates ROS-induced transcription of c-myc and VEGF in KSHV latently infected cells causing KS cytokine PDGF [32]. Thus, PDGF–PDGFR interactions, in addition to VEGF–VEGF-R2, could indeed be major paracrine mediators of KSHV vGPCR paracrine oncogenesis via a Rac1–ROS pathway (Fig. 1). In fact, PDGFR was found to be the main target in AIDS–KS phase I using the multi-tyrosine kinase inhibitor Gleevec/imatinib, which in addition to Bcr-Abl is a powerful inhibitor of c-kit and PDGFR [33]. Accordingly, we found that both imatinib and sunitinib, a PDGFR and VEGFR inhibitor were able to inhibit mouse KS tumors. This further confirms the therapeutic value of targeting this paracrine activation cascade in KSHV-induced KS and the preclinical value of our mouse mECK36 KSHV tumor model (LEC, QM, PJG and EAM unpublished).
Translational research to target KSHV latent and lytic oncogenic infection
The “paracrine oncogenesis” hypothesis predicts that agents able to either target lytically or latently infected cells could have therapeutic applications. PEL is considered a model of KSHV oncogenic latency [34]. Moreover, it was shown that latent KSHV genes, such as viral FLICE inhibitor (vFLIP) through its ability to cause constitutive NFkB activation, are essential for lymphoma survival [34]. Together with the laboratories of Izidore Lossos and Juan Carlos Ramos, we developed a direct xenograft model of PEL KSHV lymphoma in SCID mice. Using this system, we showed the higher efficacy of proteasome inhibitor bortezomib/Velcade to target latently infected PELs in vitro and in vivo by lytic induction and c-myc mediated apoptosis [35]. We next decided to study the effects of bortezomib (BTZ) in the context of a new approach based on combining agents that cause lytic herpesviral reactivation with agents that would exacerbate cell killing. As a KSHV inducer, we employed the FDA-approved HDAC inhibitor suberoylanilidehydroxamic acid (SAHA), a powerful herpesviral reactivator that was tested in combination with bortezomib. This approach showed that the combination of BTZ and SAHA was most effective in inducing the KSHV lytic cycle with massive apoptosis, which lead to an increased survival in treated PEL-bearing mice through a mechanism involving p53 acetylation and stabilization [36]. One of the major concerns of this viral reactivation therapies would be the production of infectious virus in the context of immunosuppression. We showed that the SAHA–BTZ combination was able to induce the lytic cycle of the virus and at the same time, was able to totally abrogate late lytic gene expression and virus production by massively inducing cell apoptosis; thus making this novel a new anti-KSHV concept specially suitable to treat PEL in the AIDS setting [36]. In order to test this kind of approach in the KS setting, our laboratory recently developed yet another animal model of KSHV-induced KS. To make it suitable for testing anti-viral approaches, this new animal model was not based on a Bac KSHV, but instead it was based on recombinant infectious KSHV (KSHVr219). We were able to infect with rKSHV219 freshly obtained mEC cells that in SCID/NOD mice induced tumors showing the production of mature KSHV virion particles. We also showed that in this setting, lytic replication could be induced by SAHA. The importance of showing the development of tumorigenic cells by direct infection of primary mEC, is that it opens the possibility to potentially employ cells from any genetically engineered mice, thus opening new avenues not only for anti-viral testing but also for viral pathogenesis research (BMA, QM and EAM unpublished).
Another recent collaborative project with the Dr. Theodore Lampidis Lab led to the discovery of a new mechanism whereby sugar analogs can be employed as anti-KSHV oncogenesis agents. This lab discovered that 2-deoxyglucose/2-deoxymannose (2DG) is an inhibitor of both glycolysis and N-linked glycosylation [37]. 2DG is preferentially targeted to tumor cells that have increased glucose uptake and metabolism. Thus, 2DG, which has shown prowess in recent Phase I clinical trials [38], is thought to kill cancer cells in hypoxia that are sustained by glycolysis. In addition, inhibition of N-glycosylation causes accumulation of misfolded glycoproteins in ER leading to ER stress and the unfolded protein response (UPR) triggering phosphorylation of eukaryotic initiation factor 2α (eIF2α), which results in inhibition of protein synthesis in order to maintain ER and cellular homeostasis [37]. Herpesviral lytic replication with massive viral glycoprotein production is a powerful inducer of ER stress [39]. However, herpesviruses have evolved molecular strategies to avoid UPR [40]. We speculated that exogenous activation of ER stress by 2DG inhibition of N-glycosylation would overwhelm this mechanism and lead to UPR and eIF2α phosphorylation, which is also the major target of IFNα-induced dsRNA-dependent kinase PKR [40]. We employed three systems of KSHV replication and pathogenesis as well as the lytic mouse gammaherpesvirus (MHV-68) system that we implemented in collaboration with Samita Andreansky. We found that 2DG powerfully blocks viral replication, lytic gene expression, viral DNA replication and the expression of host pathogenic factors such as PDGF and VEGF [40]. Using molecular analysis and mannose competition, we showed that (1) the glycolysis inhibitor FDG could not reproduce the inhibition, (2) inhibition of viral replication and reactivation occurred through a mannose reversible mechanism and (3) inhibition occurred with simultaneous UPR activation and eIF2α phosphorylation [40]. We concluded that 2DG was inhibiting viral replication via induction of the UPR [40]. The fact that cancer cells and KSHV-infected cells upregulate glucose uptake further suggests a mechanism for increasing targeting of 2DG to infected cells that may favor its therapeutic activity in KS. In addition, 2DG anti-angiogenic activity further suggests that our mechanism could find broad application to viral-induced cancers, particularly hepatitis C virus-induced hepatocellular carcinoma, which is known to have a major UPR-driven oncogenic component [40].
Concluding remarks
In the last years through interactions with the excellent environment at the UMMSM Dept of Microbiology and Immunology, our laboratory has consolidated our research geared to uncover viral mechanisms of oncogenesis in the hope that they will illuminate the road to novel therapeutic approaches. We are expanding our scope through the Miami Center for AIDS Research and its AIDS Malignancies Scientific Working Group to collaborate with the Hussman Institute of Genetics, with AIDS Malignancies Miami Core site of Sylvester Comprehensive Cancer Center, and with strong programmatic support from NCI and its Office of HIV/AIDS Malignancies (OHAM), the AIDS Cancer Specimen Resource (ACSR) and the AIDS Malignancies Consortium (AMC). We can now harness the power of new genomic approaches such as genome-wide association screenings to study genetic predisposition to KS. We can now employ next-generation sequencing techniques to fully understand viral and host contributions to the AIDS–KS oncogenome and molecular evaluation of KS presentation and clinical responses. These approaches aided with the unique tools of our laboratory to study KSHV oncogenesis in cell and animal models and the exceptional support of our collaborators are paving the way for implementing new therapeutic approaches to prevent, diagnose and treat KSHV-induced AIDS malignancies.
Acknowledgments
The research described in this review was funded through NIH/PHS Grants to E.A.M. AI39192, CA75918 and CA136387, by NCI/OHAM supplements to the Miami CFAR Grant 5P30AI073961, by American Cancer Society RPG-99-207-01-MBC and by the Sylvester Comprehensive Cancer Center.
Biography

Enrique A. Mesri
References
- 1.Boshoff C, Chang Y. Kaposi’s sarcoma-associated herpesvirus: a new DNA tumor virus. Annu Rev Med. 2001;52:453–70. doi: 10.1146/annurev.med.52.1.453. [DOI] [PubMed] [Google Scholar]
- 2.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10(10):707–19. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casper C The increasing burden of HIV-associated malignancies in resource-limited regions. Annu Rev Med. 2011;62:157–70. doi: 10.1146/annurev-med-050409-103711. [DOI] [PubMed] [Google Scholar]
- 4.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276–80. [PubMed] [Google Scholar]
- 5.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Medicine. 1995;332(18):1186–91. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 6.Cesarman E, Mesri EA. Virus-associated lymphomas. Curr Opin Oncol. 1999;11(5):322–32. [DOI] [PubMed] [Google Scholar]
- 7.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192): 1865–9. [DOI] [PubMed] [Google Scholar]
- 8.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, et al. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein–Barr virus. Blood. 1996;88(7): 2648–54. [PubMed] [Google Scholar]
- 9.Mesri EA, Cesarman E, Arvanitakis L, Rafii S, Moore MA, Posnett DN, et al. Human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J Exp Med. 1996;183(5):2385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci USA. 1996;93(25): 14862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganem D KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Investig. 2010; 120(4):939–49. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385(6614):347–50. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- 13.Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, et al. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391(6662):86–9. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- 14.Munshi N, Ganju RK, Avraham S, Mesri EA, Groopman JE. Kaposi’s sarcoma-associated herpesvirus-encoded G protein-coupled receptor activation of c-jun amino-terminal kinase/stress-activated protein kinase and lyn kinase is mediated by related adhesion focal tyrosine kinase/proline-rich tyrosine kinase 2. J Biol Chem. 1999;274(45):31863–7. [DOI] [PubMed] [Google Scholar]
- 15.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, et al. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60(17):4873–80. [PubMed] [Google Scholar]
- 16.Cesarman E, Mesri EA, Gershengorn MC. Viral G protein-coupled receptor and Kaposi’s sarcoma: a model of paracrine neoplasia? J Exp Med. 2000;191(3):417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuppers R The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 18.Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, Dias S, et al. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell. 2003;3(2):131–43. [DOI] [PubMed] [Google Scholar]
- 19.Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191(3):445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen KK, Manfra DJ, Grisotto MG, Martin AP, Vassileva G, Kelley K, et al. The human herpes virus 8-encoded chemokine receptor is required for angioproliferation in a murine model of Kaposi’s sarcoma. J Immunol. 2005;174(6):3686–94. [DOI] [PubMed] [Google Scholar]
- 21.Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3(1): 23–36. [DOI] [PubMed] [Google Scholar]
- 22.Montaner S, Sodhi A, Servitja JM, Ramsdell AK, Barac A, Sawai ET, et al. The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood. 2004;104(9):2903–11. doi: 10.1182/blood-2003-12-4436. [DOI] [PubMed] [Google Scholar]
- 23.Montaner S, Sodhi A, Ramsdell AK, Martin D, Hu J, Sawai ET, et al. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi’s sarcoma. Cancer Res. 2006;66(1):168–74. doi: 10.1158/0008-5472.CAN-05-1026. [DOI] [PubMed] [Google Scholar]
- 24.Sodhi A, Chaisuparat R, Hu J, Ramsdell AK, Manning BD, Sausville EA, et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10(2): 133–43. [DOI] [PubMed] [Google Scholar]
- 25.Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005;352(13):1317–23. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 26.Yarchoan R Key role for a viral lytic gene in Kaposi’s sarcoma. N Engl J Med. 2006;355(13):1383–5. doi: 10.1056/NEJMcibr063911. [DOI] [PubMed] [Google Scholar]
- 27.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, et al. Efficient infection by a recombinant Kaposi’s sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol. 2002;76(12):6185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Browning PJ, Sechler JM, Kaplan M, Washington RH, Gendelman R, Yarchoan R, et al. Identification and culture of Kaposi’s sarcoma-like spindle cells from the peripheral blood of human immunodeficiency virus-1-infected individuals and normal controls. Blood. 1994;84(8):2711–20. [PubMed] [Google Scholar]
- 29.Mutlu AD, Cavallin LE, Vincent L, Chiozzini C, Eroles P, Duran EM, et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell. 2007;11(3):245–58. doi: 10.1016/j.ccr.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275(5306):1649–52. [DOI] [PubMed] [Google Scholar]
- 31.Ma Q, Cavallin LE, Yan B, Zhu S, Duran EM, Wang H, et al. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi’s sarcoma. Proc Natl Acad Sci USA. 2009;106(21):8683–8. doi: 10.1073/pnas.0812688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Q, Cavallin LE, Leung HJ, Chiozzini C, Goldschmidt-Clermont PJ, Mesri EA. A role for virally induced reactive oxygen species in Kaposi’s sarcoma herpesvirus tumorigenesis. Antioxid Redox Signal. 2013;18(1):80–90. doi: 10.1089/ars.2012.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koon HB, Bubley GJ, Pantanowitz L, Masiello D, Smith B, Crosby K, et al. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(5):982–9. doi: 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
- 34.Cesarman E, Mesri EA. Pathogenesis of viral lymphomas. Cancer Treat Res. 2006;131:49–88. [DOI] [PubMed] [Google Scholar]
- 35.Sarosiek KA, Cavallin LE, Bhatt S, Toomey NL, Natkunam Y, Blasini W, et al. Efficacy of bortezomib in a direct xenograft model of primary effusion lymphoma. Proc Natl Acad Sci USA. 2010;107(29):13069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, Lossos IS, et al. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Investig. 2013;123(6):2616–28. doi: 10.1172/JCI64503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, et al. Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6(11):3049–58. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- 38.Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(2):523–30. doi: 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
- 39.Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79(11):6890–9. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leung HJ, Duran EM, Kurtoglu M, Andreansky S, Lampidis TJ, Mesri EA. Activation of the unfolded protein response by 2-deoxy-d-glucose inhibits Kaposi’s sarcoma-associated herpesvirus replication and gene expression. Antimicrob Agents Chemother. 2012;56(11):5794–803. doi: 10.1128/AAC.01126-12. [DOI] [PMC free article] [PubMed] [Google Scholar]