

ABSTRACT
Live-cell fluorescence imaging of methanogenic archaea has been limited due to the strictly anoxic conditions required for growth and issues with autofluorescence associated with electron carriers in central metabolism. Here, we show that the fluorescence-activating and absorption-shifting tag (FAST) complexed with the fluorogenic ligand 4-hydroxy-3-methylbenzylidene-rhodanine (HMBR) overcomes these issues and displays robust fluorescence in Methanococcus maripaludis. We also describe a mechanism to visualize cells under anoxic conditions using a fluorescence microscope. Derivatives of FAST were successfully applied for protein abundance analysis, subcellular localization analysis, and determination of protein-protein interactions. FAST fusions to both formate dehydrogenase (Fdh) and F420-reducing hydrogenase (Fru) displayed increased fluorescence in cells grown on formate-containing medium, consistent with previous studies suggesting the increased abundance of these proteins in the absence of H2. Additionally, FAST fusions to both Fru and the ATPase associated with the archaellum (FlaI) showed a membrane localization in single cells observed using anoxic fluorescence microscopy. Finally, a split reporter translationally fused to the alpha and beta subunits of Fdh reconstituted a functionally fluorescent molecule in vivo via bimolecular fluorescence complementation. Together, these observations demonstrate the utility of FAST as a tool for studying members of the methanogenic archaea.
IMPORTANCE Methanogenic archaea are important members of anaerobic microbial communities where they catalyze essential reactions in the degradation of organic matter. Developing additional tools for studying the cell biology of these organisms is essential to understanding them at a mechanistic level. Here, we show that FAST, in combination with the fluorogenic ligand HMBR, can be used to monitor protein dynamics in live cells of M. maripaludis. The application of FAST holds promise for future studies focused on the metabolism and physiology of methanogenic archaea.
KEYWORDS: archaea, FAST, fluorescence microscopy
INTRODUCTION
Methanogenic archaea (methanogens) are responsible for producing the majority of methane on Earth and are model organisms for studying cellular processes in the Archaea. Several organisms such as Methanococcus maripaludis, Methanosarcina spp., and Methanothermobacter spp. have been used extensively in genetic or biochemical studies to understand the physiology and metabolism of methanogens (1–3). However, robust tools for direct cell visualization via fluorescence microscopy to allow protein quantification, analysis of protein-protein interactions, and spatiotemporal characterization of cellular proteins have been lacking. To date, studies employing fluorescence microscopy on methanogens have been restricted to the enumeration or identification of cells via fluorescence in situ hybridization (FISH) or visualization by F420-based autofluorescence (4, 5). A strict requirement for growth under anoxic conditions as well as background autofluorescence due to the presence of the oxidized electron carrier coenzyme F420 (excitation peak centered at 420 nm and emission peak centered at 480 nm [6]) have prevented the use of most fluorescent protein tags. Green fluorescent protein (GFP) and other proteins within the GFP family require oxygen for fluorophore maturation and fail to perform under anaerobic growth conditions (7). Alternative oxygen-independent probes such as flavin mononucleotide (FMN) binding fluorescent proteins (e.g., light-oxygen-voltage [LOV]-based fluorescent proteins) typically have a weak fluorescence intensity and often exhibit emission at around 450 nm, overlapping the excitation/emission spectra of F420 (8). Immunofluorescence strategies using antibody-reporter conjugates typically involve aerobic steps and/or the permeabilization of cells using harsh detergents, which precludes live-cell imaging of anaerobes. Another consequence of aerobic preparation of methanogenic organisms is that it causes the rapid oxidation of coenzyme F420, which leads to increased autofluorescence.
Recently, an oxygen-independent fluorescent protein reporter known as the fluorescence-activating and absorption-shifting tag (FAST) has been developed and adapted to select anaerobic organisms (9–11). FAST is an engineered variant of photoactive yellow protein with a molecular mass of 14 kDa (9). Like GFP, FAST can be translationally fused to the N or C terminus of a gene of interest. By itself, FAST does not exhibit fluorescence; however, upon the addition of a fluorogenic ligand (fluorogen), the two form a complex producing a fluorescent product. Multiple fluorogens consisting of 4-hydroxybenzylidene rhodanine (HBR) derivatives exist, with each having unique excitation and emission properties upon binding to FAST, and variants of FAST that specifically bind a subset of fluorogens have been developed, allowing two-color live-cell imaging (12, 13). HBR derivatives are membrane permeable and bind reversibly and specifically to FAST (9).
There are several reasons why FAST is a particularly attractive molecular tool for studying obligate anaerobes such as methanogens. Most importantly, fluorescence is oxygen independent, and the membrane permeability of the ligand allows live-cell imaging. FAST complexes are inherently bright and emit fluorescence across a wide spectrum, avoiding the limitations of reporters such as FMN binding proteins. FAST fluorescence is reversible and quantitative, allowing the direct measurement of relative protein abundance (9, 14). Additionally, splitFAST was recently developed to visualize protein-protein interactions through bimolecular fluorescence complementation (BiFC) (15). Finally, as a small, translationally fused tag, FAST can be used to observe protein localization in live cells.
In this study, we demonstrate the successful use of the FAST toolkit in M. maripaludis, demonstrating a functional fluorescence-based system for microscopic imaging of a methanogen. We developed a platform for microscopy under anoxic conditions, allowing the visualization of live cells. Combining these advances, we observed robust and quantifiable fluorescence of differentially expressed proteins in cells grown with either H2 or formate as an electron donor, BiFC using the multisubunit formate dehydrogenase (Fdh), and two different examples of protein localization. This was accomplished using the original FAST protein, also referred to as FAST1, as well as a highly fluorescent, tandem variant. FAST-based fluorescence microscopy expands existing tools for studying the cell biology of M. maripaludis and should be broadly applicable to other methanogens with established protocols for heterologous protein expression.
RESULTS
Expression of FAST1 in M. maripaludis.
To test the functionality of FAST, a codon-optimized FAST1 gene (see Table S2 in the supplemental material) (16) was expressed in M. maripaludis on the replicating vector pLW40 under the control of the Methanococcus voltae histone promoter (17). Under these conditions, heterologously expressed protein can reach up to 1% of the total cellular protein (18). 4-Hydroxy-3-methylbenzylidene-rhodanine (HMBR), a FAST fluorogen, was added to stationary-phase cultures (optical density at 600 nm [OD600] = ~0.9) to a final concentration of 10 μM (based on the manufacturer’s recommendations) before transfer to a 96-well dark plate for quantification on a microplate reader. HMBR fluorescence was measured at 540 nm (excitation with 481 nm). Cultures that were not treated with HMBR exhibited little autofluorescence, while cells containing both FAST1 and HMBR exhibited a significant increase in fluorescence (Fig. 1A). The addition of HMBR to wild-type (WT) cells did not lead to a significant increase in fluorescence compared to cultures expressing FAST1 without HMBR addition.
FIG 1.

M. maripaludis expressing FAST is fluorescent upon HMBR addition. (A) Fluorescence intensities of M. maripaludis strains cultivated in McCas medium with H2 as the electron donor for growth. HMBR was added to a final concentration of 10 μM. Relative fluorescence units were determined by normalizing emission readings from a microplate reader against the baseline autofluorescence of the sample without fluorogen. Values were also normalized to the OD600 of the culture. (B) Titration of HMBR in cells expressing FAST1 or the tandem variant tdFAST2 grown in McCas medium with H2. Values were normalized to the OD600 of the culture. Data are averages and standard deviations from triplicate measurements.
To further optimize FAST1, we assessed the autofluorescence of wild-type cells during different stages of growth. We found that cells exhibited the lowest levels of autofluorescence prior to reaching an OD600 of ~0.40 or ~1.0 in medium supplemented with formate or H2 as the electron donor for growth, respectively (Fig. S1). We hypothesize that the increased autofluorescence at a higher OD600 is due to the accumulation of oxidized F420 when electron donors become growth limiting (19). We also noted that autofluorescence was generally higher in cultures grown on H2 and lower in cultures grown on formate; therefore, formate-grown cells were used in subsequent experiments when possible. Additionally, we tested whether altering the concentrations of HMBR could further optimize fluorescence over the background. In general, increasing concentrations of HMBR led to increased fluorescence (Fig. 1B).
HMBR is generally regarded as nontoxic. To verify if this is true for M. maripaludis, we assessed growth after exposing cells to HMBR. M. maripaludis expressing FAST1 on the plasmid pLW40 was grown in liquid overnight, and aliquots were then prepared anaerobically in a chamber by adding HMBR to a final concentration of 20 μM, the highest concentration used in this study. After a 30-min incubation, these cells were transferred to sterile medium and assessed for growth. There was no apparent difference in the lag period or growth rate compared to the controls that were not exposed to HMBR (Fig. S2). For all subsequent experiments, a concentration of 10 μM HMBR was used according to the manufacturer’s recommendations unless otherwise indicated.
Anoxic microscopy with a microscope housed inside an anoxic chamber.
Due to the oxygen sensitivity of M. maripaludis, live-cell imaging requires strict anoxia. To overcome this limitation, we developed a platform to perform fluorescence microscopy using an Echo Revolve R4 hybrid microscope inside a Coy anaerobic chamber (Fig. 2A). The microscope utilizes a computer tablet camera in place of an eyepiece, and all manipulations can be performed using a touch screen with a capacitive stylus. Using this system, HMBR addition, culture mounting, and imaging can all be performed without introducing oxygen. The microscope was operated in the upright orientation for all experiments.
FIG 2.

Anoxic microscopy of M. maripaludis expressing FAST1. (A) The anoxic fluorescence microscope used in all experiments. (B) Images of wild-type M. maripaludis and a strain expressing FAST1 from the replicating vector pLW40. Bars = 2 μm.
Wild-type M. maripaludis and the strain expressing FAST1 were examined both with and without fluorogen treatment (Fig. 2B). Only FAST1-expressing cells showed a noticeable fluorescence gain upon HMBR addition. Because preparing samples in the anoxic chamber involves transferring cells from the high H2 (or formate) concentrations of a culture tube to the low-hydrogen atmosphere of the chamber (3%), cells were imaged immediately after preparation to mitigate the possibility of increasing autofluorescence from F420 oxidation.
Evaluation of FruA localization and abundance during growth with H2 or formate.
The F420-reducing hydrogenase (Fru) catalyzes the reversible reduction of coenzyme F420 to F420H2 using H2 as an electron donor and is the primary source of F420H2 for methanogenesis (20, 21). The large subunit of the hydrogenase, FruA, was selected as a test case for analyzing a FAST1 translational fusion. Fru is abundant in the cell, shows differential expression with increased abundance when formate is supplied as an electron donor for growth, and is thought to associate with the cell membrane (21–23). Using allelic replacement mutagenesis (1), two strains with FAST1 translationally fused to either the N terminus or C terminus of FruA were created. Both strains were analyzed during early exponential growth with either H2 or formate as the electron donor.
The FAST1-FruA construct displayed a pattern of fluorescence consistent with the known membrane localization of Fru (21–23); however, this differed significantly from the pattern observed for the FruA-FAST1 fusion construct (Fig. 3A). Fluorescence in the C-terminal fusion was uniform across the cell, while the N-terminal fusion exhibited distinctly higher fluorescence along the outer perimeter of the cell. The different fluorescence patterns observed between the two FruA constructs are likely due to the proteolytic cleavage of the nascent peptide, which is required for the maturation of [Ni-Fe] hydrogenases (24). During maturation, Fru is likely proteolytically cleaved at the C terminus, resulting in the loss of the FAST1 tag from FruA-FAST1 fusions and the retention of the tag in FAST1-FruA fusions (Fig. 3B). This cleaved-FruA-FAST1-expressing strain likely retains FAST1 in the cytoplasm, leading to uniform cellular fluorescence, while the mature, processed, and nonfluorescent hydrogenase localizes to the membrane.
FIG 3.

Cellular localization and expression of N- and C-terminal translational fusions of FAST1 to FruA. (A) Images of N- and C-terminal FAST fusions to FruA. Bars = 2 μm. Arrows indicate the locations of fluorescent foci associated with the cell membrane. (B) Hypothesized maturation of the FruA peptide in the strain expressing fruA-FAST1. Maturation of FruA cleaves FAST off the mature hydrogenase. (C) Fluorescence intensities of FAST1-FruA cells grown in medium with either formate (184 cells) or H2 (236 cells) as the sole electron donor. Values were obtained by averaging the fluorescence intensities of single cells from each sample via microscopy. Relative fluorescence was normalized to correct for autofluorescence in the absence of fluorogen. Different circle colors represent data points from separate replicates, and mean values from all averaged cells in a replicate are represented by large circles. Data are averages and standard deviations from five independent cultures.
The FAST1-FruA fusion strain was further analyzed for differences in protein abundance between cells grown in medium containing H2 and those grown in medium containing formate. Several transcriptomic and proteomic studies have suggested that Fru is more abundant in cultures grown under conditions where H2 concentrations limit growth or when formate is provided as the sole electron donor (25–28). The FAST1-FruA fusion strain was examined by anoxic fluorescence microscopy during early exponential growth (OD600 = 0.2 to 0.4). As described above, cultures were processed in the absence of oxygen, treated with final concentrations of 10 μM fluorogen, and placed on a glass slide for immediate viewing. To control for autofluorescence, light intensity was measured on a per-cell basis in the absence of fluorogen, and the mean intensities of cells after fluorogen addition were normalized to the autofluorescence baseline. Cells grown utilizing formate as the sole electron donor had 1.76-fold-higher fluorescence than cells grown with H2 (Fig. 3C), consistent with increased levels of Fru protein when H2 concentrations are low.
Quantifying the expression of fdhAB with splitFAST and BiFC.
Fdh is a multisubunit protein that is essential for growth on formate (29, 30); therefore, we selected Fdh for further validation of FAST1 in measurements of protein abundance. Additionally, as FAST1 is amenable to analysis using BiFC to study protein-protein interactions (15), we generated FdhAB fusion constructs containing split versions of the FAST1 protein. FAST1 can be expressed as two nonfunctional pieces, NFAST (composed of the N-terminal 114 amino acids of FAST1) and CFAST (composed of the subsequent 10 amino acids). With splitFAST, two interacting proteins can be tagged with NFAST and CFAST, and if they colocalize, the pieces will reconstitute in the cell to form a fully functioning protein and fluoresce upon HMBR addition. M. maripaludis contains two isoforms of Fdh (31). Fdh1 was selected for analysis because strains lacking fdh1 are incapable of growth with formate (29, 30), and transcriptomic and proteomic studies suggest that, like Fru, Fdh1 is more abundant in cultures grown under conditions where H2 concentrations limit growth or when formate is provided as the sole electron donor (25–28).
An M. maripaludis strain with translational fusions encoding FdhA-NFAST and FdhB-CFAST was generated by allelic replacement of the endogenous genes. We additionally generated control strains containing either FdhA1-NFAST and Mtd-CFAST (Mtd is the F420-dependent methylenetetrahydromethanopterin dehydrogenase) or FdhB1-CFAST and Mtd-NFAST. Mtd was chosen as a negative control for these studies as Mtd and Fdh are not known to interact, and the genes encoding these two proteins exhibit similar patterns of expression (25, 28). Strains containing FdhA1-NFAST and FdhB1-CFAST, FdhA1-NFAST and Mtd-CFAST, or Mtd-NFAST and FdhB1-CFAST were analyzed by anoxic fluorescence microscopy. Relative fluorescence was measured on a per-cell basis. The average fluorescence intensity was significantly higher in the strain containing FdhA1-NFAST and FdhB1-CFAST, and both control strains with Mtd fusion constructs displayed similar levels of background fluorescence (Fig. 4A and B).
FIG 4.

BiFC and fluorescence of Fdh1 with splitFAST. (A) Fluorescence intensities of cells expressing various splitFAST constructs. The negative-control strains expressing fdhA-NFAST and mtd-CFAST (168 cells) and mtd-NFAST and fdhB-CFAST (160 cells) were compared to the strain expressing fdhA-NFAST and fdhB-CFAST (190 cells). Data were obtained using microscopy and are means and standard deviations from samples collected at least in triplicate. (B) Fluorescence intensities of cells expressing fdhA-NFAST and fdhB-CFAST grown in medium with either formate (190 cells) or H2 (183 cells) as the sole electron donor. Different circle colors represent data points from separate replicates, and mean values from all averaged cells in a replicate are represented by large circles. Data were obtained using microscopy and are means and standard deviations from quadruplicate samples.
To further validate the use of FAST to measure relative protein abundance across growth conditions, the FdhA1-NFAST- and FdhB1-CFAST-containing strain was analyzed for fluorescence differences between H2- and formate-grown cells. In cells grown with formate, the fluorescence intensity was 2.87-fold higher, consistent with the increased Fdh abundance when H2 concentrations are low (26) (Fig. 4C).
Use of tandem FAST2 to observe the cellular localization of FlaI.
In an attempt to further validate FAST for protein localization, we targeted the archaellum (archaeal flagellum), which displays a polar localization in intact cells (32). In M. maripaludis, the major membrane-associated components of the archaellum are the anchor (FlaJ) and its associated ATPase (FlaI). Initial attempts to visualize archaella using FAST1 translational fusions were unsuccessful, and we hypothesized that this was due to low fluorescence intensity. To address this, we incorporated a modified version of FAST that has a lower dissociation constant for the fluorogen (FAST2) as a tandem reporter (tandem FAST2 [tdFAST2]) (33). This alternative reporter has been shown to achieve higher fluorescence in both bacterial and eukaryotic cells (10, 33).
To test fluorescence, a codon-optimized tdFAST2 was transformed into M. maripaludis under the control of the M. voltae histone promoter on the replicating plasmid pLW40neo. FAST1, tdFAST2, and the WT were grown to an OD600 of 0.9, and fluorescence was analyzed using a microplate reader and black, flat-bottom, 96-well plates across several concentrations of HMBR (Fig. 1B). Strains expressing tdFAST2 displayed significant increases in fluorescence over cells expressing FAST1. When normalized to the OD600 and controlling for the inherent fluorescence background, microplate reader assays showed that cells expressing tdFAST2 exhibited a 1.9- to 2.1-fold increase in fluorescence over FAST1 upon HMBR addition.
A strain expressing FlaI translationally fused to tdFAST2 was generated. These cells were viewed by fluorescence microscopy in an anoxic chamber as described above except that they were treated with 20 μM HMBR. Robust fluorescence was observed associated with the cell membrane and localized to a single focus, consistent with a polar localization of archaella (Fig. 5).
FIG 5.
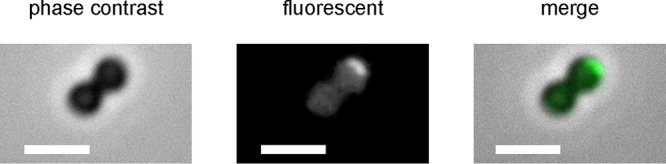
Localization of FlaI tagged with tdFAST2. Images are of early-exponential-phase cells treated with 20 μM HMBR. Bars = 2 μm.
DISCUSSION
Several features of FAST proteins make them ideal for studies in anaerobic organisms. These include the minimal manipulation required to achieve robust fluorescence, a suite of fluorogens with excitation/emission maxima across the visible spectrum (12), and several variant proteins (FAST1, FAST2, tdFAST, and splitFAST, etc.) to facilitate a variety of studies (13, 15, 33). The use of FAST to assess differential protein abundance across growth conditions, protein localization, and protein-protein interactions in live cells of M. maripaludis represents a significant advance over previous studies that were limited to using FISH or autofluorescence to assess methanogen abundance (4, 5).
FAST proteins function with a variety of fluorogenic ligands. For example, binding of the HBR derivative 4-hydroxy-3,5-dimethoxybenzylidene rhodanine (HBR-3,5DOM) shifts the properties of FAST1 such that excitation/emission wavelengths are 518/600 nm (34). While HBR-3,5DOM resulted in functional fluorescent protein in M. maripaludis, the overall fluorescence was lower than that with HMBR (see Fig. S3 in the supplemental material), and we generally observed higher background fluorescence from the culture medium, so HBR-3,5DOM was not used further. Autofluorescence was also generally higher with cultures grown on H2-containing medium, which led us to collect samples using cells grown on formate, when possible. However, we note that increased autofluorescence in H2 medium is not a significant impediment to data collection as we could still assess differences in relative expression across growth conditions (Fig. 3 and 4). Autofluorescence was likely due to H2 limitation as H2 diffusion is outpaced by cellular consumption at a higher density; under H2 limitation, levels of oxidized, fluorescent F420 significantly increase (19). Additionally, the removal of cultures from a shaking incubator during sample preparation and transfer to the anoxic chamber can impact H2 diffusion, requiring rapid sample preparation and processing. Autofluorescence was less of an issue in cultures grown with formate, likely because formate is soluble in aqueous medium, so diffusion does not limit growth.
FAST could be further optimized for use in M. maripaludis with the use of protein variants. A previous study with FAST2 and tdFAST2 in mammalian cell culture showed that these variants could achieve 1.7-fold- and 3.8-fold-higher fluorescence, respectively, than with the original FAST1 (33). We found a more modest increase for tdFAST2 in M. maripaludis of ~2-fold. FAST2 has an identical quantum yield, a very similar molar absorption value, and a low dissociation constant compared to FAST1 (33); therefore, it is likely that the increased fluorescence with tdFAST2 was a result of the tandem nature of the reporter. The performance of tdFAST2 in M. maripaludis was consistent with observations in Eubacterium limosa where a 1.5-fold increase in brightness was observed (10).
The expression of FAST proteins in M. maripaludis could be accomplished using a high-expression vector or under the control of native promoters on the genome. Generally, total fluorescence was lower when FAST was expressed from native promoters, and cellular fluorescence varied with the use of different promoters. As a result, expression analysis was carried out on single cells using a microscope. For each fused gene, differences in fluorescence were reflective of observed differences in mRNA and protein abundances in previous transcriptomic and proteomic studies (25–28). For Fru, previous studies found that proteins were ~1.7-fold more abundant when formate was provided as the sole electron donor for growth or when H2 concentrations were growth limiting. The FAST-based fluorescence pattern observed here is consistent with these data; the mean fluorescence intensities for Fru were 1.76-fold higher when formate was provided as an electron donor. For Fdh, previous studies suggested that these proteins were 2- to 3-fold more abundant when cultures were grown on formate (35), consistent with the 2.87-fold increase in fluorescence for formate-grown cells observed here. Together, these data validate the use of FAST protein fusions to measure relative differences in gene expression and protein abundance.
Protein localization applications typically require a high fluorescence signal to spatially resolve cellular features. While autofluorescence can complicate these analyses, we successfully visualized the subcellular localization of two proteins that were hypothesized to associate with the cell membrane. Archaella in M. maripaludis are associated with the cell pole, and tagging FlaI with tdFAST2 resulted in a single discrete focus characteristic of the hypothesized localization pattern. Localization of Fru to the cell membrane was previously inferred from immunogold labeling of this protein in M. voltae (22) and from biochemical studies where Fru activity was enriched in membrane fractions (21). Here, we provide a third line of evidence for the membrane association of Fru. It remains unclear whether membrane association is required for Fru activity in vivo.
Protein fluorescence can be used to characterize protein-protein interactions using dual-reporter constructs via fluorescence resonance energy transfer or using a single reporter via BiFC. We used splitFAST to tag both subunits of Fdh and observed robust fluorescence above the background through BiFC. While modest increases in fluorescence above the background were also observed in control strains expressing either NFAST or CFAST fused to Mtd, the fluorescence from NFAST and CFAST fused to FdhA and FdhB was significantly higher than that in either control experiment. The high background in control strains was likely due to the fact that Mtd is most abundant when cultures are grown with formate (25, 26), and BiFC can occur through transient interactions between abundant proteins.
We have demonstrated the utility of FAST for protein analysis in M. maripaludis. Combined with anoxic microscopy, FAST allows live-cell imaging, protein localization analysis, determination of protein-protein interactions, and expression analysis. These tools should be broadly applicable to other methanogenic archaea with established methods for heterologous protein expression and allelic replacement. Alternative fluorogens and FAST reporter proteins may further expand the utility of FAST in these organisms. The application of anoxic fluorescence reporters presented here expands the already robust toolkit for molecular biology studies in the methanogenic archaea.
MATERIALS AND METHODS
Strains, medium, and growth conditions.
Strains in this study are listed in Table S1 in the supplemental material. M. maripaludis was grown on McCas medium at 37°C with agitation, except that sodium sulfide was replaced with ammonium sulfide for liquid medium (36, 37). For growth in liquid medium, cultures were grown in 5-mL volumes in Balch tubes. When H2 was supplied as the electron donor for growth, Balch tubes were pressurized to 280 kPa with an 80% H2–balance CO2 gas mix. When formate was supplied as the sole electron donor for growth, McCas-formate medium was used at 37°C without agitation (38), and Balch tubes were pressurized to 210 kPa with an 80% N2–balance CO2 gas mix. For growth on agar plates, 1.5% Noble agar was used, and cultures were grown in an anaerobic incubation vessel pressurized to 140 kPa with an 80% H2–balance CO2 gas mix as described previously (36). When necessary, the following antibiotics were added to medium at the indicated concentrations: neomycin (1 mg mL−1), puromycin (2.5 μg mL−1), and 6-azauracil (0.25 mg mL−1).
To assess the toxicity of HMBR, M. maripaludis strains expressing FAST1 on pLW40 were grown overnight, and 495-μL aliquots were transferred to microcentrifuge tubes in a Coy anaerobic chamber. Five microliters of double-distilled water (ddH2O), dimethyl sulfoxide (DMSO), or 3 mM HMBR (dissolved in DMSO) was added, and cells were left to incubate at room temperature for 30 min. After incubation, cells were inoculated into McCas medium, and growth was monitored by measuring the optical density at 600 nm.
Plasmid construction and transformation of Escherichia coli.
Plasmids and primers are listed in Table S1 in the supplemental material. To generate FAST constructs, a codon-optimized version of the gene was synthesized by Integrated DNA Technologies. The codon-optimized sequences were determined using the JCat codon optimizer (16) with default parameters and with M. maripaludis selected as the organism. To generate the tdFAST2 construct, it was necessary to alter the sequence of the gene to prevent the loss of the tandem sequence through homologous recombination. To accomplish this, the N-terminal copy of FAST2 was codon optimized in JCat using M. maripaludis as the selected organism, and the C-terminal copy was codon optimized in JCat using Methanocaldococcus jannaschii. The nucleotide sequences of the FAST1 and tdFAST2 genes used in this study can be found in Table S2. To further prevent homologous recombination within the tdFAST2 sequence, the 21 bp that separate the N-terminal and C-terminal copies were manually edited to reduce similarity to the 33-bp linker that was consistently used for tagged constructs. In this case, the second most prevalent codon was chosen as determined by the Kazusa database (39).
To generate in-frame FAST constructs, genomic regions flanking either the N terminus or the C terminus of the gene of interest were amplified by PCR using primers suitable for downstream assembly using Gibson assembly (40). Codon-optimized FAST1 or tdFAST2 with an additional 33-bp linker sequence encoding a linker peptide (9) was also amplified by PCR. PCR products were assembled into XbaI/NotI-digested pCRuptneo (38). pCRuptneo contains features for propagation in Escherichia coli (origin of replication and ampicillin resistance gene) and for selection (neomycin selection) and counterselection (uracil phosphoribosyltransferase) in methanogens. For expression on pLW40 (17), FAST1 or tdFAST2 was PCR amplified and placed into the vector by Gibson assembly at the NsiI and AscI restriction sites under the control of the M. voltae histone promoter.
Gibson-assembled constructs were transferred to E. coli DH5α by electroporation. E. coli transformants were selected on lysogeny broth agar medium containing ampicillin (50 μg mL−1). Plasmids were extracted using the PureLink Quick plasmid miniprep kit (Invitrogen) before transfer to M. maripaludis. All constructs were sequence verified by Sanger sequencing at the University of Minnesota Genomics Center.
Transformation of M. maripaludis.
All strains used in this study were generated in an M. maripaludis background lacking the gene for uracil phosphoribosyltransferase (Δupt mutant) (36). DNA was introduced into M. maripaludis using either a natural transformation method or a polyethylene glycol (PEG)-mediated transformation method (1, 41). For splitFAST constructs, compound, sequential transformations were performed using the integrative vector pCRuptneo. NFAST and CFAST sequences were assembled by amplifying a truncated portion of the codon-optimized FAST1 sequence. Each splitFAST strain was transformed once with either NFAST or CFAST, subjected to selection and counterselection to remove the integrated pCRuptneo vector, and then transformed again to incorporate the second tag.
Cultures for natural transformation were grown in McCas medium using a 2% (vol/vol) inoculum and H2 as the electron donor. After growth overnight (~18 h) to stationary phase (OD600 of ~1), cultures were moved into a room-temperature Coy anaerobic chamber (3% H2–10% CO2 atmosphere). Plasmid DNA that was preequilibrated for 1 h in the anaerobic chamber was mixed with 0.5 mL fresh McCas medium and added to the culture using a syringe. The culture-plasmid mixture was pressurized to 280 kPa with an 80% H2–balance CO2 gas mix and incubated at 37°C with agitation for 4 h. After outgrowth, 0.2 mL of culture material was transferred to medium containing an antibiotic (neomycin for pCRuptneo or puromycin for pLW40) to select for transformants. After growth on neomycin, integrative vectors were allowed to resolve by growth overnight without selection. Mutants were isolated by plating onto 6-azauracil-containing McCas agar, and the resulting colonies were screened by PCR.
PEG-mediated transformations were performed according to protocols described previously (1, 41), with all steps performed under anoxic conditions. Briefly, cultures were grown to an OD600 of ~0.7 before washing in transformation buffer (TB) (50 mM Tris, 350 mM sucrose, 380 mM NaCl, 1 mM MgCl2 [pH 7.5]). Washed cells were resuspended in 0.375 mL of TB, and ~5 μg of plasmid DNA was added before the addition of 0.225 mL of a PEG solution (40% [wt/vol] PEG 8000 in TB). After a 1-h incubation, cells were washed twice in McCas medium, pressurized to 280 kPa with an 80% H2–balance CO2 gas mix, and incubated at 37°C with agitation for at least 4 h. After outgrowth, cultures were treated in the same way as for the natural transformation method.
Fluorescence quantification using a SpectraMax M2e plate reader.
HMBR was purchased from The Twinkle Factory (https://www.the-twinkle-factory.com/) and solubilized in DMSO to a concentration of 2 mM as a stock solution for all experiments. M. maripaludis cells were grown in McCas medium with H2 to stationary phase (OD600 = ~0.9) before analysis. The M. maripaludis liquid culture was directly aliquoted into wells of a black, flat-bottom, 96-well plate (catalog number P96FL; Bioassay Systems) with 10 μM HMBR (unless otherwise indicated in the text) to a final volume of 200 μL. Cells were incubated with HMBR for 60 s according to the manufacturer’s instructions before readings. Fluorescence was measured in a SpectraMax M2e plate reader (Molecular Devices) and analyzed using SoftMax Pro 7 software. The excitation wavelength was set to 481 nm, and the emission wavelength was set to 540 nm, utilizing autoemission cutoff settings. Each sample was normalized to the baseline reading of the same sample without fluorogen addition to correct for autofluorescence. Samples were further normalized on a per-cell basis using the OD600 of the cells prior to plate preparation.
Samples prepared with HBR-3,5DOM were treated by utilizing the same methodology as the one for samples treated with HMBR, with the addition of a wash step. To reduce the high levels of autofluorescence in the medium, cells were pelleted aerobically via centrifugation at 15,500 × g for 2 min and resuspended to their original volume with sterile ddH2O.
Microscopy.
Imaging was performed in a Coy-type anaerobic chamber with an environment of 3% H2–balance N2. The chamber also contained a 20-cm by 20-cm tray with Drierite desiccant to control humidity. All imaging was performed using an Echo Revolve R4 hybrid microscope operated in the upright orientation with a high-resolution condenser (numerical aperture [NA], 0.85; working distance, 7 mm). Images were taken with the high-gain setting on, and exposure settings were modified depending on the genetic construct. Cells were viewed using a 40× fluorite lens objective (0.75 NA and 0.51 mm) or a 100× fluorite oil-phase lens objective (1.30 NA and 0.2 mm). Fluorescence imaging was carried out using standard fluorescein isothiocyanate (FITC) filter sets.
To minimize autofluorescence, culture tubes were kept in the incubator until they were ready for immediate imaging. Samples were prepared by aliquoting the liquid culture into microcentrifuge tubes and subsequently adding HMBR in DMSO to the final concentrations specified. Aliquots of 5 μL were added to glass slides, and glass coverslips were placed over them. All steps were performed under anoxic conditions.
Quantification of fluorescence microscopy data using ImageJ.
For expression analysis, 16-bit .tiff images of both a phase-contrast and a fluorescence channel were used to analyze a single field of view utilizing a 40× objective. The FIJI package of ImageJ was used (version 2.1.0/1.53j; Java version 1.8.0_172). A mask was generated using the phase-contrast image by generating an 8-bit image using the threshold tool. The watershed and fill holes binary features were used as appropriate. The pixels of the resulting image in which cells were located were assigned a value of 1 and pixels elsewhere were assigned a value of 0 through the use of the division function. This image was multiplied with the corresponding fluorescence image to create a composite image with a 32-bit float, and the mean fluorescence intensity per cell was determined using the particle analyzer feature. An average of averages was obtained from the compiled list of mean fluorescence intensities. Relative fluorescence units were determined by measuring the gain of fluorescence in cells after HMBR treatment. The average autofluorescent background was determined on a per-sample basis using the methods listed above and was subtracted from the values obtained after the addition of fluorogen. For protein localization analysis, 16-bit .tiff images of both a phase-contrast and a fluorescence channel were used to analyze a single field of view utilizing a 100× objective. For figures, a median filter was applied for visual clarity, and data are presented using the SuperPlotsOfData Web application (42).
Statistical analysis.
Data are presented as means ± standard deviations. Statistical analysis was completed by two-tailed Student’s t test using Microsoft Excel software. P values for Fdh splitFAST against negative controls were determined using one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test for multiple-comparison significance in R (version 3.6.2) (43). Significant differences were considered when P values were <0.05.
Data availability.
Images collected and analyzed in this study have been archived and are freely available at the Data Repository for the University of Minnesota (DRUM) at https://doi.org/10.13020/qwp0-1824.
ACKNOWLEDGMENTS
We thank Thomas Hanson for suggesting FAST as a potential reporter system and providing HMBR for preliminary experiments.
This work was sponsored by a Young Investigator Program award from the Army Research Office, grant number W911NF-19-1-0024.
Footnotes
Supplemental material is available online only.
Contributor Information
Kyle C. Costa, Email: kcosta@umn.edu.
Julie A. Maupin-Furlow, University of Florida
REFERENCES
- 1.Sarmiento FB, Leigh JA, Whitman WB. 2011. Genetic systems for hydrogenotrophic methanogens. Methods Enzymol 494:43–73. 10.1016/B978-0-12-385112-3.00003-2. [DOI] [PubMed] [Google Scholar]
- 2.Leigh JA, Albers S-V, Atomi H, Allers T. 2011. Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol Rev 35:577–608. 10.1111/j.1574-6976.2011.00265.x. [DOI] [PubMed] [Google Scholar]
- 3.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 4.Kumar S, Dagar SS, Mohanty AK, Sirohi SK, Puniya M, Kuhad RC, Sangu KPS, Griffith GW, Puniya AK. 2011. Enumeration of methanogens with a focus on fluorescence in situ hybridization. Naturwissenschaften 98:457–472. 10.1007/s00114-011-0791-2. [DOI] [PubMed] [Google Scholar]
- 5.Lambrecht J, Cichocki N, Hübschmann T, Koch C, Harms H, Müller S. 2017. Flow cytometric quantification, sorting and sequencing of methanogenic archaea based on F420 autofluorescence. Microb Cell Fact 16:180. 10.1186/s12934-017-0793-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Purwantini E, Mukhopadhyay B. 2009. Conversion of NO2 to NO by reduced coenzyme F420 protects mycobacteria from nitrosative damage. Proc Natl Acad Sci USA 106:6333–6338. 10.1073/pnas.0812883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heim R, Prasher DC, Tsien RY. 1994. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA 91:12501–12504. 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley AM, Petersen J, Roe AJ, Douce GR, Christie JM. 2015. LOV-based reporters for fluorescence imaging. Curr Opin Chem Biol 27:39–45. 10.1016/j.cbpa.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Plamont M-A, Billon-Denis E, Maurin S, Gauron C, Pimenta FM, Specht CG, Shi J, Quérard J, Pan B, Rossignol J, Moncoq K, Morellet N, Volovitch M, Lescop E, Chen Y, Triller A, Vriz S, Le Saux T, Jullien L, Gautier A. 2016. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci USA 113:497–502. 10.1073/pnas.1513094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaiz M, Ludwig G, Bengelsdorf FR, Dürre P. 2021. Production of the biocommodities butanol and acetone from methanol with fluorescent FAST-tagged proteins using metabolically engineered strains of Eubacterium limosum. Biotechnol Biofuels 14:117. 10.1186/s13068-021-01966-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Streett HE, Kalis KM, Papoutsakis ET. 2019. A strongly fluorescing anaerobic reporter and protein-tagging system for Clostridium organisms based on the fluorescence-activating and absorption-shifting tag protein (FAST). Appl Environ Microbiol 85:e00622-19. 10.1128/AEM.00622-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myasnyanko IN, Gavrikov AS, Zaitseva SO, Smirnov AY, Zaitseva ER, Sokolov AI, Malyshevskaya KK, Baleeva NS, Mishin AS, Baranov MS. 2021. Color tuning of fluorogens for FAST fluorogen-activating protein. Chemistry 27:3986–3990. 10.1002/chem.202004760. [DOI] [PubMed] [Google Scholar]
- 13.Tebo AG, Moeyaert B, Thauvin M, Carlon-Andres I, Böken D, Volovitch M, Padilla-Parra S, Dedecker P, Vriz S, Gautier A. 2021. Orthogonal fluorescent chemogenetic reporters for multicolor imaging. Nat Chem Biol 17:30–38. 10.1038/s41589-020-0611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monmeyran A, Thomen P, Jonquière H, Sureau F, Li C, Plamont M-A, Douarche C, Casella J-F, Gautier A, Henry N. 2018. The inducible chemical-genetic fluorescent marker FAST outperforms classical fluorescent proteins in the quantitative reporting of bacterial biofilm dynamics. Sci Rep 8:10336. 10.1038/s41598-018-28643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tebo AG, Gautier A. 2019. A split fluorescent reporter with rapid and reversible complementation. Nat Commun 10:2822. 10.1038/s41467-019-10855-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. 2005. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526–W531. 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodsworth JA, Leigh JA. 2006. Regulation of nitrogenase by 2-oxoglutarate-reversible, direct binding of a PII-like nitrogen sensor protein to dinitrogenase. Proc Natl Acad Sci USA 103:9779–9784. 10.1073/pnas.0602278103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner WL, Whitman WB. 1999. Expression vectors for Methanococcus maripaludis: overexpression of acetohydroxyacid synthase and beta-galactosidase. Genetics 152:1439–1447. 10.1093/genetics/152.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Poorter LMI, Geerts WJ, Keltjens JT. 2005. Hydrogen concentrations in methane-forming cells probed by the ratios of reduced and oxidized coenzyme F420. Microbiology (Reading) 151:1697–1705. 10.1099/mic.0.27679-0. [DOI] [PubMed] [Google Scholar]
- 20.Hendrickson EL, Leigh JA. 2008. Roles of coenzyme F420-reducing hydrogenases and hydrogen- and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. J Bacteriol 190:4818–4821. 10.1128/JB.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baron SF, Ferry JG. 1989. Purification and properties of the membrane-associated coenzyme F420-reducing hydrogenase from Methanobacterium formicicum. J Bacteriol 171:3846–3853. 10.1128/jb.171.7.3846-3853.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muth E. 1988. Localization of the F420-reducing hydrogenase in Methanococcus voltae cells by immuno-gold technique. Arch Microbiol 150:205–207. 10.1007/BF00425164. [DOI] [Google Scholar]
- 23.Baron SF, Williams DS, May HD, Patel PS, Aldrich HC, Ferry JG. 1989. Immunogold localization of coenzyme F420-reducing formate dehydrogenase and coenzyme F420-reducing hydrogenase in Methanobacterium formicicum. Arch Microbiol 151:307–313. 10.1007/BF00406556. [DOI] [Google Scholar]
- 24.Lacasse MJ, Zamble DB. 2016. [NiFe]-hydrogenase maturation. Biochemistry 55:1689–1701. 10.1021/acs.biochem.5b01328. [DOI] [PubMed] [Google Scholar]
- 25.Costa KC, Yoon SH, Pan M, Burn JA, Baliga NS, Leigh JA. 2013. Effects of H2 and formate on growth yield and regulation of methanogenesis in Methanococcus maripaludis. J Bacteriol 195:1456–1462. 10.1128/JB.02141-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Q, Wang T, Hendrickson EL, Lie TJ, Hackett M, Leigh JA. 2009. Quantitative proteomics of nutrient limitation in the hydrogenotrophic methanogen Methanococcus maripaludis. BMC Microbiol 9:149. 10.1186/1471-2180-9-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrickson EL, Liu Y, Rosas-Sandoval G, Porat I, Söll D, Whitman WB, Leigh JA. 2008. Global responses of Methanococcus maripaludis to specific nutrient limitations and growth rate. J Bacteriol 190:2198–2205. 10.1128/JB.01805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hendrickson EL, Haydock AK, Moore BC, Whitman WB, Leigh JA. 2007. Functionally distinct genes regulated by hydrogen limitation and growth rate in methanogenic archaea. Proc Natl Acad Sci USA 104:8930–8934. 10.1073/pnas.0701157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costa KC, Lie TJ, Xia Q, Leigh JA. 2013. VhuD facilitates electron flow from H2 or formate to heterodisulfide reductase in Methanococcus maripaludis. J Bacteriol 195:5160–5165. 10.1128/JB.00895-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lupa B, Hendrickson EL, Leigh JA, Whitman WB. 2008. Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. Appl Environ Microbiol 74:6584–6590. 10.1128/AEM.01455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poehlein A, Heym D, Quitzke V, Fersch J, Daniel R, Rother M. 2018. Complete genome sequence of the Methanococcus maripaludis type strain JJ (DSM 2067), a model for selenoprotein synthesis in archaea. Genome Announc 6:e00237-18. 10.1128/genomeA.00237-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarrell KF, Stark M, Nair DB, Chong JPJ. 2011. Flagella and pili are both necessary for efficient attachment of Methanococcus maripaludis to surfaces. FEMS Microbiol Lett 319:44–50. 10.1111/j.1574-6968.2011.02264.x. [DOI] [PubMed] [Google Scholar]
- 33.Tebo AG, Pimenta FM, Zhang Y, Gautier A. 2018. Improved chemical-genetic fluorescent markers for live cell microscopy. Biochemistry 57:5648–5653. 10.1021/acs.biochem.8b00649. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Plamont M-A, Sladitschek HL, Rodrigues V, Aujard I, Neveu P, Le Saux T, Jullien L, Gautier A. 2017. Dynamic multicolor protein labeling in living cells. Chem Sci 8:5598–5605. 10.1039/c7sc01364g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood GE, Haydock AK, Leigh JA. 2003. Function and regulation of the formate dehydrogenase genes of the methanogenic archaeon Methanococcus maripaludis. J Bacteriol 185:2548–2554. 10.1128/JB.185.8.2548-2554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fonseca DR, Abdul Halim MF, Holten MP, Costa KC. 2020. Type IV-like pili facilitate transformation in naturally competent archaea. J Bacteriol 202:e00355-20. 10.1128/JB.00355-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore BC, Leigh JA. 2005. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. J Bacteriol 187:972–979. 10.1128/JB.187.3.972-979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA. 2010. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci USA 107:11050–11055. 10.1073/pnas.1003653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura Y, Gojobori T, Ikemura T. 2000. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res 28:292. 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 41.Tumbula DL, Makula RA, Whitman WB. 1994. Transformation of Methanococcus maripaludis and identification of a Pst I-like restriction system. FEMS Microbiol Lett 121:309–314. 10.1111/j.1574-6968.1994.tb07118.x. [DOI] [Google Scholar]
- 42.Goedhart J. 2021. SuperPlotsOfData—a Web app for the transparent display and quantitative comparison of continuous data from different conditions. Mol Biol Cell 32:470–474. 10.1091/mbc.E20-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.R Core Development Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S3; Tables S1 and S2. Download jb.00120-22-s0001.pdf, PDF file, 0.1 MB (146.3KB, pdf)
Data Availability Statement
Images collected and analyzed in this study have been archived and are freely available at the Data Repository for the University of Minnesota (DRUM) at https://doi.org/10.13020/qwp0-1824.