

ABSTRACT
Candida albicans causes debilitating, often azole-resistant, infections in patients with chronic mucocutaneous candidiasis (CMC). Amphotericin B (AMB) resistance is rare, but AMB use is limited by parenteral administration and nephrotoxicity. In this study, we evaluated cochleated AMB (CAMB), a new oral AMB formulation, in mouse models of oropharyngeal candidiasis (OPC) and vulvovaginal candidiasis (VVC) and in patients with azole-resistant CMC. OPC and VVC were modeled in Act1−/− mice, and mucosal tissue fungal burden was assessed after once-daily treatment with CAMB, vehicle, or AMB-deoxycholate (AMB-d). Four patients with azole-resistant CMC enrolled in a phase 2 CAMB dose-escalation study. The primary endpoint was clinical improvement at 2 weeks followed by optional extension for long-term CMC suppression to assess safety and efficacy. CAMB-treated mice had significantly reduced tongue and vaginal fungal burdens compared to vehicle-treated mice and exhibited comparable fungal burden reduction relative to AMB-d-treated mice. All CAMB-treated patients reached clinical efficacy by 2 weeks, three at 400 mg twice daily and one at 200 mg twice-daily dosing. All patients continued to the extension phase, with three having sustained clinical improvement of OPC and esophageal candidiasis (EC) for up to 60 months. One patient had a relapse of esophageal symptoms at week 24 and was withdrawn from further study. Clinical responses were not seen for onychomycosis or VVC. CAMB was safe and well-tolerated, without any evidence of nephrotoxicity. In summary, oral CAMB reduced tongue and vaginal fungal burdens during murine candidiasis. A proof-of-concept clinical trial in human CMC showed efficacy with good tolerability and safety. This study has been registered at ClinicalTrials.gov under identifier NCT02629419.
KEYWORDS: amphotericin B, chronic mucocutaneous candidiasis, cochleated, mouse model, mucosal candidiasis, phase 2 trial
INTRODUCTION
Chronic antifungal drug administration is often required as prophylaxis and/or treatment in patients who suffer from chronic mucocutaneous candidiasis (CMC) caused by HIV/AIDS or various inborn errors of immunity, such as Hyper-IgE syndrome caused by dominant negative STAT3 mutations (STAT3 DN; Job’s syndrome), autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) syndrome caused by AIRE mutations, STAT1 gain-of-function (GOF), and other monogenic disorders with impaired interleukin-17 (IL-17) receptor signaling (1–6). The long-term management of CMC is frequently associated with the acquisition of antifungal resistance, primarily against triazole antifungal agents (7–12).
In patients infected with azole-resistant Candida strains, amphotericin B (AMB) derivatives can be used given their fungicidal activity and the rarity of acquired AMB resistance (12). However, AMB use requires parenteral administration and causes significant side effects such as nephrotoxicity, which worsens with longer durations of therapy (13). Cochleated AMB (CAMB, currently known as MAT2203) is a lipid-nanocrystal formulation designed for targeted oral delivery of AMB. CAMB is a spiral-shaped phospholipid-cation precipitate that primarily consists of phosphatidylserine and calcium. The lipid bilayer of cochleates is rolled into a spiral, with no internal aqueous space. This structure provides protection against degradation in the gastrointestinal tract, while allowing for systemic and targeted delivery of CAMB and intracellular AMB accumulation within macrophages (14–18). Animal studies of CAMB administration have demonstrated systemic absorption of AMB with comparable clinical efficacy to that of AMB deoxycholate (AMB-d) against invasive candidiasis, aspergillosis, sporotrichosis, and cryptococcosis, but with greatly reduced toxicity (19–23). A recent phase 1 ascending-dose trial of CAMB administered at 1 to 2 g per day in 4 to 6 divided doses for up to 7 days demonstrated that CAMB was well-tolerated with only mild gastrointestinal symptoms, without renal toxicity or other severe laboratory abnormalities (24). An ongoing phase 2 clinical trial (EnACT) is aiming to evaluate the safety, tolerability, and efficacy of CAMB in HIV-infected patients with cryptococcal meningitis (https://clinicaltrials.gov/, NCT04031833).
However, whether CAMB protects against CMC has not been examined thus far. Here, we determined the in vivo efficacy of CAMB in mouse models of OPC and VVC in IL-17 receptor signaling-deficient Act1−/− mice. In addition, we performed a proof-of-concept phase 2 open-label dose-escalation study in patients with CMC infections that were intolerant or resistant to azole antifungal drugs to assess the safety, tolerability, and efficacy of CAMB.
RESULTS
CAMB exhibits comparable efficacy with AMB-d in mouse models of OPC and VVC.
CAMB has been previously reported to protect mice from mortality in the mouse model of invasive candidiasis, as does AMB-d (20, 22). To verify these findings, we assessed the efficacy of CAMB in the mouse model of invasive candidiasis and found that it exhibited comparable protection from mortality relative to AMB-d in WT mice infected systemically with C. albicans (Fig. 1b). Moreover, CAMB administration in Act1−/− mice infected orally with C. albicans resulted in significantly reduced tongue tissue fungal burden at day 5 postinfection. Notably, AMB-d demonstrated comparable reduction in tongue tissue fungal burden relative to CAMB during OPC (Fig. 1c). Similarly, CAMB treatment of Act1−/− mice with VVC resulted in a significant decrease in vaginal tissue fungal burden at day 5 postinfection compared to vehicle treatment and comparable efficacy with AMB-d (Fig. 1c). Of note, CAMB or AMB-d treatment did not affect fungal burden in the vaginal fluid (Fig. S1 in the supplemental material). Taken together, these mouse studies demonstrate that CAMB exhibits similar in vivo efficacy to AMB-d in mouse models of invasive candidiasis, OPC, and VVC.
FIG 1.

Cochleated amphotericin B (CAMB) exhibits comparable efficacy with AMB deoxycholate (AMB-d) in mouse models of invasive (IC), oropharyngeal (OPC), and vulvovaginal candidiasis (VVC). (a) Mice were treated daily with CAMB via oral gavage, AMB-d intraperitoneally, or the vehicle control starting at day 1 postinfection. (b) C. albicans was injected intravenously in C57BL/6 mice and survival was monitored (n = 5 per group). (c) Act1−/− mice were infected with C. albicans in models of OPC (n = 4 to 11 per group) and VVC (n = 4 to 8 per group). At day 5 postinfection, the mice were euthanized and tongue (for OPC) or vaginal tissue (for VVC) was harvested to quantify fungal burden. *, P < 0.05; **, P < 0.01 as determined using a log-rank test (b), one-way ANOVA with Tukey’s multiple-comparison test (panel c, OPC), or Kruskal-Wallis test with Dunn’s multiple-comparison test (panel c, VVC). Data are summary of one (b) or two independent experiments (c).
Patient characteristics.
Four patients with CMC were enrolled in the phase 2 clinical study (Table 1). All four were women ranging in age from 42 to 47 years. Three had STAT3 DN and manifested classic clinical features, including eczema, recurrent sinopulmonary infections, CMC, and connective tissue and skeletal abnormalities. All three had been on long-standing triazole therapy with posaconazole and had persistent thrush with azole-resistant Candida strains, with C. albicans in all three patients, and one patient with Candida glabrata as well (Table 2). Two patients also had a history of onychomycosis involving multiple nails, one patient had a history of concurrent C. albicans chronic VVC, and one patient had a history of concurrent C. albicans esophagitis. The other patient (Patient 3) did not have a known genetic etiology of CMC but had persistent azole-resistant C. albicans esophagitis which necessitated prolonged courses of echinocandin therapy due to treatment failures with fluconazole, itraconazole, and posaconazole (Table 2).
TABLE 1.
Characteristics of the CMC patients included in this studya
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age (yrs) at enrollment | 43 | 42 | 46 | 47 |
| Gender | Female | Female | Female | Female |
| Ethnicity | Caucasian | Hispanic | Caucasian | Caucasian |
| Underlying CMC syndrome | STAT3 DN | STAT3 DN | Esophageal candidiasis | STAT3 DN |
| Duration of infection (yrs) | 41 | 41 | 27 | 40 |
| OPC | + | + | – | + |
| EC | – | – | + | – |
| VVC | + | – | – | – |
| Primary type of candidiasis | OPC | OPC | EC | OPC |
| Other infections | Staphylococcus aureus SSTIs, bacterial sinopulmonary infections, chronic pulmonary Pseudomonas | S. aureus SSTIs, bacterial sinopulmonary infections, S. aureus paraspinal abscess | Varicella pneumonia during pregnancy, bacterial pneumonia | S. aureus SSTIs, bacterial sinopulmonary infections, pulmonary chronic E. coli, pulmonary aspergillosis, osteomyelitis (organism unknown) |
CMC, chronic mucocutaneous candidiasis; STAT3, signal transducer and activator of transcription 3; STAT3 DN, Hyper-IgE syndrome caused by dominant negative STAT3 mutations (Job’s syndrome); OPC, oropharyngeal candidiasis; EC, esophageal candidiasis; VVC, vulvovaginal candidiasis; SSTI, skin and soft tissue infection.
TABLE 2.
MICs of Candida strains to AMB and other antifungal agentsa
| Characteristic | MICs (mg/L) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | ||||||||||||||
| 1 | 2 | 3 | 4 | |||||||||||
| Isolation site | Oral | Vagina | Oral | Oral | Eso | Toe | Oral | Fingernail | Oral | Oral | Eso | Eso | Oral | Oral |
| Isolated species | Ca | Ca | Ca | Ca | Ca | Ca | Ca | Ch | Ca | Cg | Ca | Cg | Ca | Ca |
| Time of analysis | Baseline | On CAMB therapy | Baseline | On CAMB therapy | Baseline | End of CAMB therapy | Baseline | End of CAMB therapy | ||||||
| Antifungal agent | ||||||||||||||
| AMB | 1 | 0.25 | 0.5 | 1 | 0.25 | 0.25 | 0.25 | 4 | 0.5 | 0.25 | 0.5 | 1 | 0.5 | 0.5 |
| Anidulafungin | 0.5 | 0.03 | 0.5 | <0.015 | <0.015 | <0.015 | <0.015 | 0.12 | 0.03 | <0.015 | 0.015 | 0.03 | <0.015 | 0.03 |
| Caspofungin | 2 | 0.12 | 2 | 0.03 | 0.06 | 0.03 | 0.015 | 0.25 | 0.03 | 0.06 | 0.015 | 0.03 | 0.12 | 0.03 |
| Micafungin | 0.5 | 0.06 | 1 | 0.015 | 0.015 | 0.015 | 0.015 | 0.25 | <0.008 | 0.015 | <0.008 | 0.015 | 0.015 | 0.03 |
| Fluconazole | 128 | 256 | 128 | 32 | 16 | 64 | 32 | 128 | 8 | 64 | 8 | 32 | 64 | 32 |
| Posaconazole | 2 | 8 | >8 | 2 | 1 | 8 | 2 | 1 | 0.25 | 2 | 0.25 | 2 | >8 | 2 |
| Voriconazole | 2 | 4 | 4 | 1 | 2 | 1 | 1 | 1 | 0.12 | 2 | 0.12 | 0.5 | 2 | 1 |
MIC, minimum inhibitory concentration; AMB, amphotericin B; CAMB, cochleated amphotericin B; Ca, Candida albicans; Ch, Candida haemulonii; Cg, Candida glabrata; Eso, esophagus.
CAMB clinical efficacy.
Dose escalation was well tolerated by all patients. Patient 2 met the clinical response criteria to stop escalation at 200 mg twice-daily CAMB dosing. Patients 1, 3, and 4 reached clinical response criteria at the maximum CAMB dose of 400 mg twice daily (Fig. 2). Patient 1 primarily had OPC and met clinical response criteria with a 57% decrease in clinical severity score at 2 weeks of 400 mg twice-daily dosing. In contrast, this patient’s VVC did not meet the clinical severity score endpoint at 2 weeks (Fig. 2). Patient 2 primarily had OPC which met clinical response criteria with a 57% decrease in clinical severity score at 2 weeks of 200 mg twice-daily dosing. In Patient 3, the esophagus was the primary infection site, as documented by an upper endoscopy which revealed diffuse Candida esophagitis at baseline. This patient met clinical response criteria at 2 weeks of 200 mg twice-daily dosing, with amelioration of Candida esophagitis symptoms and a 71% decrease in clinical severity score (Fig. 2). A repeat upper endoscopy after 3.5 weeks of maximum-dose CAMB therapy showed fewer white plaques with some plaques remaining only in the lower esophagus. Patient 4 primarily had OPC and met clinical response criteria with a 50% decrease in clinical severity score at 2 weeks of 400 mg twice-daily dosing. None of the patients achieved complete remission from symptoms during the initial phase of the study.
FIG 2.

Clinical efficacy of CAMB in patients with azole-resistant chronic mucocutaneous candidiasis (CMC). (a) Shown are the clinical severity scores for corresponding manifestations of CMC at baseline and during CAMB treatment, at the primary endpoint evaluation. (b) Evolution of clinical score over time following initiation of CAMB treatment.
All four patients enrolled in the extension phase of the study. Three of them continued up to 60 months with sustained clinical improvement, despite persistent mild thrush upon examination for Patients 1 and 4, and persistent VVC for Patient 1. Patients 2 and 4 achieved complete resolution of signs and symptoms of OPC at 5 and 9.5 months, respectively, but the complete response was transient, with some symptoms returning while therapy was ongoing. There was no clinical response in onychomycosis throughout the study and extension phases for Patients 1 and 4. Patient 3 had a relapse of esophageal symptoms at week 24 of CAMB treatment, with Candida plaques visualized on an endoscopy. C. albicans and C. glabrata were confirmed with culture and consistent pathology, leading to the patient’s withdrawal from the study to allow a change in antifungal therapy. Patient 2 discontinued CAMB therapy after 3.5 years due to a new C. albicans skin infection on her feet and Candida haemulonii onychomycosis, leading to a change in antifungal therapy. Patient 4 discontinued CAMB therapy after 2.5 years due to a change in therapy for worsening thrush and fungal skin infections after increasing needs for broad-spectrum antibiotics and corticosteroids. Patient 1 discontinued CAMB after 5 years due to persistent thrush and study nonadherence, leading to study closure. None of the patients developed increased MICs of the mucosal Candida strains, which would be suggestive of AMB resistance (Table 2), further attesting to the known low likelihood of acquired AMB resistance by Candida.
Fungal cultures during CAMB treatment.
All four patients had moderate Candida growth on semiquantitative fungal cultures at the primary infection sites at study initiation. After 2 weeks of maximum-dose CAMB therapy, only Patient 3 exhibited a decrease in semiquantitative culture growth, and after 4 weeks of maximum dose therapy, three patients showed decreased culture growth. Decreases in culture growth did not appear to be sustained; at 6 months of CAMB therapy, three patients had moderate growth despite improved clinical symptoms. Of the four patients, only Patient 1 presented with VVC. During CAMB treatment, we observed no decline in Patient 1’s vaginal Candida culture growth.
CAMB pharmacokinetic studies.
Following the first single dose of 100 mg on day 1, the median peak AMB plasma concentration (maximum concentration of drug in serum [Cmax]) was 28.4 ng/mL (27.02 to 52.06 ng/mL) occurring at a median Tmax (time to maximum concentration of drug in serum) of 16 h (4 to 16 h) (Fig. 3a). Median AMB exposure (area under the curve over 24 hours [AUC0–24]) was 507 ng · hr/mL (371.92 to 773.14 ng · hr/mL). Elimination showed similar variability to the other parameters, with a median clearance and elimination half-life of 201.67 L/hr (129.34 to 268.88) and 17.15 h (8.83 to 23.37), respectively.
FIG 3.
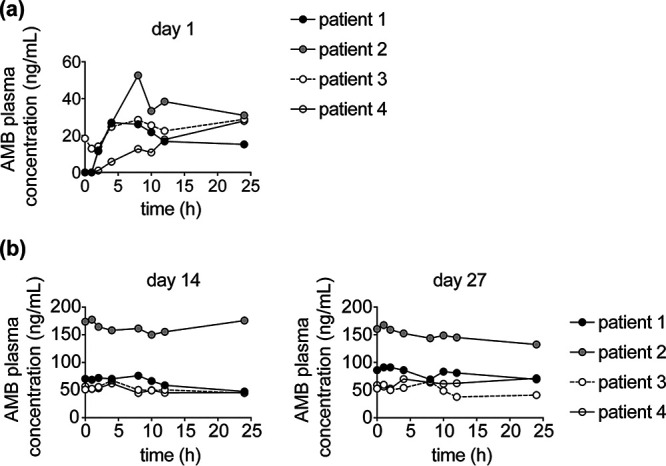
Pharmacokinetic data of CAMB administration in patients with CMC. (a and b) Plasma concentrations of amphotericin B (in ng/mL) measured over serial time points, as indicated, (a) at day 1 after initiation at 100 mg, (b) at day 14 after continuation at 200 mg/day, and at day 27 after the first dose of the 400 mg twice-daily CAMB dosing for Patients 1, 3, and 4 or after continuation of 200 mg twice daily for Patient 2.
All four participants demonstrated accumulation following 14 days of continuous dosing at 100 mg twice daily. Median steady-state peak concentrations (Cmax,ss) had risen to 71.92 ng/mL (62.15 to 177.66 ng/mL), while exposure had increased more than 2.5-fold to a median AUC0–24,ss of 1,357.05 (1,168.05 to 3,914.31 ng · hr/mL) (Fig. 3b). Three study participants escalated their doses to 400 mg twice daily, while patient two stayed at 200 mg twice daily, resulting in marginal increases in exposures and sustained plasma concentrations in the 50 to 100 ng/mL range by day 27 (Fig. 3c). Two study participants had urine and fecal pharmacokinetic sampling conducted on days 2, 28, and 40. The fraction of drug excreted in the urine and feces was markedly low (<1.25%). The biodistribution and mass balance of AMB in humans is complex. For example, liposomal AMB significantly alters the excretion and mass balance of AMB. The ability of liposomes to sequester drugs in circulating liposomes and within deep tissue compartments may account for these differences (25).
CAMB safety.
CAMB was well tolerated by all four patients. No signs of renal, hepatic, or hematologic toxicity were noted throughout the study period, including throughout the extension phase of up to 60 months (Fig. 4). Self-limited diarrhea was noted by patient 1 during resumption of 400 mg twice-daily dosing following a brief treatment interruption. No serious adverse events related to the study drug were observed.
FIG 4.

Long-term administration of CAMB in patients with CMC does not cause renal impairment or anemia. (a) Serum levels of creatinine and potassium (left panel) and hemoglobin (right panel) over time after initiation of CAMB treatment. Horizontal dashed lines depict the normal range of creatinine values, and horizontal dotted lines depict the normal range of potassium (left panel) and hemoglobin (right panel) values. (b) Serum levels of erythropoietin after initiation of CAMB treatment in patient 1 (day 1,214 from CAMB initiation), patient 2 (days 148, 203, and 574 from CAMB initiation), and patient 4 (days 28, 88, 220, 674, and 796 from CAMB initiation). Horizontal dotted lines depict the normal range of erythropoietin.
DISCUSSION
In this murine study, we initially evaluated the preclinical efficacy of CAMB against the two most common clinical manifestations of mucosal candidiasis in the setting of IL-17 receptor signaling deficiency in Act1−/− mice. We found that oral administration of CAMB significantly reduced the mucosal tissue fungal burden relative to vehicle treatment during OPC and VVC, at levels comparable to those achieved with parenteral administration of AMB-d. These data extend the previously reported comparable in vivo efficacy of CAMB and AMB-d in a mouse model of invasive candidiasis (20, 22), which we confirmed in our study. Future studies will be needed to determine whether CAMB also protects against OPC and VVC caused by Candida strains other than SC5314, including azole- and echinocandin-resistant strains and clinical strains harvested from the mucosal surfaces of patients, which are associated with more efficient long-term mucosal colonization (26–28). Furthermore, studies aimed at assessing longer treatment durations for CAMB and comparing its efficacy more broadly with other antifungal agents, such as echinocandins, will be useful.
We also assessed the safety, tolerability, and efficacy of CAMB in a phase 2 open-label dose-escalation study in four patients with CMC infections that were intolerant or resistant to azole antifungal drugs. Patients with inborn errors of immunity that manifest with CMC often develop azole-resistant mucosal candidiasis, and when that occurs, the oral treatment options available are very limited. Although AMB resistance is rare in Candida, the administration of parenteral formulations of AMB is hindered by the adverse effects of long-term use, particularly nephrotoxicity. Therefore, long-term administration of an oral formulation of CAMB as a treatment and/or primary or secondary prophylaxis without the associated renal toxic effects is particularly attractive for these patients.
We found that CAMB dose escalation was well-tolerated in all four evaluated patients and led to systemic absorption of AMB, with sustained plasma levels between 50 and 200 ng/mL depending on the patient. Administration of CAMB resulted in clinical efficacy in both OPC and EC by 2 weeks of treatment with 200 or 400 mg twice-daily dosing. No clinical efficacy was seen with 100 mg twice-daily dosing. The VVC response was less apparent initially compared to the OPC response. In addition, no clinical response was observed for onychomycosis throughout the study period for the two affected patients. Two patients had emergence of new Candida infection sites while on CAMB therapy. Patient 2 had a fingernail nail infection with an AMB-resistant (minimum inhibitory concentration [MIC], 4.0 μg/mL) C. haemulonii strain after trauma from a dog bite as well as an AMB-susceptible (MIC, 0.25 μg/mL) C. albicans infection in the toenails and surrounding skin. Patient 4 had emergence of tinea corporis-like lesions, with skin scrapings positive for hyphae and culture positive for an AMB-susceptible (MIC, 0.5 μg/mL) C. albicans strain. The improved efficacy of CAMB for treating OPC and EC compared to skin, nail, and VVC lesions raises the possibility of a topical antifungal effect versus decreased penetration into the nail bed, skin, and vaginal secretions, at least with the dosing that was used in this study.
All patients continued onto the extension phase of the CAMB clinical trial and received the drug for up to 60 months with sustained clinical improvement of OPC and EC, although a relapse of esophageal symptoms was noted in one patient who discontinued treatment. Importantly, long-term administration of CAMB was safe with no development of renal toxicity, hypokalemia, or anemia, which are well-established adverse effects with AMB-d and liposomal formulations of AMB (13, 29). In keeping with the lack of nephrotoxicity, no significant urine secretion of AMB was noted in our patients. Moreover, renal injury underlies AMB-induced anemia, which is caused by impaired erythropoietin production (29). Notably, serum erythropoietin levels were not decreased in our CAMB-treated patients. Taken together, our proof-of-concept clinical study shows promising clinical efficacy of CAMB in azole-resistant mucosal candidiasis and indicates that long-term administration can be well tolerated in humans. The ongoing EnACT clinical trial will help shed further light on the pharmacokinetics (PK) and efficacy of CAMB in humans in the setting of HIV-associated cryptococcal meningitis. Future studies will also be needed to determine the preclinical and clinical efficacy of CAMB compared to that of AMB-d and/or lipid formulations of AMB (liposomal AMB) and/or echinocandins in candidiasis and other life-threatening invasive fungal infections in which AMB plays a significant therapeutic role, such as mucormycosis, fusariosis, disseminated forms of histoplasmosis, coccidioidomycosis, blastomycosis, and leishmaniasis. Because CAMB was well tolerated, investigation into increased doses for CMC should be considered, as in the ongoing EnACT clinical trial, which may improve the clinical efficacy for VVC, skin, and nail candidal disease.
MATERIALS AND METHODS
Mice.
The 8 to 12-week-old female mice were maintained under specific pathogen-free conditions in ventilated cages. The mouse studies were performed following the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, under the auspices of protocol LCIM-14E, approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases (NIAID). Wild-type (WT) C57BL/6 mice were purchased from Taconic Biosciences. Act1−/− (also known as Ciks−/− or Traf3ip2−/−) mice, which lack IL-17 receptor signaling and are highly susceptible to OPC (30, 31), were obtained from Ulrich Siebenlist, NIAID.
Mouse models of invasive candidiasis, OPC, and VVC.
The C. albicans strain SC5314 was used in the mouse experiments (MIC for AMB, 0.25 μg/mL) and was prepared as previously described (6). For invasive candidiasis experiments, WT C57BL/6 mice were infected by a lateral tail vein injection of 106 Candida blastospores, as described previously (32). For OPC and VVC experiments, Act1−/− mice were inoculated sublingually or vaginally, and mucosal tissue fungal burdens were determined as previously described (6, 33). CAMB was administered via oral gavage at 25 mg/kg/day in mice with invasive candidiasis and OPC and has previously been shown to be effective in a mouse model of cryptococcosis (21). CAMB was administered via oral gavage at 25 or 83.5 mg/kg/day in mice with VVC, and the two doses displayed similar efficacy. AMB-d was administered intraperitoneally at 2 mg/kg/day in mice with invasive candidiasis and at 25 mg/kg/day in mice with OPC or VVC. The first dose of CAMB or AMB-d was administered 24 h after inoculation of mice with C. albicans (Fig. 1a), a time point when mucosal infection is already well established, as shown previously (34–36). For OPC and VVC experiments, mice were treated daily for 4 days, and the fungal burden was determined at day 5 postinfection, which is an established time point for assessing mucosal fungal burden in these models (33–36). For invasive candidiasis, mice were treated for the entire experimental duration of 14 days, as shown in Fig. 1a.
Preparation of CAMB and vehicle control formulations.
The lipid nanocrystal used for AMB drug delivery was formulated following the method previously described by Santangelo et al. (22). Briefly, a basic solution of AMB (22.75 g; Sigma-Aldrich, St. Louis, MO) in 0.1 N NaOH (707 mL) was added, dropwise while stirring, to an aqueous suspension of liposomes (106 g lipoid PS P50X in 3.533 liters of 50 mM phosphate buffer [pH 7.4]), followed by the addition, dropwise while stirring, of calcium chloride (1.0 M, 194.8 mL) to form the CAMB suspension containing 1.8 mg AMB/mL. The final formulation step involved removal of the supernatant followed by the addition of methylcellulose (0.3%) as a suspending agent, with adjustment to a final concentration of 5 mg AMB/mL. During the course of this study, the CAMB formulation was improved by the addition of sweeteners and flavor enhancers. Additionally, the AMB/mL concentration was increased to 20 mg/mL by spray-drying the original suspension and resuspending, thereby reducing the dosing volume. Preparation of CAMB for the clinical trial followed a similar protocol but involved larger CAMB volumes.
Clinical study design and participants.
The clinical study was designed as an open-label trial with dose-titration to assess the efficacy, safety, tolerability, and pharmacokinetics of CAMB in patients with mucocutaneous candidiasis who were refractory or intolerant to standard non-parenteral antifungal treatment. Dose escalation was followed by an extension phase for continued assessment of safety and efficacy. Patients were recruited who had at least 5 days of persistent OPC, EC, and/or VVC with an infecting Candida strain with documented azole resistance within the preceding 6 months of CAMB administration, and/or were intolerant to standard non-parenteral antifungal drugs or had a lack of improvement or worsening of OPC after treatment with appropriately dosed oral azole therapy.
Patients aged 18 to 75 years old were recruited largely from cohorts followed on NIAID protocols with inborn errors of immunity predisposing them to CMC, including STAT3 DN, STAT1 GOF, APECED, and others. Patients were enrolled on a NIH institutional review board-approved protocol (Clinicaltrials.gov, NCT02629419) and provided written informed consent in accordance with the Declaration of Helsinki. Patients were excluded if they exhibited underlying organ dysfunction, including renal and hepatic dysfunction, or hypokalemia (see Inclusion and Exclusion Criteria in Supplementary File 1). The dose escalation included 14-day clinical evaluation periods with two potential dose escalations. Dosages included 100 mg twice-daily, 200 mg twice-daily, and 400 mg twice-daily (Fig. 5). Dose selection was based on the results of a previous phase 1 clinical study (CAM-102) which demonstrated that CAMB was well tolerated with only mild gastrointestinal adverse events after a single dose of either 200 or 400 mg. Because increased gastrointestinal events were seen in the 800-mg single-dose group in that phase 1 study, twice-daily dosing of 400 mg for a total daily dose of 800 mg was selected for the current phase 2a study to improve tolerability and maximize treatment effect. On the first day of each dose, the patients received a single dose and a PK analysis of blood was performed 24 h later. Starting on day 2, dosing was twice-daily and clinical response and safety laboratory assessments were performed on days 4, 7, and 14. At the end of each dosing period, the clinical investigator made a determination, based on clinical response and tolerability, to either continue dosing the patient for an additional 14 days at the same dosage or to escalate the dosage up to two times for an additional 14 days each time (Fig. 5). A long-term, progressive extension phase of study treatment was offered to patients who responded clinically to therapy and had no safety or tolerability concerns.
FIG 5.
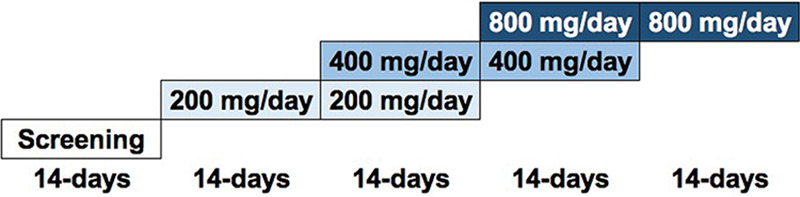
Schematic representation of the CAMB dose escalation in the phase 2 clinical study.
The study drug was supplied as an oral suspension and was mixed prior to administration. The patients were asked to swish the suspension in their mouths before swallowing if OPC was present, and then to refrain from eating or drinking for 30 min following the dose. The drug volumes were 20 mL for 100-mg dosing, 40 mL for 200-mg dosing, and 80 mL for 400-mg dosing, administered twice daily. A change in concentration was made to the CAMB formulation 4 years into the study from 5 mg/mL to 20 mg/mL which affected patients 1, 2, and 4, resulting in a reduction in the administered volume to either 10 mL (400 mg dose) or 20 mL (800 mg dose) twice daily.
Study definitions.
Dose escalation decisions were based on the clinical response status and tolerability of CAMB. A clinical responder was defined as a patient who achieved complete clinical cure or clinical improvement. For OPC, complete clinical cure was defined as an absence of thrush plaques and absent or minimal symptoms. Clinical improvement was defined as a partial resolution (≥50%) of pretreatment (baseline) signs and symptoms on the clinical severity score (see Supplementary File 1). For EC, patients were encouraged to undergo an upper endoscopy at the end of therapy or within 4 weeks of clinical improvement, and clinical cure was defined as an absence of plaques on endoscopy and absent or minimal symptoms, whereas clinical improvement was defined as a partial resolution (≥50%) of pretreatment (baseline) symptoms or signs on the clinical severity score. Clinical cure of VVC was defined as an absence of signs and symptoms of vaginitis, and clinical improvement was defined as a ≥50% reduction of the clinical severity score from the baseline (see Supplementary File 1).
Study outcomes.
The primary objective was a clinical response to CAMB treatment of OPC, EV, or VVC after 14 days with the highest titrated dosage. This time point, although early for evaluating a sustained clinical response, can effectively assess an initial response to antifungal therapy and can have a significant impact on improving overall quality of life, as has been shown previously, where partial and complete clinical responses to voriconazole were evaluated in HIV/AIDS patients with fluconazole-resistant OPC/EC at days 7 and 14 (37). For patients with more than infection site (such as OPC and VVC), the score for the site marked as primary made the determination, although both sites were scored for clinical response. Secondary objectives included assessment of the safety and tolerability of CAMB, the response in semiquantitative fungal cultures after completion of 14 days at the maximum dosage (i.e., no growth, one colony, scant, light, moderate, or heavy growth), and assessment of plasma PK after a single dose and after 14 and 27 days of the starting dosage and subsequent dose titrations. An exploratory objective was the assessment of long-term safety and efficacy for those patients who continued on to the extension phase of the study.
Adverse events were graded and recorded according to the NIAID Division of Microbiology and Infectious Diseases Adult Toxicity Table. Specific endpoints included the rate of treatment-limiting toxicity, the incidence of nephrotoxicity (increase of more than 100% of baseline serum creatinine), and the incidence of hypokalemia (≤3 mmol/L) during or within the 3 weeks of completion of CAMB.
Statistical analyses.
Ordinary one-way analysis of variance or Kruskal-Wallis tests with Tukey’s or Dunn’s multiple-comparison tests were used, where appropriate, to compare mucosal tissue fungal burdens in mouse models of OPC and VVC among the treatment groups. The log-rank (Mantel-Cox) test was used to compare mouse survival following invasive candidiasis among the treatment groups. GraphPad Prism v8 was used for statistical analyses. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
These studies were performed as part of a Cooperative Research and Development Agreement between the National Institutes of Health (NIH) and Matinas Biopharma Nanotechnologies, Inc. This work was also supported in part by the Intramural Research Program of the NIH National Institute of Allergy and Infectious Diseases (NIAID).
T.M. and R.M. are employees of Matinas Biopharma Nanotechnologies, Inc., which is the producer of CAMB. M.S.L. and A.F.F. received funding support from Matinas Biopharma for these studies. No other authors have conflicts of interest to declare.
Footnotes
Supplemental material is available online only.
Contributor Information
Michail S. Lionakis, Email: lionakism@mail.nih.gov.
Alexandra F. Freeman, Email: freemaal@mail.nih.gov.
REFERENCES
- 1.Lionakis MS, Netea MG, Holland SM. 2014. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med 4:a019638. 10.1101/cshperspect.a019638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC. 2008. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776. 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova J-L. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68. 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova J-L. 2012. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 12:616–622. 10.1097/ACI.0b013e328358cc0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferre EMN, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, Browne SK, Hsu AP, Rampertaap S, Swamydas M, Collar AL, Kong HH, Lee C-CR, Chascsa D, Simcox T, Pham A, Bondici A, Natarajan M, Monsale J, Kleiner DE, Quezado M, Alevizos I, Moutsopoulos NM, Yockey L, Frein C, Soldatos A, Calvo KR, Adjemian J, Similuk MN, Lang DM, Stone KD, Uzel G, Kopp JB, Bishop RJ, Holland SM, Olivier KN, Fleisher TA, Heller T, Winer KK, Lionakis MS. 2016. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 1:e88782. 10.1172/jci.insight.88782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, Chauss D, Harrison OJ, Alejo J, Williams DW, Pittaluga S, Lee C-CR, Bouladoux N, Swamydas M, Hoffman KW, Greenwell-Wild T, Bruno VM, Rosen LB, Lwin W, Renteria A, Pontejo SM, Shannon JP, Myles IA, Olbrich P, Ferré EMN, Schmitt M, Martin D, Barber DL, Solis NV, Notarangelo LD, Serreze DV, Matsumoto M, Hickman HD, Murphy PM, Anderson MS, Lim JK, Holland SM, Filler SG, Afzali B, Belkaid Y, Moutsopoulos NM, Lionakis MS, Genomics and Computational Biology Core . 2021. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371:eaay5731. 10.1126/science.aay5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Break TJ, Desai JV, Healey KR, Natarajan M, Ferre EMN, Henderson C, Zelazny A, Siebenlist U, Yates CM, Cohen OJ, Schotzinger RJ, Perlin DS, Garvey EP, Lionakis MS. 2018. VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J Antimicrob Chemother 73:2089–2094. 10.1093/jac/dky170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Break TJ, Desai JV, Natarajan M, Ferre EMN, Henderson C, Zelazny AM, Siebenlist U, Hoekstra WJ, Schotzinger RJ, Garvey EP, Lionakis MS. 2018. VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans. J Antimicrob Chemother 73:151–155. 10.1093/jac/dky170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox R, Neal KR, Leen CL, Ellis ME, Mandal BK. 1991. Fluconazole resistant candida in AIDS. J Infect 22:201–204. 10.1016/0163-4453(91)91767-R. [DOI] [PubMed] [Google Scholar]
- 10.Rautemaa R, Richardson M, Pfaller M, Koukila-Kähkölä P, Perheentupa J, Saxén H. 2007. Decreased susceptibility of Candida albicans to azole antifungals: a complication of long-term treatment in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) patients. J Antimicrob Chemother 60:889–892. 10.1016/0163-4453(91)91767-r. [DOI] [PubMed] [Google Scholar]
- 11.Moorhouse AJ, Rennison C, Raza M, Lilic D, Gow NAR. 2016. Clonal strain persistence of Candida albicans Isolates from chronic mucocutaneous candidiasis patients. PLoS One 11:e0145888. 10.1371/journal.pone.0145888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rautemaa R, Richardson M, Pfaller MA, Perheentupa J, Saxén H. 2008. Activity of amphotericin B, anidulafungin, caspofungin, micafungin, posaconazole, and voriconazole against Candida albicans with decreased susceptibility to fluconazole from APECED patients on long-term azole treatment of chronic mucocutaneous candidiasis. Diagn Microbiol Infect Dis 62:182–185. 10.1016/j.diagmicrobio.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Wong-Beringer A, Jacobs RA, Guglielmo BJ. 1998. Lipid formulations of amphotericin B: clinical efficacy and toxicities. Clin Infect Dis 27:603–618. 10.1086/514704. [DOI] [PubMed] [Google Scholar]
- 14.Aigner M, Lass-Florl C. 2020. Encochleated amphotericin B: is the oral availability of amphotericin b finally reached? J Fungi (Basel) 6:66. 10.3390/jof6020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipa-Castro A, Nicolas V, Angelova A, Mekhloufi G, Prost B, Chéron M, Faivre V, Barratt G. 2021. Cochleate formulations of Amphotericin b designed for oral administration using a naturally occurring phospholipid. Int J Pharm 603:120688. 10.1016/j.ijpharm.2021.120688. [DOI] [PubMed] [Google Scholar]
- 16.Perlin DS. 2004. Amphotericin B cochleates: a vehicle for oral delivery. Curr Opin Invest Drugs 5:198–201. [PubMed] [Google Scholar]
- 17.Shende P, Khair R, Gaud RS. 2019. Nanostructured cochleates: a multi-layered platform for cellular transportation of therapeutics. Drug Dev Ind Pharm 45:869–881. 10.1080/03639045.2019.1583757. [DOI] [PubMed] [Google Scholar]
- 18.Zarif L. 2005. Drug delivery by lipid cochleates. Methods Enzymol 391:314–329. 10.1016/S0076-6879(05)91018-5. [DOI] [PubMed] [Google Scholar]
- 19.Batista-Duharte A, Lastre M, Romeu B, Portuondo DL, Téllez-Martínez D, Manente FA, Pérez O, Carlos IZ. 2016. Antifungal and immunomodulatory activity of a novel cochleate for amphotericin B delivery against Sporothrix schenckii. Int Immunopharmacol 40:277–287. 10.1016/j.intimp.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Delmas G, Park S, Chen ZW, Tan F, Kashiwazaki R, Zarif L, Perlin DS. 2002. Efficacy of orally delivered cochleates containing amphotericin B in a murine model of aspergillosis. Antimicrob Agents Chemother 46:2704–2707. 10.1128/AAC.46.8.2704-2707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu R, Hollingsworth C, Qiu J, Wang A, Hughes E, Xin X, Konrath KM, Elsegeiny W, Park Y-D, Atakulu L, Craft JC, Tramont EC, Mannino R, Williamson PR. 2019. Efficacy of oral encochleated amphotericin B in a mouse model of cryptococcal meningoencephalitis. mBio 10:e00724-19. 10.1128/mBio.00724-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santangelo R, Paderu P, Delmas G, Chen Z-W, Mannino R, Zarif L, Perlin DS. 2000. Efficacy of oral cochleate-amphotericin B in a mouse model of systemic candidiasis. Antimicrob Agents Chemother 44:2356–2360. 10.1128/AAC.44.9.2356-2360.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zarif L, Graybill JR, Perlin D, Najvar L, Bocanegra R, Mannino RJ. 2000. Antifungal activity of amphotericin B cochleates against Candida albicans infection in a mouse model. Antimicrob Agents Chemother 44:1463–1469. 10.1128/AAC.44.6.1463-1469.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skipper CP, Atukunda M, Stadelman A, Engen NW, Bangdiwala AS, Hullsiek KH, Abassi M, Rhein J, Nicol MR, Laker E, Williams DA, Mannino R, Matkovits T, Meya DB, Boulware DR. 2020. Phase I EnACT trial of the safety and tolerability of a novel oral formulation of amphotericin B. Antimicrob Agents Chemother 64:e00838-20. 10.1128/AAC.00838-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. 2002. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother 46:828–833. 10.1128/AAC.46.3.828-833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schönherr FA, Sparber F, Kirchner FR, Guiducci E, Trautwein-Weidner K, Gladiator A, Sertour N, Hetzel U, Le GTT, Pavelka N, d'Enfert C, Bougnoux M-E, Corti CF, LeibundGut-Landmann S. 2017. The intraspecies diversity of C. albicans triggers qualitatively and temporally distinct host responses that determine the balance between commensalism and pathogenicity. Mucosal Immunol 10:1335–1350. 10.1038/mi.2017.2. [DOI] [PubMed] [Google Scholar]
- 27.McDonough LD, Mishra AA, Tosini N, Kakade P, Penumutchu S, Liang S-H, Maufrais C, Zhai B, Taur Y, Belenky P, Bennett RJ, Hohl TM, Koh AY, Ene IV. 2021. Candida albicans isolates 529L and CHN1 Exhibit stable colonization of the murine gastrointestinal tract. mBio 12:e0287821. 10.1128/mBio.02878-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman D, Mistry M, Thavaraj S, Challacombe SJ, Naglik JR. 2007. Murine model of concurrent oral and vaginal Candida albicans colonization to study epithelial host-pathogen interactions. Microbes Infect 9:615–622. 10.1016/j.micinf.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacGregor RR, Bennett JE, Erslev AJ. 1978. Erythropoietin concentration in amphotericin B-induced anemia. Antimicrob Agents Chemother 14:270–273. 10.1128/AAC.14.2.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pisitkun P, Claudio E, Ren N, Wang H, Siebenlist U. 2010. The adaptor protein CIKS/ACT1 is necessary for collagen-induced arthritis, and it contributes to the production of collagen-specific antibody. Arthritis Rheum 62:3334–3344. 10.1002/art.27653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verma AH, Richardson JP, Zhou C, et al. 2017. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol 2:eaam8834. 10.1126/sciimmunol.aam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lionakis MS, Lim JK, Lee C-CR, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun 3:180–199. 10.1159/000321157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yano J, Fidel PL Jr.. 2011. Protocols for vaginal inoculation and sample collection in the experimental mouse model of Candida vaginitis. J Vis Exp 8:3382. 10.3791/3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL. 2009. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299–311. 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conti HR, Bruno VM, Childs EE, Daugherty S, Hunter JP, Mengesha BG, Saevig DL, Hendricks MR, Coleman BM, Brane L, Solis N, Cruz JA, Verma AH, Garg AV, Hise AG, Richardson JP, Naglik JR, Filler SG, Kolls JK, Sinha S, Gaffen SL. 2016. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20:606–617. 10.1016/j.chom.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clemons KV, Spearow JL, Parmar R, Espiritu M, Stevens DA. 2004. Genetic susceptibility of mice to Candida albicans vaginitis correlates with host estrogen sensitivity. Infect Immun 72:4878–4880. 10.1128/IAI.72.8.4878-4880.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegener P, Troke PF, Fätkenheuer G, Diehl V, Ruhnke M. 1998. Treatment of fluconazole-resistant candidiasis with voriconazole in patients with AIDS. AIDS 12:2227–2228. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods and Fig. S1. Download aac.00308-22-s0001.pdf, PDF file, 0.1 MB (112.1KB, pdf)