

Abstract
The hepatitis B virus (HBV) polymerase synthesizes the viral DNA genome from the pre-genomic RNA (pgRNA) template through reverse transcription. Initiation of viral DNA synthesis is accomplished via a novel protein priming mechanism, so named because the polymerase itself acts as a primer, whereby the initiating nucleotide becomes covalently linked to a tyrosine residue on the viral polymerase. Protein priming, in turn, depends on specific recognition of the packaging signal on pgRNA called epsilon. These early events in viral DNA synthesis can now be dissected in vitro as described here.
The polymerase is expressed in mammalian cells and purified by immunoprecipitation. The purified protein is associated with host cell factors, is enzymatically active, and its priming activity is epsilon dependent. A minimal epsilon RNA construct from pgRNA is co-expressed with the polymerase in cells. This RNA binds to and co-immunoprecipitates with the polymerase. Modifications can be made to either the epsilon RNA or the polymerase protein by manipulating the expression plasmids. Also, the priming reaction itself can be modified to assay for the initiation or subsequent DNA synthesis during protein priming, the susceptibility of the polymerase to chemical inhibitors, and the precise identification of the DNA products upon their release from the polymerase. The identity of associated host factors can also be evaluated. This protocol closely mirrors our current understanding of the RNA binding and protein priming steps of the HBV replication cycle, and it is amenable to modification. It should therefore facilitate both basic research and drug discovery.
Keywords: Hepatitis B virus, Polymerase, Reverse transcriptase, Protein priming, RNA binding
1. Introduction
Hepatitis B virus encodes one enzymatic protein, a multifunctional polymerase (called variously P, HP, Pol, and RT). The hepatitis B virus polymerase consists of four domains: the terminal protein (TP) domain at the N-terminus, a spacer region, the reverse transcriptase (RT) domain, and the C-terminal RNase H domain. During its replication cycle, HBV polymerase synthesizes DNA from both RNA and DNA templates, and has the interesting characteristic of acting as its own primer. The TP domain contains a tyrosine residue at position 63 (Y63) which covalently attaches to the nascent DNA through a phosphotyrosyl bond [1–3].
The polymerase is encoded on the viral pre-genomic RNA (pgRNA), which is transcribed by RNA Pol II in host cell nuclei from viral DNA [4]. The pgRNA serves as messenger RNA for translation of viral proteins, and also acts as the template for reverse transcription to produce progeny viral DNA. The pgRNA contains a packaging signal near the 5′ end, which is a 61-nucleotide stemloop structure called epsilon (ε RNA) to which the RT binds. This ε RNA contains an internal bulge on the hairpin with the sequence UUC [5]. This is copied by the polymerase, first to the initial dGMP, and then to dAMP nucleotides at the 5′ end of the viral minus-strand DNA. During viral replication, this small dGAA oligonucleotide then moves to the 3′ end of the RNA, facilitating a switch of templates so a nearly full-length DNA copy can be made from the pgRNA. This RNA is degraded during minus-strand DNA synthesis, and the plus-strand DNA is then copied. The result is a partially double-stranded circular DNA that is not covalently closed, but is covalently attached to the polymerase on the 5′ end of the minus strand. This relaxed circular DNA (rcDNA) is what is found in infectious viral particles of HBV [6, 7].
The priming assay described in this protocol can be used to test the early steps of DNA synthesis—both priming initiation and downstream synthesis of the nascent minus-strand of DNA [8–10]. Since dGMP is the obligatory initiating nucleotide, it can be used alone in a priming reaction to test initiation. Other nucleotides can be added to test subsequent steps in synthesis. This simple setup can facilitate drug screening. The inclusion of a nucleoside or non-nucleoside inhibitor in the reaction can reveal if the drug affects these early steps of viral DNA synthesis. For example, the guanidine analog RT inhibitor, entecavir, was shown to act competitively (with respect to dGTP) to block priming in a competitive manner and is incorporated into the DNA product [9]. This guanidine analog thus blocks the initiation step of priming, which normally requires dGTP. Interestingly, clevudine—a thymidine analog—was also able to inhibit priming initiation as well as elongation, which requires dATPs. It was further shown that clevudine triphosphate is not incorporated into the DNA product, but instead acts as a non-nucleoside inhibitor, unlike any other nucleoside analog inhibitors [9]. In addition to these findings, the priming assay described herein may help reveal the mechanisms of the next generation of HBV RT inhibitors.
One interesting function of the polymerase is what has been described as a novel protein-primed terminal transferase activity [11]. When manganese instead of magnesium is used as the enzyme cofactor, there is a loss of template dependence. The polymerase will synthesize DNA by random incorporation of nucleotides, but favors TTP incorporation for making DNA strands. It is not known how relevant this function is in vivo during an infection.
An important step during viral replication is the removal of the RT from the rcDNA genome. The protein must be removed in order to close the circular DNA covalently [12–14]. The covalently closed circular DNA (cccDNA) is the root source of virus production, since the cell uses it to make all viral RNAs. It is unknown how the polymerase is removed from rcDNA, but one candidate is the human enzyme tyrosyl DNA phosphodiesterase 2 (Tdp2) [15, 16]. Whether or not it is used during actual infections, it can be used in vitro and is employed here to evaluate the synthesized DNA by cleaving the DNA from the HBV RT enzyme [8–11].
The current protocol is basically a two component system: the HBV polymerase and an epsilon-containing RNA construct. Both can be mutagenized by manipulating the plasmids used to express them. Once co-transfected in mammalian cell culture, the HBV polymerase and ε RNA will associate. During HBV replication, both protein priming and the subsequent DNA synthesis occur inside the viral nucleocapsid. However, in this reduced system which expresses no capsid protein, only RNA binding and limited amounts of protein priming occur while inside the transfected cells. Upon purification from the cells, the polymerase is competent to carry out protein-primed DNA synthesis in vitro using the ε RNA bound to it. Also, in vitro transcribed ε RNA can be added to HBV polymerase which is purified alone (no ε RNA expressed with it in cell culture) to determine the level of RNA binding activity. This in vitro RNA binding assay is another way to evaluate this critical association.
The entire pgRNA is not expressed, in part because pgRNA contains two copies of the epsilon sequence, however only the 5′ copy is functional as a packaging signal in vivo. The construct used here produces an mRNA that is 330 bases in length (not counting the poly(A) tail which would be added by the cell), as opposed to the 3.5 kb full length pgRNA.
The purified HBV polymerase in this system is associated with host cell factors, some of which are chaperone proteins which facilitate polymerase function [17–19]. A lack of necessary host factors may be the reason that no in vitro translated human HBV RT has been shown to be active enzymatically in protein priming. These facilitating host factors have been characterized in part, but other host factors may be found and studied using the current protocol.
For years the ability to measure the amount of HBV DNA has been available in a DNA synthesis assay, which consists of purifying capsids and detecting the DNA contained in these nucleocapsids, usually by Southern blot. Similarly, an RNA packaging (encapsidation) assay is a similar procedure, but detects viral RNA, usually by northern blot. Although these protocols accurately reflect the activity of the viral polymerase, they cannot evaluate the initial steps in DNA synthesis. The current protocol satisfies a need to molecularly dissect these early DNA synthesis steps which are critical to the viral replication cycle and may be inhibited by drug compounds.
The use of the solid-phase agarose beads facilitates purification and buffer exchange, and relatively large amounts of purified protein can be obtained. Modifications can be made to the RT or the ε RNA to examine critical features of these partners and how these features affect their interaction. The ability to test several enzymatic roles of the HBV polymerase, and test their inhibition through drug screening, makes this procedure an important and novel method to study this important virus.
2. Materials
2.1. HBV Polymerase Expression and Purification (See Note 1)
HEK 293T cells.
DMEM/F12 (1:1) medium. Complete medium is supplemented with 10 % FBS, 100 U/mL penicillin and 10 μg/mL streptomycin.
pcDNA-3FHP: Expresses a triple FLAG-tagged HBV polymerase (HP) under the human cytomegalovirus (CMV) promoter in the pcDNA3 (Invitrogen) backbone. The plasmid was constructed by fusing in-frame three copies of the FLAG epitope tag to the N-terminus of the HBV polymerase coding sequence [8]. Strain ayw (GenBank accession number X59795.1) was used, which is phylogenetically HBV genotype D as in pCMV-HBV [20].
pCMV-HE: For ε RNA expression in human cells, pCMV-HE was produced by substituting a 0.5 kb fragment of the CMV promoter plus HBV sequence from 1801–1993 from pCMV-HBV (NdeI to XbaI) for the CMV and T7 promoter in pcDNA3 (NdeI to XbaI fragment) [8]. pCMV-HE will produce, upon RNA Pol II transcription in transfected mammalian cells, a capped and polyadenylated HBV RNA initiating at the authentic pgRNA initiation site (1814) and containing the 5′ DR1 (1822–1832) and ε RNA sequences (1845–1905) (see Note 2).
Calcium phosphate transfection kit (Clontech).
Anti-FLAG M2 antibody (Sigma).
Protein A/G agarose beads.
Low retention pipet tips.
Diethylpyrocarbonate (DEPC) (see Note 3).
RNaseZap or similar RNase removal product (Ambion).
Dithiothreitol (DTT), 1 M, make fresh for each use, keep RNase-free (see Note 4).
β-mercaptoethanol, 12.8 M, keep RNase-free (see Note 5).
Complete EDTA-free protease inhibitor, 25× concentration, store prepared solution for up to 3 months at −20 °C, keep RNase-free (Roche).
Complete protease inhibitor, 25× concentration, store prepared solution for up to 3 months at −20 °C, keep RNase-free (Roche).
E-64 protease inhibitor, 2 mM, store prepared solution for up to 9 months at −20 °C, keep RNase-free.
Leupeptin protease inhibitor, 1 mg/mL, store prepared solution for up to 6 months at −20 °C, keep RNase-free.
Phenylmethanesulfonyl fluoride (PMSF) protease inhibitor, 200 mM in isopropanol, store at room temperature for up to 9 months, keep RNase-free (see Note 6).
RNasin Plus (Promega) or RNaseOUT (Thermo Fisher) RNase inhibitors.
RNase-free Tris–HCl, 1 M, pH 7.0 (Ambion).
RNase-free NaCl 5 M (Ambion).
KCl, 1 M, keep RNase-free.
Glycerol, 80 %, keep RNase-free.
NP-40, 10 %, keep RNase-free.
EDTA, 0.5 M, pH 8.0, store at room temperature, keep RNase-free.
TN buffer: 50 mM Tris–HCl pH 7.0, 100 mM NaCl. For 50 mL, combine 2.5 mL of 1 M Tris–HCl pH 7.0, 1 mL of 5 M NaCl, and 46.5 mL of DEPC-treated water (see Note 7). Store at room temperature.
10× RNase-free PBS (Ambion). Prepare 1× PBS with DEPC-treated water, keep RNase-free.
1× PBS with protease inhibitors: 28 μM E-64, 5 μg/mL leupeptin, 1 mM PMSF. To 50 mL of 1× PBS, add 350 μL of 2 mM E-64, 125 μL of 1 mg/mL leupeptin, and 125 μL of 200 mM PMSF. Prepare just before use and do not store, keep RNase-free.
FLAG lysis buffer: 50 mM Tris–HCl pH 7.0, 100 mM NaCl, 50 mM KCl, 10 % glycerol, 1 % NP-40, 1 mM EDTA pH 8.0. For 50 mL, combine 2.5 mL of 1 M Tris–HCl pH 7.0, 1 mL of 5 M NaCl, 2.5 mL of 1 M KCl, 6.25 mL of 80 % glycerol, 5 mL of 10 % NP-40, 100 μL of 0.5 M EDTA pH 8.0, and 32.65 mL of DEPC-treated water. Store at room temperature, keep RNase-free.
FLAG lysis buffer with inhibitors: FLAG lysis buffer plus 1× Complete protease inhibitor cocktail, 10 mM β-mercaptoethanol, 2 mM DTT, 1 mM PMSF, 250 U/mL RNase inhibitor. To 1 mL of FLAG lysis buffer, add 40 μL of 25× Complete protease inhibitor cocktail, 0.78 μL of 12.8 M β-mercaptoethanol, 2 μL of 1 M DTT, 5 μL of 200 mM PMSF, and 6.25 μL of 40 U/μL RNase inhibitor. Prepare just before use, use on ice, do not store, keep RNase-free.
FLAG wash buffer with inhibitors: FLAG lysis buffer plus 28 μM E-64, 5 μg/mL leupeptin, 1 mM PMSF, 10 mM β-mercaptoethanol, 2 mM DTT, 10 U/μL RNase inhibitor. To 1 mL of FLAG lysis buffer, add 14 μL of 2 mM E-64, 5 μL of 1 mg/mL leupeptin, 5 μL of 200 mM PMSF, 0.78 μL of 12.8 M β-mercaptoethanol, 2 μL of 1 M DTT, and 0.25 μL of 40 U/μL RNase inhibitor. Prepare just before use, use on ice, do not store, keep RNase-free.
2× SDS lysis buffer: 125 mM Tris–HCl pH 6.8, 20 % glycerol, 4.6 % SDS, 0.1 % bromophenol blue. Store at room temperature. For 50 mL, combine 6.94 mL of 1 M Tris–HCl pH 6.8, 11.1 mL of glycerol, 25.56 mL of 10 % SDS, 1.1 mL of 5 % bromophenol blue, and 5.28 mL of distilled water. Before use, add one-tenth volume 12.8 M β-mercaptoethanol.
2.2. In Vitro and In Vivo RNA Binding (See Note 8)
MAXIscript SP6 kit (Ambion).
[α-32P] UTP (10 mCi/mL, 3000 Ci/mmol) (PerkinElmer).
Quick Spin columns for radiolabeled RNA purification (Sephadex G-25, fine) (Roche).
RIPA buffer: 50 mM Tris–HCl pH 7.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 0.05 % NP-40. For 50 mL, combine 2.5 mL of 1 M Tris–HCl pH 7.0, 1.5 mL of 5 M NaCl, 100 μL of 0.5 M EDTA pH 8.0, 250 μL of 10 % NP-40, and 45.65 mL of DEPC-treated water. Store at room temperature, keep RNase-free.
RIPA buffer with inhibitors: RIPA buffer plus 2 mM DTT, 1 mM PMSF, 1× Complete protease inhibitor cocktail, 1 U/μL RNase inhibitor. To 1 mL of RIPA buffer add 2 μL of 1 M DTT, 5 μL of 200 mM PMSF, 40 μL of 25× Complete protease inhibitor cocktail, and 25 μL of 40 U/mL RNase inhibitor. Prepare just before use, use on ice, do not store, keep RNase-free.
RIPA wash buffer: RIPA buffer plus 2 mM DTT, 1 mM PMSF, 28 μM E-64, 5 μg/mL leupeptin, and 10 U/mL RNase inhibitor. To 1 mL of RIPA buffer add 2 μL of 1 M DTT, 5 μL of 200 mM PMSF, 14 μL of 2 mM E-64, 5 μL of 1 mg/mL leupeptin, and 0.25 μL of 40 U/mL RNase inhibitor. Prepare just before use, use on ice, do not store, keep RNase-free.
Formamide gel loading buffer II (Ambion).
10× TBE: 0.89 M Tris, 0.89 M boric acid, 20 mM EDTA. For 1 L, add 108 g of Tris base, 55 g of boric acid, and 40 mL of 0.5 M EDTA pH 8.0. Fill to 1 L with water.
1× TBE: 89 mM Tris, 89 mM boric acid, 2 mM EDTA. For 1 L, add 100 mL of 10× TBE and 900 mL of water.
ULTRAhyb solution (Ambion).
20× SSC pH 7.0: 3 M NaCl, 0.3 M sodium citrate. For 1 L, add 175.3 g NaCl and 88.2 g sodium citrate. Adjust pH and fill to 1 L with water.
Low stringency wash buffer: 2× SSC, 0.1 % SDS. For 500 mL, add 50 mL of 20× SSC, 2.5 mL of 20 % SDS, and 447.5 mL of water.
High stringency wash buffer: 0.1× SSC, 0.1 % SDS. For 500 mL, add 2.5 mL of 20× SSC, 2.5 mL of 20 % SDS, and 495 mL of water.
2.3. In Vitro Protein Priming, Transferase Activity, and Tdp2 Cleavage (See Note 8)
[α-32P] dATP (10 mCi/mL, 3000 Ci/mmol) (PerkinElmer).
[α-32P] dCTP (10 mCi/mL, 3000 Ci/mmol) (PerkinElmer).
[α-32P] dGTP (10 mCi/mL, 3000 Ci/mmol) (PerkinElmer).
[α-32P] TTP (10 mCi/mL, 3000 Ci/mmol) (PerkinElmer).
100 mM deoxynucleotide triphosphate set (dNTPs) (Invitrogen).
1 M MgCl2, store at −20 °C, keep RNase-free.
1 M MnCl2, store at −20 °C, keep RNase-free (see Note 9).
10× TMgNK buffer: 200 mM Tris–HCl pH 7.0, 150 mM NaCl, 100 mM KCl, 40 mM MgCl2. For 500 μL, combine 315 μL DEPC-treated water, 100 μL of 1 M Tris–HCl pH 7.0, 15 μL of 5 M NaCl, 50 μL of 1 M KCl, and 20 μL of 1 M MgCl2. Store at −20 °C, keep RNase-free.
TMgNK priming buffer: 1× TMgNK, 1× Complete EDTA-free protease inhibitor cocktail, 4 mM DTT, 1 U/μL RNase inhibitor, 1 mM PMSF. For 100 μL, combine 82.6 μL DEPC-treated water, 10 μL of 10× TMgNK, 4 μL of 25× Complete EDTA-free protease inhibitor cocktail, 0.4 μL of 1 M DTT, 2.5 μL of 40 U/μL RNase inhibitor and 0.5 μL of 200 mM PMSF. Prepare just before use, use on ice, do not store, keep RNase-free.
10× TMnNK buffer: 200 mM Tris–HCl pH 7.0, 150 mM NaCl, 100 mM KCl, 20 mM MnCl2. For 500 μL, combine 325 μL DEPC-treated water, 100 μL of 1 M Tris–HCl pH 7.0, 15 μL of 5 M NaCl, 50 μL of 1 M KCl, and 10 μL of 1 M MnCl2. Store at −20 °C, keep RNase-free.
TMnNK priming buffer: 1× TMnNK, 1× Complete EDTA-free protease inhibitor cocktail, 4 mM DTT, 1 U/μL RNase inhibitor, 1 mM PMSF. For 100 μL, combine 82.6 μL DEPC-treated water, 10 μL of 10× TMnNK, 4 μL of 25× Complete EDTA-free protease inhibitor cocktail, 0.4 μL of 1 M DTT, 2.5 μL of 40 U/μL RNase inhibitor, and 0.5 μL of 200 mM PMSF. Prepare just before use, use on ice, do not store, keep RNase-free.
TNK buffer: 20 mM Tris–HCl pH 7.0, 15 mM NaCl, 10 mM KCl. For 50 mL, combine 1 mL of 1 M Tris–HCl pH 7.0, 150 μL of 5 M NaCl, 500 μL of 1 M KCl, and 48.35 mL of DEPC-treated water, store at room temperature, keep RNase-free.
TNK wash buffer: TNK buffer plus 28 μM E-64, 1 mM PMSF, 5 μg/mL leupeptin, and 10 mM β-mercaptoethanol. To 50 mL of TNK buffer, add 700 μL of 2 mM E-64, 250 μL of 200 mM PMSF, 250 μL of 1 mg/mL leupeptin, and 39.06 μL of 12.8 M β-mercaptoethanol. Prepare just before use, use on ice, do not store, keep RNase-free.
Tyrosyl DNA phosphodiesterase 2 (Tdp2/TTRAP) (Abnova). Enzyme concentration was 0.23 μg/μL but may vary by lot, suspended in 50 mM Tris–HCl pH 8.0 and 10 mM reduced glutathione. Store at −80 °C.
Tdp2 mock buffer: 50 mM Tris–HCl pH 8.0 and 10 mM reduced glutathione. Store at −20 °C.
2× Tdp2 buffer: 50 mM Tris–HCl pH 8, 260 mM KCl, 2 mM DTT, 20 mM MgCl2. For 1 mL, combine 50 μL of 1 M Tris–HCl pH 8.0, 260 μL of 1 M KCl, 2 μL of 1 M DTT, 20 μL of 1 M MgCl2, and 668 μL of DEPC-treated water (see Note 7). Make fresh and do not store.
1× Tdp2 wash buffer: 1× Tdp2 buffer, 28 μM E-64, 1 mM PMSF, 5 μg/mL leupeptin. For 1 mL, combine 500 μL of 2× Tdp2 buffer, 476 μL of DEPC-treated water, 14 μL of 2 mM E-64, 5 μL of 200 mM PMSF, and 5 μL of 1 mg/mL leupeptin. Make fresh, use on ice, and do not store.
1× Tdp2 reaction mix: 1× Tdp2 buffer, 1× Complete EDTA-free protease cocktail inhibitor, with Tdp2 enzyme or mock buffer. For 100 μL, combine 50 μL of 2× Tdp2 buffer, 4 μL of 25× Complete EDTA-free protease inhibitor cocktail, 26 μL of DEPC-treated water, and 20 μL Tdp2 enzyme (or Tdp2 mock buffer). Make fresh and do not store.
3. Methods
3.1. Cell Culture and Transfection
Human embryonic kidney (HEK) HEK293T cells are maintained in complete DMEM/F12 (1:1) media in a humidified cell culture incubator at 37 °C, 5 % CO2 (see Notes 10 and 11).
Passage cells 1 day before transfection, plating approximately 1.25 × 106 cells per 6 cm dish, or an amount that yields 60–90 % confluence the next day (see Note 12).
Change medium 2–3 h before transfection.
Transfect each plate with half pCDNA-3FHP and half pCMV-HE (by weight) using calcium phosphate transfection (see Note 13). Use 10 μg total weight of DNA for 6 cm dishes, 20 μg for 10 cm dishes, and 50 μg for 15 cm dishes. Include any desired controls (see Notes 14 and 15).
Calculate how much sterile water would be needed to have a total volume of 500 μL total for a 6 cm dish, 1 mL total for a 10 cm dish, or 2.5 mL for a 15 cm dish (when combining water, DNA, the calcium solution, and the 2× HBS phosphate solution).
Add in the following order: sterile water, DNA, and calcium chloride (calcium chloride volume is 31 μL for 6 cm, 62 μL for 10 cm, or 155 μL for 15 cm dishes).
To the DNA-calcium tube, add 2× HBS dropwise (250 μL for 6 cm, 500 μL for 10 cm, or 1.25 μL for 15 cm dishes) while agitating the receiving tube by flicking or agitating on a vortexer which is set low enough that no splashing occurs.
After 5–20 min, apply the transfection reagent dropwise onto labeled plates.
Incubate at 37 °C, 5 % CO2 for 8 h to overnight with transfection reagent. Wash cells once with 1× PBS and apply fresh medium.
Allow cells to grow for 2 days, at which point the cells are lysed according to the procedure below. Alternatively the cells can be frozen at −80 °C (see Note 16).
3.2. Protein and RNA Purification
Two days after transfection, the anti-FLAG antibody is bound to agarose beads, the cells are lysed, and then the lysate is combined with the beads overnight to immunoprecipitate FLAG-tagged HBV polymerase and any associated RNA or host factors (Fig. 1a).
Fig. 1.
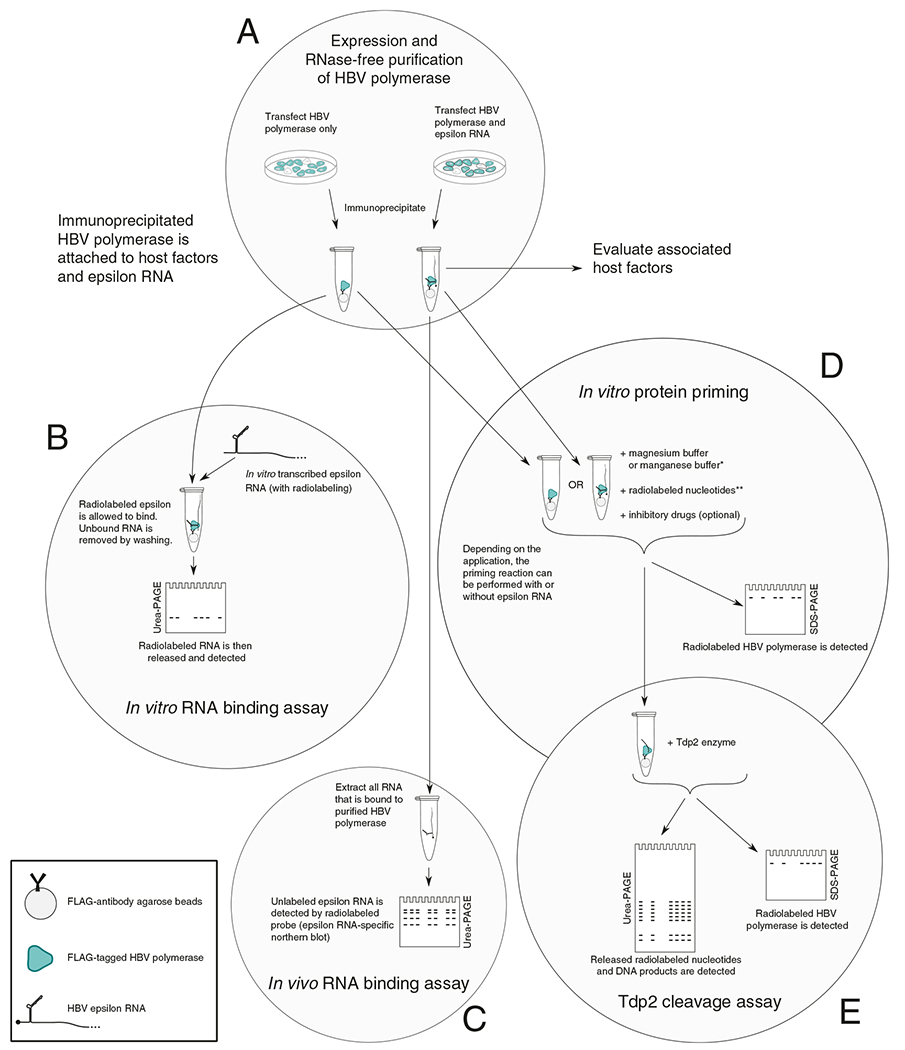
Protein priming and RNA binding protocol diagram. (a) The HBV polymerase can be purified (with or without epsilon RNA) by binding the FLAG-tagged polymerase to agarose beads, which are pre-bound with anti-FLAG antibodies. (b) An in vitro synthesized RNA is added to HBV polymerase (purified without epsilon) to perform the in vitro RNA binding assay. (c) The epsilon RNA which is co-purified with the polymerase can be evaluated for the in vivo RNA binding assay. (d) The purified polymerase, which is enzymatically active, can synthesize new DNA by adding nucleotides and buffer in the in vitro priming assay. Due to the protein priming activity, the polymerase itself is covalently labeled with the nucleotides and can be detected by autoradiography. (e) Alternatively, the DNA product from the priming reaction can be released from the polymerase by Tdp2 (the Tdp2 cleavage assay). *The priming assay, when using the manganese buffer, can also evaluate HBV polymerase’s terminal transferase activity. This novel activity is epsilon independent but also protein-primed. **The epsilon RNA template is copied beginning with an initial dGAA oligonucleotide. Therefore the initiation is dependent on dGTP, whereas the continued polymerization is dependent on dATP, and then other nucleotides
The purified protein, expressed with or without ε RNA, is the basis for all subsequent protocols. The beads, which contain the immunoprecipitated HBV polymerase and all associated proteins and RNA, are the starting material for the in vitro and in vivo RNA binding assays, the protein priming assay, the transferase activity, and release of the DNA product by the Tdp2 cleavage assay (Fig. 1).
Wash steps are performed by adding the indicated wash buffer, resuspending the beads by inverting the tube, centrifugation at 350 × g at 4 °C for 2 min, and removing the wash buffer. Any RNase-free filtered tips can be used for wash steps; however, perform all pipet transfers of the beads themselves with low-retention tips to reduce bead adherence to the pipet tip.
3.2.1. Prepare FLAG Ab-Bound Beads
Resuspend the immobilized protein A/G beads by inverting the bottle several times. Transfer 20 μL of the bead suspension per 6 cm plate (50 μL per 10 cm plate, 125 μL per 15 cm plate) into a single tube, which will be split into bead groups later.
Pellet the beads by centrifugation and remove the storage buffer. Wash the beads three times with TN buffer. Wash by adding the buffer to resuspend the beads, then centrifuge and remove the buffer.
After washing, resuspend the beads in TN buffer at half the original bead volume but at least 200 μL.
Bind anti-FLAG IgG Ab onto the washed beads by adding 2.8 μL of anti-FLAG antibody per each 6 cm dish (7 μL per 10 cm dish, or 17.5 μL per 15 cm dish).
Rotate at room temperature for 3–4 h. Proceed to cell lysis during incubation.
After the anti-FLAG antibody is bound to the beads, spin and remove the unbound antibody (see Note 17). Wash the beads three times with 500 μL FLAG lysis buffer (without inhibitors) (see Note 18). When adding the wash buffer the final time, resuspend the beads with the low retention tips, and divide equal volumes into separate tubes for each condition used in the transfection. Place the tubes on ice.
3.2.2. RNase-Free Cell Lysis
Wash cells once with 2 mL 1× PBS per 6 cm plate (4 mL per 10 cm plate, or 10 mL per 15 cm plate) (see Note 19).
Wash cells once with 2 mL cold 1× PBX with protease inhibitors per 6 cm plate (4 mL per 10 cm plate, or 10 mL per 15 cm plate) (see Note 20).
To each 6 cm plate, add 0.4 mL cold FLAG lysis buffer with inhibitors (1 mL to 10 cm plates, or 2.5 mL to 15 cm plates). Free cells from the dish by scraping with a cell scraper or spraying with the buffer from a pipet tip.
Collect cells from the same treatment condition into a single chilled tube, and rotate for 20 min at 4 °C.
Spin lysate at 4 °C for 10 min at maximum speed in a microcentrifuge ~18,000 × g (see Note 21).
Transfer the supernatant to a chilled tube of prepared Ab-bound beads (see Note 22).
Rotate the Ab-bound beads and cell lysate supernatant at 4 °C overnight to allow the immunoprecipitation to occur.
The next day, spin beads at 4 °C for 2 min at 350 × g. Remove unbound supernatant (see Note 23).
Wash the beads five times with 500 μL FLAG wash buffer. When adding the wash buffer the final time, resuspend the beads with the low retention tips, and divide equal volumes into separate tubes for each assay to be performed (see Notes 24 and 25).
Store bead aliquots at −80 °C (see Note 26). Approximate bead volume is 10 μL per tube (see Note 27).
3.2.3. Downstream Analysis of Immunopurified HBV Polymerase Complexes
For western blotting or protein staining (see Note 28) such as Coomassie blue or silver stain: Keeping the samples on ice throughout, remove the wash buffer, and add 20 μL 2× SDS lysis buffer to the ~10 μL beads.
Boil for 5 min, vortex, boil another 5 min, and place the samples back on ice. Spin briefly (~4000 × g for one second) to remove condensed liquid.
Mix the samples and load 20 μL on 9 % SDS-PAGE gel (see Note 29).
If analyzing/troubleshooting each step, load lysate supernatant (30 μL lysate + 30 μL 2× SDS lysis buffer, load 30 μL) and load insoluble pellet (add 50 μL TE, vortex to resuspend pellet, then add 200 μL 2× SDS lysis buffer, mix, boil, load 50 μL).
At this point, the gel can be used for staining or transferred to a membrane for western blotting (see Note 30).
3.3. In Vitro and In Vivo RNA Binding Assays
The purified HBV polymerase can be tested for its capacity to bind in vitro transcribed ε RNA. Any unbound RNA is removed by washing and the bound RNA is detected by autoradiography (Fig. 1b).
3.3.1. In Vitro Transcription of 32P-Radiolabeled ε RNA and In Vitro RNA Binding
Radiolabeled ε RNA is transcribed using the SP6 MAXIscript kit components (see Note 31). For a 20 μL reaction add 4 μL of DEPC-treated water; 1 μL each of 10 mM ATP, CTP, and GTP; 2 μL of 0.5 mM UTP (diluted from 10 mM stock 1:20 with DEPC-treated water); 1 μL of 1 μg/μL template DNA; 3 μL of SP6 enzyme; and 5 μL of [α-32P] UTP (see Note 32).
Incubate at 37 °C for 5 h total. Halfway through the incubation, spin briefly (~4000 × g for one second) to collect condensate.
After incubation, add 1 μL DNase and incubate at 37 °C for 30 min.
Inactivate the DNase by incubating at 70 °C for 15 min.
Labeled samples can be stored at −80 °C (see Note 33).
Combine each aliquot of immunoprecipitated HBV polymerase-bound beads (see Note 34) with 0.5 μg in vitro-transcribed 32P-labeled ε RNA (approximately 1–3 μL) and 20 μL RIPA buffer with inhibitors [19] (see Note 35).
Incubate for 3 h at room temperature with shaking to allow HBV polymerase to bind the ε RNA (see Note 36).
Save the supernatant, which contains any unbound components at −80 °C.
Wash the beads five times with 200 μL RIPA wash buffer. Samples may be stored at −80 °C for later processing (see Note 37).
After removing the final wash buffer, elute bound materials by adding 60 μL of 2× SDS lysis buffer and boiling 5 min. Vortex and boil 5 more minutes. Load 30 μL of sample onto a 15 % SDS-PAGE gel (see Note 38).
Cut the gel horizontally at approximately 50 kDa. The top portion of the gel which contains the HBV polymerase protein can be visualized by western blotting using the anti-FLAG antibody. The bottom portion of the gel containing the 32P-labeled ε RNA can be dried and directly exposed to film to detect the labeled ε RNA, which was bound to (and then disassociated from) the purified RT.
3.3.2. In Vivo RNA Binding Assay
Any RNA associated with the purified HBV polymerase is extracted from the bead sample. An ε RNA-specific probe is used to detect the amount of binding (Fig. 1c).
Using immunoprecipitated HBV polymerase (see Note 34), perform a TRIzol extraction of 20 μL lysate (see Note 22) and the ~10 μL bead aliquot (see Note 39). Resuspend the extracted RNA samples in a final volume of 20 μL DEPC-treated water. Add 20 μL of formamide gel loading buffer II to each sample and heat to 95 °C for 5 min by boiling or in a thermal cycler. Load the hot samples onto a 6 % acrylamide 8 M urea-PAGE gel in TBE. Run the gel at 300 V until bromophenol blue dye front comes off (approximate time is 2 h).
Rinse gel for 20 min in 1× TBE, rinse a nylon membrane in water for 5 min then in 1× TBE for 5 min.
For electrophoretic transfer, run below 300 mA (6–7 V) for 1 h.
While the transfer is running, preheat ULTRAhyb solution to 68 °C to dissolve precipitated material.
Cross-link the RNA to the membrane by exposing to UV transilluminator.
Prehybridize for at least 30 min at 68 °C. Use 10 mL ULTRAhyb buffer per 100 cm2 of membrane.
Add prepared ε RNA-specific radiolabeled probe (see Note 40) to the ULTRAhyb and allow the probe to hybridize 2 h to overnight.
Make sure wash buffers have no precipitate, if so, heat to 37 °C. Preheat high stringency wash to 68 °C.
Wash blot twice in room temperature low stringency wash solution (20 mL per 100 cm2 blot) with agitation for 5 min, dispose of washes in 32P waste.
Wash twice for 15 min at 68 °C with high stringency wash solution with agitation. Dispose of washes in 32P waste.
Seal radiolabeled blot in plastic wrap and expose to film or a phosphorimager screen.
3.4. In Vitro Protein Priming Assay
For priming, the buffer and labeled nucleotides are added to the beads, and priming and/or DNA synthesis can then occur. Due to the use of HBV polymerase itself as the primer, it becomes a radiolabeled protein, which can be visualized by autoradiography. The labeled RT is run on 9 % SDS-PAGE gel, and the gel is dried and exposed to film (Fig. 1d).
To a bead aliquot (approximately 10 μL beads and 10 μL residual buffer), add 19 μL of either the TMgNK (see Note 41) or TMnNK (see Notes 42 and 43) priming buffer.
Add 1 μL of radiolabeled nucleotides or 1 μL of a 100 μM solution of unlabeled nucleotides (see Note 44). Additionally, you may include nucleoside analogs or other RT inhibitors (see Note 45).
Pellet the beads at 350 × g for 1 min. Remove supernatant to radioactive waste (see Note 47).
Wash each sample once with 500 μL of TNK wash buffer. Remove the supernatant to radioactive waste.
Add 20 μL 2× SDS lysis buffer. Boil for 5 min, vortex, and boil for another 5 min. Spin briefly to collect condensate (~4000 × g for 1 s).
Load 20 μL of each sample on a 9 % SDS-PAGE gel.
Place the gel onto a piece of filter paper and cover with plastic wrap. Dry for 3 h on a vacuum gel drying apparatus at 75 °C (see Note 48).
Expose the dried gel to a film or phosphorimager screen.
3.5. Tdp2 Cleavage Assay
Tdp2 mediates the release of DNA which is covalently linked to HBV polymerase, and the released nucleotides and labeled polymerase are detected by autoradiography (Fig. 1e).
Perform the priming assay as described above (see Note 42). Complete a priming assay with sufficient aliquots of protein so that each condition can be mock treated and Tdp2 treated (see Note 49).
Wash the priming reaction three times with 500 μL TNK wash buffer.
Wash twice with 100 μL 1× Tdp2 wash buffer.
Add 10 μL of Tdp2 reaction mix (which includes the Tdp2 enzyme or mock buffer) to each sample.
Incubate at 37 °C for 1 h and with shaking (see Note 50).
After the incubation, spin for 1 min at 350 × g.
Separate the supernatant (released nucleotides) into a new tube and save the bead pellet (protein). Samples may be stored at −80 °C until testing.
For testing the supernatant (released nucleotides): Add 5 μL 2× formamide gel loading buffer II to a 5 μL aliquot. Heat to 95 °C for 5 min by boiling or with a thermal cycler. Load all 10 μL of the samples while still hot onto a 20 % acrylamide 8 M urea-PAGE gel in 1× TBE (see Note 51). Expose the gel to film or a phosphorimager screen.
For testing the bead pellet (protein): Add 20 μL of 2× SDS lysis buffer, boil 5 min, vortex, and boil another 5 min. Spin briefly to collect condensate (~4000 × g for 1 s), and load 10 μL of each sample on a 9 % SDS-PAGE gel. Expose the gel to a film or phosphorimager screen.
Acknowledgments
This work was supported by a Public Health Service grant (R01 AI043453 to J.H.) and a National Research Service Award grant (T32 CA 60395 to D.C. and S.J.) from the National Institutes of Health.
Footnotes
1. All autoclavable buffers that come into contact with HBV polymerase during experimentation should be autoclaved to avoid polymerase degradation (the polymerase tends to be very sensitive to degradation). After 2 months of storage and use of autoclaved reagents, re-autoclave the buffers.
2. Polyadenylation of this ε RNA can occurs at the strong bovine growth hormone (BGH) poly(A) site on pcDNA3, 223 nucleotides downstream from the native HBV poly(A) site. The native site (1915–1919) is used infrequently since it is inherently weak due to its proximity to the 5′ cap. Both RNA species can be seen on RNA gels.
3. DEPC-treated water or other molecular biology grade nuclease-free water should be used.
4. When using powdered chemicals under RNase-free conditions, standard (not certified RNase-free) weigh boats and disposable spatulas may be used, but a new package should be used which is kept separate and only for RNA work. When possible, weighing should be done in the tube and not on a weigh boat. If desired, certified RNase-free boats or spatulas are available.
5. When using commercially available liquids under RNase-free conditions, an unopened bottle is labeled for RNA work and then only used for such. RNA work should be done in a dedicated area, or an area cleaned by an RNase-removal product. Clean pipets with 70 % ethanol or RNase removal products before beginning work. Use dedicated tip boxes, and keep samples in storage boxes set apart for RNA work.
6. PMSF is always added last, and never placed on ice with other components, since it will precipitate out of solution at colder temperatures.
7. Some buffers need not be RNase-free for the purpose of their use, but as a matter of continuity and good practice they are made with DEPC-treated water and kept RNase-free.
8. Non-isotopic labeling was not tested, but could be a viable option.
9. Manganese solutions are slightly pink. Freeze thaw cycles tend to turn the solutions a dark brown. This coloration appears to have little effect on the priming reaction.
10. Although all experiments were performed using this cell line, any human or mammalian cell line that supports transfection could potentially work.
11. Passage number appeared to play an important role in HBV polymerase expression. After reviving a batch of HEK293T cells, the cells were passaged for at least 3 weeks before transfection. Cells were then frozen down at this stage for later revival. If cells were transfected a week after reviving, HBV polymerase expression tended to be lower. HEK293T cells were passaged and used for transfection to express HBV polymerase for 3 months after reviving. If cells were used for transfection after this period, RT expression decreases dramatically.
12. The number and size of plates can be determined empirically and according to need. A good starting point may be single 10 cm dishes (yield is five bead aliquots), three 6 cm dishes (all transfected with the same plasmids and pooled together when lysed, yield is six bead aliquots), or larger plates when an increased amount of purified protein is desired. The lysis protocol is written for 6, 10, and 15 cm dishes, but could be scaled. An advantage of pooling together smaller plates is to reduce variability of transfection efficiency. A good practice is to plate several extra plates in preparation for transfection, and choose the best plates to transfect, discarding any plates that are not at the same confluency, etc. as the other plates.
13. Any transfection method could potentially work. Transfection efficiency can approach 90 % with HEK 293T cells using the calcium phosphate method.
14. The ability to use wild-type or mutant plasmids is an advantage of this protocol. Both the HBV polymerase and/or the HBV epsilon RNA construct can be altered by introducing mutations on the plasmids.
15. A separate plate transfected in parallel with a GFP-expressing plasmid may be included. This GFP plate can be used to gauge the transfection efficiency. It can also be lysed with the other plates to control for nonspecific binding to the beads.
16. When freezing the plates, perform the PBS wash as shown in step 1 of the lysis procedure. Leave the plates tilted for 1 min and remove all PBS. Freeze the plates in a single layer (not stacked) in a −80 °C freezer, and once chilled they can be stacked. When beginning with step 2, place the plates on ice to continue the lysis procedure.
17. If desired, the unbound antibody can be saved and used in anti-FLAG western blotting to determine HBV polymerase levels in the transfected cell lysates.
18. The wash steps may be performed with a wash buffer that includes protease and RNase inhibitors; however, since these antibody-bound beads are only exposed to samples that already contain these inhibitors, it is unnecessary.
19. HEK 293T cells may exhibit weak attachment to cell culture plates. Wash by spraying the liquid slowly onto the side, not the bottom of the plate, being very careful not to detach the cells during the wash steps. If cells do become detached, they can be recovered by pelleting the suspended cells—centrifuge at 1000 × g at 4 °C for 5 min.
20. Transfer cells out of cell culture hood before or after the first wash. Note that this and all subsequent steps are on ice, and are performed under RNase-free conditions.
21. This NP-40-based lysis contains mainly cytoplasm in the supernatant and nuclei in the pellet.
22. Before transferring all of the supernatant to the antibody-bound beads, it is very helpful to save ~100 μL of supernatant and the insoluble pellet at −80 °C. These can be stored in a single or separate tubes. The pellet and/or supernatant can be used if additional analysis and troubleshooting are needed. Analysis of the lysate supernatant is recommended for the ε RNA northern blot (the in vivo RNA binding assay), where it is useful to run RNA extracted from both lysate supernatant and beads on the same gel.
23. The unbound supernatant can be saved at −80 °C for troubleshooting the immunoprecipitation.
24. During the centrifugation steps, prepare and label tubes for bead aliquots and store the empty tubes on ice for a cold transfer. The number of bead aliquot tubes needed may vary, but include at a minimum one tube for a western blot to verify RT expression, and one for a northern blot to evaluate the amount of ε RNA bound to the beads. Other tubes may be used for magnesium priming, manganese priming, the Tdp2 assay, or other assays.
25. After the final wash, it is not necessary to remove the wash buffer before freezing. However, before any downstream assay remove as much wash buffer as you are comfortable removing without disturbing the beads in a manner that leaves the same volume in all samples, approximately 10 μL beads and 10–20 μL wash buffer.
26. Purified HBV polymerase is stable at −80 °C for years, although it is recommended that samples be used promptly after expression.
27. The expression of HBV polymerase varies from experiment to experiment, ranging from approximately 20–100 ng per 6 cm dish.
28. HBV polymerase is bound to host proteins which can be detected by staining or western blot. Expression of HBV polymerase itself can be verified by an anti-FLAG western blot.
29. The acrylamide percentage can be adjusted depending on what proteins are to be detected; 9 % is apt for the large RT protein, which is approximately 94 kDa.
30. The same anti-FLAG antibody used in the immunoprecipitation should be used for western blotting to evaluate HBV polymerase levels in the transfected cells. A suggested amount is a 1:2000 dilution of primary antibody, and a 1:20,000 dilution of anti-mouse secondary antibody.
31. The template used is a DNA oligonucleotide coding for the ε RNA sequence annealed to an SP6 promoter [19]. Other constructs such as plasmid-based expression constructs should work as well.
32. Negative controls may include immunoprecipitated HBV polymerase that is deficient in RNA binding or RNA constructs that cannot bind HBV polymerase [5, 10].
33. If desired, transcribed labeled RNA can be visually verified on a 1.5 % agarose gel in 1× TAE under RNase-free conditions. Use an appropriate RNA ladder to evaluate size.
34. The RNA binding assay, when performed in an in vitro manner, would use beads from a single-plasmid transfection of HBV polymerase alone (and not a co-transfection of polymerase and ε RNA). The in vivo RNA binding assay uses co-transfected HBV polymerase and ε RNA.
35. Inhibitory compounds can be tested with this assay. They should be added prior to addition of 32P-labeled ε RNA.
36. Place a vortexer behind a shield, set between 2 and 3. The beads should be moving and not sitting at the bottom of the tube. Another method is to shake by hand every 5–10 min to keep the beads suspended.
37. As controls for the washing procedure, you may desire to save the first and fifth wash to run alongside the washed samples.
38. Controls may include the input ε RNA (before binding) at a 1:100 dilution, the unbound ε RNA at a 1:100 dilution, and the washes (see Note 37). To prepare these controls, add 30 μL of 2× SDS lysis buffer to 30 μL of the indicated sample. Boil 5 min, vortex, and boil 5 more minutes. Load 30 μL onto a 15 % SDS-PAGE gel.
39. This will reveal the amount of ε RNA co-immunoprecipitated with the HBV polymerase. This is referred to as an in vivo RNA binding assay. It is useful to run both beads and lysate supernatant on the same gel to reveal the amount of ε RNA produced in transfected cells compared to the amount bound by the RT.
40. The same pCMV-HE plasmid used to transfect can be used to make an ε RNA-specific antisense probe. The probe is a 1389 bp fragment purified from an NcoI digest under RNase-free conditions. This purified fragment is used in an SP6 in vitro transcription using radiolabeled [α-32P] UTP.
41. Priming occurs in a template-directed manner when using magnesium as the enzyme cofactor. The template on the ε RNA is UUC, which is copied as a dGAA trinucleotide. Therefore, if using a single nucleotide with magnesium priming, it should be dGTP.
42. For manganese-dependent transferase activity, the HBV RT prefers TTP, and this radiolabeled nucleotide will give the strongest signal. TTP is also ideal for the Tdp2 cleavage assay.
43. When including manganese as the enzyme cofactor, the RT can perform a transferase reaction, which is random DNA synthesis that is not necessarily template directed. The manganese-dependent transferase reaction can thus be performed with beads from HBV polymerase alone (from a single-plasmid transfection), as opposed to beads from a co-transfection expressing both HBV polymerase and ε RNA. When performing transferase activity, HBV polymerase will synthesize homo- and hetero-oligomeric or polymeric DNA strands up to several hundred nucleotides in length.
44. These should be the triphosphate form of the nucleotide or drug compound. When inhibitors are in the triphosphate form, they can be evaluated for their ability to terminate polymerization. Termination can also be tested by using dideoxynucleotide triphosphates (ddNTPs) which lack a 3′ –OH.
45. Any inhibitory compound should be added before addition of nucleotides.
46. A series of two reactions can be performed instead of a single 4 h reaction. When performing two reactions, incubate the first reaction for 2 h, then spin and remove supernatant. Wash twice with TNK wash buffer, and apply the second round of nucleotides or other compounds in fresh TMgNK or TMnNK buffer. Incubate a further 2 h and continue the protocol. For example, to study the addition of the dAMP in the synthesis of the dGAA oligomer, add unlabeled dGTP in the first reaction, then [α-32P] dATP—this will evaluate if polymerization occurs after the initiation step. In another example, nucleoside inhibitors can be added in the first round, then [α-32P] dATP—this will evaluate if the incorporated drug can inhibit polymerization.
47. At this point, the priming products may be visualized directly as the labeled polymerase protein or alternatively, the labeled nucleotides or DNA oligomers can be released from the polymerase by Tdp2 cleavage and subsequently visualized.
48. The drying step could potentially be skipped if a gel drying apparatus is not available. In this case, keep the gel well sealed to avoid drying, such that the gel does not shrink and move relative to the film or phosphorimager screen during exposure.
49. Testing the protein is only useful in the context of the mock-treated sample. There should be a decrease in signal in the Tdp2-treated protein when compared to the mock-treated protein. This is due to the radiolabeled nucleotides being cleaved from the RT.
50. Place a vortexer behind a shield in a 37 °C incubator, set between 2 and 3. The beads should be moving and not sitting at the bottom. Another method is to incubate in a water bath and shake by hand every 5–10 min to keep the beads suspended.
51. Pre-run gels for 30 min and flush out wells before loading. For a sequencing-style gel (40 × 16 cm with 1 mm spacers), run at 2500 V for approximately 2 h, or until the bromophenol blue dye front has migrated approximately 2/3 the length of the gel. For a smaller gel (16 × 17 cm with 2 mm spacers), run at 300 V for approximately 2 h, or until the bromophenol blue dye front has migrated approximately 2/3 the length of the gel.
References
- 1.Zoulim F, Seeger C (1994) Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol 68(1):6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang GH, Seeger C (1992) The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71(4):663–670 [DOI] [PubMed] [Google Scholar]
- 3.Lanford RE, Notvall L, Lee H, Beames B (1997) Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J Virol 71(4):2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rall LB, Standring DN, Laub O, Rutter WJ (1983) Transcription of hepatitis B virus by RNA polymerase II. Mol Cell Biol 3(10):1766–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu J, Boyer M (2006) Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J Virol 80(5):2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J (2011) Secretion of genome-free hepatitis B virus—single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7(9):e1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu J, Seeger C (2015) Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5(7):a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones SA, Boregowda R, Spratt TE, Hu J (2012) In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J Virol 86(9):5134–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones SA, Murakami E, Delaney W, Furman P, Hu J (2013) Non-competitive inhibition of hepatitis B virus reverse transcriptase protein priming and DNA synthesis by the nucleoside analog clevudine. Antimicrob Agents Chemother 57(9):4181–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones SA, Clark DN, Cao F, Tavis JE, Hu J (2014) Comparative analysis of hepatitis B virus polymerase sequences required for viral RNA binding, RNA packaging, and protein priming. J Virol 88(3):1564–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones SA, Hu J (2013) Protein-primed terminal transferase activity of hepatitis B virus polymerase. J Virol 87(5):2563–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao W, Hu J (2007) Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81(12):6164–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT (2007) Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81(22):12472–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, Mao R, Block TM, Guo JT (2010) Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84(1):387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui X, McAllister R, Boregowda R, Sohn JA, Ledesma FC, Caldecott KW, Seeger C, Hu J (2015) Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS One 10(6):e0128401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Königer C, Wingert I, Marsmann M, Rösler C, Beck J, Nassal M (2014) Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci U S A 111(40):E4244–E4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Toft DO, Seeger C (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 16(1):59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu J, Seeger C (1996) Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A 93(3):1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Flores D, Toft D, Wang X, Nguyen D (2004) Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78(23):13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fallows DA, Goff SP (1995) Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J Virol 69(5):3067–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]