

Abstract
Transient receptor potential vanilloid 4 (TRPV4), a member of the TRP superfamily, is a broadly expressed, cell surface-localized cation channel that is activated by a variety of environmental stimuli. Importantly, TRPV4 has been increasingly implicated in the regulation of cellular morphology. Here we propose that TRPV4 and the cytoskeletal remodeling small GTPase RhoA together constitute an environmentally sensitive signaling complex that contributes to pathological cell cytoskeletal alterations during neurological injury and disease. Supporting this hypothesis is our recent work demonstrating direct physical and bidirectional functional interactions of TRPV4 with RhoA, which can lead to activation of RhoA and reorganization of the actin cytoskeleton. Furthermore, a confluence of evidence implicates TRPV4 and/or RhoA in pathological responses triggered by a range of acute neurological insults ranging from stroke to traumatic injury. While initiated by a variety of insults, TRPV4–RhoA signaling may represent a common pathway that disrupts axonal regeneration and blood–brain barrier integrity. These insights also suggest that TRPV4 inhibition may represent a safe, feasible, and precise therapeutic strategy for limiting pathological TRPV4–RhoA activation in a range of neurological diseases.
Keywords: cytoskeleton, peripheral neuropathy, RhoA, spinal cord injury, stroke, TRPV4
INTRODUCTION: TRPV4 MODULATES CELL PHYSIOLOGY IN RESPONSE TO ENVIRONMENTAL STIMULI
In complex biological systems and living tissues, the ability of cells to detect external stimuli is essential for maintaining homeostasis in the face of dynamic environmental conditions. Signal transduction from the outside environment into the cell interior often involves plasma-membrane-embedded cellular “sensors” and their associated intracellular interacting protein complexes. These complexes transduce extracellular stimuli into intracellular signaling cascades, which in turn regulate downstream pathways that affect a wide range of cellular processes, including cell growth and migration, cell–cell junctional integrity, gene transcription and translation, and programmed cell death.[1]
Transient receptor potential vanilloid 4 (TRPV4) is an example of a cellular sensor with critical functions in many cell and tissue types. A member of the TRP family of ion channels, TRPV4 is a nonselective, Ca2+-permeable cation channel that was first described in 2000 by the research groups of Schultz[2] and Liedtke.[3] TRPV4 is expressed at the cell surface of a variety of cell types and has been linked to a broad range of physiological processes: central osmoregulation, osmosensation in the bladder and kidney, pain signaling in keratinocytes and sensory neurons, vascular tone, epithelial and endothelial barrier integrity, and bone remodeling, among many others.[4,5] Within these tissues, TRPV4 functions to transduce a range of environmental signals into intracellular signaling cascades. TRPV4 is a mechanosensitive channel that responds to shear stress, mechanical cell stretch, and cell swelling, and it is also sensitive to moderate heat and endogenous chemical stimuli such as epoxyeicosatrienoic acid (EET) metabolites, glycerophospholipids, and anandamides.[6,7] Although the intracellular effects of TRPV4 are diverse and incompletely understood, converging evidence has highlighted a critical function of TRPV4 in influencing dynamics of the cytoskeleton and cellular morphology. Furthermore, the importance of TRPV4 for cell physiology has been highlighted by the discovery of a range of dominantly inherited diseases caused by mutations in TRPV4.[8–13] These so-called TRPV4 channelopathies result from gain-of-function missense mutations within various functional domains of the TRPV4 protein, which give rise to a spectrum of neuromuscular diseases and distinct diseases of connective tissue and bone.[14]
TRPV4 is primarily expressed at the plasma membrane where it forms a homotetrameric ion channel. Each TRPV4 subunit within the tetramer consists of six transmembrane domains, a C-terminal intracellular domain, and a large cytosolic N-terminal domain that contains the ankyrin repeat domain (ARD)[15] (Figure 1). The ARD is thought to serve a scaffolding function central to the observed effects of TRPV4 on the cytoskeleton.[16] The ARD is also adjacent to a proline-rich region (PRR), which is involved in protein–protein interactions,[17,18] and a phosphoinositide binding domain (PBD), which can interact with the plasma membrane. The PBD’s interaction with the lipid membrane also exerts potent regulatory control of TRPV4 responses to thermal and hypotonic stimuli.[19] Notably, TRPV4 mutations that cause neurological disease primarily cluster in the N-terminal ARD that is believed to support a protein scaffolding function of TRPV4, highlighting the importance of this domain for TRPV4 function.[20]
FIGURE 1.
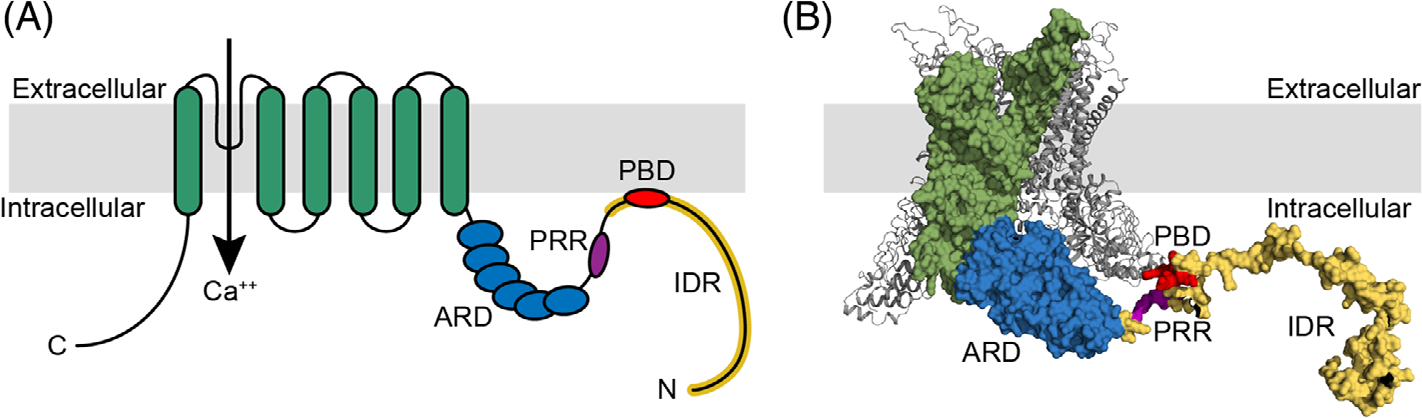
Structure of transient receptor potential vanilloid 4 (TRPV4). (A) Schematic of TRPV4 structural domains including transmembrane domains (green) with the ion channel pore between S5 and S6, the ankyrin repeat domain (ARD, blue), the proline-rich region (PRR, purple), PIP2 binding domain (PBD, red), and the intrinsically disorder region (IDR, yellow). (B) Crystal structure of xenopus tropicalis TRPV4 demonstrating structural domains highlighted in (A).
Activation of TRPV4 by various stimuli leads to opening of the channel pore and influx of extracellular cations, particularly calcium, which initiate diverse downstream intracellular signaling cascades.[21] For example, TRPV4-mediated calcium influx been shown to affect signaling via ATP/pannexin,[22,23] nitric oxide,[24,25] phospholipase A2,[26] ERK1/2,[27] CaMKII,[28,29] and IP3,[30,31] among many others.[26] Notably, many of these signaling cascades converge on pathways that regulate cytoskeletal remodeling machinery. For example, in trabecular meshwork cells of the eye, stretch-induced TRPV4 activation leads to phosphorylation of focal adhesion kinases and cytoskeletal stress fiber formation.[32,33] In vascular endothelial cells, TRPV4 -mediated calcium influx activates phosphoinositide 3-kinase, which in turn promotes cytoskeletal remodeling during angiogenesis and tumor-driven migration.[34–36]
Classically, the function of TRPV4 in regulating the cytoskeleton has been attributed to its activity as a calcium conduit and activation of calcium-dependent signaling cascades. However, accumulating evidence suggests that TRPV4 can also regulate the cytoskeleton independent of channel activity via protein–protein interactions. For example, prior work in C. elegans has demonstrated ion channel-independent functions of the invertebrate TRPV4 ortholog OSM-9.[37] In addition, immunoprecipitation studies have suggested that TRPV4 interacts with tubulin and actin as well as cytoskeletal signaling molecules PKCε and CaMKII, and these interactions are independent of Ca2+ influx.[28] TRPV4 can also exert effects on the cytoskeleton through its interactions with PACSINs (protein kinase C and casein kinase substrate in neurons), also known as syndapins.[17,18] These proteins have diverse functions in binding to and deforming curved membranes, regulating endocytosis, and recruiting cytoskeletal remodeling machinery.[17,18,38] Thus, TRPV4 exerts control of the cytoskeleton through both modulation of intracellular calcium levels and through scaffolding of cytoskeletal modulating proteins. Recent work has further highlighted the ion channel-independent scaffolding function of TRPV4 and its powerful role in driving changes in cell shape and morphology.
TRPV4–RhoA COMPLEX REGULATES THE CYTOSKELETON
We recently identified RhoA as a TRPV4 binding partner that is regulated by both calcium influx and ion channel-independent interactions.[39] These results suggest a dual regulatory mechanism yet to be described for other TRPV4 protein–protein interactions. RhoA is an abundant and ubiquitously expressed small GTPase that functions as a master regulator of the actin cytoskeleton. It acts as a molecular switch by cycling between an inactive GDP-bound state and an active GTP-bound state in which it binds and activates downstream cytoskeletal modulating proteins, including Rho-associated coiled-coil containing kinase (ROCK), mDia, ADF/cofilin, and nonmuscle myosin. Classically, active RhoA binds the negative regulatory region of ROCK, thereby promoting phosphorylation of the ROCK target proteins myosin-light chain (MLC), MLC kinase, and LIM kinases. Phosphorylation of these proteins results in increased myosin II ATPase activity, formation of actin stress fibers, actin bundling, and changes in cell morphology[40] (Figure 2(A)). Like other small GTPases, the activation of RhoA is dynamically regulated by guanine-nucleotide exchange factors (GEFs), which facilitate GTP binding leading to activation of RhoA, and GTPase activating protein (GAPs), which hydrolyze GTP to GDP and inactivate RhoA.[41] RhoA activity is further modulated by interactions with guanine-nucleotide dissociation inhibitors (GDIs), which bind RhoA and lock it in the GDP-bound, inactive, state.[41]
FIGURE 2.
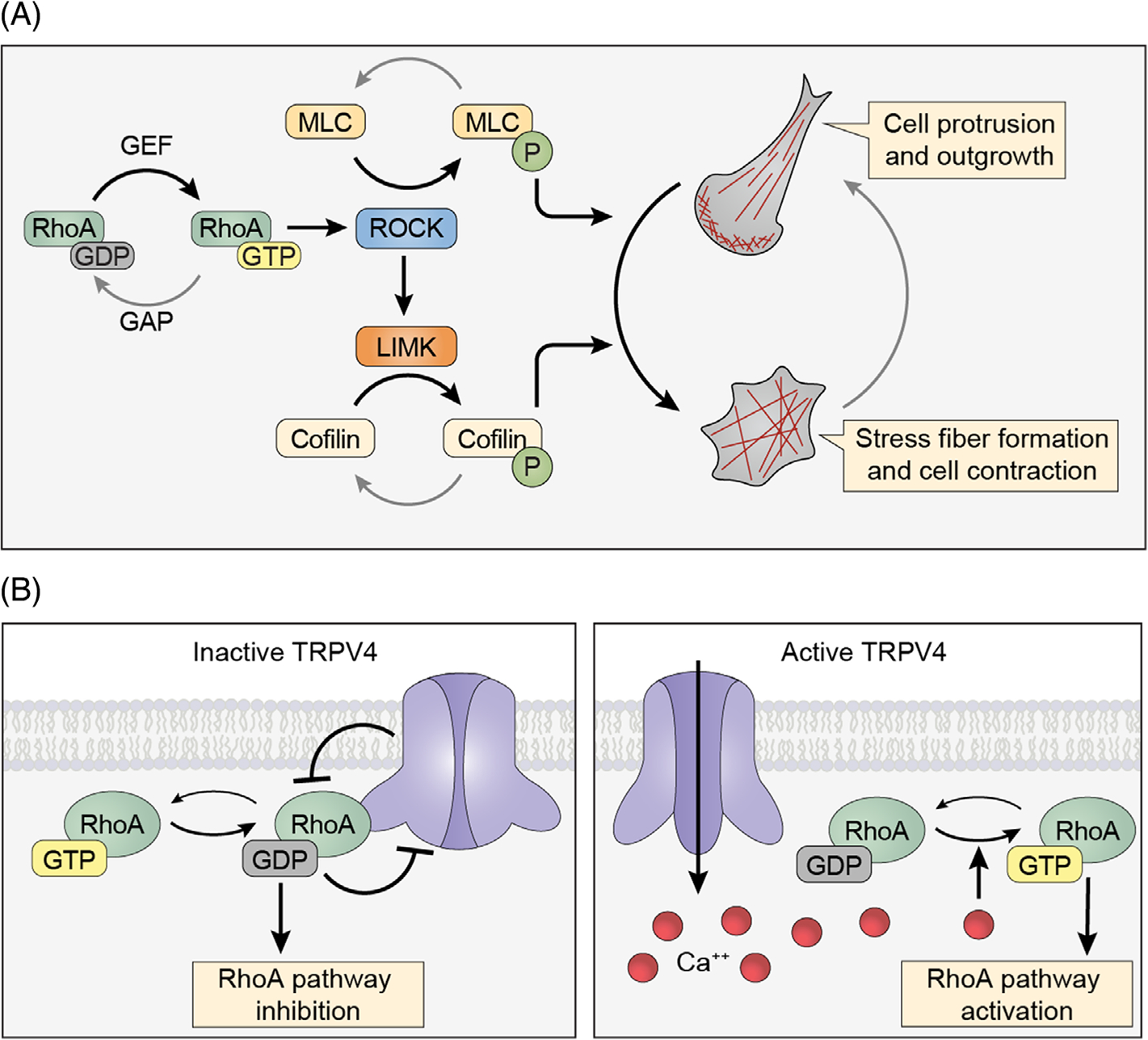
Transient receptor potential vanilloid 4 (TRPV4) and RhoA-dependent pathways regulate the actin cytoskeleton. A. Schematic depicting the activity cycle of RhoA and downstream effectors. RhoA is activated by guanine nucleotide exchange factors (GEFs), which promote GTP loading, and inactivated by GTPase activating proteins (GAPs), which promote hydrolysis of GTP to GDP. Active RhoA leads to activation of ROCK, which promotes phosphorylation of myosin light chain (MLC) and activation of nonmuscle myosin. ROCK also activates LIMK, which increases activation of ADF/Cofilin. Increased activation of RhoA-dependent signaling leads to actin stress fiber formation and cellular contraction. (B) Schematic depicting TRPV4–RhoA functional interactions. Inactive TRPV4 channels bind GDP-bound, inactive RhoA and reduce the active pool of RhoA. Activation of TRPV4 ion channel activity leads to release of RhoA, calcium-mediated RhoA activation, and resultant cytoskeletal changes
Our work demonstrated direct TRPV4–RhoA binding interactions that lead to bidirectional regulation that dynamically controls actin cytoskeletal changes. These results are consistent with a prior unbiased proteomics study that identified RhoA as a potential TRPV4 interactor.[42] Although the TRPV4 ARD has long been presumed to function as a platform for protein–protein interactions, previous studies have only demonstrated ARD interactions with calmodulin and phosphatidylinositol-4,5-bisphosphate.[43,44] We found that the switch and interswitch regions of inactive, GDP-bound RhoA directly bind to the TRPV4 ARD. In addition, we demonstrated that RhoA binding to TRPV4 suppressed ion channel activity, and conversely, TRPV4 binding to RhoA inhibited RhoA activation, likely by sequestering inactive RhoA in a manner similar to RhoGDI[45] (Figure 2(B)). Expression of TRPV4 in the presence of an antagonist led to dramatic stimulation of neurite-like processes from cultured cells, and formation of these cellular processes correlated with reduced activation of RhoA. Together, these findings demonstrate the ability of TRPV4 to promote actin cytoskeletal outgrowth and link this function with TRPV4-mediated inhibition of RhoA signaling.
Apart from modulating RhoA through sequestration and inhibition, we also found that TRPV4 could activate RhoA through ion channel activity and calcium influx. Stimulation of TRPV4 and increased intracellular calcium triggered rapid, time-locked activation of RhoA, followed by actin stress fiber formation and cytoskeletal contraction. Interestingly, TRPV4–RhoA complexes became dissociated in response to hypotonic stress, indicating that certain environmental stimuli are able to trigger release of RhoA from the inhibitory effect of TRPV4. Our results are in alignment with prior work showing stimulation of RhoA activity by calcium[46,47] and TRPV4-mediated activation of RhoA-dependent pathways.[36] Indeed, other recent studies have shown that TRPV4-mediated calcium influx increases RhoA activation and downstream ROCK activity as well as F-actin levels in endometrial cancer cells, endothelial cells, and trabecular meshwork cells of the eye.[32,36,48] Consistent with a central role for ROCK, our unpublished results demonstrate that inhibition of ROCK with the specific inhibitor Y-27632 blocks TRPV4-calcium-mediated cytoskeletal changes in HEK293T cells despite calcium-dependent RhoA activation (Figure 3). Together, these results point to a critical role for TRPV4 in regulating RhoA activation, downstream ROCK activation, and resulting cytoskeletal changes in response to extracellular stimuli. Interestingly, there is also precedent for regulation of RhoA and related Rho GTPases by other TRP channels, suggesting that TRPV4–RhoA regulation may be one aspect of a more general role for TRP channel-mediated regulation of the cytoskeleton.[49] Importantly, TRPV4 channel-mediated regulation of RhoA may have implications for pathological pathways activated in a range of human diseases.
FIGURE 3.
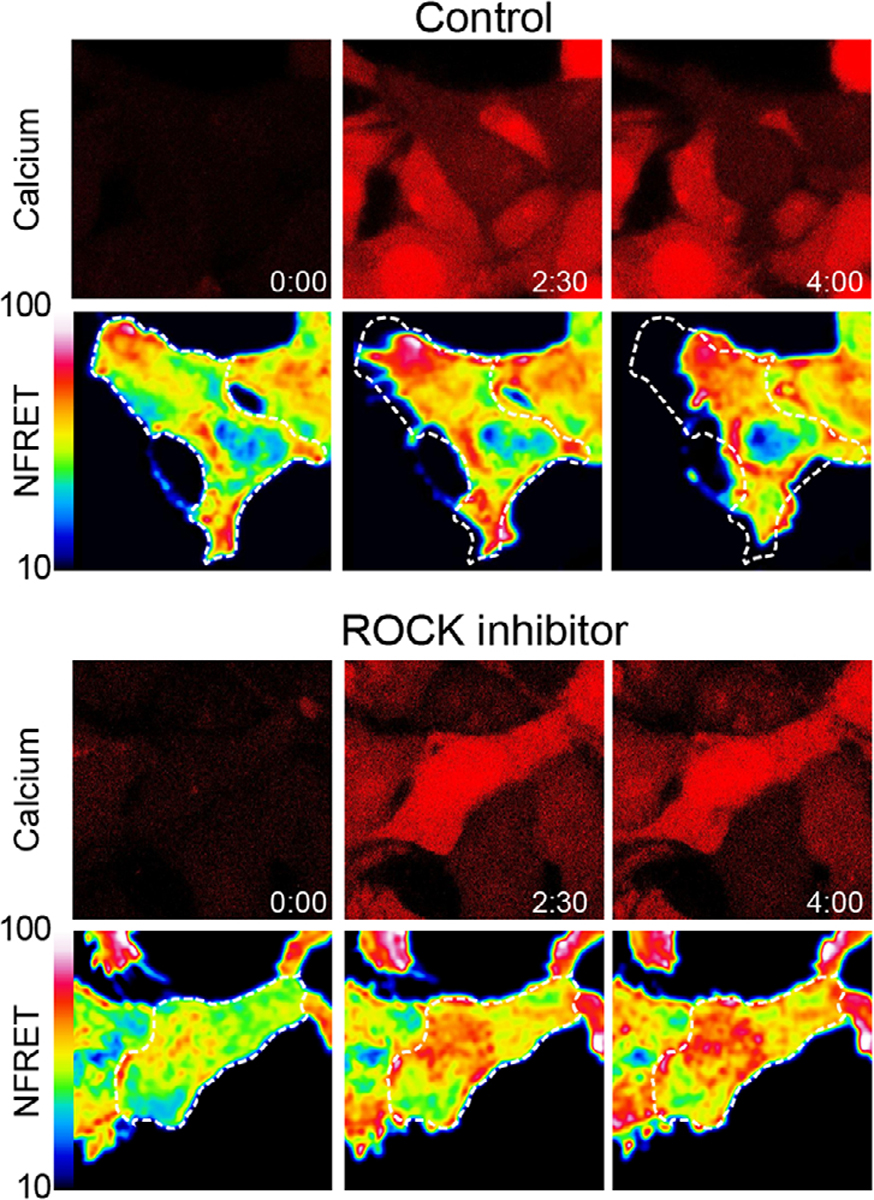
Stimulation of transient receptor potential vanilloid 4 (TRPV4) leads to RhoA activation and ROCK-dependent cytoskeletal changes. Time-lapse confocal microscopy images of stable, inducible TRPV4-expressing HEK293T cells (T-Rex-TRPV4) expressing a RhoA FRET biosensor (RhoA2G-mVenus-mTFP) and loaded with Cal590 calcium indicator demonstrate dynamics of calcium influx, RhoA activation, and cytoskeletal changes after stimulation of TRPV4. In the top panel, addition of the TRPV4 agonist GSK1016790A (100 nM) leads to calcium influx and increased RhoA activation (indicated by increased normalized FRET (NFRET)) followed by cellular contraction over 4 min. In the bottom panel, addition of the ROCK inhibitor Y-26732 (10 μM) blocks cytoskeletal changes downstream of TRPV4-mediated RhoA activation. Detailed methods were described previously[39]
PATHOLOGICAL RhoA ACTIVATION IN NEUROLOGICAL DISEASES
RhoA has been implicated in the pathogenesis of multiple neurological diseases, suggesting that maintenance of the unique properties of the nervous system requires precise RhoA regulation. RhoA plays particularly important roles in neurons and in vascular endothelial cells of the nervous system.[50–52] In general, RhoA activation within axonal growth cones of developing neurites drives actin cytoskeletal changes that restrain neurite outgrowth and/or promote neurite retraction.[53] In the context of neuronal injury, RhoA and ROCK activation inhibit axonal regeneration and subsequent reinnervation of target tissues. In mouse models of spinal cord injury (SCI), for example, levels of RhoA increased at the site of injury, and RhoA inhibition promoted motor recovery.[54–56] In animal models of traumatic brain injury (TBI), activation of RhoA and ROCK signaling promoted dendritic spine retraction, whereas inhibition of RhoA signaling pathways led to improved motor and cognitive outcomes.[57]
RhoA also contributes to neurological disease by modulating the barrier function of neural vascular endothelial cells. The blood–brain barrier is formed by endothelial cells that line the blood vessels of the brain and restrict the movement of ions, molecules, and cells from the blood into the surrounding neural tissue. Critical components of this barrier are tight junctions and adherens junctions between endothelial cells, which tether neighboring cells together and provide a physical barrier that limits the diffusion of blood constituents across the endothelial layer.[58] RhoA and ROCK regulate and maintain these junctions via modulation of the actin cytoskeleton that anchors these protein complexes.[59] Disruption of blood–brain barrier permeability has been identified as a pathological hallmark of a wide range of neurological disease states including those associated with acute neurological injury, such as stroke, TBI, and SCI; demyelinating diseases such as multiple sclerosis; neurodegenerative diseases such as Alzheimer’s disease; and chronic neurological sequelae of COVID-19 infection.[58,60,61] In neurological conditions associated with pathological blood–brain barrier disruption, such as ischemic stroke, RhoA signaling pathways are upregulated within brain vascular endothelial cells at sites of neurovascular insult.[62,63] In a mouse model of TBI, inhibition of RhoA via TIMP1-induced FAK phosphorylation and attenuated blood–brain barrier permeability and loss of junctional proteins.[64] A similar rescue of blood–brain barrier permeability and junctional protein expression by RhoA/ROCK inhibition was demonstrated in a mouse model and an in vitro brain endothelial cell model of inflammatory blood–brain barrier disruption using lipopolysaccharide.[65] Furthermore, in a rat model of SCI, inhibition of ROCK ameliorated blood–spinal cord barrier disruption and improved motor outcomes.[66] These results were attributed to inhibition of ROCK-mediated activation of the actin cytoskeletal modulating activities of ADF/cofilin and myosin phosphatase. Recent work has also suggested that RhoA activation downstream of the SARS CoV-2 spike protein contributes to blood–brain barrier disruption.[67]
Given the central role of RhoA signaling in neuropathological states, pharmacological approaches to limit RhoA activity have been examined in disease models and in humans, albeit with mixed success. In humans, the ROCK inhibitor netarsudil is approved for the treatment of refractory glaucoma.[68] In animal models, studies of RhoA pathway inhibition have most often utilized the bacterial endotoxin C3 transferase (a Rho GTPase inhibitor that targets RhoA as well as RhoB and RhoC) and ROCK inhibitors Y-27632 and fasudil (HA-1077). In addition to animal studies, fasudil has also been used in several clinical trials for a range of neurological diseases.[69–72] Targeting RhoA or ROCK signaling can improve some aspects of pathogenicity in disease models, but it can also exacerbate others.[73] These results are not surprising given the myriad essential roles of RhoA in multiple cell types and tissues. For example, a recent study of a mouse model of SCI found that while inhibition of RhoA activity in neurons promoted regeneration, specific deletion of RhoA within astrocytes was detrimental.[52] As opposed to therapies directly targeting RhoA, strategies to specifically inhibit pathological RhoA activation in relevant tissues and/or cell types could theoretically avoid the pitfalls of more generalized inhibition. We propose that TRPV4 may be such a therapeutic target.
TRPV4 AS A REGULATOR OF PATHOLOGICAL RhoA ACTIVATION IN THE NERVOUS SYSTEM
Strikingly, many of the same neurological diseases associated with excessive RhoA and ROCK signaling are also associated with increased TRPV4 activation. Particular examples of coincident RhoA and TRPV4 activation have come from animal models of stroke, TBI, and SCI. These studies have highlighted the importance of endothelial cell TRPV4 function in blood–brain and blood–spinal cord barrier integrity in these conditions.[74–77] These findings are consistent with single-cell and bulk RNA sequencing studies of nervous tissue from humans and mice showing that TRPV4 is primarily expressed in endothelial cells of the neurovasculature.[78–82] In contrast, there is surprisingly little evidence for widespread expression of TRPV4 in human and mouse neurons.[78,83] In the mouse spinal cord, unbiased expression profiling has demonstrated TRPV4 expression in a small subpopulation of sensory neurons, but very little expression in alpha motor neurons.[79,84] However, given that small numbers of clustered ion channels can elicit robust signaling events,[85–87] the absence of high-level expression does not necessarily rule out an important functional role of TRPV4 in neurons. Studies to evaluate the role of TRPV4 in neurons, in some cases using exogenous expression, have shown that TRPV4 ion channel function can influence neurite outgrowth, similar to RhoA.[28] TRPV4 colocalized with actin-rich structures within neuronal growth cones, and ion channel activation led to rapid changes in morphology of actin-rich lamellipodia and filopodia.[28] Activation of TRPV4 in cultured neurons caused disruption of axonal integrity with varicosity formation and neurite retraction.[29,88] In a Drosophila model of TRPV4 neuropathy, expression of gain-of-function TRPV4 mutations led to axonal and dendritic degeneration, consistent with TRPV4-mediated inhibition of neurite outgrowth.[29] Importantly, degeneration of dendrites and axons in this model could be rescued by inhibition of Rho1, the fly homolog of RhoA, further linking TRPV4-mediated cytoskeletal changes with RhoA signaling.[39]
While these studies suggest a potential role for TRPV4 and RhoA in neurons, the extent to which TRPV4 is expressed within neurons of the brain, spinal cord, and peripheral nerves remains debatable. In contrast, a functional role for TRPV4 in neural vascular endothelial cells is more robustly established. Like RhoA, TRPV4 has been implicated in maintaining endothelial barrier function and affecting blood–brain and blood–spinal cord barrier permeability. In rodent models of SCI, endothelial cell TRPV4 was found to be upregulated in response to injury in conjunction with disruption of the blood–spinal cord barrier.[74] Furthermore, in mice subjected to SCI, knockout of TRPV4 prevented blood–spinal cord barrier breakdown and degradation of tight junction proteins with associated improvement in behavioral outcomes. Similarly, a mouse model of hemorrhagic stroke demonstrated upregulation of TRPV4 expression and concurrent RhoA pathway activation with the corresponding disruption of blood–brain barrier integrity.[76] In this model, inhibition of TRPV4 reduced RhoA and ROCK-dependent stress fiber formation resulting in preserved blood–brain barrier integrity. Moreover, in a mouse model of post-ischemic brain injury, TRPV4 knockout decreased loss of tight junction proteins and blood–brain barrier disruption in ischemic tissue, further highlighting the importance of TRPV4 activity in the response to neurovascular injury.[75] Importantly, whether TRPV4 and RhoA play a similar role in regulating blood–nerve barriers of the peripheral nervous system has yet to be established. Furthermore, in addition to a direct role in regulating cytoskeletal changes in endothelial cells, TRPV4 channel activation can also trigger inflammatory signaling cascades that may further exacerbate tissue injury and contribute to pain sensation, as is reviewed elsewhere.[89–91]
Together, animal models show a benefit of TRPV4 channel inhibition in the context of neurological injuries in which RhoA is known to be activated and detrimental. Furthermore, the association of TRPV4 and RhoA in the same pathological conditions and recent data demonstrating linked TRPV4–RhoA signaling cascades suggests that TRPV4 may be an important regulator of pathological RhoA activation. Thus, by acting as a sensor of mechanical disruption and/or injury, TRPV4 may be a critical and pharmacologically tractable node in pathological RhoA activation across a wide range of neurological insults (Figure 4).
FIGURE 4.
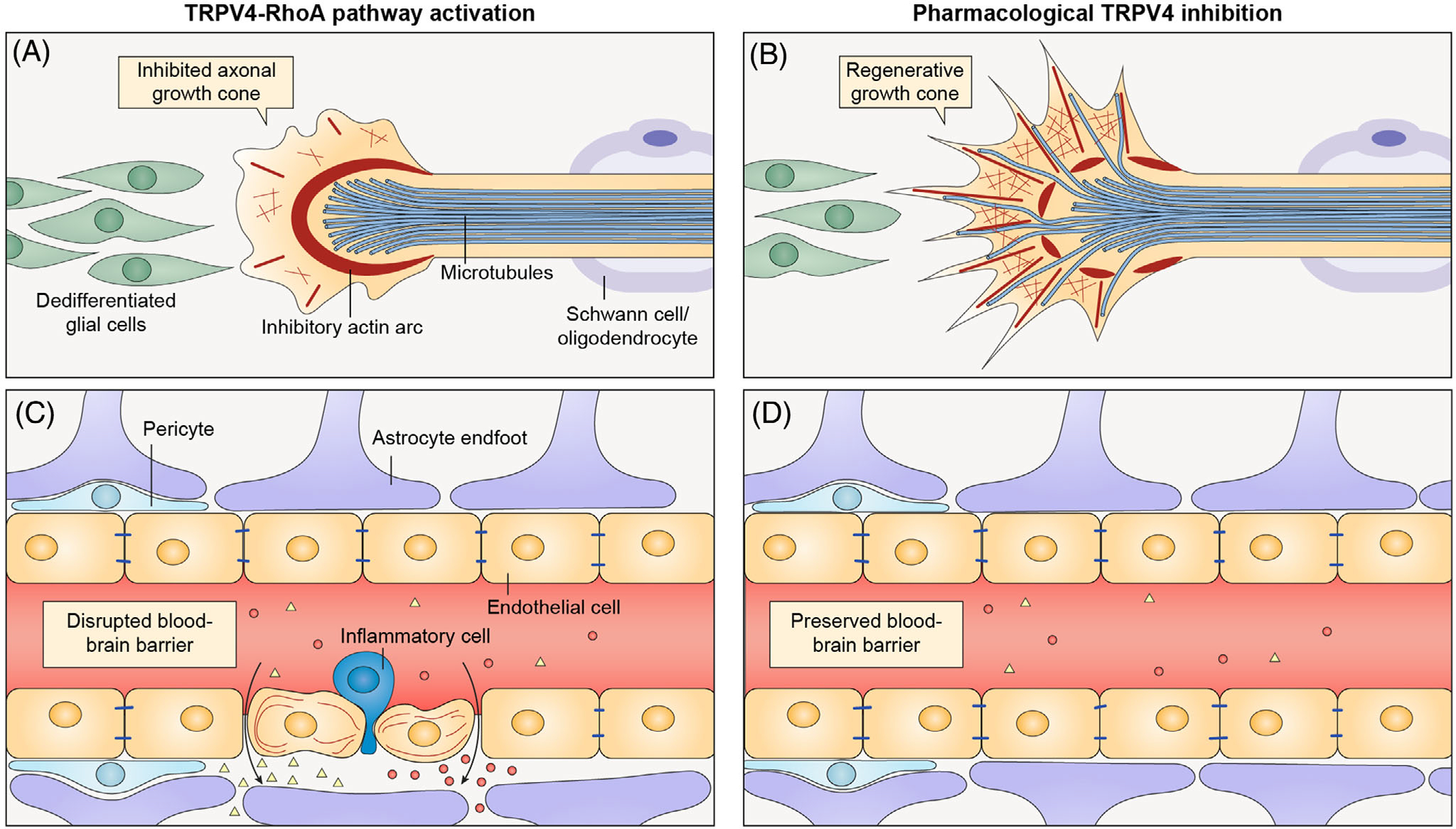
Schematic of the potential role of transient receptor potential vanilloid 4 (TRPV4) inhibition in limiting neurological injury. (A) In injured neurons, activation of TRPV4 and RhoA leads to ROCK-dependent bundling of actin at the base of the axonal growth cone, thereby restricting microtubule entry into the growth cone. (B) Inhibition of TRPV4 could potentially reduce RhoA activation and create a regeneration-permissive environment within the axonal growth cone. Specific TRPV4–RhoA functions in glia require further study. (C) In injured vascular endothelial cells of the brain and spinal cord, TRPV4 activation leads to RhoA-dependent disruption of the blood–brain barrier with subsequent extravasation of blood constituents and inflammatory cells, which worsen local tissue injury. (D) Inhibition of TRPV4 could potentially reduce RhoA-dependent blood–brain barrier breakdown, thereby limiting the extent of surrounding tissue injury
TRPV4 INHIBITION IS A PROMISING THERAPEUTIC ROUTE TO TREAT NEUROLOGICAL DISEASE
Since the discovery of TRP channels as mechanically gated ion channels important for sensory function,[92] there has been substantial interest in potential pharmacologic applications of small molecules and tissue-specific delivery strategies for targeting these channels.[93,94] Importantly, while gain of function mutations in TRPV4 cause human disease, TRPV4 knockout does not display any overt deleterious phenotypes in mice.[95–97] Several naturally occurring pharmacological compounds targeting TRPV4 have been discovered, and several other synthetic compounds have been developed. Small-molecule antagonists include nonselective inhibitors such as ruthenium red, and more selective inhibitors such as HC-067047, GSK2193874, GSK205, and the orally bioavailable GSK2798745.[98–100] These inhibitors have been successfully used in vivo without obvious side effects, and GSK2798745 has been tested in Phase II clinical trials for pulmonary edema and chronic cough (NCT03372603).[101] While these trials did not meet efficacy endpoints for these disease indications, the antagonist drug was well tolerated without any significant adverse effects (NCT02119260).[101] This drug is currently being tested in a Phase I clinical trial for diabetic macular edema (NCT04292912), motivated by work implicating TRPV4 in the regulation of blood–retinal barrier integrity.[102]
Pharmacological TRPV4 antagonists have been successful at inhibiting several neuropathological processes in animal models of neurological disease.[76,103–106] However, to more firmly establish the connections between TRPV4 activation and RhoA signaling as well as the therapeutic potential of pharmacological targeting of TRPV4, further studies are needed. In animal models of neurological injury, such as stroke, TBI, and SCI, activation or inhibition of TRPV4 needs to be combined with more detailed assessment of RhoA pathway activation. In vitro, Rho activation assays, RhoA biosensors, and immunohistochemistry for active RhoA mediators can be readily utilized to correlate RhoA activation with TRPV4 channel activity. In vivo approaches to monitor RhoA pathway activity are more challenging, but could utilize immunohistochemistry for activated RhoA or phosphorylation of RhoA targets such as MLC, myosin phosphatase (MYPT1), and ADF/cofilin, as well as a RhoA–FRET-based biosensor mouse.[107–109] These approaches could be coupled with in vivo experiments utilizing genetically encoded calcium indicators, such as GCaMP, to monitor TRPV4 ion channel activity in real time.[109]
Understanding TRPV4–RhoA contributions to disease will also require more detailed assessment of cell-type-specific activation of downstream pathways. While global RhoA knockout is lethal, cell-specific knockout in endothelial cells has been tested using Cre-recombinase approaches.[110] Similarly, the generation of tissue-specific TRPV4 knockout mice would allow for direct interrogation of TRPV4 function in potentially relevant cell types, including endothelial cells, glia, and neurons. Such approaches will be critical to elucidating cell-specific contributions of TRPV4 in various nerve injury paradigms, as well as providing clarity regarding whether TRPV4 and RhoA regulate blood–nerve barriers in the peripheral nervous system in a similar fashion to what has been demonstrated in the central nervous system. This issue is particularly relevant given that studies of TRPV4 in different peripheral nerve injury models have produced somewhat conflicting results. For example, pharmacologic TRPV4 inhibition was found to be beneficial in mouse models of chemotherapy-induced neuropathy[111,112] and temporomandibular pain,[113] whereas global knockout of TRPV4 worsened outcomes in a mouse model of peripheral nerve transection.[114] The latter result may relate to disruption of Schwann cell-specific TRPV4 functions as there is evidence of a beneficial role for TRPV4 in regulating cytoskeletal changes that support glial migration to sites of inflammation and injury.[115] Together, these results and unanswered questions underscore the need to better define cell type-specific functions of TRPV4 and RhoA as well as biodistribution and nervous system penetration of TRPV4-acting drugs.
An additional consideration is the tissue-specific expression pattern of TRPV4, both in health and in response to neurological injury. While the lack of specificity of available TRPV4 antibodies has limited immunohistochemical approaches to define cellular expression, assessments of TRPV4 transcript and protein levels have demonstrated upregulation in response to brain, nerve, and SCI.[74,77,113] Further work using complementary methods, such as TRPV4 reporter mice,[116] will be vital to deriving a more complete picture of TRPV4 expression within relevant cell types under normal and disease conditions. Such knowledge could guide the implementation of tissue-specific TRPV4 inhibition strategies to target the most relevant cell types in a given pathological state.[94]
In addition to defining cell type-specific contributions of TRPV4 and RhoA function, it will be critical to define temporal aspects of pathological TRPV4–RhoA pathway activation in various neurological disease states. As neurological injury triggers a complex cascade of pathological events evolving over disparate timescales, from hours to days to weeks,[117] there may be discrete windows during which the negative contribution of TRPV4–RhoA activation is maximal and in which therapeutic intervention would be most beneficial. Elucidation of these temporal aspects of pathological TRPV4 function will likely require careful longitudinal assessment of the impacts of pharmacological TRPV4 inhibition in animal models of disease.
CONCLUSIONS
In this review, we highlight accumulating evidence that activation of TRPV4 and RhoA is linked to pathological events that drive actin cytoskeletal change in response to neurological injury. This collective work supports a model in which TRPV4 acts as a critical gatekeeper of RhoA-mediated cytoskeletal changes in multiple pathological states. As a cell surface-expressed ion channel that can be modulated by available drugs, TRPV4 is an attractive and tractable pharmacological target that may allow precise inhibition of pathological RhoA activation associated with nerve injury and blood–brain barrier disruption. Driven by this important insight, experimental approaches to precisely delineate TRPV4 expression patterns, kinetics of activation, and downstream pathological RhoA activation may illuminate a range of therapeutic opportunities for currently untreatable neurological insults.
ACKNOWLEDGMENTS
This work was supported by K08 NS102509 (B.A.M.), American Academy of Neurology Neuroscience Research Training Fellowship (B.A.M.), NIH (NINDS) R01 NS115475 (C.J.S), NIH (NINDS) R35 NS122306 (C.J.S.), and Muscular Dystrophy Association 629305 (C.J.S). We thank Natalia Nedelsky for manuscript editing and graphical assistance.
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: NS102509, NS115475, R35 NS122306; American Academy of Neurology, Grant/Award Number: 0078966; Muscular Dystrophy Association, Grant/Award Number: 629305
Abbreviations:
- ARD
ankyrin repeat domain
- EET
epoxyeicosatrienoic acid
- GAP
GTPase activating protein
- GDI
guanine-nucleotide dissociation inhibitor
- GEF
guanine-nucleotide exchange factor
- MLC
myosin-light chain
- MYPT
myosin phosphatase
- PBD
phosphoinositide binding domain
- PRR
proline-rich region
- ROCK
Rho-associated coiled-coil containing kinase
- SCI
spinal cord injury
- TBI
traumatic brain injury
- TRPV4
transient receptor potential vanilloid
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Janmey PA (1998). The cytoskeleton and cell signaling: Component localization and mechanical coupling. Physiological Reviews, 78, 763–781. [DOI] [PubMed] [Google Scholar]
- 2.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, & Plant TD (2000). OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nature Cell Biology, 2, 695–702. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Andrejšali, Hudspeth AJ, Friedman JM, & Heller S (2000). Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell, 103, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grace MS, Bonvini SJ, Belvisi MG & McIntyre P (2017). Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacology & Therapeutics, 177, 9–22. [DOI] [PubMed] [Google Scholar]
- 5.Liedtke W (2005). TRPV4 as osmosensor: A transgenic approach. Pflugers Archiv: European Journal of Physiology, 451, 176–180. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Wang Zi-L, Yeo M, Zhang Q-J, López-Romero AE, Ding H-P, Zhang X, Zeng Q, Morales-Lázaro SL, Moore C, Jin Y-Ai, Yang H-He, Morstein J, Bortsov A, Krawczyk M, Lammert F, Abdelmalek M, Diehl AM, Milkiewicz P, … Liedtke W (2021). Epithelia-sensory neuron cross talk underlies cholestatic itch induced by Lysophosphatidylcholine. Gastroenterology, 161, 301–317.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White JPM, Cibelli M, Urban L, Nilius B, Mcgeown JG, & Nagy I (2016). TRPV4: Molecular conductor of a diverse orchestra. Physiological Reviews, 96, 911–973. [DOI] [PubMed] [Google Scholar]
- 8.Auer-Grumbach M, Olschewski A, Papić L, Kremer H, Mcentagart ME, Uhrig S, Fischer C, Fröhlich E, Bálint Z, Tang Bi, Strohmaier H, Lochmüller H, Schlotter-Weigel B, Senderek J, Krebs A, Dick KJ, Petty R, Longman C, Anderson NE, … Guelly C (2010). Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nature Genetics, 42, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng H-X, Klein CJ, Yan J, Shi Y, Wu Y, Fecto F, Yau H-J, Yang Yi, Zhai H, Siddique N, Hedley-Whyte ET, Delong R, Martina M, Dyck PJ, & Siddique T (2010). Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nature Genetics, 42, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landouré G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, Inada H, Shi Y, Taye AA, Kong L, Munns CH, Choo SS, Phelps CB, Paudel R, Houlden H, Ludlow CL, Caterina MJ, Gaudet R, Kleta R, Fischbeck KH, & Sumner CJ (2010). Mutations in TRPV4 cause Charcot–Marie–Tooth disease type 2C. Nature Genetics, 42(2), 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krakow D, Vriens J, Camacho N, Luong P, Deixler H, Funari TL, Bacino CA, Irons MB, Holm IA, Sadler L, Okenfuss EB, Janssens A, Voets T, Rimoin DL, Lachman RS, Nilius B, & Cohn DH (2009). Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia. Kozlowski type and metatropic dysplasia. American Journal of Human Genetics, 84(3), 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, Lachman RS, Wilcox WR, Reyno S, Quadrelli R, Vaglio A, Owsianik G, Janssens A, Voets T, Ikegawa S, Nagai T, Rimoin DL, Nilius B, & Cohn DH (2008). Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nature Genetics, 40, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mah W, Sonkusare SK, Wang T, Azeddine B, Pupavac M, Carrot-Zhang J, Hong K, Majewski J, Harvey EJ, Russell L, Chalk C, Rosenblatt DS, Nelson MT, & Séguin C (2016). Gain-of-function mutation in TRPV4 identified in patients with osteonecrosis of the femoral head. Journal of Medical Genetics, 53, 705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCray BA, Schindler A, Hoover-Fong JE, & Sumner CJ (Seattle 1993–2022). Autosomal dominant TRPV4 disorders. In Adam MP, Ardinger HH, Amemiya A, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM (Eds.), GeneReviews®. University of Washington. [PubMed] [Google Scholar]
- 15.Garcia-Elias A, Mrkonjić S, Jung C, Pardo-Pastor C, Vicente R & Valverde MA (2014). The TRPV4 channel. In Mammalian Transient Receptor Potential (TRP) Cation Channels. Handbook of Experimental Pharmacology, Springer, Berlin, Heidelberg, Berlin, Heidelberg, (Vol. 222, pp. 293–319). [DOI] [PubMed] [Google Scholar]
- 16.Nilius B, & Voets T (2013). The puzzle of TRPV4 channelopathies. Embo Reports, 14, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuajungco MP, Grimm C, Oshima K, D’hoedt D, Nilius B, Mensenkamp AR, Bindels RJM, Plomann M, & Heller S (2006). PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. Journal of Biological Chemistry, 281, 18753–18762. [DOI] [PubMed] [Google Scholar]
- 18.Goretzki B, Glogowski NA, Diehl E, Duchardt-Ferner E, Hacker C, Gaudet R, & Hellmich UA (2018). Structural basis of TRPV4 N terminus interaction with Syndapin/PACSIN1–3 and PIP2. Structure (London, England), 26, 1583–1593.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Elias A, Mrkonjić S, Pardo-Pastor C, Inada H, Hellmich UA, Rubio-Moscardó F, Plata C, Gaudet R, Vicente R, & Valverde MA (2013). Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. PNAS, 110, 9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inada H, Procko E, Sotomayor M, & Gaudet R (2012). Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry, 51, 6195–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahman M, Mukherjee S, Sheng W, Nilius B, & Janssen LJ (2016). Electrophysiological characterization of voltage-dependent calcium currents and TRPV4 currents in human pulmonary fibroblasts. American Journal of Physiology-Lung Cellular and Molecular Physiology, 310, L603–L614. [DOI] [PubMed] [Google Scholar]
- 22.Turovsky EA, Braga A, Yu Y, Esteras N, Korsak A, Theparambil SM, Hadjihambi A, Hosford PS, Teschemacher AG, Marina N, Lythgoe MF, Haydon PG, & Gourine AV (2020). Mechanosensory signaling in astrocytes. The Journal of Neuroscience, 40, 9364–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daneva Z, Ottolini M, Chen YL, Klimentova E, Kuppusamy M, Shah SA, Minshall RD, Seye CI, Laubach VE, Isakson BE, & Sonkusare SK (2021). Endothelial pannexin 1–TRPV4 channel signaling lowers pulmonary arterial pressure in mice. eLife, 10, e67777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE & Sonkusare SK Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (TRPV4) channel cooperativity and endothelial function in small pulmonary arteries. Journal of the American Heart Association, 6, e007157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sukumaran SV, Singh TU, Parida S, Reddy Narasimha, Ch E, Thangamalai R, Kandasamy K, Singh V, & Mishra SK (2013). TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacological Research, 78, 18–27. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Kan H, He C, Zhang X, Yang Z, Jin J, Zhang P, & Ma X (2018). TRPV4 activates cytosolic phospholipase A2 via Ca2+-dependent PKC/ERK1/2 signalling in controlling hypertensive contraction. Clinical and Experimental Pharmacology and Physiology, 45, 908–915. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Fang Q, Wang Z, Zhang JY, Macleod AS, Hall RP, & Liedtke WB (2016). Transient receptor potential vanilloid 4 ion channel functions as a pruriceptor in epidermal keratinocytes to evoke histaminergic itch. Journal of Biological Chemistry, 291, 10252–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goswami C, Kuhn J, Heppenstall PA, & Hucho T (2010). Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells. Plos One, 5, e11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woolums BM, Mccray BA, Sung H, Tabuchi M, Sullivan JM, Ruppell KT, Yang Y, Mamah C, Aisenberg WH, Saavedra-Rivera PC, Larin BS, Lau AR, Robinson DN, Xiang Y, Wu MN, Sumner CJ, & Lloyd TE (2020). TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca2+. Nature Communication, 11, 2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longden TA, Mughal A, Hennig GW, Harraz OF, Shui Bo, Lee FK, Lee JC, Reining S, Kotlikoff MI, König GM, Kostenis E, Hill-Eubanks D, & Nelson MT (2021). Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Science Advances, 7, eabh0101.(30), [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heathcote HR, Lee MD, Zhang X, Saunter CD, Wilson C & McCarron JG (2019). Endothelial TRPV4 channels modulate vascular tone by Ca2+-induced Ca2+ release at inositol 1,4,5-trisphosphate receptors. British Journal of Pharmacology, 176(17), 3297–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakk M, & Križaj D (2021). TRPV4-Rho signaling drives cytoskeletal and focal adhesion remodeling in trabecular meshwork cells. American Journal of Physiology-Cell Physiology, 320, C1013–C1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel PD, Chen Y-L, Kasetti RB, Maddineni P, Mayhew W, Millar JC, Ellis DZ, Sonkusare SK & Zode GS (2021). Impaired TRPV4-eNOS signaling in trabecular meshwork elevates intraocular pressure in glaucoma. PNAS 118(16), e2022461118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, & Ingber DE (2009). TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circulation Research, 104, 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pla F, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, Paria B, Munaron L, & Ambudkar IS (2012). TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene, 31, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thoppil RJ, Cappelli HC, Adapala RK, Kanugula AK, Paruchuri S, & Thodeti CK (2016). TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget, 7, 25849–25861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindy AS, Parekh PK, Zhu R, Kanju P, Chintapalli SV, Tsvilovskyy V, Patterson RL, Anishkin A, Van Rossum DB, & Liedtke WB (2014). TRPV channel-mediated calcium transients in nociceptor neurons are dispensable for avoidance behaviour. Nature Communication, 5, 4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quan A, & Robinson PJ (2013). Syndapin––a membrane remodelling and endocytic F-BAR protein. The FEBS Journal, 280(21), 5198–5212. [DOI] [PubMed] [Google Scholar]
- 39.Mccray BA, Diehl E, Sullivan JM, Aisenberg WH, Zaccor NW, Lau AR, Rich DJ, Goretzki B, Hellmich UA, Lloyd TE, & Sumner CJ (2021). Neuropathy-causing TRPV4 mutations disrupt TRPV4–RhoA interactions and impair neurite extension. Nature Communication, 12, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riento K, & Ridley AJ (2003). ROCKs: Multifunctional kinases in cell behaviour. Nature Reviews Molecular Cell Biology, 4, 446–456. [DOI] [PubMed] [Google Scholar]
- 41.Spiering D & Hodgson L (2011). Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adhesion & Migration, 5, 170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doñate-Macián P, Jungfleisch J, Pérez-Vilaró G, Rubio-Moscardo F, Perálvarez-Marín A, Diez J, & Valverde MA (2018). The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nature Communication, 9, 2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phelps CB, Wang RR, Choo SS, & Gaudet R (2010). Differential Regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. Journal of Biological Chemistry, 285, 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi N, Hamada-Nakahara S, Itoh Y, Takemura K, Shimada A, Ueda Y, Kitamata M, Matsuoka R, Hanawa-Suetsugu K, Senju Y, Mori MX, Kiyonaka S, Kohda D, Kitao A, Mori Y, & Suetsugu S (2014). TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P2. Nature Communication, 5, 4994. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Mata R, Boulter E, & Burridge K (2011). The “invisible hand”: Regulation of RHO GTPases by RHOGDIs. Nature Reviews Molecular Cell Biology, 12, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kugelmann D, Rotkopf LT, Radeva MY, Garcia-Ponce A, Walter E, & Waschke J (2018). Histamine causes endothelial barrier disruption via Ca2+-mediated RhoA activation and tension at adherens junctions. Science Reports, 8, 13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakurada S, Takuwa N, Sugimoto N, Wang, Yu, Seto, M., Sasaki, Y., & Takuwa, Y. (2003). Ca2+-dependent activation of rho and rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circulation Research, 93, 548–556. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Cheng Y, Wang Z, Zhou J, Jia Y, He X, Zhao L, Dong Y, Fan Y, Yang X, Shen B, Wu X, Wang J, Xiong C, Wei L, Li X, & Wang J (2020). Calcium and TRPV4 promote metastasis by regulating cytoskeleton through the RhoA/ROCK1 pathway in endometrial cancer. Cell Death & Disease, 11, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavanderos B, Silva I, Cruz P, Orellana-Serradell O, Saldías MP, & Cerda O (2020). TRP channels regulation of Rho GTPases in brain context and diseases. Frontiers in Cell and Developmental Biology, 8, 582975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fusco L, Lefort R, Smith K, Benmansour F, Gonzalez G, Barillari C, Rinn B, Fleuret F, Fua P, & Pertz O (2016). Computer vision profiling of neurite outgrowth dynamics reveals spatiotemporal modularity of Rho GTPase signaling. Journal of Cell Biology, 212, 91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dupraz S, Hilton BJ, Husch A, Santos TE, Coles CH, Stern S, Brakebusch C, & Bradke F (2019). RhoA controls axon extension independent of specification in the developing brain. Current Biology, 29, 3874–3886.e9. [DOI] [PubMed] [Google Scholar]
- 52.Stern S, Hilton BJ, Burnside ER, Dupraz S, Handley EE, Gonyer JM, Brakebusch C, & Bradke F (2021). RhoA drives actin compaction to restrict axon regeneration and astrocyte reactivity after CNS injury. Neuron, 109, 3436–3455.e9. [DOI] [PubMed] [Google Scholar]
- 53.Fujita Y, & Yamashita T (2014). Axon growth inhibition by RhoA/ROCK in the central nervous system. Frontiers in Neuroscience, 8, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubreuil CI, Winton MJ, & Mckerracher L (2003). Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. Journal of Cell Biology, 162, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madura T, Yamashita T, Kubo T, Fujitani M, Hosokawa Ko, & Tohyama M (2004). Activation of Rho in the injured axons following spinal cord injury. EMBO Reports, 5, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boato F, Hendrix S, Huelsenbeck SC, Hofmann F, Große G, Djalali S, Klimaschewski L, Auer M, Just I, Ahnert-Hilger G, & Höltje M (2010). C3 peptide enhances recovery from spinal cord injury by improved regenerative growth of descending fiber tracts. Journal of Cell Science, 123, 1652–1662. [DOI] [PubMed] [Google Scholar]
- 57.Mulherkar S, Firozi K, Huang W, Uddin MD, Grill RJ, Costa-Mattioli M, Robertson C, & Tolias KF (2017). RhoA-ROCK inhibition reverses synaptic remodeling and motor and cognitive deficits caused by traumatic brain injury. Science Reports, 7, 10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sweeney MD, Sagare AP, & Zlokovic BV (2018). Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews Neurology, 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radeva MY, & Waschke J (2018). Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiologica (Oxford, England), 222, e12860. [DOI] [PubMed] [Google Scholar]
- 60.Park JC, Baik SH, Han S-Ho, Cho HJ, Choi H, Kim HJ, Choi H, Lee W, Kim DK, & Mook-Jung I (2017). Annexin A1 restores Aβ1–42-induced blood–brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell, 16, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balcom EF, Nath A, & Power C (2021). Acute and chronic neurological disorders in COVID-19: Potential mechanisms of disease. Brain, 144, 3576–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu Na, Stetler RA, Zhang F, Liu X, Leak RK, Keep RF, Ji X, & Chen J (2016). Rapid endothelial cytoskeletal reorganization enables early blood–brain barrier disruption and long-term ischaemic reperfusion brain injury. Nature Communication, 7, 10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson CL, Srivastava K, Sprigg N, Bath PMW, & Bayraktutan U (2014). Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. Journal of Neurochemistry, 129, 816–826. [DOI] [PubMed] [Google Scholar]
- 64.Tang J, Kang Y, Huang L, Wu L, & Peng Y (2020). TIMP1 preserves the blood–brain barrier through interacting with CD63/integrin 1 complex and regulating downstream FAK/RhoA signaling. Acta Pharmaceutica Sinica B, 10, 987–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng S, Zou Li, Wang H, He R, Liu Ke, & Zhu H (2018). RhoA/ROCK-2 pathway inhibition and tight junction protein upregulation by catalpol suppresses lipopolysaccaride-induced disruption of blood–brain barrier permeability. Molecules (Basel, Switzerland), 23, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang S, & Cao Y (2021). The ROCK inhibitor Y-27632 ameliorates blood-spinal cord barrier disruption by reducing tight junction protein degradation via the MYPT1-MLC2 pathway after spinal cord injury in rats. Brain Research, 1773, 147684. [DOI] [PubMed] [Google Scholar]
- 67.Deore BJ, Tran KA, Andrews AM, Ramirez SH, & Galie PA (2021). SARS-CoV-2 spike protein disrupts blood–brain barrier integrity via RhoA activation. Journal of Neuroimmune Pharmacology, 16, 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Villegas NC, & Lee W-S (2021). Effectiveness of netarsudil as an additional therapy for glaucoma in patients already on maximally tolerated medical therapy. Clinical Ophthalmology, 15, 4367–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vesterinen HM, Currie GL, Carter S, Mee S, Watzlawick R, Egan KJ, Macleod MR, & Sena ES (2013). Systematic review and stratified meta-analysis of the efficacy of RhoA and Rho kinase inhibitors in animal models of ischaemic stroke. Systematic Reviews, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mulherkar S, & Tolias KF (2020). RhoA-ROCK signaling as a therapeutic target in traumatic brain injury. Cells, 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R, Mcdonald DA, Rorrer A, Gallione C, Pytel P, Liao JK, Marchuk DA, & Awad IA (2017). RhoA kinase inhibition with fasudil versus simvastatin in murine models of cerebral cavernous malformations. Stroke; A Journal of Cerebral Circulation, 48, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lingor P, Weber M, Camu W, Friede T, Hilgers R, Leha A, Neuwirth C, Günther R, Benatar M, Kuzma-Kozakiewicz M, Bidner H, Blankenstein C, Frontini R, Ludolph A, & Koch JC (2019). ROCK-ALS: Protocol for a randomized, placebo-controlled, double-blind phase IIa trial of safety, tolerability and efficacy of the Rho kinase (ROCK) inhibitor fasudil in amyotrophic lateral sclerosis. Frontiers in Neurology, 10, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guiler W, Koehler A, Boykin C, & Lu Q (2021). Pharmacological modulators of small GTPases of rho family in neurodegenerative diseases. Frontiers in Cellular Neuroscience, 15, 661612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar H, Lim C. Su, Choi H, Joshi HP, Kim K-T, Kim Y. Ho, Park C-K, Kim HM, & Han In-Bo (2020). Elevated TRPV4 levels contribute to endothelial damage and scarring in experimental spinal cord injury. Journal of Neuroscience, 40, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanaka K, Matsumoto S, Yamada T, Yamasaki R, Suzuki M, Kido MA, & Kira J-I (2020). Reduced post-ischemic brain injury in transient receptor potential vanilloid 4 knockout mice. Frontiers in Neuroscience, 14, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao H, Zhang K, Tang R, Meng H, Zou Y, Wu P, Hu R, Liu X, Feng H, & Chen Y (2018). TRPV4 blockade preserves the blood–brain barrier by inhibiting stress fiber formation in a rat model of intracerebral hemorrhage. Frontiers in Molecular Neuroscience, 11, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu K-T, Huang T-C, Tsai Ya-H, & Yang Yi-L (2017). Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-cotransporter-induced brain edema after traumatic brain injury. Journal of Neurochemistry, 140, 718–727. [DOI] [PubMed] [Google Scholar]
- 78.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, Van De Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, … Jones AR (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature, 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blum JA, Klemm S, Shadrach JL, Guttenplan KA, Nakayama L, Kathiria A, Hoang PT, Gautier O, Kaltschmidt JA, Greenleaf WJ, & Gitler AD (2021). Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons. Nature Neuroscience, 24, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, & Nathans J (2018). Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife, 7, e36187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, & Nathans J (2018). Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife, 7, e36187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, & Betsholtz C (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature, 554, 475–480. [DOI] [PubMed] [Google Scholar]
- 83.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, De Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, & Mccarroll SA. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell, 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Russ DE, Cross RBP, Li Li, Koch SC, Matson KJE, Yadav A, Alkaslasi MR, Lee DI, Le Pichon CE, Menon V, & Levine AJ (2021). A harmonized atlas of mouse spinal cord cell types and their spatial organization. Nature Communication, 12, 5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pfeiffer P, Egorov AV, Lorenz F, Schleimer J-H, Draguhn A, & Schreiber S (2020). Clusters of cooperative ion channels enable a membrane-potential-based mechanism for short-term memory. eLife, 9, e49974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sato D, Hernández-Hernández G, Matsumoto C, Tajada S, Moreno CM, Dixon RE, O’dwyer S, Navedo MF, Trimmer JS, Clancy CE, Binder MD, & Santana LF (2019). A stochastic model of ion channel cluster formation in the plasma membrane. Journal of General Physiology, 151, 1116–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baratchi S, Keov P, Darby WG, Lai A, Khoshmanesh K, Thurgood P, Vahidi P, Ejendal K, & Mcintyre P (2019). The TRPV4 agonist GSK1016790A regulates the membrane expression of TRPV4 channels. Frontiers in Pharmacology, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gu Y, Jukkola P, Wang Q, Esparza T, Zhao Yi, Brody D, & Gu C (2017). Polarity of varicosity initiation in central neuron mechanosensation. Journal of Cell Biology, 216, 2179–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Michalick L, & Kuebler WM (2020). TRPV4-A missing link between mechanosensation and immunity. Frontiers in Immunology, 11, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scheraga RG, Southern BD, Grove LM, & Olman MA (2020). The role of TRPV4 in regulating innate immune cell function in lung inflammation. Frontiers in Immunology, 11, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peng S, Poole DP, & Veldhuis NA (2022). Mini-review: Dissecting receptor-mediated stimulation of TRPV4 in nociceptive and inflammatory pathways. Neuroscience Letters, 770, 136377. [DOI] [PubMed] [Google Scholar]
- 92.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, & Julius D (1997). The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature, 389, 816–824. [DOI] [PubMed] [Google Scholar]
- 93.Koivisto A-P, Belvisi MG, Gaudet R & Szallasi A (2021). Advances in TRP channel drug discovery: From target validation to clinical studies. Nature Reviews. Drug Discovery, 21(1), 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Z, Ukidve A, Kim J, & Mitragotri S (2020). Targeting strategies for tissue-specific drug delivery. Cell, 181, 151–167. [DOI] [PubMed] [Google Scholar]
- 95.Lee H (2005). Altered thermal selection behavior in mice lacking transient receptor potential vanilloid 4. Journal of Neuroscience, 25, 1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mizuno A, Matsumoto N, Imai M, & Suzuki M (2003). Impaired osmotic sensation in mice lacking TRPV4. American Journal of Physiology. Cell Physiology, 285, C96–C101. [DOI] [PubMed] [Google Scholar]
- 97.Liedtke W, & Friedman JM (2003). Abnormal osmotic regulation in trpv4−/− mice. PNAS, 100, 13698–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, Mcnamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, & Voets T (2010). Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proceedings of the National Academy of Sciences, 107, 19084–19089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin Pu, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, … Willette RN (2012). An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Science Translational Medicine, 4, 159ra148. [DOI] [PubMed] [Google Scholar]
- 100.Moore C, Cevikbas F, Pasolli HA, Chen Y, Kong W, Kempkes C, Parekh P, Lee SH, Kontchou N-A, Yeh I, Jokerst NM, Fuchs E, Steinhoff M, & Liedtke WB (2013). UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. PNAS, 110, E3225–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goyal N, Skrdla P, Schroyer R, Kumar S, Fernando D, Oughton A, Norton N, Sprecher DL, & Cheriyan J (2019). Clinical pharmacokinetics, safety, and tolerability of a novel, first-in-class TRPV4 ion channel inhibitor, GSK2798745, in healthy and heart failure subjects. American Journal of Cardiovascular Drugs, 19, 335–342. [DOI] [PubMed] [Google Scholar]
- 102.Orduña Ríos M, Noguez Imm R, Hernández Godínez NM, Bautista Cortes AM, López Escalante DD, Liedtke W, Martínez Torres A, Concha L, & Thébault S (2019). TRPV4 inhibition prevents increased water diffusion and blood–retina barrier breakdown in the retina of streptozotocin-induced diabetic mice. Plos One, 14, e0212158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu M, Liu X, Wang L, Wang Yu, Dong F, Wu J, Qu X, Liu Y, Liu Z, Fan H, & Yao R (2018). TRPV4 inhibition improved myelination and reduced glia reactivity and inflammation in a cuprizone-induced mouse model of demyelination. Frontiers in Cellular Neuroscience, 12, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hochstetler AE, Smith HM, Preston DC, Reed MM, Territo PR, Shim JW, Fulkerson D, & Blazer-Yost BL (2020). TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight, 5, 137646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Z, Zhou L, An D, Xu W, Wu C, Sha S, Li Y, Zhu Y, Chen A, Du Y, & Chen L (2019). TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death & Disease, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jie P, Lu Z, Hong Z, Li L, Zhou L, Li Y, Zhou R, Zhou Y, Du Y, Chen L, & Chen L (2016). Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Molecular Neurobiology, 53, 8–17. [DOI] [PubMed] [Google Scholar]
- 107.Nobis M, Herrmann D, Warren SC, Kadir S, Leung W, Killen M, Magenau A, Stevenson D, Lucas MC, Reischmann N, Vennin C, Conway JRW, Boulghourjian A, Zaratzian A, Law AM, Gallego-Ortega D, Ormandy CJ, Walters SN, Grey ST, … Timpson P (2017). A RhoA-FRET biosensor mouse for intravital imaging in normal tissue homeostasis and disease contexts. Cell Reports, 21, 274–288. [DOI] [PubMed] [Google Scholar]
- 108.Nobis M, Herrmann D, Warren SC, Strathdee D, Cox TR, Anderson KI, & Timpson P (2020). Shedding new light on RhoA signalling as a drug target in vivo using a novel RhoA-FRET biosensor mouse. Small GTPases, 11, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Inoue M (2021). Genetically encoded calcium indicators to probe complex brain circuit dynamics in vivo. Neuroscience Research, 169, 2–8. [DOI] [PubMed] [Google Scholar]
- 110.Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, Masedunskas A, Weigert R, Chavakis T, Adams RH, Offermanns S, Mochizuki N, Zheng Yi, & Gutkind JS (2015). RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nature Communication, 6, 6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alessandri-Haber N (2004). Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. Journal of Neuroscience, 24, 4444–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boehmerle W, Huehnchen P, Lee SLL, Harms C, & Endres M (2018). TRPV4 inhibition prevents paclitaxel-induced neurotoxicity in preclinical models. Experimental Neurology, 306, 64–75. [DOI] [PubMed] [Google Scholar]
- 113.Chen Y, Williams SH, Mcnulty AL, Hong Ji. H, Lee SH, Rothfusz NE, Parekh PK, Moore C, Gereau RW, Taylor AB, Wang F, Guilak F, & Liedtke W (2013). Temporomandibular joint pain: A critical role for Trpv4 in the trigeminal ganglion. Pain, 154, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Feng X, Takayama Y, Ohno N, Kanda H, Dai Yi, Sokabe T, & Tominaga M (2020). Increased TRPV4 expression in non-myelinating Schwann cells is associated with demyelination after sciatic nerve injury. Commun Biol, 3, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nishimoto R, Derouiche S, Eto K, Deveci A, Kashio M, Kimori Y, Matsuoka Y, Morimatsu H, Nabekura J, & Tominaga M (2021). Thermosensitive TRPV4 channels mediate temperature-dependent microglia movement. PNAS 118(17), e2012894118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luo J, Qian A, Oetjen LK, Yu W, Yang Pu, Feng J, Xie Z, Liu S, Yin S, Dryn D, Cheng J, Riehl TE, Zholos AV, Stenson WF, Kim BS, & Hu H (2018). TRPV4 channel signaling in macrophages promotes gastrointestinal motility via direct effects on smooth muscle cells. Immunity, 49, 107–119.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rust R, & Kaiser J (2017). Insights into the dual role of inflammation after spinal cord injury. Journal of Neuroscience, 37, 4658–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.