

Abstract
Langerhans cell histiocytosis (LCH) is a potentially fatal condition characterized by granulomatous lesions with characteristic clonal mononuclear phagocytes (MNPs) harboring activating somatic mutations in mitogen-activated protein kinase (MAPK) pathway genes, most notably BRAFV600E. We recently discovered that the BRAFV600E mutation can also affect multipotent hematopoietic progenitor cells (HPCs) in multisystem LCH disease. How the BRAFV600E mutation in HPCs leads to LCH is not known. Here we show that enforced expression of the BRAFV600E mutation in early mouse and human multipotent HPCs induced a senescence program that led to HPC growth arrest, apoptosis resistance and a senescence-associated secretory phenotype (SASP). SASP, in turn, promoted HPC skewing toward the MNP lineage, leading to the accumulation of senescent MNPs in tissue and the formation of LCH lesions. Accordingly, elimination of senescent cells using INK-ATTAC transgenic mice, as well as pharmacologic blockade of SASP, improved LCH disease in mice. These results identify senescent cells as a new target for the treatment of LCH.
Histiocytoses are a group of conditions characterized by the accumulation of histiocytes in various tissues. Histiocytes, now more commonly known as mononuclear phagocytes (MNPs), consist of heterogeneous cells of myeloid origin that include dendritic cells and macrophages. Specifically, LCH results from the accumulation of CD207+CD1a+ cells that resemble Langerhans cells1. LCH occurs mostly in children with an incidence of 8.9 in 106 children per year, similar to pediatric Hodgkin’s lymphoma2. LCH can arise in a variety of settings from single bone lesions to life-threatening disseminated disease that affects the spleen, liver and bone marrow (BM). Front-line chemotherapy fails in more than 50% of patients with multisystem disease, highlighting the need for new therapies for this group of patients3.
Activating somatic mutations in MAPK pathway genes, most notably BRAFV600E, have been identified in 85% cases of LCH, specifically in the pathogenic CD207+ cells within LCH lesions4–7. Importantly, we and others8–10 recently discovered that the BRAFV600E mutation can also be identified in CD34+ BM HPCs in patients with systemic LCH disease, which led to the reclassification of multifocal LCH as a myeloid neoplasia3,7,11. These new results revealed a new conundrum in LCH pathophysiology: how does the somatic BRAFV600E mutation in multipotent progenitor (MPP) cells lead to LCH lesions?
Here we explored the mechanisms by which the BRAFV600E mutation could potentially drive a multipotent hematopoietic progenitor to give rise to LCH lesions. Strikingly, we found that enforced expression of the BRAFV600E mutation in early mouse and human multipotent HPCs induced a senescence program that led to HPC growth arrest, apoptosis resistance and SASP induction. This SASP, in turn, promoted multipotent HPC skewing toward the MNP lineage, away from other hematopoietic lineages. BRAFV600E-induced senescence persisted in differentiated MNPs that accumulated in tissues, where SASP induction and sustained MNP survival contributed to the formation of LCH lesions. Accordingly, we found that genetic elimination of senescent cells using INK-ATTAC (Ink4a/Arf or Cdkn2a–apoptosis through targeted activation of caspase) transgenic mice, as well as pharmacologic blockade of SASP, improved LCH disease burden in mice. These results transform our understanding of LCH pathophysiology and identify senescent cells as a new target for the treatment of LCH.
Results
Expression of the BRAFV600E mutation in mouse multipotent HPCs is sufficient to drive to LCH lesion formation.
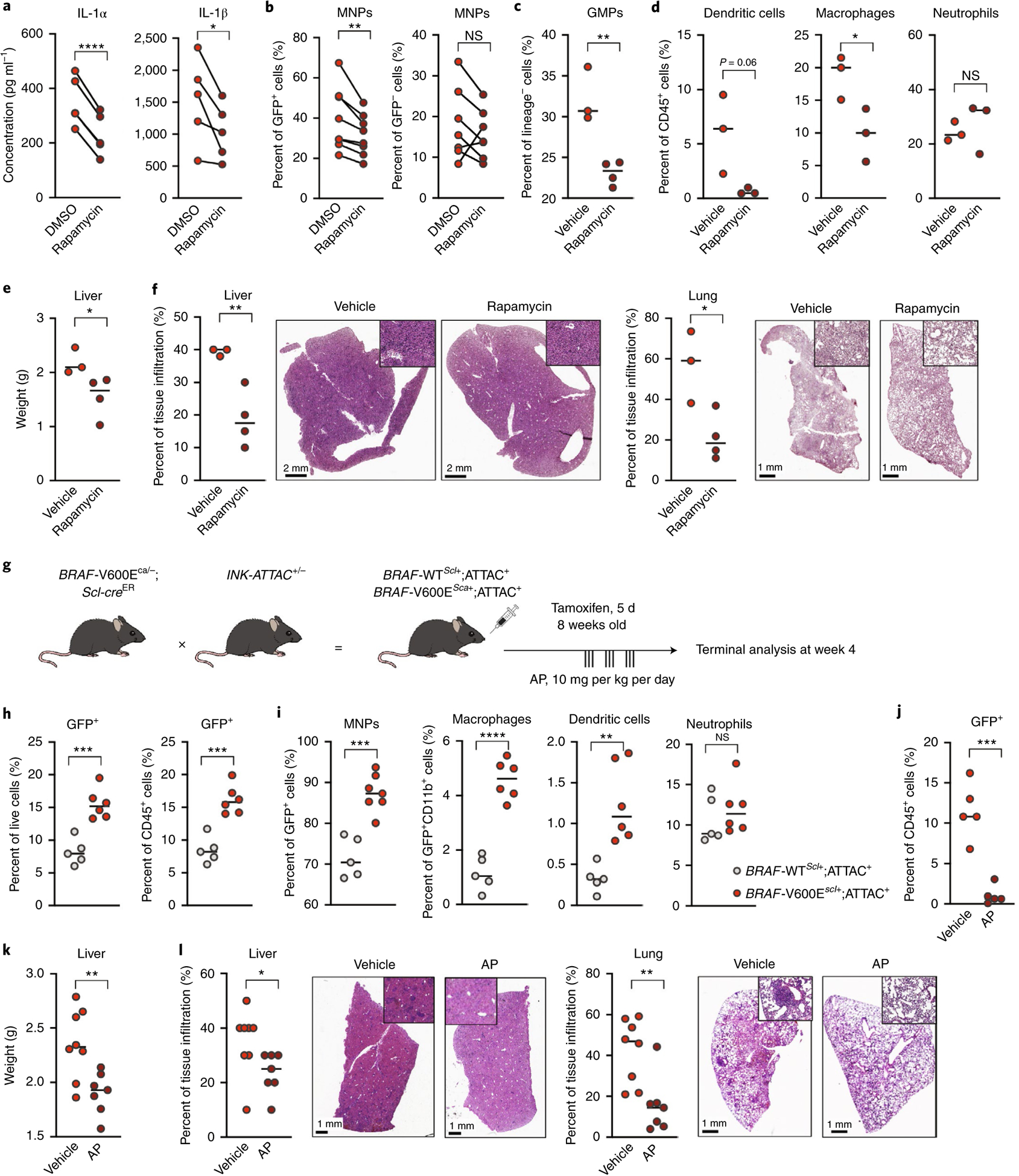
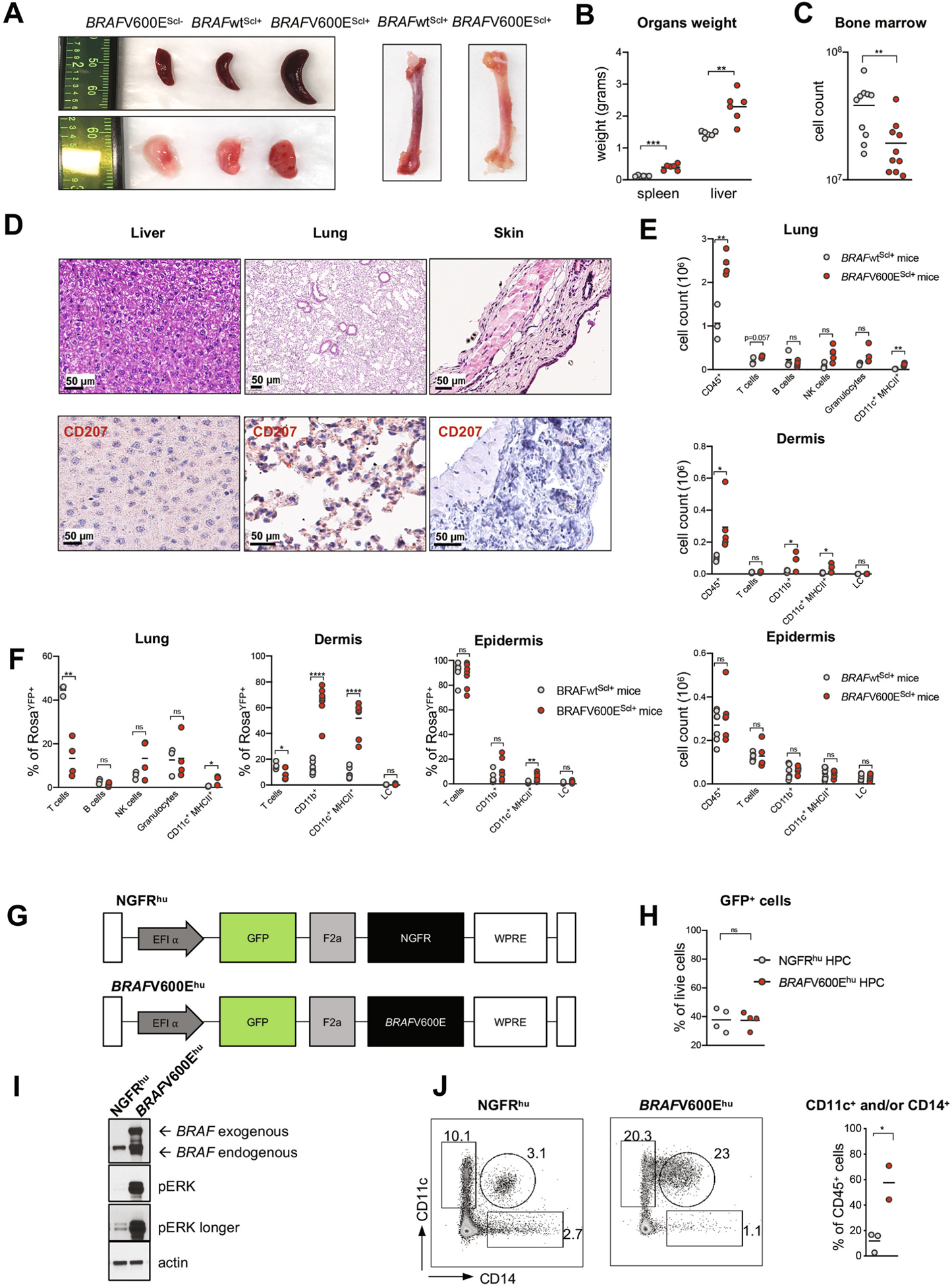
To examine whether BRAF-V600E+ HPCs can drive LCH lesions, we enforced expression of the BRAFV600E mutation in cells expressing Scl (also known as Tal1), a molecule expressed specifically in long-term and short-term hematopoietic stem cells (HSCs) in mice12. We genetically engineered somatic mosaicism for a BRAFV600E allele linked to a yellow fluorescent protein (YFP) in HSCs using tamoxifen-inducible targeting in Scl-creER mice, which we named BRAF-V600EScl+ and used BRAF-wild-type (WT)Scl+ mice as control littermates (Fig. 1a). Thus, RosaYFP+ cells carried the BRAFV600E mutation in BRAF-V600EScl+ mice, whereas RosaYFP+ cells in BRAF-WTScl+ mice did not carry the mutation but underwent Rosa26-locus driven Cre recombination. Pulsed administration of tamoxifen led to YFP expression in 30% of HSCs, providing the opportunity to compare BRAF-V600E+(YFP+)Scl+ cells to BRAFWT(YFP−)Scl+ cells within the same mouse (Fig. 1b). Strikingly, BRAF-V600EScl+ mice developed substantial organomegaly, granulomatous bone lesions and BM aplasia within 4 weeks of tamoxifen administration, while BRAF-WTScl+ control littermates did not (Fig. 1c and Extended Data Fig. 1a–c). Histological analysis of hematoxylin and eosin (H&E)-stained formalin-fixed paraffin-embedded (FFPE) tissue sections revealed an accumulation of histiocytes within granulomatous lesions, multinucleated giant cells and an inflammatory infiltrate in the liver, lungs and dermises of BRAF-V600EScl+ mice (Fig. 1d and Extended Data Fig. 1d). Immunohistochemistry analysis revealed that many histiocytes expressed CD207 (Fig. 1d). Flow cytometry analysis confirmed the significant increase in immune cell infiltration and the expansion of the MNP compartment within the immune cell infiltrate (Fig. 1f and Extended Data Fig. 1e). Importantly, in the skin, MNPs mostly accumulated in the dermis and were absent from the epidermis (Fig. 1f), mimicking the dermotropism of the human disease. Flow cytometry analysis also revealed the preferential expansion of MNPs within the BRAF-V600EYFP+ immune cell compartment in tissues known as targets of LCH lesions (Fig. 1e and Extended Data Fig. 1f).
Fig. 1 |. Expression of the BRAFV600E mutation in mouse and human multipotent HPCs is sufficient to lead to LCH lesions.
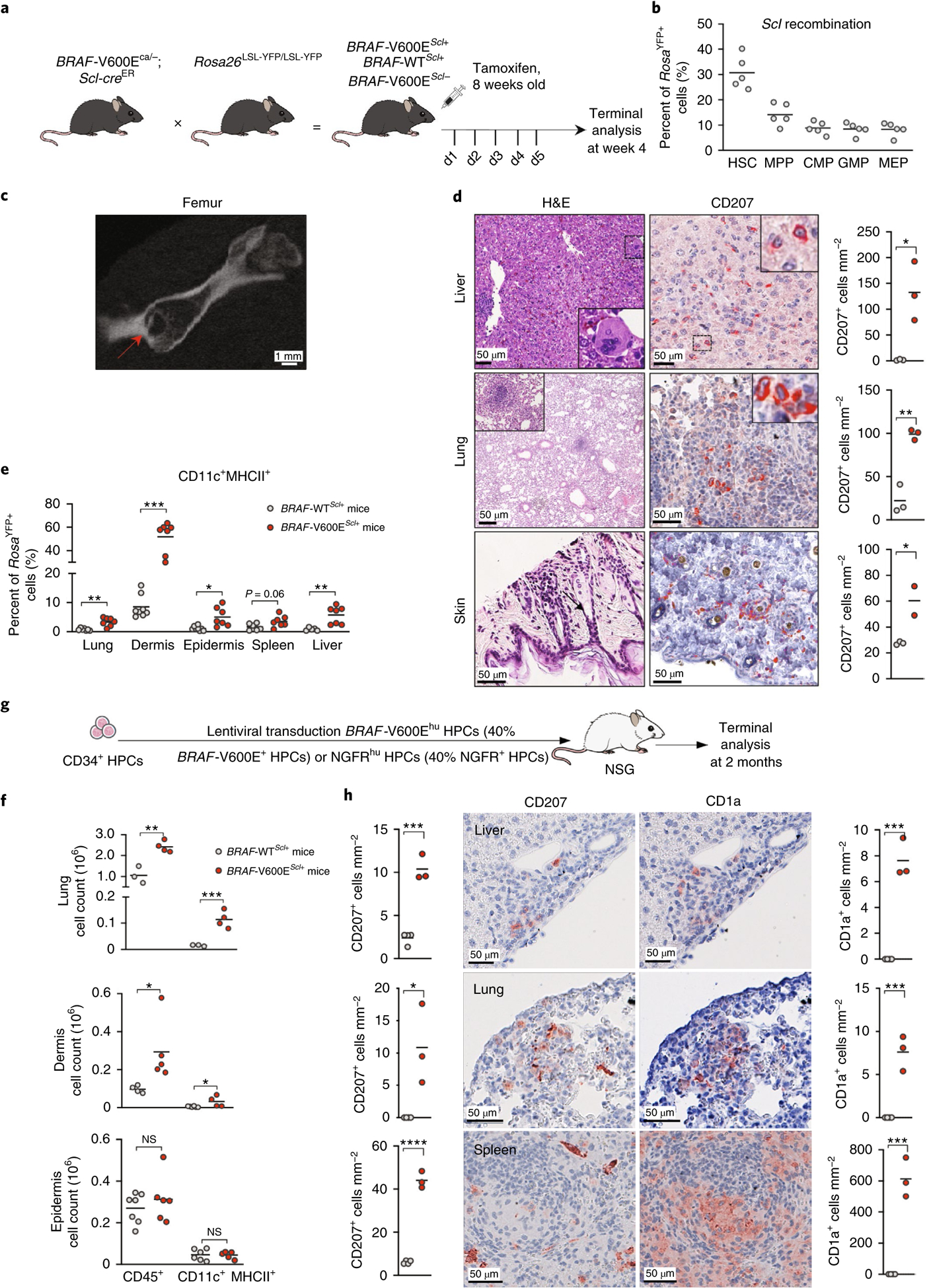
a, Cartoon shows the experimental breeding scheme used to generate BRAF-V600EScl+ mice and control littermates. d1–5 indicates day 1–5. b, Percentage of RosaYFP+ cells among purified hematopoietic progenitors in BRAF-WTScl+ mice 4 weeks after tamoxifen injections (n = 5 mice). CMP, common myeloid progenitor; MEP, megakaryocytic erythroid progenitor. c, Representative computerized tomography image of a femur from a BRAF-V600EScl+ animal. The red arrow shows a pathognomonic granuloma. d, H&E staining and CD207 immunohistochemistry staining of tissues isolated from BRAF-V600EScl+ mice. We note the presence of multinucleated giant cells and granuloma-like lesions. The graph represents the number of CD207+ cells per mm2 in tissue sections isolated from animals (n = 3 mice per group). e, Percentage of the CD11c+MHCII+ MNP population among RosaYFP+ cells in organs of BRAF-V600EScl+ mice and control littermates. Data are representative of three experiments (n = 7 mice). f, Absolute numbers of total CD45+ immune cells and CD11c+MHCII+ MNPs that populate the lung and the skin of BRAF-V600EScl+ mice and control littermates. Data are representative of three experiments (n = 3–7 mice). NS, not significant. g, Cartoon shows the scheme used to generate the humanized LCH mouse. h, Liver, lung and spleen tissue sections isolated from humanized mice reconstituted with BRAF-V600Ehu HPCs were stained with anti-CD207 and anti-CD1a antibodies. Graphs represent the number of CD207+ or CD1a+ cells per mm2 in tissue sections isolated from animals (n = 3–4 mice). Data are represented as mean ± s.e.m.; statistical significance was analyzed by unpaired two-sided t-tests and is indicated by *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. These data are representative of three independent experiments.
Humanized mice reconstituted with human CD34+ HPCs expressing the BRAFV600E mutation develop LCH-like disease.
To determine whether expression of the BRAFV600E mutation in human HPCs could also lead to LCH disease, we generated a lentiviral vector expressing the GFP reporter together with the BRAFV600E mutation (BRAF-V600Ehu) or the NGFR truncated gene (nerve growth factor receptor (NGFR)hu) under the EF1A1 promoter (Extended Data Fig. 1g). HPC transduction efficiency, around 40%, led to sustained extracellular signal-regulated kinase (ERK) phosphorylation in BRAF-V600Ehu but not in NGFRhu HPCs (Extended Data Fig. 1h,i). We reconstituted sublethally irradiated NOD-severe combined immunodeficient (SCID) gamma (NSG) mice with human CD34+ cord blood HPCs transduced with this lentiviral vector (Fig. 1g). NSG mice deteriorated within 2 months following reconstitution with BRAF-V600EhuCD34+ HPCs but not NGFRhuCD34+ HPCs and developed an expansion of CD11c+CD14+ MNP cells circulating in the blood (Extended Data Fig. 1j), accompanied by an accumulation of CD1a+CD207+ cells in the liver, lung and spleen, pathognomonic of LCH disease (Fig. 1h). These results, together with our data showing that BRAF-V600EScl+ mice develop LCH-like lesions, establish that expression of the BRAFV600E mutation in mouse and human early HPCs is sufficient to drive LCH lesion formation.
BRAFV600E HPCs differentiate predominantly toward the MNP lineage.
To explore how the BRAFV600E mutation in early HPCs drives the specific accumulation of MNPs in the periphery, we measured the differentiation potential of BRAF-V600E+YFP+ HPCs in BRAF-V600EScl+ mice. Strikingly, we found that levels of BRAF-V600E+ HSCs and MPPs were reduced, while levels of BRAF-V600E+ granulocyte–macrophage progenitors (GMPs) were significantly expanded in the BM compared to those in BRAFWT progenitors (Fig. 2a and Extended Data Fig. 2a), suggesting that the BRAF-V600E+ HPCs failed to expand and were instead biased toward the myeloid lineage. The expansion of BRAF-V600E+ GMPs was associated with a remarkable expansion of BM BRAF-V600E+ monocytes, macrophages and dendritic cells, whereas BM BRAF-V600E+ neutrophils did not expand (Fig. 2b and Extended Data Fig. 2b). To directly examine whether the BRAFV600E mutation skewed HPC commitment to the MNP lineage, we cultured purified HPCs from BRAF-V600EScl+ or BRAF-WTScl+ mice in methylcellulose and measured HPC progeny using a colony-forming unit (CFU) assay that reflects differentiation into committed progenitors. Consistent with the lineage bias observed in vivo, we found that BRAFV600E HPCs preferentially differentiated into CFUs consisting of macrophage progenitors (CFU-M), while numbers of granulocytic progenitors (CFU-G) and erythroid progenitors, also called burst-forming unit erythroids (BFU-E), were reduced (Fig. 2c).
Fig. 2 |. Expression of the BRAFV600E mutation in multipotent hematopoietic progenitors enforces their differentiation toward the MNP lineage.
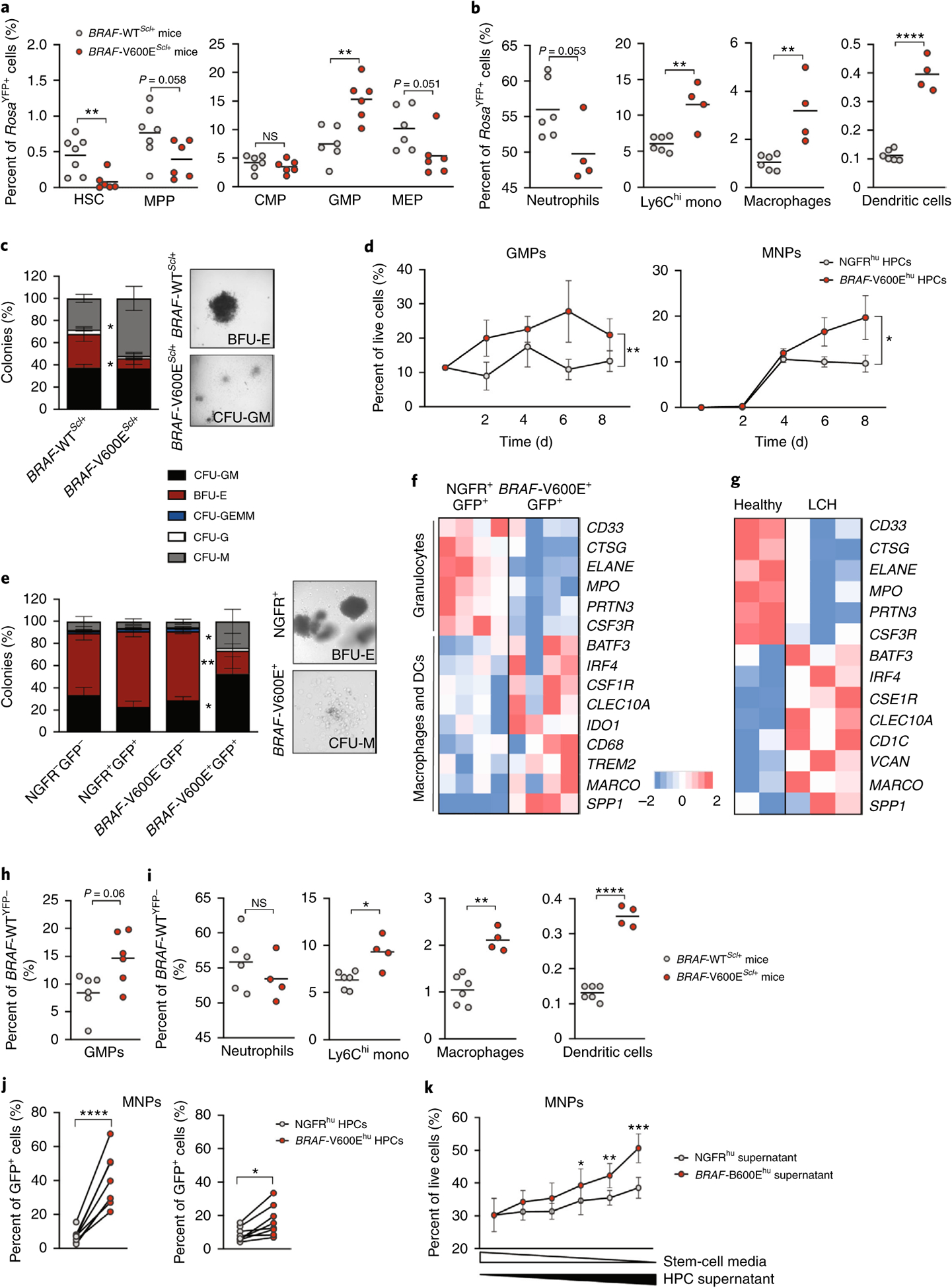
a–c, BRAF-V600EScl+ mice were generated as described in Fig. 1a, and, in these mice, RosaYFP+ cells mark BRAF-V600E+ cells, while RosaYFP− cells mark BRAF-V600E− cells. In BRAF-WTScl+ control littermates, RosaYFP+ cells mark those that underwent a Cre recombination as well. a, Graphs show the percentage of HSCs, MPPs, common myeloid progenitors, granulocytic myeloid progenitors and megakaryocytic erythroid progenitors among RosaYFP+ cells in the BM of BRAF-V600EScl+ mice and BRAF-WTScl+ control littermates (n = 6–7 mice). b, Percentages of neutrophils, Ly6Chi monocytes (mono), macrophages and dendritic cells among RosaYFP+ cells in the BM of BRAF-V600EScl+ mice and BRAF-WTScl+ control littermates (n = 4–6 mice). c, CFU assays were performed on purified lineage-negative BM cells isolated from BRAF-V600EScl+ mice or BRAF-WTScl+ control littermates (n = 3 mice) cultured in the presence of methylcellulose, with representative phase contrast photomicrographs of colonies. GEMM, granulocytes, erythrocytes, megakaryocytes, monocytes, megakaryocytes. d–f, Purified CD34+ HPCs transduced with BRAFV600E (BRAF-V600Ehu) or NGFR control (NGFRhu) lentiviral vectors were cultured in stem cell medium for 8 d. d, Graphs show the percentages of GMPs (defined as CD34+CD38+CD10−CD123hi/intCD45RA+/−)34 and the percentages of MNPs (defined as CD66b−CD11c+MHCII+) among live cells (n = 3 donors). e, CFU assays in methylcellulose performed on purified BRAF-V600E−GFP− and BRAF-V600E+GFP+ cells isolated from HPCs transduced with the BRAFV600E lentiviral vector or purified NGFR+GFP+ or NGFR−GFP− cells isolated from HPCs transduced with the NGFR lentiviral vector as described in Extended Data Fig. 3 (n = 6 donors). Representative phase contrast photomicrographs of colonies are shown. f, Human CD34+ HPCs transduced with BRAFV600E or the control NGFR lentiviral vector were cultured for 7 d in stem cell medium and analyzed using microarray sequencing. Heatmap representation of genes involved in granulopoiesis and the macrophage–dendritic cell (DC) lineage is shown (n = 4 independent donors per group). g, Purified CD34+ BM cells were isolated from a healthy pediatric donor (in duplicate) and from three patients with LCH and BM mononuclear cells known to be BRAF-V600E+, and these cells were analyzed using bulk RNA-seq. Heatmap representation of genes involved in granulopoiesis and the macrophage–dendritic cell lineage is shown. h,i, BRAF-V600EScl+ mice and BRAF-WTScl+ control littermates were generated as described in Fig. 1a. The percentages of GMPs (h), neutrophils, Ly6Chi monocytes, macrophages and dendritic cells (i) among BRAFWT;RosaYFP− cells in the BM of BRAF-V600EScl+ mice and BRAF-WTScl+ control littermates is shown. j, Human CD34+ HPCs were transduced with BRAFV600E or NGFR lentiviral constructs and cultured in stem cell medium. Seven days later, the percentages of CD11c+ and/or CD14+ MNP cells among BRAFV600E (GFP+) or BRAFWT (GFP−) cells were measured by flow cytometry. Because the transduction efficiency was around 40% for both vectors, GFP+ cells mark BRAF-V600E+ or NGFR+ cells, while GFP− cells mark BRAFWT or NGFR− cells among HPCs transduced with the BRAFV600E or NGFR lentiviral construct, respectively (n = 8 independent donors). k, Healthy human CD34+ cord blood HPCs were cultured in stem cell medium (concentration is represented by the white triangle) in addition to supernatant isolated from HPCs transduced with the BRAFV600E lentiviral construct or the NGFR control (concentration is represented by the black triangle). The graph shows the percentage of HPC differentiation into CD14+ MNPs analyzed by flow cytometry after 5 d of culture (n = 4 independent donors), analyzed by paired t-tests. Data are represented as mean ± s.e.m.; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (unpaired two-sided t-tests, two-way ANOVA test for d and paired two-sided t-tests for j,k). Data are representative of at least three independent experiments.
We also examined whether the BRAFV600E mutation affected human HPC lineage commitment. Consistent with the results obtained from murine BRAF-V600EScl+ HPCs, we found that human BRAF-V600Ehu HPCs preferentially differentiated into the MNP lineage (Fig. 2d). Remarkably, using the same CFU assay applied in mice, we found that human BRAF-V600E+ HPCs differentiated preferentially into CFU-GM and CFU-M colonies but not into CFU-G colonies, whereas NGFR+ HPCs retained a pluripotent differentiation potential (Fig. 2e and Extended Data Fig. 2c). Gene expression profiling of human BRAF-V600E+GFP+ and control NGFR+GFP+ HPCs 7 d after culture in stem cell medium revealed enrichment for an MNP signature in BRAF-V600E+ HPCs that included the expression of classical dendritic cell genes (BATF3, IRF4, CLEC10A) as well as that of macrophage genes (CSF1R, CD68, SPP1), while HPC differentiation into granulocytes was strongly reduced compared to that in control NGFR+ HPCs (Fig. 2f).
To further confirm the MNP skewing of BRAF-V600E+ HPCs in patients with LCH, we performed RNA-seq of purified CD34+ HPCs isolated from the BM of patients with LCH (Supplementary Table 1). Similar to the signature of BRAFV600E- transduced HPCs, CD34+ HPCs isolated from patients with LCH showed reduced expression of genes associated with granulopoiesis (ELANE, MPO, PRTN3, CSF3R) and an increased expression of genes involved in dendritic cell–macrophage commitment (BATF3, IRF4, CSF1R, CLEC10A) compared to that in CD34+ HPCs isolated from an age-matched individual (Fig. 2g). In sum, these data establish that the BRAFV600E mutation directs mouse and human HPCs to differentiate into the MNP lineage, away from the lymphoid and the granulocytic lineage.
Skewing of BRAF-V600E+ HPCs into the MNP lineage is driven by both cell-intrinsic and cell-extrinsic cues.
To examine whether the enhanced MNP differentiation of BRAF-V600E+ HPCs was cell intrinsic or a result of external cues, we measured the expansion of BRAF-V600E+ and BRAFWT HPCs within the same animal. Strikingly, while we failed to observe an expansion of GMPs in BRAF-WTScl+ mice, there was an expansion of BRAF-V600E+ and BRAFWT GMPs within the same BRAF-V600EScl+ mice (Fig. 2h), associated with an increase in BM BRAF-V600E+ and BRAFWT MNPs but not an increase in neutrophils (Fig. 2i). The expansion of BM BRAFWT MNPs in BRAF-V600EScl+ mice but not in BRAF-WTScl+ mice suggests that MNP skewing may not be driven only by cell-intrinsic cues.
To examine whether similar cell-extrinsic cues can also drive human HPC differentiation into MNPs, we took advantage of the fact that HPC transduction efficiency with BRAFV600E or NGFR lentiviral vectors did not exceed 40% (Extended Data Fig. 1g) to compare, within the same culture, the differentiation of BRAF-V600E+GFP+ and BRAF-V600E−GFP− HPCs as well as that of NGFR+GFP+ and NGFR−GFP− HPCs. Similar to our results in mice, BRAF-V600E−GFP− cells co-cultured with BRAF-V600E+GFP+ cells had an enhanced MNP differentiation potential, although at a reduced scale compared to that of BRAF-V600E+GFP+ HPC counterparts (Fig. 2j). This contrasts with our earlier results showing that purified human BRAF-V600E−GFP− HPCs cultured alone in CFU assays did not have an MNP differentiation advantage (Fig. 2e and Extended Data Fig. 2c). This suggests that secreted molecules produced by BRAF-V600E+GFP+ HPCs contributed to driving MNP differentiation of healthy HPCs. To directly measure whether the BRAF-V600E+ HPC secretome skewed the differentiation of BRAFWT HPCs toward the MNP lineage, we cultured human CD34+ HPCs in stem cell medium in the presence or absence of supernatant isolated from human BRAF-V600E+ HPCs or NGFR+ HPCs. Consistent with our hypothesis, we observed that healthy HPCs cultured in the presence of supernatant obtained from human BRAF-V600E+ HPC cultures were more prone to differentiate into MNPs than healthy HPCs cultured in the presence of supernatant from NGFR+ HPCs and that the enhanced MNP differentiation potential was dependent on the dose of supernatant from added BRAF-V600E+ HPCs (Fig. 2k).
The BRAFV600E mutation drives mouse and human hematopoietic progenitors into oncogene-induced senescence.
The realization that the BRAFV600E mutation limited the expansion of HSCs (Fig. 2a) prompted us to further assess the proliferation potential of BRAFV600E-mutant HPCs using a competitive reconstitution assay in which purified BRAF-V600E+ HPCs or BRAFWT HPCs (isolated from CD45.2+ animals) were injected together with host HPCs (CD45.1+) at a 2:1 ratio into sublethally irradiated CD45.1+ recipients. Strikingly, we found that, at 4 and 8 weeks after transplantation, the number of engrafted BRAF-V600E+ HPCs was strongly reduced compared to that of BRAFWT HPCs (Fig. 3a and Extended Data Fig. 3a), thus revealing a proliferative disadvantage of BRAFV600E HPCs compared to BRAFWT HPCs.
Fig. 3 |. The BRAFV600E mutation drives mouse and human hematopoietic progenitors into senescence.
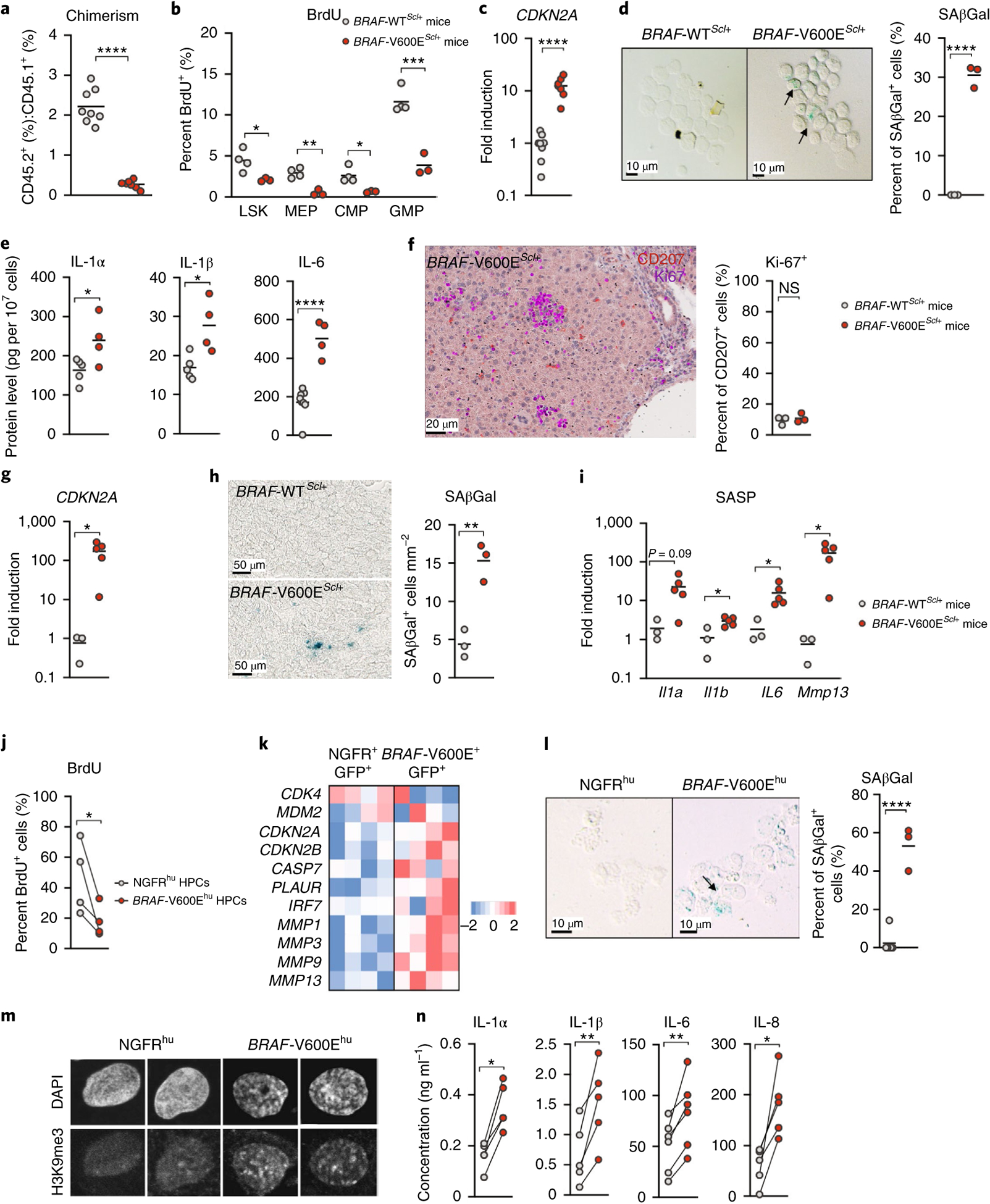
a, Lineage-negative BM cells isolated from either BRAF-V600EScl+CD45.2+ or BRAF-WTScl+CD45.2+ mice were injected intravenously together with lineage-negative CD45.1+ BM cells into lethally irradiated CD45.1+ mice at a 2:1 ratio (CD45.2+:CD45.1+). Graph shows the ratio of CD45.2+:CD45.1+ cells circulating in the blood in each group, measured 4 weeks after transplantation using flow cytometry (n = 8 animals). b–e, BM cells were isolated from BRAF-V600EScl+ and BRAF-WTScl+ control littermates and analyzed for the percentage of BrdU expression in lineage-negative Sca-1+c-Kit+ cells (LSK), megakaryocytic erythroid progenitors, common myeloid progenitors and GMPs after seven daily BrdU injections (1 mg per day) (n = 3–4 mice) (b); Cdkn2a expression from purified lineage-negative BM cells measured by quantitative PCR with reverse transcription (RT–qPCR) (normalized to actin expression) (n = 8 mice) (c); and SAβGal activity of purified lineage-negative BM cells (d). The graph shows the percentage of SAβGal+ cells among total cells (n = 3 mice per group). e, IL-1α, IL-1β and IL-6 protein levels in the BM supernatant, detected by ELISA, are shown (n = 4–6 mice per group). f–i, Liver tissue sections were obtained from BRAF-V600EScl+ and BRAF-WTScl+ control littermates and analyzed for Ki-67+ (violet) cells among liver infiltrating CD207+ LCH cells (red) (f). Graphs show the percentage of Ki-67+ cells among CD207+ cells in liver sections (n = 3 mice per group), Cdkn2a expression by RT–qPCR (normalized to actin expression) (n = 3–5 mice per group) (g) and SAβGal activity (h). i, Graph shows the number of SAβGal+ cells per mm2 in liver sections (n = 3 mice per group) and expression of Il1a, Il1b, Il6 and Mmp13 by RT–qPCR normalized to actin expression (n = 3–5 per group). j–n, Human CD34+ cord blood HPCs were transduced with BRAFV600E (BRAF-V600Ehu) or NGFR control (NGFRhu) lentiviral vectors, cultured in stem cell medium and analyzed for BrdU expression after daily pulses of BrdU from day 10 to day 13 (n = 4 different donors) (j), gene expression of senescence-associated genes using microarray expression profiling 7 d after transduction (n = 4 donors) (k) and SAβGal activity 9 d after transduction (l). l, The percentage of SAβGal+ cells among total cells is indicated (n = 4 donors). m, Trimethylation at lysine 9 of histone 3 (H3K9me3) and 4,6-diamidino-2-phenylindole (DAPI) immunofluorescence co-staining of NGFRhu and BRAF-V600Ehu HPCs 9 d after transduction; images are representative of senescence-associated-heterochromatin foci. n, IL-1α, IL-1β, IL-6 and IL-8 levels in the culture supernatant 7 d after HPC transduction (n = 4–6 donors). Data are represented as mean ± s.e.m.; statistical significance analyzed by unpaired and paired (for j,n) two-sided t-tests is indicated by *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Data are representative of at least two independent experiments.
The BRAFV600E mutation was previously described to drive cellular senescence in human nevus lesions13. Cellular senescence classically occurs secondary to DNA damage or because of oncogene activation and results in cell cycle arrest driven by cell cycle regulators, such as p16INK4a, and prolonged survival driven by the anti-apoptotic proteins BCL-xL and BCL-2. Senescent cells also form heterochromatin foci and express senescence-associated β-galactosidase (SAβGal), as well as exhibiting a SASP (reviewed in refs.14,15), which leads to the production of a wide range of inflammatory molecules such as interleukin (IL)-1, IL-6, IL-8 and several matrix metalloproteinases.
Strikingly, we found that BRAF-V600EScl+ HPCs were unable to cycle, as shown by the low BrdU incorporation potential, which likely accounts for their poor ability to engraft compared to that of HPCs from BRAF-WTScl+ mice in a competitive reconstitution assay (Fig. 3b). HPCs isolated from BRAF-V600EScl+ mice were also enlarged compared to HPCs from BRAF-WTScl+ mice, a common feature of senescent cells (Extended Data Fig. 3b); expressed high levels of Cdkn2a transcripts, which code for the cell cycle regulator p16INK4a (Fig. 3c); were positive for SAβGal (Fig. 3d); and produced SASP-associated proteins (Fig. 3e), all features consistent with a senescent state. LCH cells isolated from peripheral tissues of BRAF-V600EScl+ mice were also in a senescent state, as shown by reduced Ki-67 expression (Fig. 3f), expression of Cdkn2a (Fig. 3g), SAβGal activity (Fig. 3h) and the production of SASP-associated proteins (Fig. 3i). These results suggest that the BRAFV600E mutation induced a senescence program in HPCs, which was maintained in differentiated LCH cells.
The BRAFV600E mutation in human HPCs led to an initial phase of proliferation (Extended Data Fig. 3c), followed by a marked cell cycle arrest in vitro (Fig. 3j and Extended Data Fig. 3d). Human BRAFV600E-transduced HPCs exhibited canonical markers of cellular senescence, including enlarged cellular size (Extended Data Fig. 3e), positivity for SAβGal staining (Fig. 3l), senescence-associated heterochromatin foci (Fig. 3m) and increased production of SASP cytokines (Fig. 3n and Extended Data Fig. 3f). Gene expression profiling of transduced human BRAF-V600E+GFP+ HPCs confirmed the senescence signature16 (Fig. 3k), and LCH lesions that formed in humanized mice reconstituted with human BRAFV600E CD34+ cells also had increased SAβGal activity and high p16INK4a levels (Extended Data Fig. 3g,h).
Importantly, CD34+ BM cells isolated from patients with LCH also expressed a senescence signature, including increased expression of CDKN2A, CDKN2C, CDKN2D, CD9, MDM2 and genes encoding matrix metalloproteinases, which was absent from CD34+ BM cells isolated from age-matched healthy individuals (Fig. 4a and Extended Data Fig. 4a).
Fig. 4 |. The BRAFV600E mutation induces a senescence program in human LCH lesions.
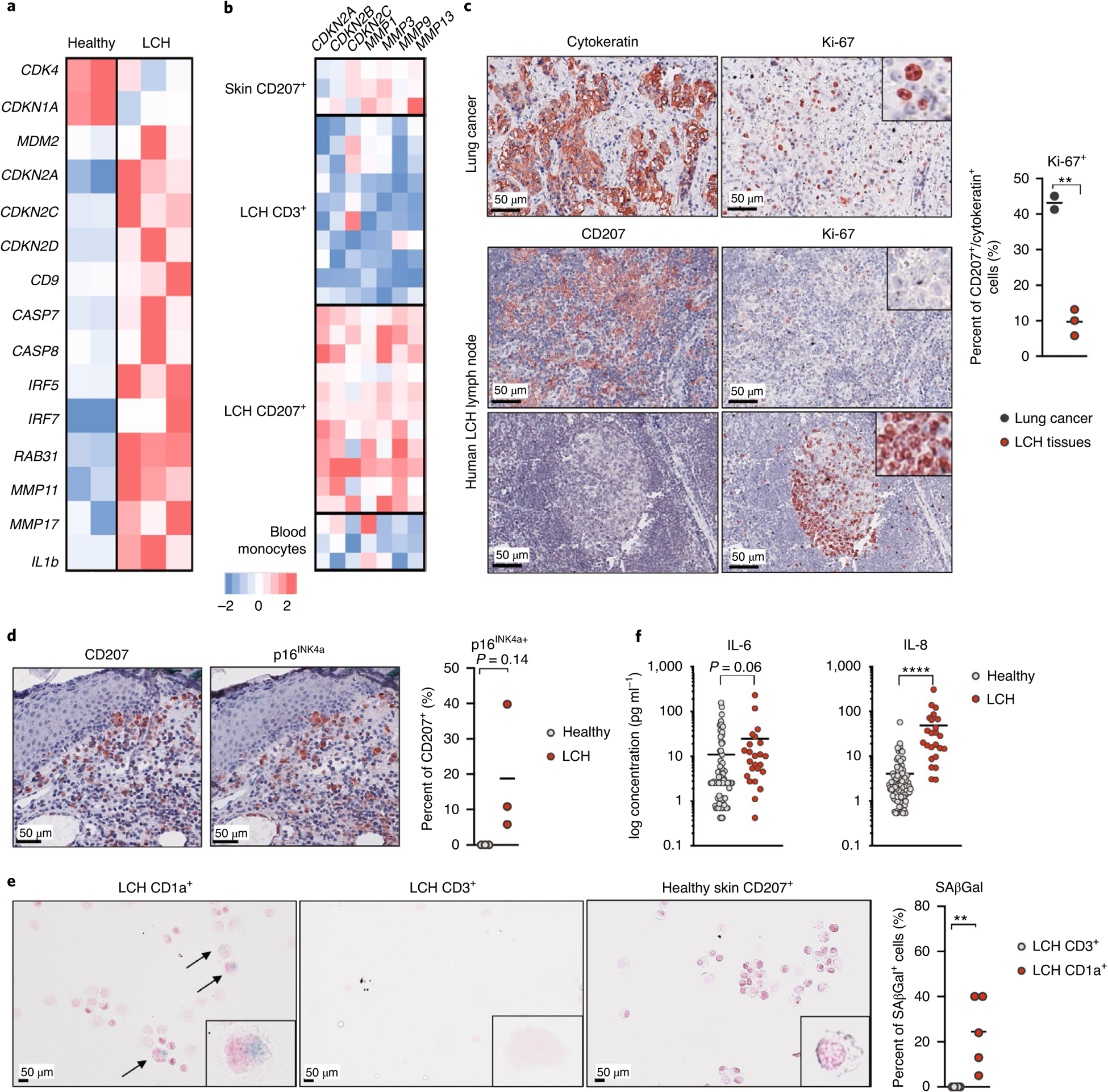
a, Heatmap representation of senescence-associated gene expression, measured by RNA-seq in purified CD34+ BM cells isolated from three patients with LCH and one pediatric healthy donor (performed in duplicate). b, Skin CD207+ cells were isolated from healthy skin (n = 3 patients), CD3+ and CD207+ cells were purified from LCH lesions (n = 11 patients), and circulating CD14+ cells were isolated from three healthy pediatric donors, and these cells were analyzed for gene expression profiles using microarrays. The expression profile of CDKN2A, CDKN2B, CDKN2C, MMP1, MMP3, MMP9 and MMP13 is shown. c, FFPE sections of LCH lymph node lesions from three different patients were immunostained for CD207 and Ki-67, and FFPE lung cancer sections (n = 2 patients) were immunostained for cytokeratin and Ki-67. Images at the top show the high proliferation index of lung cancer. Images at the bottom show the same LCH donor with LCH in a lymph node. The middle panel shows a low proliferative index of the LCH as compared to that in the bottom panel of an uninvolved lymphoid follicle with its typical proliferative activity in the polarized germinal center. The graph represents the percent of Ki-67+ cells among CD207+ cells in LCH tissue or among cytokeratin-positive cells in lung cancers. d, Skin tissue sections isolated from patients with LCH and healthy age-matched donors were stained for CD207 and p16INK4a using immunohistochemistry. One representative image is shown. The graph shows the percentage of p16INK4a-positive cells among CD207+ cells (n = 3 patients for each group). e, SAβGal activity in human LCH lesions. CD207+ cells and CD3+ cells were sorted from LCH lesions (n = 5 patients), healthy skin and healthy tonsils, and these cells were stained for SAβGal activity. The graph shows the percent of SAβGal+ cells among CD207+ cells and CD3+ cells, highlighting that the staining occurred mostly in LCH cells. f, Graphs show IL-6 and IL-8 protein levels in the plasma of patients with LCH and age-matched healthy donors (n = 24 patients with multisystem LCH, n = 89 healthy pediatric donors). Data are represented as mean ± s.e.m.; statistical significance analyzed by unpaired two-sided t-tests is indicated by *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Purified CD207+ cells from human LCH lesions expressed high levels of CDKN2A, CDKN2B and CDKN2C transcripts and high SASP transcript levels (MMP1, MMP3, MMP9, MMP13) (Fig. 4b). LCH cells from LCH lesions expressed low levels of or no Ki-67 (Fig. 4c), while they expressed high levels of p16INK4a protein and had high SAβGal activity (Fig. 4d,e and Extended Data Fig. 4b,c). In addition, we found high IL-6 and IL-8 cytokine levels in the plasma of patients with multisystem LCH disease (Fig. 4f).
A neurodegenerative syndrome (LCH-ND) characterized by progressive ataxia and learning and behavior difficulties arises in some patients with LCH and remains one of the most difficult-to-treat complications of LCH. We previously demonstrated the presence of BRAF-V600E+ perivascular microglial-like cells along with BRAF-V600E+ mononuclear cells circulating in the blood of patients with LCH-ND. To determine whether senescence could also contribute to LCH-ND pathophysiology, we examined brain autopsy sections of a patient with fatally progressive ND-LCH17. Strikingly, we found multifocal aggregates of enlarged BRAF-V600E+ cells expressing monocyte–macrophage markers (CD14, CD163, CD33) and overexpressing the senescent marker p16INK4a, aggregating in areas of the white matter (Extended Data Fig. 4d,e). These cells also lacked the microglial marker (P2RY12, Extended Data Fig. 4f) and accumulated in perivascular regions, suggesting that senescent circulating MNPs that were recruited from the blood to the brain parenchyma, rather than brain-resident microglial cells, might have contributed to neurodegeneration in this patient.
In sum, these data suggest that the BRAFV600E mutation induces a senescence program in HPCs that persisted in differentiated LCH cells that accumulate in the peripheral tissues and brain parenchyma.
Pharmacological blockade of the mTOR pathway inhibits SASP induction and limits MNP differentiation of BRAF-V600E+ HPCs.
Our results above, suggesting that the secretome produced by BRAF-V600E+ HPCs contributed to the skewing of HPCs toward the MNP lineage, prompted us to examine whether SASP inhibition could help rescue HPC multilineage differentiation potential. The SASP includes a wide range of cytokines that can contribute to MNP skewing, and, therefore, targeting a specific cytokine might not be sufficient to obtain a therapeutic benefit. SASP induction in senescent cells is thought to be driven by the sustained activation of the mammalian target of rapamycin (mTOR) pathway18,19. Thus, we asked whether rapamycin, a potent mTOR inhibitor, could reduce MNP differentiation skewing of BRAF-V600E+ senescent HPCs by its ability to blunt the pro-inflammatory phenotype of senescent cells.
We first confirmed that inhibition of the mTOR pathway was sufficient to reduce the production of inflammatory cytokines by BRAF-V600E+ senescent HPCs in vitro (Fig. 5a). Importantly, and in line with our hypothesis, SASP inhibition significantly reduced BRAF-V600E+ HPC skewing into MNPs in vitro (Fig. 5b). Rapamycin treatment also partially reduced excess MNP differentiation from BRAF-V600E−GFP− HPCs co-cultured with senescent BRAF-V600E+GFP+ HPCs, further establishing that mTOR inhibition reduced the release of SASP-associated molecules driving HPC-sustained MNP differentiation potential (Fig. 5b). Of note, we did not observe any difference in expression of genes involved in the mTOR–phosphatidylinositol 3-kinase (PI3K) pathway (Extended Data Fig. 5a).
Fig. 5 |. SASP inhibition and eradication of senescent cells improve LCH outcome.
a,b, BRAF-V600Ehu-transduced CD34+ HPCs were cultured in stem cell medium and treated at day 1 and day 4 with rapamycin (10 nM) or dimethylsulfoxide (DMSO). a, Graphs show IL-1α and IL-1β protein levels in the supernatant of CD34+ HPCs 7 d after transduction (n = 4–5 independent donors). b, Percentages of MNPs (CD11c+ and/or CD14+) among total GFP+BRAF-V600E+ cells and among total GFP−BRAF-V600E− cells within cultures of BRAFV600E-transduced HPCs (BRAF-V600Ehu) that were treated with rapamycin or DMSO diluent were quantified by flow cytometry after 7 d of culture (n = 7 donors). c–e, BRAF-V600EScl+ mice were treated with rapamycin (0.5 mg per kg of body weight per day) or vehicle control for 10 d. Percentage of GMPs (defined as DAPI−lineage−c-Kit+Sca−CD16/CD32+CD34hi/int) among lineage-negative BM cells (c) and percentages of dendritic cells, macrophages and neutrophils among total CD45+ BM cells were measured using flow cytometry (n = 3–4 mice) (d). e, Liver weight in each group (n = 3–4 mice). Data are representative of two experiments. f, The percentage of tissue infiltration in the liver and lung is shown with representative images of H&E staining of tissues isolated from BRAF-V600EScl+ mice. g, Cartoon shows the experimental breeding scheme and treatment strategy used to generate BRAF-V600EScl+;ATTAC+ mice and control littermates. h,i, BRAF-V600EScl+;ATTAC+ mice and BRAF-WTScl+;ATTAC+ control littermates were treated with tamoxifen for 5 d and sacrificed 4 weeks later. h, The percentage of senescent p16INK4a-overexpressing cells was assessed by flow cytometry in the BM from BRAF-V600EScl+;ATTAC+ mice and control mice (n = 5–6 mice). Graphs show the percentage of GFP+ cells among live cells and among CD45+ cells, suggesting that senescent cells were exclusively hematopoietic cells. i, The nature of senescent p16INK4a-positive cells was assessed by flow cytometry. Graphs show the percentage of CD11b+ myeloid cells among GFP+ senescent cells and the percentage of macrophages, dendritic cells and neutrophils among GFP+CD11b+ myeloid senescent cells in BRAF-V600EScl+;ATTAC+ mice and control animals, highlighting that senescent cells in BRAF-V600EScl+;ATTAC+ mice are almost exclusively myeloid cells and that among them there is an accumulation of macrophages and dendritic cells, rather than neutrophils (n = 5–6 mice). j–l, Following tamoxifen injections, BRAF-V600EScl+;ATTAC+ mice and BRAF-WTScl+;ATTAC+ control littermates were treated with AP or vehicle for 3 weeks and sacrificed 4 weeks after tamoxifen injections. j, The percentage of senescent p16INK4a-overexpressing cells in the BM from untreated and treated BRAF-V600EScl+;ATTAC+ mice (n = 5) was assessed by flow cytometry. The graph shows the percentage of GFP+ cells among CD45+ cells. k, Liver weight in each group (n = 8). l, Percentages of tissue infiltration in the liver and lung are shown with representative images of H&E staining of tissues isolated from untreated and treated BRAF-V600EScl+;ATTAC+ mice (n = 8). Data are representative of three experiments and are represented as mean ± s.e.m.; statistical significance analyzed by unpaired and paired (for a,b) two-sided t-tests is indicated by *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Data are representative of at least two independent experiments.
Importantly, administration of rapamycin to BRAF-V600EScl+ mice reduced the accumulation of BM GMPs and MNPs, while BM neutrophils were not affected by the treatment (Fig. 5c,d and Extended Data Fig. 5b). Rapamycin administration reduced organomegaly in BRAF-V600EScl+ animals (Fig. 5e) and the inflammatory infiltrate in tissues (Fig. 5f), improving LCH disease. Of note, rapamycin did not induce apoptosis of BRAF-V600E+ cells (Extended Data Fig. 5c). While we cannot exclude the possibility that improved LCH disease symptoms in treated BRAF-V600EScl+ animals may also be linked to a direct effect of rapamycin on T cells infiltrating LCH lesions, the ability of rapamycin to inhibit the release of inflammatory cytokines from BRAFV600E HPCs and the MNP skewing induced by secreted factors from BRAF-V600E+ HPCs, in addition to rapamycin inhibition of GMP and MNP accumulation in the BM of BRAF-V600EScl+ animals, emphasize the strong contribution of mTOR-driven SASP to the increased MNP differentiation observed in mice and human BRAFV600E HPCs.
Elimination of senescent cells improves the clinical outcome of LCH-bearing mice.
To measure the exact contribution of senescence to LCH pathogenesis, we took advantage of mice expressing the INK-ATTAC transgene that includes an open reading frame coding for enhanced GFP and the FK506-binding protein–caspase 8 (FKBP–Casp8) fusion protein, expressed under the promoter of the Cdkn2a gene, which encodes the p16INK4a protein20. These mice enable Cdkn2a-p16INK4a+ senescent cells to be visualized based on GFP expression and conditionally deleted upon administration of AP20187 (AP), a synthetic drug that induces the dimerization of membrane-bound myristoylated FKBP–Casp8, leading to the apoptosis of Casp8-expressing cells.
INK-ATTAC mice were crossed with BRAF-V600Eca/ca;Scl-creER (BRAF-V600EScl+;ATTAC+) or BRAF-WTca/ca;Scl-creER mice (BRAF-WTScl+;ATTAC+) as a control group. Both groups received tamoxifen for 5 d to induce LCH lesions, and we measured the accumulation and composition of senescent cells in the BM (Fig. 5g and Extended Data Fig. 5d). In line with our previous results, we observed an accumulation of GFP+ senescent cells in the BM of BRAF-V600EScl+;ATTAC+ animals compared to that in BRAF-WTScl+;ATTAC+ control littermates (Fig. 5h). The majority of BM GFP+ senescent cells were skewed toward the MNP lineage and led to enhanced accumulation of senescent macrophages and dendritic cells in BRAF-V600EScl+;ATTAC+ mice compared to that in BRAF-WTScl+;ATTAC+ mice (Fig. 5i). BRAF-V600EScl+;ATTAC+ mice were then treated with AP to delete senescent cells for 3 weeks. AP treatment cleared GFP+ cells from the BM of BRAF-V600EScl+;ATTAC+ mice (Fig. 5j). Importantly, clearance of GFP+ senescent cells from BRAF-V600EScl+;ATTAC+ mice reduced organomegaly of LCH-bearing mice (Fig. 5k) and reduced the immune infiltrate in liver and lung tissues (Fig. 5l), showing that senescent cells are responsible for LCH pathophysiology.
Finally, to further examine whether pharmacological elimination of senescent cells can improve LCH outcome, we used ABT-263, an inhibitor of the anti-apoptotic proteins BCL-2 and BCL-xL, both of which are highly expressed in senescent cells. While not specific to senescent cells and potentially active in non-senescent neoplastic cells21, ABT-263 was shown to improve the clinical outcome of several senescence-associated diseases22–24. We previously showed that inhibition of BCL-xL can efficiently eliminate LCH cells25. We administered ABT-263 to BRAF-V600EScl+ animals or BRAF-WTScl+ control littermates for 3 weeks (Extended Data Fig. 5e). The diluent for ABT-263 prevents its systemic administration, and gavage of both ABT-263 and the diluent control are quite toxic to mice, making it difficult to measure the beneficial outcome of ABT-263 in treated mice. Nonetheless, and in accordance with the ex vivo experiment, we found specific clearance of BRAF-V600E+ cells (Extended Data Fig. 5f), reduced organomegaly (Extended Data Fig. 5g) and reduced immune lung infiltrate (Extended Data Fig. 5h) in BRAF-V600EScl+ mice treated with ABT-263 that survived. These results suggest that ABT-263 could provide a new clinical strategy to treat patients with LCH either alone or in combination with inhibitors of the BRAF kinase.
Discussion
Using genetically engineered mouse models, we demonstrated that expression of the BRAFV600E mutation in mouse and human HPCs is sufficient to drive the formation of LCH lesions with a tissue distribution comparable to that of human systemic LCH. We also showed that mouse and human HPCs with enforced BRAFV600E mutation, as well as primary human CD34+ hematopoietic progenitors isolated from patients with LCH, are in a senescent state and that BRAFV600E-induced senescence in hematopoietic progenitors contributes to LCH pathophysiology. Finally, using the INK-ATTAC transgene, we confirmed the accumulation of senescent cells in the BM and demonstrated that depletion of senescent cells in LCH-bearing mice improves disease outcome.
We previously generated a mouse model in which one allele of BRAFV600E was induced under the Cd11c (Itgax) promoter expressed by MNP progenitors and mature dendritic cells8. In that model, mice developed LCH-like disease in the spleen, lung and liver but only minimal disease in the skin and bones, which was surprising, as human LCH lesions frequently involve skin and bone tissues8. In the current study, we showed that, when the BRAFV600E mutation was expressed in pluripotent hematopoietic progenitors upstream of the MNP progenitors (BRAF-V600EScl+ mice), mice also developed skin lesions in the dermis that mimicked human LCH lesions, as well as granulomatous lesions, indicating that expression of the BRAFV600E mutation at the HSC level drives a more clinically relevant LCH phenotype than the disease that forms upon induction of the BRAFV600E mutation at the MNP progenitor stage.
We showed here that expression of the BRAFV600E oncogene in early hematopoietic progenitors promotes the induction of a senescence program and that senescence contributes to shaping the LCH phenotype. We also demonstrated that BRAFV600E HPCs have a much lower proliferative capacity in vitro and in vivo, which likely explains their decreased ability to engraft in competitive transplant assays when compared to WT HPCs (Fig. 3a). Instead, they are more prone to differentiate into pathogenic BRAFV600E senescent MNPs, which persist in tissues for prolonged periods of time, where they continue to release SASP-driven cytokines. The senescence program sheds light on key features of LCH disease, such as (1) the accumulation of poorly proliferative MNPs driven by the expression of the senescent-associated cell cycle inhibitor p16INK4a and (2) the large immune infiltrate, with subsequent fibrotic injuries (that is, to the central nervous system, including the pituitary gland and hepatic bile ducts) that accumulate in LCH lesions and lead to LCH morbidities, which we show are driven by SASP+ senescent LCH cells. In addition to inducing senescence, our previous findings, that BRAFV600E expression downregulates C–C motif chemokine receptor (CCR)7 expression in LCH cells and traps them in peripheral tissues, may further contribute to local pathogenic LCH cell accumulation25.
While deletion of CDKN2A or CDKN2B has not been described in LCH lesions, clonal deletion of CDKN2A or CDKN2B was described in Langerhans cell sarcoma26, a rare and highly aggressive disease defined by potent histiocyte proliferation with cytologic atypia and increased mitotic index. These results suggest that BRAFV600E-induced senescence likely protects LCH cells from malignant transformation, similar to what was observed in melanocytic nevi.
The nature of the inflammatory infiltrate in LCH lesions, while well known and widely described27, has remained poorly understood, prompting several groups to search for a microbial origin of LCH28,29. Here we showed that it is instead the SASP program that drives the release of inflammatory cytokines in BM HPCs and differentiated cells in the periphery. We also showed that senescence-driven cytokine release by BM HPCs drives the induction of an MNP differentiation program. The release of inflammatory cytokines not only promotes the differentiation of HPCs into MNP cells but also likely contributes to the development of granulomatous lesions in peripheral tissues, common in LCH lesions, and subsequent fibrotic injury. mTOR inhibition by rapamycin was shown to reduce SASP expression18,19. In accordance with these studies, we found that mTOR inhibition reduced HPC skewing toward the MNP lineage and improved disease phenotype in vivo. Altogether, our results showing that SASP contributes to enhanced myeloid cell differentiation in the BM could explain the myeloid-biased hematopoiesis observed in aged patients (reviewed in ref.30) and, if confirmed, could suggest that mTOR inhibition in HPCs could help restore immune balance and homeostasis in older patients.
Systemic LCH remains a difficult-to-treat condition; BRAF or MEK kinase inhibition therapies can improve clinical outcome, although this approach is not curative in most cases, as most patients recur upon treatment interruption31–33. Our study strongly suggests that mTOR inhibitors and senolytics may provide a critical alternative to cytotoxic chemotherapy and chronic MAPK pathway inhibition for the treatment of patients with LCH, including those with LCH-ND.
Altogether, these results reveal the contribution of BRAFV600E-driven senescence to LCH pathophysiology and identify senescent cells as an important target for the treatment of systemic LCH.
Methods
Animal experiments.
All animal experiments performed in this study were approved by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. BRAF-V600EScl+, BRAF-WTScl+, BRAF-V600EScl− and BRAF-WTScl− mice were created by crossing BRAF-V600Eca/ca mice (provided by M.W. Bosenberg, Yale University35) with mice expressing an inducible Cre recombinase under the control of the Scl promoter (provided by Y. Fujiwara, Harvard Medical School, Howard Hughes Medical Institute12). BRAF-V600Eca/−; Scl-creER mice were then crossed with Rosa26YFP/YFP mice (C57BL/6 background; Jackson Laboratory). To induce the Cre recombinase, BRAF-V600EScl+, BRAF-WTScl+ or BRAF-V600EScl− control littermates were injected intraperitoneally with tamoxifen for 5 consecutive days (5 mg, day 1; 2 mg, days 2 and 3; 1 mg, days 4 and 5). All animals were housed under specific pathogen-free conditions and sacrificed at the indicated time points. All experiments were controlled using Cre-positive littermates negative for the BRAFV600E construct or littermates negative for the Cre recombinase transgene construct. BRAFV600E or BRAFWT chimeras were generated by transplantation of 1–3 million whole BM cells flushed from the femurs of 8-week-old BRAF-V600EScl+ or BRAF-WTScl+ mice following tamoxifen injections into lethally irradiated (650 cGy × 2) CD45.1 mice aged 8–12 weeks (C57BL/6 background, Charles River Laboratories). Mice were kept on sulfamethoxazole–trimethoprim (STI Pharma) for 3 weeks. Mice were allowed to recover for 2 weeks after transplantation, before initiation of drug treatments.
BRAF-V600EScl+;ATTAC+ and BRAF-WTScl+;ATTAC+ mice were generated by crossing litters from BRAF-V600Eca/ca;Scl-creER mice with ATTAC+/− mice (kindly provided by L. Galuzzi, Weill Cornell Medical College University). The Cre recombinase was induced as described earlier.
NSG mice were purchased from Jackson Laboratory (BALB/c background) and were housed under specific pathogen-free conditions. Humanized mice were generated by transplanting 200,000 human CD34+ cells previously transduced for 16–24 h with the BRAFV600E or NGFR lentiviral vector into 8-week-old NSG mice sublethally irradiated with 250 cGy.
Human specimens.
Human tissues used in these studies were collected, stored and processed according to protocols approved by the Baylor College of Medicine Institutional Review Board. Healthy control tissues were obtained from the Biorepository and Pathology CoRE facility (K. Donovan) at Mount Sinai School of Medicine. Cord blood was obtained from the New York Blood Center, National Cord Blood Program.
Lentiviral vector construction.
BRAFV600E and NGFR transgenes were cloned into lentiviral expression vectors upstream of a 2A peptide linked to GFP for in vivo and in vitro analysis of transduced cells using standard cloning procedures. Briefly, PCR products were amplified from the pcDNA3 plasmid (provided by P.I.P.) using high-fidelity Taq polymerase. PCR products were purified using the QIAquick PCR purification kit (Qiagen), and restriction enzymes SalI and XbaI were used to digest the vector and PCR products. PCR products were ligated with the expression vector using NEB Quick Ligase, and plasmids were verified by Sanger sequencing.
Lentiviral vector production and titration.
Lentiviral vectors were produced as previously described36. Briefly, 293T cells were seeded approximately 24 h before transfection in a 15-cm plate and incubated at 37 °C with 5% CO2. The following day, cells were transfected with a third-generation packaging system (pVSV, pMDLg/pRRE, pRSV-REV) and the appropriate transfer plasmid using calcium phosphate. The medium was changed 14 h later. Another 30 h later, the supernatant containing the vector was collected, passed through a 0.22-μm filter and ultracentrifuged (20,000 r.p.m. for 2 h) to concentrate the vector 300–500-fold. The vector particles were resuspended in sterile PBS, aliquoted and stored at −80 °C. Vector titration was performed on 293T cells using the limiting dilution method.
Cord blood processing and transduction.
Mononuclear cells from cord blood were isolated via a Ficoll-Paque PLUS (GE Healthcare) gradient. Mononuclear cells were washed and resuspended in FACS buffer (PBS without Ca2+ and Mg2+, supplemented with 2% heat-inactivated FBS and 5 mM EDTA). CD34+ cells were purified using a Human CD34 MicroBead kit from Miltenyi Biotec (130-100-453).
CD34+ human HPCs were resuspended at a concentration of 1 million cells per ml in StemSpan medium SFEM II (Stemcell Technologies, 09605) supplemented with 100 ng ml−1 SCF, 100 ng ml−1 FLT3L and 50 ng ml−1 TPO (so-called stem cell medium). Two hundred thousand cells were plated in a 48-well plate, and 5 μl lentiviral construct (either BRAFV600E or NGFR) was added to the medium at an MOI of 50. Cells were incubated at 37 °C with 5% CO2. Supplemented StemSpan medium was added 24 h after the initiation of transduction, and cells were kept in culture for up to 14 d. GFP-transduced cells were analyzed for myeloid and lymphoid surface marker expression (see antibody list) starting 2 d after transduction.
Flow cytometry.
Adult skin (from the ears), lung, liver, spleen and femurs were dissected from mice at the age of 12–16 weeks. Adult skin was first incubated overnight at 4 °C in Dispase II solution (Roche) (2.5 mg ml−1 in PBS) with the dermal side facing down. The dermis was separated mechanically from the epidermis, and both tissues were digested in a solution of collagenase D (Roche) (1 mg ml−1) and DNase I (Roche) (1 mg ml−1) in RPMI (Corning) with 10% FBS for 1 h at 37 °C. After digestion, the dermis and epidermis were homogenized using an 18G needle. The cell suspension was filtered through a 70-μm cell strainer into a flow cytometry tube.
For the spleen and the lung, tissues were enzymatically digested in a solution of collagenase D (Roche) (1 mg ml−1) and DNase I (Roche) (1 mg ml−1) in HBSS (Corning) for 30 min at 37 °C, followed by mechanical trituration with an 18G blunt-tipped syringe and filtration through a 70-μm filter.
Single-cell suspensions from livers were obtained after digestion with collagenase IV (C5138, Sigma) in HBSS (Corning) at 37 °C for 45 min. Non-parenchymal cells were enriched by centrifugation in 35% Percoll (17-0891-01, GE Healthcare) for 30 min at 1,300 r.c.f.
BM single-cell suspensions were obtained after flushing out the BM from the femur using a 27G needle and then were incubated with ACK lysis buffer (420301, BioLegend) for 3 min at room temperature. Blood was drawn from the liver sinus and treated twice with ACK lysis buffer for 5 min at room temperature.
For flow cytometry, cells were stained in FACS buffer with flow cytometry monoclonal antibodies for 20 min at 4 °C.
Murine HPCs were sorted as DAPI−lineage−c-Kit+Sca1− singlets, and murine LSK+ cells were sorted as DAPI−lineage−c-Kit+Sca1+ singlets. Human transduced HPCs were sorted as DAPI−, GFP+ or GFP− singlets.
Flow cytometry was performed using a Fortessa analyzer (BD), and sorting was performed using a FACSAria II (BD) or an LSR II (BD). Flow cytometry data analysis was performed using FlowJo (TreeStar) software. Antibodies used are listed in Table 1.
Table 1 |.
Primary antibodies used for flow cytometry
| Markers | Reactivity | Clone | Company | Dilution |
|---|---|---|---|---|
| CD16/CD32 | Mouse | 93 | eBioscience | 1:200 |
| Sca1 | Mouse | D7 | eBioscience | 1:200 |
| c-Kit | Mouse | 2B8 | eBioscience | 1:200 |
| FLT3L | Mouse | A2F10 | eBioscience | 1:100 |
| CD34 | Mouse | RAM34 | eBioscience | 1:100 |
| CD11b | Mouse | M1/70 | eBioscience | 1:100 |
| Ter119 | Mouse | TER-119 | eBioscience | 1:200 |
| Gr1 | Mouse | RB6–8C5 | eBioscience | 1:200 |
| B220 | Mouse | RA3–6B2 | eBioscience | 1:200 |
| CD115 | Mouse | AFS98 | eBioscience | 1:200 |
| CD45 | Mouse | 30-F11 | BioLegend | 1:200 |
| Ly6G | Mouse | 1A8 | BioLegend | 1:200 |
| MHCII | Mouse | M5/114.15.2 | eBioscience | 1:200 |
| CD11c | Mouse | N418 | Invitrogen | 1:200 |
| Ly6C | Mouse | A2–21 | eBioscience | 1:200 |
| F4/80 | Mouse | BM8 | BioLegend | 1:200 |
| Siglec F | Mouse | E50:2440 | BD Biosciences | 1:200 |
| CD24 | Mouse | M1/69 | BD Biosciences | 1:200 |
| CD103 | Mouse | 2E7 | BioLegend | 1:200 |
| EpCAM | Mouse | G8.8 | eBioscience | 1:200 |
| CD4 | Mouse | RM4.5 | eBioscience | 1:200 |
| CD8 | Mouse | 5H10 | Invitrogen | 1:200 |
| CD3 | Mouse | 145–2C11 | BD Biosciences | 1:200 |
| NK1.1 | Mouse | PK136 | eBioscience | 1:200 |
| CD45 | Human | 2D1 | eBioscience | 1:200 |
| CD34 | Human | 581 | BioLegend | 5:100 |
| CD38 | Human | HIT2 | BD Biosciences | 3:100 |
| CD123 (IL3Rα 6) | Human | 6H6 | BioLegend | 1:100 |
| CD11c | Human | B-ly6 | BD Horizon | 1:100 |
| CD1a | Human | HI149 | BioLegend | 1:100 |
| CD14 | Human | MHCD1405 | Invitrogen | 1:100 |
| CD207 | Human | IM3577 | Beckman Coulter | 1:100 |
Multiplexed immunohistochemical consecutive staining on a single slide.
Tissues were fixed with 4% formaldehyde for 24 h and embedded in paraffin. Five-mm-thick FFPE tissue sections on glass slides were baked at 37 °C overnight, deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol. Next, tissue sections were incubated in retrieval solution (pH 6 or 9) for antigen retrieval at 95 °C for 30 min. Tissue sections were incubated in 3% hydrogen peroxide and in serum-free protein block solution (Dako, X0909) before adding the primary antibody for 1 h at room temperature. After signal amplification using the EnVison+ System HRP Labeled Polymer anti-mouse (Dako, K4001) or anti-rabbit (Dako, K4003) antibodies, chromogenic revelation was performed using 3-amino-9-ethylcarbazole (Vector Laboratories, SK4200). Slides were counterstained with hematoxylin, mounted with a glycerol-based mounting medium (Dako, C0563) and scanned for digital imaging (Hamamatsu NanoZoomer S60 Whole Slide Scanner). Next, the same slides were successively stained as described previously37. Primary antibodies used are listed in Table 2.
Table 2 |.
Primary antibodies used for multiplexed immunohistochemical consecutive staining
| Marker | Reactivity | Reference | Clone | Antigen retrieval | Dilution | Incubation |
|---|---|---|---|---|---|---|
| Langerin | Human | Novocastra | 12D6 | pH 9 | 1:50 | 1 h at RT |
| CD1a | Human | Dako | O10 | pH 6 | 1:50 | 1 h at RT |
| Ki-67 | Human | Ventana | 30–09 | pH 9 | RTU | 1 h at RT |
| CD207 | Mouse | eBioscience | eBioRMUL.2 | pH 9 | 1:200 | Overnight at 4°C |
| Ki-67 | Mouse | Bethyl | IHC-00375 | pH 9 | 1:250 | Overnight at 4°C |
| p16INK4a | Human | Santa Cruz Biotechnology | Sc-759 | pH 9 | 1:50 | 1 h at RT |
| p16INK4a | Human | Ventana | BioHistomarker | CC1 | N/A | |
| CD33 | Human | Leica | PWS44 | CC1 | 1:100 | |
| CD163 | Human | Novocastra | 10D6 | CC1 | 1:200 | |
| CD14 | Human | Abcam | 7 | CC1 | 1:10 | |
| BRAF V600E | Human | Ventana | VE1 | CC1 | 1:50 | |
| P2RY12 | Human | Sigma | N/A | Target Retrieval Solution (Dako) | 1:1,000 |
CC1, cell conditioning 1; N/A, not available; RT, room temperature; RTU, ready to use.
Images were analyzed (automatic analysis) using QuPath software (https://qupath.github.io/).
Senescence-associated β-galactosidase staining.
Cells or frozen tissues were stained for SAβGal using the Senescence Detection kit (BioVision, K320-250) according to the manufacturer’s instructions. Senescent cells were identified as blue-stained cells by light microscopy. Images were taken using a Hamamatsu NanoZoomer S60 Whole Slide Scanner. Images were analyzed using QuPath software (https://qupath.github.io/).
Western blot.
Cells were lysed with lysis buffer (50 mM Tris, pH 7.5, 1% NP-40, 150 mM NaCl, 10% glycerol, 1 mM EDTA, supplemented with protease inhibitor cocktail tablets (Roche)). After a 5-min incubation on ice, lysates were centrifuged at 15,000 r.p.m. for 10 min, and supernatants were collected. Protein concentrations were quantified using BCA (Pierce). Proteins were separated with NuPAGE 4–12% Bis-Tris gels (Novex) and transferred to nitrocellulose membranes (10600006, Amersham) according to standard protocols. Membranes were immunoblotted overnight with primary antibodies against pERK (4370L, Cell Signaling), BRAF (9002, Santa Cruz) and β-actin (5125S, Cell Signaling), followed by secondary anti-rabbit IgG antibody (7074S, Cell Signaling). Chemiluminescent signals were detected on X-ray film.
Methylcellulose assays.
To determine the differentiation potential of murine progenitors, femurs from BRAF-V600EScl+ or BRAF-WTScl+ control littermates were collected. Lineage-negative BM cells were obtained with a lineage cell depletion kit (MACS, Miltenyi Biotec, 130-090-858). One thousand lineage-negative BM cells (or sorted lineage-negative RosaYFP+ BM cells, depending on the experiment) were resuspended in IMDM (Gibco) supplemented with 2% FBS, plated in 1 ml methylcellulose medium (MethoCult M3434, Stemcell Technologies) and kept in culture for 14 d. Numbers and types of CFUs were assessed at day 14.
To determine the differentiation potential of human progenitors, 500 transduced CD34+ HPCs were sorted according to GFP expression levels 3 d after transduction, resuspended in IMDM (Gibco) supplemented with 2% FBS and plated in 1 ml methylcellulose medium (MethoCult H4434, Stemcell Technologies) for 14 d. Numbers and types of CFUs were assessed at day 14.
Gene expression profiling using microarrays and RNA-seq.
One hundred thousand human transduced HPCs were sorted according to GFP expression levels directly into Arcturus PicoPure Extraction Buffer (Applied Biosystems, 12317-03) 7 d after transduction with BRAFV600E or NGFR lentiviral constructs. Total RNA was processed from the sorted cells according to the PicoPure RNA Isolation kit protocol (Molecular Devices). RNA concentration and quality were verified using the 6000 Pico Chip (Agilent) at the Baylor College of Medicine Microarray Facility. cDNA amplification was performed with the WT-Ovation Pico System, according to the manufacturer’s protocol (NuGen). Fragmented and biotinylated cDNA samples for the gene chip studies were generated using the FL-Ovation cDNA Biotin Module version 2 (NuGEN).
For Fig. 4b, total RNA was isolated from sorted subpopulations from peripheral blood and lesion specimens according to the Arcturus PicoPure RNA Isolation kit protocol (Applied Biosystems). RNA quality was verified using the Pico Chip at the Baylor College of Medicine Microarray Facility (all RIN > 6). cDNA amplification was performed using the Ovation Pico WTA version 2 system according to the manufacturer’s protocol (NuGEN). Fragmented and biotinylated cDNA was hybridized to the GeneChip Human Transcriptome Array 2.0 according to the manufacturer’s procedures (Affymetrix). Raw data from all samples were normalized using the SST-RMA algorithm implemented in the Affymetrix Expression Console. Differentially expressed genes were identified using BRB-ArrayTools version 4.5 (version 4.5, Biometric Research Branch, NCI) with a multivariate permutation test with a confidence level of false discovery rate assessment at 80% and the maximum allowed proportion of false-positive proteins at 0.1.
Human CD34+ BM cells from patients with LCH were sorted directly into TRIzol LS Reagent (Ambion, Life Technologies, 10296028) and stored at −80 °C. RNA was extracted according to the RNeasy Mini kit protocol (Qiagen). RNA (1 ng) was converted to cDNA using the SMART-Seq version 4 Ultra Low Input RNA kit (Clontech). RNA-seq libraries were prepared using the Low Input Library Prep kit version 2 (Clontech). Sequencing was performed using the Illumina NextSeq 500 system. Transcript abundance was quantified with the Ensembl GRCh38 cDNA reference using Kallisto version 0.43.0.
Quantitative PCR.
Conventional reverse transcription was performed using the RNA to cDNA EcoDry Premix (639548, Takara Bio) in accordance with the manufacturer’s instructions. qPCR was performed on a CFX384 Touch Real-Time PCR detection system (Bio-Rad) using EXPRESS SYBR GreenER master mix (11784200, Invitrogen) as follows: one cycle at 95 °C (10 min) and 40 cycles of 95 °C (15 s) and 58 °C (1 min). The expression of actin was used to normalize expression. The average threshold cycle number for each tested mRNA was used to quantify the relative expression: 2(Ct(Actb)−Ct(Cdkn2a)). Primers are listed below in Table 3.
Table 3 |.
Primers used for qPCR
| 5′-actin (Actb) | CTAAGGCCAACCGTGAAA |
| 3′-actin | ACCAGAGGCATACAGGGA |
| 5′-Cdkn2a | CCCAACGCCCCGAACT |
| 3′-Cdkn2a | GCAGAAGAGCTGCTACGTGAA |
| 5′-Il1a | CCATAACCCATGATCTGGAAGAGAC |
| 3′-Il1a | GTCCACATCCTGATATATAGTTTG |
| 5′-Il1b | TGGCAACTGTTCCTGAAC |
| 3′-Il1b | GGGTCCGTCAACTTCAAA |
| 5′-Il6 | GCTACCAAACTGGATATA |
| 3′-Il6 | CCAGGTAGCTATGGTACT |
| 5′-Mmp13 | GGAGCCCTGATGTTTCCCAT |
| 3′-Mmp13 | GTCTTCATCGCCTGGACCATA |
Proliferation assays.
For in vivo assay, BRAF-V600EScl+ or control littermate mice aged 8 weeks were injected intraperitoneally with 1 mg BrdU for 7 consecutive days. Femurs were collected 24 h after the last injection and processed into single-cell suspensions for BrdU incorporation by flow cytometry analysis. Tissue preparation and BrdU staining were performed according to the APC BrdU Flow kit protocol (BD PharMingen, 557892).
For in vitro assays, BRAFV600E- or NGFR-transduced CD34+ HPCs were kept in culture in StemSpan SFEM II medium (Stemcell Technologies). Transduced CD34+ HPCs were incubated from day 10 to day 13 with 10 μM BrdU supplemented daily in cell culture medium. Transduced HPCs were collected at day 1 and analyzed for BrdU incorporation by flow cytometry using the same kit used for the in vivo experiment.
Cell Trace Violet.
Transduced CD34+ HPCs were labeled with 1 μM CFSE (Life Technologies) in PBS with 0.1% BSA for 10 min at 37 °C. Cells were analyzed 92 h after treatment with Cell Trace Violet.
Competitive transplantation assays.
Femurs from 12-week-old BRAF-V600EScl+ or BRAF-WTScl+ mice were collected 4 weeks after tamoxifen injections. Lineage-negative BM cells from either BRAF-V600EScl+ or BRAF-WTScl+ mice (both CD45.2+) were obtained using a MACS mouse lineage cell depletion kit (Miltenyi Biotec) and were injected intravenously together with lineage-negative BM cells of the host type (CD45.1+) into lethally irradiated CD45.1+ mice (1,200 rad delivered in two doses of 600 rad each, 3 h apart) at a ratio of 2:1 (200,000 CD45.2+ cells:100,000 CD45.1+ cells). Levels of blood donor chimerism were analyzed by measuring the percentage of CD45.2+ cells among CD45+ cells 4 and 8 weeks after transplantation.
Enzyme-linked immunosorbent assay.
BM was prepared by flushing two femurs with 500 μl PBS. After pelleting the cells, supernatants were used for BM extra-fluid measurements and stored at −80 °C. For detecting and quantifying proteins, commercially available ELISA kits for IL-1α (R&D Systems, DY400), IL-1β (R&D Systems, DY401) and IL-6 (R&D Systems, DY406) were used.
Supernatants from transduced CD34+ HPCs were collected at different time points (days 7, 9 and 14) and stored at −80 °C. For measurement of proteins levels, commercially available ELISA kits for IL-1α (BioLegend, 445807), IL-1β (BioLegend, 437007), IL-6 (BioLegend, 430507) and IL-8 (BioLegend, 431507) were used.
For human plasma specimens, blood was collected from pre-therapy pediatric patients with multisystem LCH and healthy pediatric controls in EDTA tubes and centrifuged at 400g for 30 min over a Ficoll gradient, and then the plasma supernatant was collected and recentrifuged for 5 min, and then purified supernatant was stored at −80 °C. None of the plasma samples analyzed underwent more than two freeze–thaw cycles. Protein levels were determined using the MAGPIX instrument (Luminex). The concentration of each analyte was measured by comparison to the protein standards. Levels of IL-6 and IL-8 were measured with the Millipore Human Cytokine/Chemokine Panel I (1:2 dilution).
Therapeutic trials.
Rapamycin in vitro.
Rapamycin (InvivoGen, tlrl-rap) was added at a concentration of 10 nM at day 1 and day 4 following transduction of CD34+ HPCs. DMSO was used as a mock treatment. GFP-transduced cells were analyzed for myeloid surface marker expression (see antibody list) at day 8 post-transduction.
Rapamycin in vivo.
Mice were treated with rapamycin or vehicle via intraperitoneal injection (diluted in 200 μl PBS per mouse) at 0.5 mg per kg of body weight per day for 10 consecutive days, starting 15 d after the generation of BRAF-WTScl+ or BRAF-V600EScl+ BM chimera. Mice were sacrificed at day 30.
AP20187 in vivo.
Mice were treated with vehicle (4% ethanol:10% polyethylene glycol 400:2% Tween-20 in distilled water) or AP (purchased from Takara Bio (635069) and diluted in 4% ethanol:10% polyethylene glycol 400:2% Tween-20 in distilled water). AP or diluent was administered every Monday, Wednesday and Friday (triweekly) for 3 consecutive weeks by gavage at 10 mg per kg of body weight per day, following tamoxifen injections.
ABT-263 (navitoclax) in vivo.
Mice were treated with diluent (10% ethanol:30% polyethylene glycol 400:60% Phosal 50 PG) or ABT-263 (diluted in 10% ethanol:30% polyethylene glycol 400:60% Phosal 50 PG). ABT-263 and vehicle were administered to mice by gavage at 50 mg per kg of body weight per day for 3 consecutive weeks.
Extended Data
Extended Data Fig. 1 |. Langerhans Cell Histiocytosis mouse models.
a-f, BRAFV600EScl+ mice and BRAFwtScl+ mice were generated as described in Fig. 1a. a, Representative images of spleen, lung, and femurs at 4 weeks post tamoxifen injections. b, Liver and spleen weights. c, Absolute number of BM cells in BRAFV600EScl+ mice and control littermates (n = 10 mice per group). d, Hematoxylin and eosin staining and CD207 immunohistochemistry staining of tissues isolated from BRAFwtScl+ mice (n = 2–3 mice per group). e, Absolute numbers of lung, dermal and epidermal immune cells in BRAFV600EScl+ mice and control littermates. Data are representative of 3 experiments (n = 3–8). f, Percentage of immune cell populations among lung and skin infiltrating RosaYFP+ cells in BRAFV600EScl+ mice and control littermates. Data are representative of 3 experiments (n = 3–8). g, Scheme of the lentiviral vector constructs used to transduce human CD34+ HPC. h, Graph shows that the transduction efficiency of BRAFV600E and NGFR lentiviral constructs in human CD34+ HPC does not exceed 40 %. i, Western blot of BRAF and phospho-ERK performed on purified BRAFV600Ehu and NGFRhu human CD34+ HPC 7 days after transduction. j, Representative flow plot showing the percentage of CD11c+ CD14+ MNP among circulating blood cells in NSG mice reconstituted with BRAFV600Ehu and NGFRhu HPC. Graph represents the percentage of CD11c and or CD14+ cells among human CD45+ cells from NSG circulating blood (n = 2–3 mice per group). Data are represented as mean ± s.e.m; statistical significance analyzed by an unpaired two-sided t-test is indicated by *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Extended Data Fig. 2 |. Myeloid skewing of BRAFV600E+ hematopoietic progenitors.
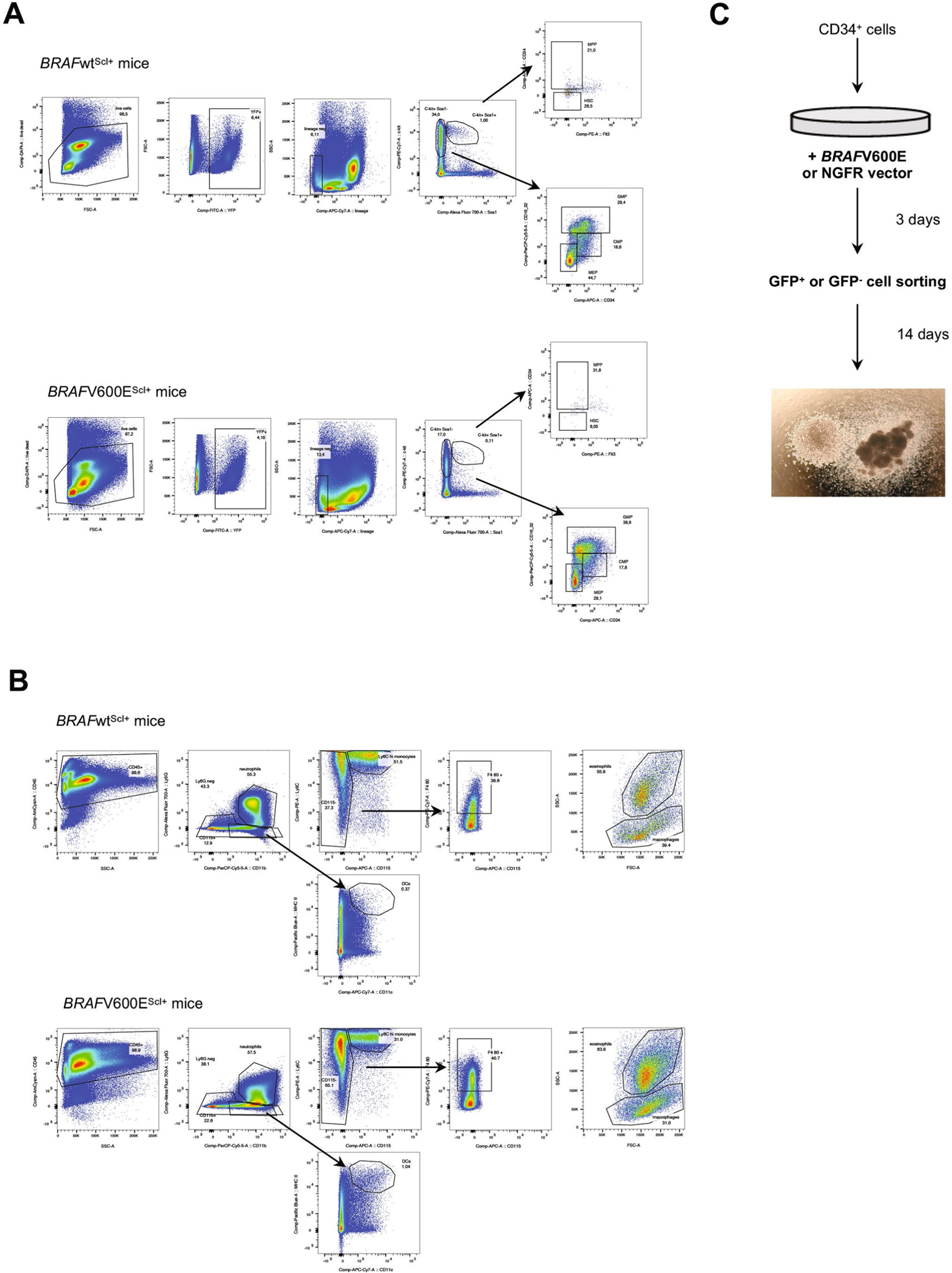
a, Flow cytometry gating strategy for hematopoietic progenitors (HSC: hematopoietic stem cell, MPP: multipotent progenitors, GMP: Granulocyte-macrophage progenitors, CMP: common myeloid progenitors, MEP: megakaryocyte-erythroid progenitors) in BRAFwtScl+ and BRAFV600EScl+ animals. b, Flow cytometry gating strategy for bone marrow myeloid cells in BRAFwtScl+ and BRAFV600EScl+ animals. c, Experimental design of the CFU assays performed in Fig. 2e is shown.
Extended Data Fig. 3 |. Senescence program in different LCH models.
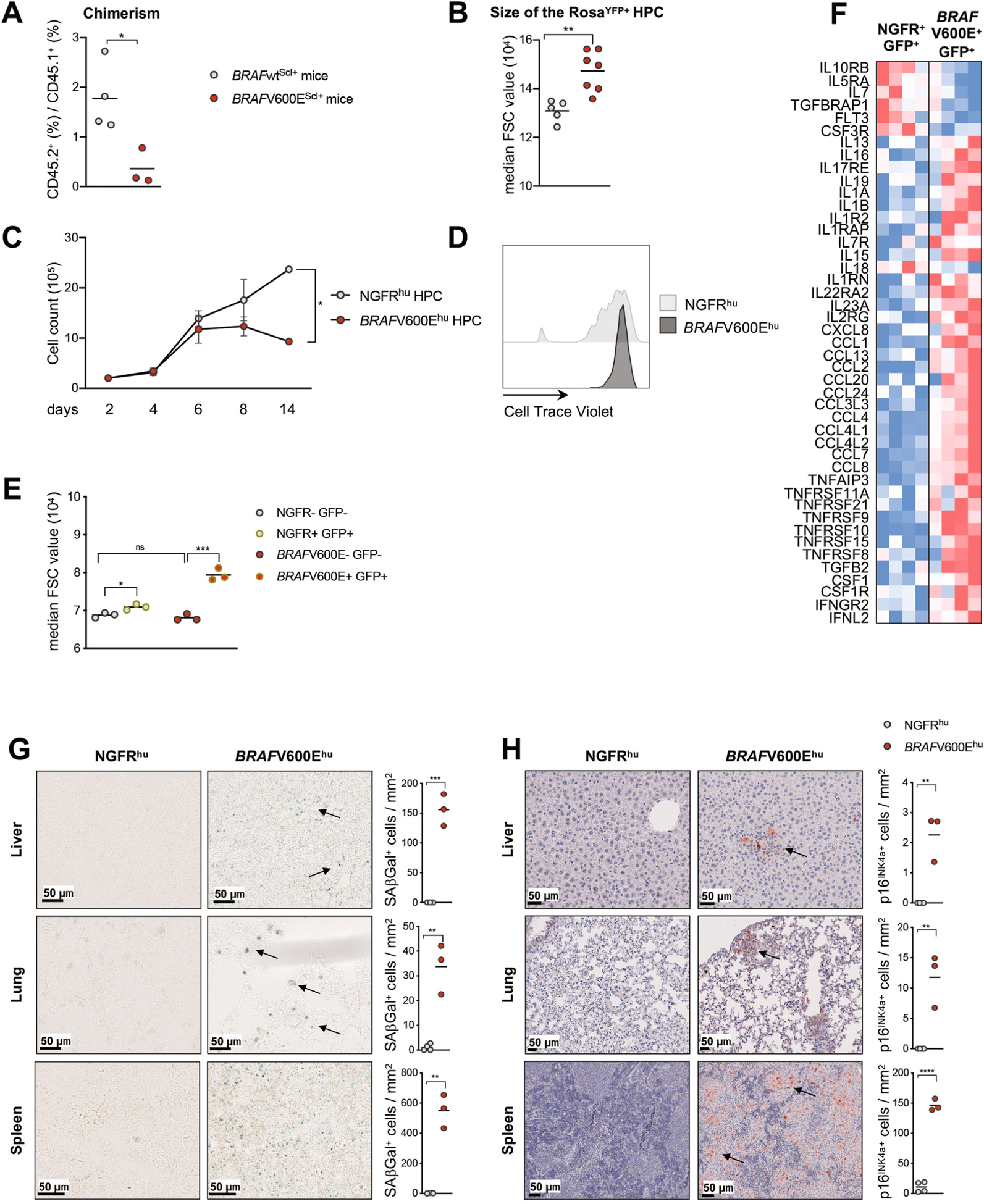
a, Lineage negative BM cells isolated from either BRAFV600EScl+ CD45.2+ or BRAFwtScl+ CD45.2+ mice were injected intravenously together with lineage negative CD45.1+ BM cells into lethally irradiated CD45.1+ mice at a 2:1 ratio (CD45.2+: CD45.1+). Graph shows the ratio of CD45.2+ / CD45.1+ in blood circulation in each group measured 8 weeks after transplantation using flow cytometry (n = 8). b, Forward scatter measurement of lineage negative RosaYFP+ cells in the BM from BRAFV600EScl+ mice and BRAFwtScl+ control littermates (n = 5–7 mice per group). c, Expansion of human CD34+ cord blood HPC transduced with NGFR (NGFRhu) or BRAFV600E (BRAFV600Ehu) lentiviral constructs and cultured in stem-cell media. Data are representative of at least 3 experiments (n = 3 donors). d, Proliferation capacity of NGFRhu and BRAFV600Ehu HPC analyzed by Cell Trace Violet dilution, 92 hours after staining. e, Forward scatter measurement of NGFRhu and BRAFV600Ehu HPC (n = 3 different donors) f, Heat map representation of SASP genes analyzed by microarray expression profiling of BRAFV600E+ GFP+ HPC and NGFR+ GFP+ HPC 7 days after transduction (n = 4 different donors). g-h, Frozen or FFPE tissue sections isolated from humanized mice reconstituted with BRAFV600Ehu and NGFRhu HPC and stained for g, SAβGal activity or (h) p16INK4a. Graph shows the number of positive cells per mm2. Data are represented as mean ± s.e.m; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (unpaired two-sided t-tests, two-way ANOVA test for Extended Data Fig. 2c).
Extended Data Fig. 4 |. Human LCH lesions show features of senescence.
a, Heat map representation of SASP associated gene expression measured by RNA sequencing in purified CD34+ BM cells isolated from three LCH patients and one age-matched healthy donor (performed as duplicates). b, Skin from LCH patient and healthy age-matched donor were stained for CD207 and p16INK4a by immunohistochemistry. c, SAβGal activity of human tissues and LCH lesions. One representative image of healthy skin, naevus and LCH lesion are shown. Graph shows the number of SAβGal positive cells per mm2 in healthy skins (n = 3), naevus (n = 1) and LCH lesions (n = 3). d, Autopsy brain sections from a patient with neurodegenerative-LCH and circulating BRAFV600E+ peripheral blood cells. The H&E image shows a temporal lobe white matter injury with increased numbers of infiltrating plump cells. The immunohistochemical characterization of the enlarged plump cells shows a positivity for CD163, CD33, CD14, BRAFVE1 and p16INK4a. (*indicates perivascular space). e, Control tissue with rare p16INK4a positivity by immunohistochemistry in meningioma and no appreciable staining in human brain (non-diseased from autopsy). f, Brain section from human patient with neurodegenerative-LCH (see Fig. 4g) dual-stained for P2RY12 (green) and CD163 (red). Spindled P2RY12+ microglial cells surround the aggregates of plump activated CD163+ macrophages. There is no dual staining. There is diminished P2RY12 staining within the CD163+ macrophage rich aggregate. Image courtesy of Dr. Julia Kofler, MD UPMC Division of Neuropathology. Data are represented as mean ± s.e.m; statistical significance analyzed by an unpaired two-sided t-test is indicated by *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Extended Data Fig. 5 |. Rapamycin and senolytic treatments improve LCH outcome.
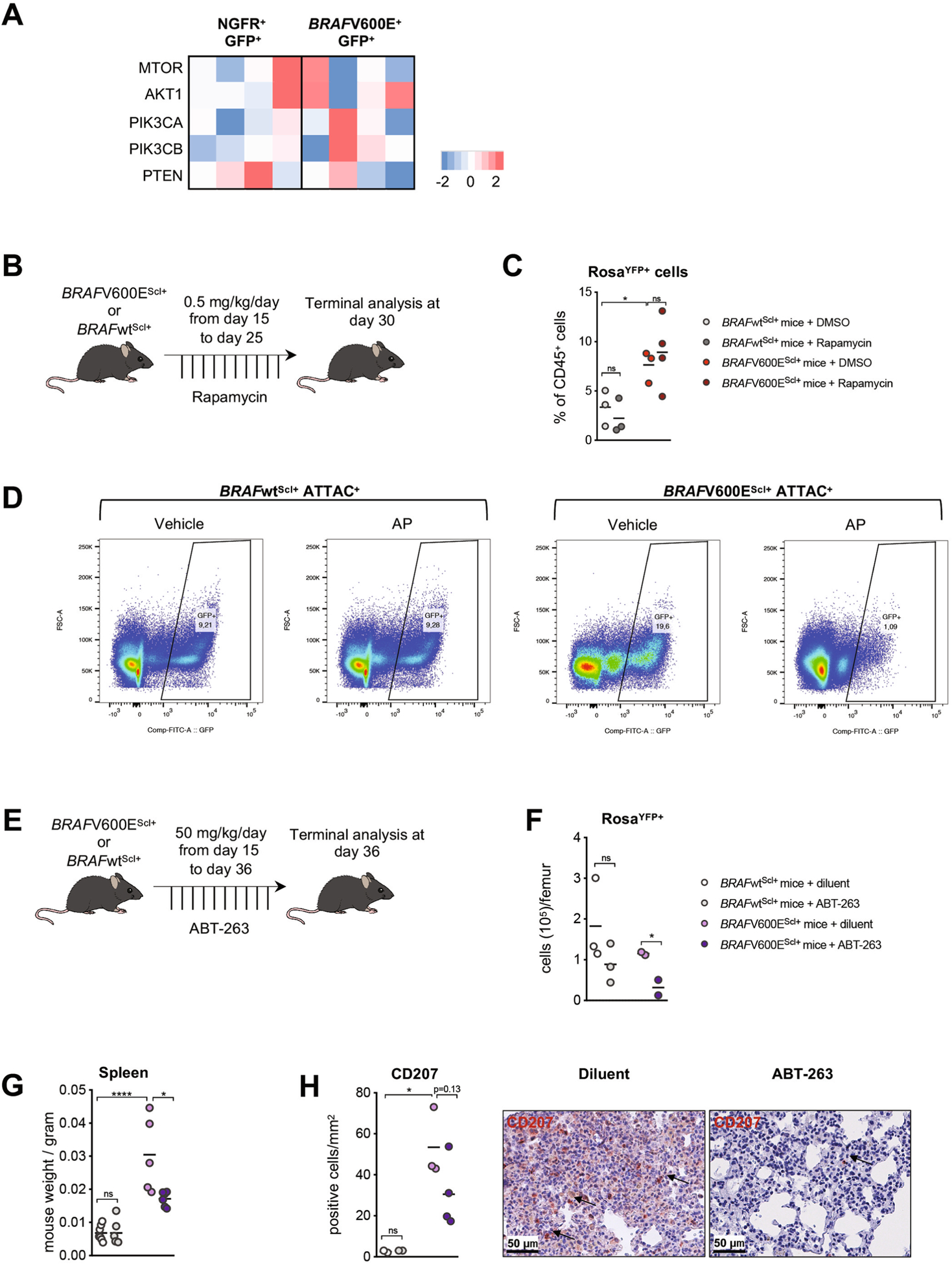
a, Human CD34+ cord blood HPC were transduced with BRAFV600E (BRAFV600Ehu) or NGFR control (NGFRhu) lentiviral vectors, cultured in stem-cell media and analyzed for gene expression of PI3K pathway genes using microarray expression profiling 7 days after transduction (n = 4 donors); b-c, BRAFV600EScl+ mice and BRAFwtScl+ control littermates were treated with rapamycin (0.5 mg/ kg body weight/ day) or DMSO for 10 days. b, Cartoon shows the experimental design to test the clinical benefit of rapamycin on LCH in mice. b, Graph shows the percentage of RosaYFP+ cells among CD45+ BM cells (n = 3–4 mice per group). d, BRAFV600EScl+ ATTAC+ mice and BRAFwtScl+ ATTAC+ control littermates were treated by tamoxifen for five days. Following tamoxifen injections, BRAFV600EScl+ ATTAC+ mice and BRAFwtScl+ ATTAC+ control littermates were treated by AP or vehicle during three weeks and sacrificed 4 weeks after tamoxifen injections. Representative flow plot showing the percentage of GFP+ cells among CD45+ live cells in the bone marrow from BRAFV600EScl+ ATTAC+ mice and BRAFwtScl+ ATTAC+ control littermates. e-h, BRAFV600EScl+ mice and BRAFwtScl+ control littermates were treated with ABT-263 (50 mg/ kg body weight/ day) or diluent for 21 days. e, Cartoon shows the experimental design to test the clinical benefit of ABT-263 on LCH in mice. f, Graph shows the absolute numbers of bone marrow RosaYFP+ cells (n = 2–3) g, Spleen weight and (h) lung infiltrating CD207+ cells isolated from mice treated with ABT-263 or diluent (n = 3–4) with representative images of lung tissue sections isolated from mice treated with ABT-263 or diluent and stained with CD207. Data are representative of 2 experiments (n = 3–4). Data are represented as mean ± s.e.m; statistical significance analyzed by an unpaired two-sided t-test is indicated by *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Supplementary Material
Acknowledgements
We thank the Biorepository and Pathology Core and the Flow Cytometry Core Facilities at the Icahn School of Medicine at Mount Sinai for their technical expertise. We also thank K. Phaik Har Lim, T.-K. Man, T. Burke and B. Scull for their help with the generation of microarray sequencing data. We thank J. Kofler for kindly providing LCH-ND pictures and performing immunostaining. We thank L. Troncoso for her help. We thank C. Woods, image application specialist, Cincinnati Children’s Hospital Medical Center, for figure preparation for human studies (Fig. 4g) and J. Kofler, UPMC Division of Neuropathology, for creating Extended Data Fig. 4e. M.M. received funding from the National Institute of Health (R01 CA154947 and R01 CA190400). C.B. received fellowships from the Fondation pour la Recherche Médicale (FDM20170638478), from l’Institut Servier and from Assistance Publique Hôpitaux de Paris (Année Recherche). C.M.W. is supported by the Swiss National Science Foundation (SNSF PostDoc Mobility Fellowship P400PM_186740) and by the Swiss Cancer League (grant for bursaries BIL KFS 4724-02-2019). We thank the research coordinators of the Histio-Lymphoma team at Texas Children’s Cancer Center for their help with patient sample collection. The TXCH Histiocytosis Program is supported by a research grant from the HistioCure Foundation. This work was supported by the Department of Defense through the Peer Reviewed Cancer Research Program under award no. W81XWH-19-1-0167 (R.C.).
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-021-01304-x.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01304-x.
Peer review information Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Joao Monteiro was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01304-x.
Data availability
Datasets supporting the findings presented in this study are available from the corresponding author upon reasonable request. Any data that can be shared will be released via a material transfer agreement. Microarray sequencing data obtained from CD34+ BM cells transduced with BRAFV600E or control NGFR lentiviral vector are listed in Supplementary Table 3. RNA-seq data obtained from CD34+ cells purified from the BM of patients with LCH and healthy donors are listed in Supplementary Table 4. Source data are provided with this paper.
References
- 1.Merad M, Ginhoux F & Collin M Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol 8, 935–947 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Stålemark H et al. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr. Blood Cancer 51, 76–81 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Allen CE, Merad M & McClain KL Langerhans-cell histiocytosis. N. Engl. J. Med 379, 856–868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badalian-Very G et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116, 1919–1923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakraborty R et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124, 3007–3015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty R et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128, 2533–2537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim KPH et al. Circulating CD1c+ myeloid dendritic cells are potential precursors to LCH lesion CD1a+CD207+ cells. Blood Adv. 4, 87–99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berres M-L et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J. Exp. Med 211, 669–683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durham BH et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 130, 176–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne P et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim–Chester disease in adults. Blood 130, 167–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berres M-L, Merad M & Allen CE Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to Histiocytosis X? Br. J. Haematol 169, 3–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Göthert JR et al. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood 105, 2724–2732 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Michaloglou C et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Segura A, Nehme J & Demaria M Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Muñoz-Espín D & Serrano M Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol 15, 482–496 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Kuilman T et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008). [DOI] [PubMed] [Google Scholar]
- 17.McClain KL et al. CNS Langerhans cell histiocytosis: common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124, 2607–2620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herranz N et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol 17, 1205–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laberge R-M et al. MTOR regulates the pro-tumorigenic senescence- associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol 17, 1049–1061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker DJ et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson WH et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet. Oncol 11, 1149–1159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bussian TJ et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogstad B et al. RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J. Exp. Med 215, 319–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xerri L et al. CDKN2A/B deletion and double-hit mutations of the MAPK pathway underlie the aggressive behavior of Langerhans cell tumors. Am. J. Surg. Pathol 42, 150–159 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Picarsic J & Jaffe R Nosology and pathology of Langerhans cell histiocytosis. Hematol. Oncol. Clin. North Am 29, 799–823 (2015). [DOI] [PubMed] [Google Scholar]
- 28.McClain K, Jin H, Gresik V & Favara B Langerhans cell histiocytosis: lack of a viral etiology. Am. J. Hematol 47, 16–20 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Jenson HB, McClain KL, Leach CT, Deng JH & Gao SJ Evaluation of human herpesvirus type 8 infection in childhood Langerhans cell histiocytosis. Am. J. Hematol 64, 237–241 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Montecino-Rodriguez E, Berent-Maoz B & Dorshkind K Causes, consequences, and reversal of immune system aging. J. Clin. Invest 123, 958–965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen Aubart F et al. Targeted therapies in 54 patients with Erdheim– Chester disease, including follow-up after interruption (the LOVE study). Blood 130, 1377–1380 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Diamond EL et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 567, 521–524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckstein OS, Visser J, Rodriguez-Galindo C & Allen CE Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133, 1691–1694 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J et al. Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat. Immunol 18, 877–888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dankort D et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 21, 379–384 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baccarini A et al. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol 21, 369–376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remark R et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci. Immunol 1, aaf6925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets supporting the findings presented in this study are available from the corresponding author upon reasonable request. Any data that can be shared will be released via a material transfer agreement. Microarray sequencing data obtained from CD34+ BM cells transduced with BRAFV600E or control NGFR lentiviral vector are listed in Supplementary Table 3. RNA-seq data obtained from CD34+ cells purified from the BM of patients with LCH and healthy donors are listed in Supplementary Table 4. Source data are provided with this paper.