

Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors which induce synthetic lethality of BRCA-mutant breast and ovarian cancers are now under active exploration for treatment of hematological neoplasms. Experimental data has revealed that DNA repair deficiencies similar to those found in BRCA mutant solid tumors function in malignant hematopoietic cells to enhance cell survival and promote therapy resistance. Preclinical studies have demonstrated that inhibition of PARP with a variety of agents can dramatically enhance the efficacy of other therapeutic approaches including cytotoxic and epigenetic chemotherapy, small molecule inhibitors (IDH and FLT3 inhibitors) and antibody drug conjugates. This has led to early stage clinical trials of multiple PARP inhibitors (PARPi) for leukemia and lymphoma patients. Despite small patient numbers, evidence of modest clinical efficacy and tolerability in combinatorial regimens support the promise of PARP inhibition as a novel therapeutic strategy, particularly in molecular subsets (MLL rearranged, FLT3/IDH1 mutant) of acute myeloid leukemia.
Keywords: PARP inhibition, DNA repair, DNA damage acute myeloid leukemia, mantle cell lymphoma, chronic lymphocytic leukemia, olaparib, talazoparib
1. Introduction
Inhibition of poly (ADP-ribose) polymerase (PARP) inhibition has been established as a successful molecular therapy for therapy of BRCA1 and BRCA2 mutant cancers. These entities contain defective homologous recombination (HR) DNA repair processes, which can be triggered via targeted approaches to undergo synthetic lethality due to the limited DNA repair capacity of these cells. To date, multiple PARPi have been FDA approved in ovarian cancer, select subsets of breast cancer, and most recently, in individuals with prostate cancer characterized by mutations in BRCA1/2 or ataxia telangiectasia–mutated (ATM) (1–3).
Although the majority of clinical studies evaluating PARP inhibition for cancer have been restricted to solid tumors, there is mounting evidence supporting the PARP inhibition as an effective therapeutic strategy for hematological malignancies. Of note, identification of cytogenetic and molecular events in acute myeloid leukemia (AML) blasts resulting in aberrant DNA repair mechanisms have piqued interest in PARP blockade as an attractive molecular approach in specific subsets of AML (4).
2. Overview of Poly (ADP-ribose) Polymerase (PARP) pathway
PARP1 and PARP2 constitute abundant nuclear proteins within the PARP protein superfamily critical for routine repair of single stranded breaks (SSB) in DNA in normal cells. PARP proteins detect SSBs and bind to the damaged DNA via the N-terminal zinc finger domain with subsequent activation of the C-terminal catalytic domain (5, 6). This catalyzed reaction hydrolyzes NAD+ and requires oxidized nicotinamide adenine dinucleotide for PARP to function. The release of nicotinamide as a byproduct leads to the creation of mono or poly (ADP-ribosyl)ation (PARylation) of multiple protein substrates. These polymers extend ADP-ribose chains to recruit additional DNA repair proteins (7, 8). PARP1 is largely responsible for the majority of total cellular PARylation activity, with PARP2 only accounting for 5–10% of total activity (9, 10). Parylation of multiple protein substrates primarily promotes the recruitment of DNA repair proteins to the site of SSBs, allowing for DNA repair. After this, PARP1 dissociates from DNA via auto-parylation at the regulatory domain (6, 11). Of note, due to its effects on parylation posttranslational modification, PARP is also critically involved in several cellular processes other than DNA damage repair, including regulation of gene transcription, cell division, and energy metabolism which may also be critical for tumor cell survival (7, 8, 10, 12–14).
3. Overview of PARP Inhibitors in Hematological Malignancies
The role of PARP1 in malignancy is well established in BRCA1/2 tumors deficient in homologous recombination where the synthetic lethality achieved by inhibiting the PARP1 dependent DNA repair pathways has led to regulatory approval in several tumor types. Until recently, PARP1/2 inhibition has not been pursued as a potential therapeutic strategy in hematological cancers because mutations in the BRCA1/2 genes are uncommon in these diseases (15). In recent years, however, characterization of the myriad roles of PARP1 with respect to regulation of gene transcription, protein stability, and modulation of chromatin structures (14, 16) has led to realization that this pathway contributes in essential ways to other key components of HR, many of which are fundamentally dysregulated in blood cancers, such as ATM, ATR, CHK1, and RAD51 (3). Moreover, evidence has emerged that DNA polymorphisms and/or aberrant transcriptional regulation may result in faulty DNA-damage response (DDR) in AML (17). PARP inhibition is also an attractive means of exploiting known defects in DNA repair in tumor cells with known chromosomal instability (18).
Multiple pharmacologic inhibitors of PARP with varying pharmacokinetic and inhibitory properties are in preclinical and clinical usage for cancer therapy and are summarized in Table 1. Here we detail the preclinical evidence supporting specific PARP inhibitory agents in hematological malignancies. We then discuss current clinical evidence and the promise of these agents in specific clinical subsets.
Table 1.
Characterization of PARP inhibitors with preclinical data in hematological malignancies
| Name of Inhibitor | Structure | IC50 | In vitro effects | In vivo effects | Stage of Development |
|---|---|---|---|---|---|
| Talazoparib (BMN673) |
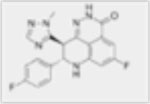
|
0.6nM | Combination with DNMTis synergistic toxicity and reduced colony formation. | Combination with DNMTis resulted in increase in overall survival and significant anti-tumor response. | Recruiting/Phase 1 |
| Olaparib (Ku-0059436) |
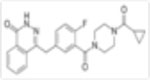
|
5nM | Anti-proliferative and apoptotic events in AML cell lines, accumulation of S phase cell cycle arrest. Synergistic anti-leukemic effects in combination with HDACi, WEE1 inhibition, GO. | Increase in overall survival in combination with AZD1775 | Phase 1 Phase 2 |
| PJ34 HCl |
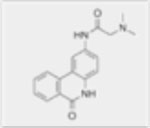
|
N/A | Dose-dependent decrease in cell viability, induces apoptosis in AML cell lines. | Alleviated hepatomegaly and splenomegaly and reduced level of C1498 cells. | N/A |
| Veliparib (ABT 888) |
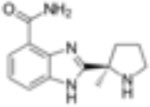
|
5nM | Anti-leukemic activity in combination with DNMTis. Synergistic anti-proliferative effects in combination with TRAIL. Enhances activity of TMZ. | Veliparib combined with Cisplatin achieved significant tumor growth inhibition. | Phase I Active not yet recruiting |
| Niraparib (MK-4827) |
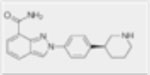
|
4nM | N/A | N/A | N/A |
| Rucaparib (AG-0146991,PF-01367338) |
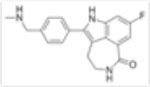
|
1nM | Increased sensitivity in AML cell lines at 0.1–100M. | Significant improvement in overall survival in combination with fluororacil. | N/A |
4. Pharmacologic PARP inhibitors
4.1. Olaparib
Olaparib (AZD-2281, Ku-0059436) is an orally bioavailable and well-tolerated poly(ADP-ribose) polymerase (PARP) inhibitor. In December 2014, olaparib was FDA approved for treatment of BRCA1 and BRCA2 mutant ovarian cancer as well as triple-negative breast cancer (2, 3). Although recent studies have indicated that olaparib binds at least four members of the PARP super family, PARP1-PARP4 (19), PARP1 and PARP2 downregulation prevents further sensitization by olaparib, indicating that these isoforms are the most pertinent PARP inhibitor targets for death ligand sensitization.
4.2. Talazoparib (BMN-673)
Talazoparib has been described in the literature as most potent PARP inhibitor due to its ability to trap PARP at the site of DNA damage (20). The cytotoxicity of PARPi has been directly correlated to their respective capacities to trap PARP1/2 at 5’-dRP lesions generated during BER steps under PARP inhibition. This tight binding of PARP1 to chromatin forms a cytotoxic DNA-PARP1 complex. This leads to the accumulation of single-stranded breaks (SSB), which in turn become double-stranded breaks (DSB) and result in cell death (5, 20). At low nanomolar concentrations (1, 2.5, 5 nM) talazoparib, traps PARP1 in chromatin extracts to the same extent as other PARPi dosed at much higher concentrations (20).
4.3. Veliparib (ABT-888)
Because of its relatively low capacity to trap PARP at the sites of damaged DNA, veliparib is classified as a weak PARP inhibitor (21). It has been demonstrated as 100-fold weaker than that of the strongest PARP inhibitor, talazoparib. When combined with DNMTis, veliparib was treated with 500nM to achieve the same capacity of trapping of PARP to chromatin, whereas talazoparib was treated with doses ranging from 1 to 5nM (20).
4.4. Rucaparib (Rubraca, AG-0146991)
Rucaparib (Clovis Oncology, Boulder, CO) is an orally bioavailable PARP inhibitor that was FDA approved in December of 2016 for patients with deleterious BRCA mutation associated advanced ovarian cancer who have undergone treatment with 2 or more chemotherapies (22).
4.5. Niraparib (Zejula, MK-4827)
Niraparib (Tesaro Inc., Waltham, MA) is an orally bioavailable PARP inhibitor that was FDA approved in March of 2017 for patients with platinum-sensitive ovarian, fallopian tube, or peritoneal cancer (23). Niraparib is administered orally once daily continuously with 3 capsules of 100mg strength (300mg total) taken at each dose.
4.6. PJ34
PJ34 hydrochloride is another potent, specific inhibitor of PARP1/2 (24) which is not yet in clinical development for cancer therapy.
5. Preclinical Studies by Disease Type
5.1. Acute Leukemia
5.1.1. Monotherapy
Acute myeloid leukemia (AML) is a clonal disorder of the hematopoietic system, characterized by the immature accumulation of myeloid cells in the bone marrow, due to the deficient development of progenitor and stem cells. Despite the progress of the modern era, clinical outcomes for AML have not significantly improved, with only 20–30% of patients achieving long-term disease-free survival (25). There remains a great clinical need for novel therapies that are more efficacious and less toxic than conventional chemotherapy.
Potent anti-leukemic effects have consistently been reported in human AML cell lines and preclinical models treated with numerous PARPi. Olaparib is perhaps the most extensively studied agent and has consistently demonstrated anti-proliferative and apoptotic effects in several preclinical models of AML at clinically achievable levels (1.0 – 10.7µM) (26). The treatment of primary AML cell lines with olaparib resulted in a dose-dependent decrease in cell survival, while normal hematopoietic cells displayed a low sensitivity to treatment (26). Olaparib exposued induced accumulation of HL60 AML cells in the S phase with increases in G2 and M populations, suggesting stalled replication. Analysis by immunofluorescence confirmed that this sensitivity to PARP inhibition resulted from defective HR DNA repair. BRCA proficiency detected in blasts not sensitive to olaparib predicted for inducible synthetically lethal interactions, and phosphorylation of histone H2AXF was associated with PARP activity (26).
The potent PARP inhibitor (PJ34) dose-dependently decreased cell viability and generated IC50 values of 23.5+/−3.9 and 35.6 +/− 5.5 μM respectively in multiple AML cell lines. Annexin V-FITC/PI staining demonstrated an increase in apoptosis of AML cells with increasing PARP inhibition, which was confirmed by lower levels of anti-apoptotic proteins Bel-2 and Bcl-xL. In vivo, an AML murine model injected with C1498-GFP cells showed efficacy of PARP-1 inhibition with treatment of PJ34, measured by its reduced level of PAR, its enzymatic product. Further, the inhibition of PARP alleviated AML hepatomegaly and splenomegaly and reduced levels of C1498 cells. Overall PARP inhibition with PJ34 prolonged survival and alleviated tumor burden in mice (24).
Rucaparib treatment of OCI-AML2 cells led to similar changes in cell cycle, DNA damage (γH2AX), and apoptosis (27). Falzacappa et al demonstrated that PARP inhibition results in slight DNA damage, stalling in the S phase of the cell cycle, and accumulation in DNA damage, pushing cells to undergo apoptosis.
5.1.2. Combination therapy
One of the most promising avenues of investigation is the combination of PARPi with epigenetic agents for acute leukemia therapy. The combination of low doses of DNA methyltransferease inhibitors (DNMTis) along with talazoparib has been shown to enhance PARP binding to chromatin, relative to either drug alone, and to induce higher frequency of DSBs and greater synergistic cytotoxicity. Colony formation of 8 of 13 primary AML patient samples was significantly reduced following treatment with decitabine and talazoparib. Low dose therapy with azacitidine and talazoparb also resulted in significant tumor reduction and increase in overall survival in AML mouse models over controls or monotherapy (20).
Because histone deacetylase inhibitors (HDACis) induce decreased non-homologous end-joining (NHEJ), one of the main pathways for DNA double-strand break repair, of PARPi and HDACi constitute attractive partners for further clinical development. The combination of talazoparib and HDACi increases PARP trapping and induction of apoptosis, most likely due to HDACi enhancing the binding of PARP1 to I-Sce1-induced sites of DSBs. HDAC inhibition at concentrations of 300nM and 600nM in cells pretreated with talazoparib at 20nM significantly increased PARP1 in chromatin within 6 h of treatment. Additionally, this combination treatment significantly increased apoptosis at 6-day treatments relative to HDACi alone. One possible explanation for this enhanced combinatorial effect is that the increased presence of PARP1 in chromatin might prevent NHEJ activity by preventing the access of NHEJ proteins to sites of DSBs (28).
The HDAC inhibitor, MS275, has been evaluated in combination with two other PARPi, in AML preclinical models. MS275 was shown to potentiate the cytotoxic effects of olaparib following 5-aza-2’CdR by disrupting base excision repair of the induced lesions (29). This combination also yielded a dramatic increase in H2AX phosphorylation consistent with inhibition of DNA repair and the ability of PARPi to exploit defects in double-strand DNA break repair in leukemic cells (29). These authors also determined that a non-cytotoxic dose (200nM, 250nM) of 5-aza-2’CdR added 48 h before combination with olaparib resulted in increased cytotoxicity, potentially by increasing amount of DNA SSB and DSB, thereby supporting a three drug regimen (DNMTi plus HDACi plus PARP inhibition) (29). In a separate study, MS275 was shown to enhanced the cytotoxicity of PJ34 in primary AML patient samples (30).
PARPi have also been employed to enhance the activity of antibody drug conjugates where the cytotoxic drug linked to the antibody construct constitutes a potent DNA damaging agent. Yamuchi et al found that the in vitro combination of gemtuzumab ozogamicin (GO, an anti-CD33 humanized monoclonal antibody linked to calicheamicin) with olaparib exerted synergistic cytotoxicity in CD33-positive HL-60 AML cells (31). Although single agent olaparib did not induce apoptosis or inhibit cell growth with concentrations as high as 10µM, various combinations led to low IC50 for GO and a combination index of 0.86 (31). Our laboratory has also evaluated the combination of IMGN779 (a novel CD33 – targeting antibody drug conjugated to a DNA methylating agent) with olaparib (32). We found that olaparib at high concentrations was capable of inducing AML cell death as a single agent, but was much more effective at low doses when combined with IMGN779 in multiple CD33+ human AML cell lines (HEL, MV-411, HL60). This regimen dramatically increased apoptosis, reduced cell viability, and induced almost complete S-phase cell cycle arrest. In addition, colony growth of AML progenitor cells established from AML patient samples (n=7) was markedly reduced following olaparib and IMGN779 therapy. Moreover, combination IMGN779 and olaparib therapy significantly decreased leukemic burden and improved overall survival in systemic human CD33+ AML xenografts(32).
5.1.3. Other novel combinations
Meng and colleagues reported that both olaparib and veliparib sensitize AML cell lines to TRAIL, the recombinant human tumor necrosis factor-- related apoptosis inducing ligand (33) (34).. The human AML cell line ML-1 was treated for 24 h with veliparib and subsequently exposed to varying doses of recombinant human TRAIL agonist monoclonal antibodies, the natural ligand of the death receptor DR5. The rationale of utilizing recombinant TRAIL is that it has potential as an antineoplastic agent, as its binding leads to initiation of the so-called death signaling complex (DISC), initiating the apoptosis cascade. The AML cells were sensitized to TRAIL but treatment by veliparib alone did not significantly induce cell death (34). The combinatorial treatment resulted in synergistic anti-proliferative effects, with the half – maximal effects achieved at 1µM treatment of veliparib.
Inhibition of WEE1 has been shown to impair homologous recombination (HR) by indirectly inhibiting BRCA2. Garcia et al reported that the combination of WEE1 inhibition (AZD1775) and olaparib induced synergistic anti-leukemic effects in AML cells in vitro and in colony formation assays with primary AML samples. AZD1775 and olaparib also prolonged survival of murine AML models (35).
Furthermore, it has been demonstrated that the PARP inhibitor veliparib can effectively enhance the activity of alkylating agent temozolomide (TMZ) (36) in AML characterized by mismatch repair deficiencies (MMR) and low O6-methylguanine-DNA methyltransferase (MGMT) activity, both processes required for DNA repair. This combination revealed potentiation of TMZ growth inhibition 3-to-21-fold in leukemia cell lines (U937, HL-60, KG1, HEL, THP1) with an IC50 ranging from 20–196 µmol. This data suggests that veliparib may be effective at potentiating TMZ in AML patients with MMR deficiencies independent of MGMT activity.
6. Evidence supporting PARPi in specific clinical subsets
6.1. Acute Leukemia
6.1.1. Isocitrate Dehydrogenase (IDH1/2) mutant AML
As AML is a heterogeneous disease, there are several specific genetic phenotypes, which may derive the greatest benefit from PARP inhibition. Isocitrate dehydrogenase 1 (IDH1) and IDH2 mutations are each present in approximately 10% of AML patients (37). The normal function of IDH1 and IDH2 is to catalyze the conversion of isocitrate to α-ketoglutarate (αKG) within the citric acid cycle. Mutations in these proteins result in the overproduction of 2-hydroxyglutarate (2HG), which functions as an oncometabolite (38). 2HG has been demonstrated to exert pleiotropic effects on cellular biology including cellular differentiation and chromatin modulation. Furthermore, it has been demonstrated that IDH1/2 mutant AML cells are deficient in repairing DNA double-strand breaks via HR (39). Briefly, these authors created an HEL cell line with stably integrated doxycycline (dox) – inducible IDH1/2 WT and IDH1/2 mutant open reading frames (39). Dox-inducible expression of IDH1 and IDH2 proteins by Western blot was confirmed, along with increased 2HG production by liquid chromatography-mass spectrometry. Subsequently these cells were exposed to ionizing radiation, and mutant IDH1 dependent radiosensitivity was observed in a short-term viability assay. The persistence of unrepaired DSBs at baseline was also observed by transient expression of mutant IDH1 and IDH2. These results suggest that IDH1/2 mutant cells harbor intrinsic DSB repair defects (39). To confirm the integrity of HR, the authors utilized plasmid reporter assays developed to compare relative DSB repair activity between different cell lines under different conditions. A marked deficiency in HR was observed in IDH1/2 mutant AML cells, with no effect of IDH1/2 on non-homologous end joining (NHEJ). Moreover, 2HG exposure across multiple cell lines with diverse genetic backgrounds demonstrated increased rates of DSBs, recapitulating the HR phenotype observed with BRCA2 deficiency, and suggesting that DNA damage is a key outcome of high oncometabolite levels. Lastly, these findings were confirmed in primary AML patient samples, which support a “BRCAness” phenotype in IDH1/2 mutants resulting in chemo/radiosensitivity and synthetic lethality by PARP inhibition.
Recently, Molenaar et al have demonstrated that pretreating patient-derived IDH1/2 mutant cells with an IDH1/2 inhibitor (AGI-5198/AGI-6780) protects these cells against subsequent chemotherapy (daunorubicin) or irradiation. IDH1/2 mutated primary AML cells boasted more γ-H2AX+ foci as compared to IDH1/2 WT cells following exposure to daunorubicin or irradiation and exhibited high sensitivity to PARPi (olaparib, talazoparib) (40). Furthermore, IDH1/2 mutant AML cells are further sensitized to cytotoxic therapy following treatment with PARP inhibitor. The combination of PARP inhibition with DNA-damaging therapy in IDH1/2 mutant cells resulted in additive effects and was attributed to decreased transcription of the DNA repair gene ATM in mutated cells vs. wildtype IDH1/2 cells, as confirmed by TCGA data. Therapy with an IDH1/2 inhibitor restored ATM expression in IDH1/2 mutant cells.
6.1.2. FMS-like Tyrosine-kinase 3 (FLT3) mutant AML
FLT3 mutations are identified in upwards of 30% of AML diagnoses and confer a poor prognosis. Maifrede and colleagues have demonstrated that AML cells expressing the FLT3 internal tandem duplication (FLT3-ITD) exhibit HR and non-homologous end-joining (NHEJ) double strand repair (41). However, following therapy with the FLT3 inhibitor quizartinib (AC220), FLT3-ITD mutant AML cells downregulate DNA repair proteins (BRCA1, BRCA2, PALB2, RAD51, and LIG4), resulting in simultaneous inhibition of HR and NEHJ. Subsequent in vitro exposure of these FLT3-ITD mutant cells to quizartinib in addition to PARPi (olaparib and talazoparib) led to accumulation of lethal DSBs, thus mirroring synthetic lethality. Furthermore, combined therapy with a PARP inhibitor and quizartinib successfully eradicated FLT3-ITD mutant quiescent and proliferating leukemia stem cells as well as human and murine leukemic progenitors. Additionally, in vivo treatment of quizartinib with talazoparib was highly effective for the treatment of FLT3-ITD mutant primary AML xenografts in mice (41).
6.1.3. Mixed-Lineage Leukemia (MLL) Acute Leukemias
Mixed-lineage leukemia (MLL) rearrangement is found in approximately 10% of AMLs and more than 30% of therapy-related AML as well as acute lymphocytic leukemia (ALLs) and biphenotypic acute leukemias (42) In AML, the presence of an MLL rearrangement generally confers an intermediate – to – poor prognosis with disease often refractory to conventional chemotherapy. Interestingly, the proliferation and differentiation of MLL-AF9 – encoding t(9;11)(p22;q23) rearranged leukemia appears to be highly dependent on mechanisms preventing the amassing of DSBs (18).
Silvia Maifrede and colleagues recently demonstrated that PARP−/− murine bone marrow cells (mBMCs) which expressed MLL – AF9 experienced reduced clonogenic activity as compared to their PARP+/+ counterparts (12). In addition, exposure of primary patient samples carrying the MLL – AF9 – encoding t(9;11)(p22;q23) to olaparib resulted in a decreased number of Lin-CD34+ immature primary leukemia cells, including leukemia stem cell – enriched Lin−CD34+CD38− CFSElow proliferating and Lin-CD34+CD38-CFSEmax quiescent cells by approximately two-fold.
Olaparib not only resulted in accumulation of potentially lethal DSBs but also prominently enhanced the sensitivity of MLL-AF9 AML cells to chemotherapy (doxorubicin) as evidenced by greater than four-fold reduction of clonogenic activity. These results were confirmed in vivo using a murine model of MLL – AF9 leukemia treated with talazaporib or doxorubicin alone versus combination therapy (12). Esposito et al demonstrated that resistance of MLL – AF9 to PARP inhibition is mediated by the homeodomain transcription factor HOXA9 (43). These researchers used a knockout of Hoxa9 to show that ablation of Hoxa9 sensitized MLL-AF9 transformed cells to olaparib, resulting in a significant suppression of colony-forming ability and induction of differentiation and senescence. In vivo, Hoxa9 deficient MLL-AF9 cells were highly sensitive to PARP inhibition with olaparib, which significantly delayed the disease latency. Furthermore, combination therapy with an inhibitor of HOXA9 (LiCl) plus olaparib resulted in dramatic suppression of growth and induction of differentiation of MLL leukemic stem cells and prolongation of survival in an in vivo model (43).
6.1.4. AML1-ETO translocated AML
Approximately 10–15% of de novo AML cases are characterized by a chromosomal translocation t(8;21) encoding RUNX1-RUNXT1 and resulting in expression of an AML1-ETO fusion protein (44). This fusion protein subsequently downregulates BRCA2 and renders these leukemic populations attractive targets for PARP inhibition (45, 46). Esposito and colleagues demonstrated that this particular translocation resulted in inherent DDR defects, and a defective HR pathway (43). They then showed that olaparib resulted in significant suppression of the colony-forming ability of transformed cells and decreased the growth of primary mouse hematopoietic cells transformed by AML1-ETO as well as colony formation abilities of primary AML1-ETO positive primary patient samples. Furthermore, this observed activity was confirmed in vivo, with single agent olaparib resulting in significantly delayed disease onset in a mouse model (43). Similar in vitro data was presented and confirmed by Bamezai et al (47). Interestingly, Di Zhou and colleagues have identified miR-181a as a potential biomarker for PARP inhibition by establishing a role of miR-181a in the downregulation of PARP1 in MLL-rearranged AML via acetylation effects (48).
6.1.5. Acute Promyelocytic Leukemia
The promyelocytic leukemia/retinoic acid receptor alpha (PML-RARα) fusion also represses one or several of the DNA-damage response (DDR) pathways, including proteins involved in base excision repair (BER) and HR (19). The PML-RARα fusion is identified in approximately 5–20% of patients with AML (49). Esposito and colleagues found that PARP inhibition by olaparib notably inhibited the growth of primary mouse hematopoietic cells transformed by PML-RARα, an ATRA resistant APL cell line (NB4-LR2), as well as PML-RARα primary patient samples (43). Xenografts with ATRA resistant APL cells demonstrated delayed disease onset when treated with single agent olaparib. Similar to the results obtained in AML1-ETO AML, PML-RARα transformed cells demonstrated inherent DDR defects along with a defect in the HR pathway.
6.1.6. Acute lymphocytic leukemia
Limited data is available regarding the efficacy of PARP inhibition in acute lymphocytic leukemia (ALL). Perhaps the best evidence comes from Falzacappa et al who evaluated increasing concentrations of rucaparib (0.1 – 100 μM/L) in an ALL (RPMI-8402) cell line with evidence of dose dependent induction of cell death and an EC50 of 30µM/L, ten fold higher than for AML cells (27). Cells were exposed to both 5FU and rucaparib (RPMI-8402: 50µmol/L, 24µmol/L respectively) exhibited further reduction in cellular viability. An ALL mouse model generated in C57BL6/Ly5.2 mice treated with 50mg/kg of the tumorigenic agent N-ethyl-N-nitrosourea (ENU) were then challenged with 5 consecutive days of rucaparib and a single dose of 75mg/kg 5FU IP on day 2. Combination therapy significantly improved overall survival (p=0.05) compared to single agents alone. Human xenotransplantation models engrafted with primary patient ALL cells and treated with similar rucaparib and 5FU combination therapy evidenced significant reductions in circulating blasts and improved overall survival (p<0.05) (27).
6.1.7. Hematological cancers deficient in DNA repair mechanisms
The DDR dysregulation caused by abnormal transcriptional networks in AML may be targeted in a synthetically lethal approach using PARP inhibition (43, 50, 51). The nuclear phosphatase and tensin homolog (PTEN), one of the most common mutated tumor suppressors, functions to regulate DNA repair and plays a crucial role in the expression of RAD51 (52). While PTEN and HR mutations are only present in a small percentage of AML, PTEN deficient cells are highly sensitive to PARP inhibition.
6.1.8. Acute Leukemia with “BRCAness” Phenotype
Kogan and colleagues have reported in recent times that exposing AML cells to low doses of DNA methyl transferase inhibitors (DNMTi) results in altering gene expression patterns of DSB repair genes, potentially generating a HR defect or “BRCAness” effect (53). This was identified utilizing expression array analysis performed on RNA extracted from multiple BRCA-intact sporadic AML cell lines (Kasumi-1, KG-1, MOLM14) treated with DNMTi (decitabine or azacitidine). one or more HR genes were regularly downregulated in all cell lines. QPCR and/or immunoblotting of HR proteins demonstrated significantly decreased expression of RAD51, BRCA1, BRCA2, or HR-related genes FEN1 or FANCD2. In six of 8 primary AML samples treated with decitabine, significantly decreased expression of RAD51, BRCA1, and BRCA2 defined a “HRD-like” or “BRCAness” phenotype that translated to sensitivity to PARPi and could serve as a gene signature predicting response to PARP inhibition. Strikingly combination treatment with decitabine and talazoparib was significantly more effective than single agent therapy in these 6 patient samples.
6.2. Lymphomas and CLL – Preclinical Studies
Different mutations prevalant in lymphoproliferative disorders can confer sensitivity to PARP inhibition. Ataxia telangiectasia mutated (ATM) is a serine/threonine protein kinase crucial to DNA damage-induced signaling and the initiation of cell cycle checkpoint signaling following to DNA damaging agents (54, 55). Inactivation of this gene has been reported in subsets of non-Hodgkin’s lymphoma, including mantle cell lymphoma (MCL) (56, 57) and chronic lymphocytic leukemia (CLL) (58, 59). Multiple investigators have demonstrated that olaparib has preclinical efficacy against ATM deficient MCL cell lines (56, 60), and that MCL cells with p53 deficiencies exhibited increased sensitivity to olaparib (61). Moreover, olaparib therapy decreased disease burden and improved overall survival in a xenograft model of ATM mutant MCL (56, 60). Maifrede and colleagues have postulated that the t(8;14)(q24;q32) chromosomal translocation encoding IGH/MYC within Burkitt lymphoma cells also induces BRCA-2 deficiency, thus enhancing sensitivity to PARP inhibition (olaparib, talazoparib) in vitro and in vivo (62).
Combination regimens of various PARPi with other anti-lymphoma agents have also been explored with varying success. Niraparib has been combined with decitabine, romidepsin or panobinostat and has resulted in in synergistic inhibition of proliferation in a panel of human lymphoma cell lines. Moreover, addition of busulfan and/or melphalan further significantly enhanced the cytotoxic effects of the triple drug combination (63). Olaparib has also been investigated together with radioimmunotherapy in Epstein-Barr virus-infected human Raji cells (64), with cytarabine in Burkitt lymphoma models (62), and with ibrutinib in MCL cell lines (65).
7. Clinical Studies
7.1. Acute Myeloid Leukemia
At the present time, PARPi is most actively being investigated for therapy of acute myeloid leukemia. Mufti and colleagues presented their findings of the PARP inhibitor talazoparib in a Phase 1 study in patients with advanced hematological malignancies, including AML (66). A total of 33 patients were enrolled (Arm 1: 21 AML, 4 myelodysplastic syndrome (MDS); Arm 2: 4 CLL, 4 MCL), with 10 women and 23 men. The median age was 71 (range 22–86) with ECOG PS 0–1, and a median number of previous regimens being 3 (range 1–7) in Arm 1 and 6 (range 1–13) in Arm 2. The dosing levels were 100, 200, 300, 450, 900, 1350 and 2000 µg/day. Dose-limiting toxicities (DLTs) included febrile neutropenia and neutropenic sepsis in 2/4 patients in the 2000 µg/day in Arm 1. The investigators reported that the most frequent drug – related AEs were fatigue (all grades 27%), neutropenia (27%), nausea (24%), infections (21%) and thrombocytopenia (12%). Stable disease was seen in 13/24 patients in Arm 1. One patient with MDS who received 24 cycles of talazoparib became red cell transfusion independent. This study confirmed that talazoparib had variable MTDs within the different disease groups and potential limited single agent activity.
Given the limited single agent activity, it is not surprising that the remaining clinical studies focus on combining PARP inhibition with a cytotoxic therapy. Veliparib has been evaluated in two studies in AML patients. The first involves combination therapy with temozolomide (67). Patients 18 years or older with relapsed or refractory AML (RR-AML); CMML-2; newly diagnosed AML arising from prior MDS, MPN or chemotherapy; or patients aged ≥ 60 years having untreated AML with adverse cytogenetic findings and/or not considered eligible for intensive therapy were enrolled in a 3+3 design. Veliparib was administered orally once on day 1 and then twice daily on days 4 – 12 of cycle 1. In subsequent cycles it was administered twice daily on days 1 – 8. Temozolomide was given once daily on days 3 – 9 in cycle 1 and days 1 – 5 in cycle 2 and beyond. A total of 48 adults with AML with a median age of 69 years were enrolled between June 2010 and February 2014. These patients were considered high risk with 56% displaying an adverse karyotype and 65% of patients having secondary AML. No DLT was observed at veliparib doses from 20 – 150 mg twice a day and temozolomide 200 mg/m2. However, 2 of 4 patients experienced a DLT at veliparib 200mg twice daily, consisting of grade 3 oropharyngeal mucositis/esophagitis lasting more than 7days, thus the MTD and RP2D were defined as temozolomide 200mg/m2 per day for 7 days along with veliparib 150 mg twice daily for 9 days. The most common non-hematologic adverse events of any grade were nausea/vomiting (35%; 40% cycle 1), fatigue (24%; 29% cycle 1), mucositis (13%; 17% cycle 1), diarrhea (10%; 10% cycle 1), and constipation (11%; 15% cycle 1). The most common serious AEs were infections (40% cycle 1; 24% all cycles); febrile neutropenia (25% cycle 1; 20% all cycles); and oropharyngeal mucosistis/esophagitis (4%), representing the DLT. Complete responses (CR) were attained in 8 of 48 patients (16.6%) with 7 of 8 achieving CR after a single cycle. Additionally, 8 patients achieved stable disease or hematologic improvement defined as decrease or absence in circulating blasts or reduction in marrow blasts. The median follow-up for the entire cohort was 5.3 months (range 0.5–47). The median OS for all patients was 5.3 months (95% CI, 3.3 – 8.3 months). Those who achieved a CR had a median OS of 20 months (95% CI, 10 – 47+ months), while those who achieved hematologic improvement or stable disease had a median OS of 9.4 months (95% CI, 6.1 – 47+ months). Five patients were able to undergo allogeneic SCT; 3 of which had achieved remission with the study treatment. Veliparib exposure and inhibition of PAR polymer formation increased dose proportionately. Responders exhibited a veliparib – induced increase in H2AX phosphorylation in CD34+ cells, and three of 4 patients with MGMT promoter methylation achieved CR.
In addition, veliparib has also been investigated with topotecan and carboplatin in patients with AML (68). Patients > 18 of age with pathologically confirmed RR-AML, newly diagnosed aggressive MPN, or aggressive CMML were eligible. In this study, veliparib was administered orally twice daily with dose escalation. Topotecan and carboplatin were administered together by intravenous continuous infusion (IV CI) over 120 hours on days 3–7 of each cycle. Three to six patients were evaluated per dose level using a standard 3+3 design. A total of 99 patients, including 34 with primary refractory AML, 35 with secondary AML, 22 with aggressive chronic myeloid neoplasms or AML arising out of aggressive chronic myeloid neoplasms and 4 refractory ALL were accrued to the trial. The MTD was 80 mg veliparib twice daily with CI IC topotecan 1.2 mg/m2/d + carboplatin 150 mg/m2/day on days 3–7. At the MTD, dosing of veliparib was lengthened from 8 days to 14 and 21 days without increased toxicity. Accordingly, the recommended phase II dose is veliparib 80 mg twice daily for up to 21 days in combination with topotecan and carboplatin as detailed above. With respect to responses noted, 33% of patients achieved objective responses (14 CR, 11 CRi and 8 PR) noted across a wide range of dose levels. In patients with no history of antecedent MPN or CMML, the response rate was 25% (19/77). However, those who had an aggressive MPN, CMML or related AML 64% responded (14/22), 11 of whom proceeded to allogeneic BMT. The median duration for response in all patients achieving a CR, CRi, or PR was 7.5 (95% CI, 5.4 – 13.1) months. Although in the subset of patients with aggressive CMML, MPN or AML associated with these disorders, the duration of response was 11.5 (95% CI, 7.5 – NA) months. Those who achieved CR, CRi, or PR experienced a median OS of 15.3 (95% CI, 10.5 – 23) months. On the other hand, patients not achieving a response were found to have a median OS of 4.2 (95% CI, 3.1 – 5.3) months. Interestingly, patients with antecedent aggressive CMML, MPN or AML with antecedent CMML/MPN had a median OS of 13.3 (95% CI, 8.2 – 33.3) months. Relapse free survival at one year was 41% (95% CI, 25–68%) in these patients, versus 12% in patients without prior MPN or CMML. Lastly, those patients with baseline DNA repair defects, as evidenced by compromised DNA damage – induced FANCD2 monoubiquitination, had improved overall survival ([HR = 0.56 (95% CI, 0.27 – 0.92)]. Of note, single agent as well as combination veliparib with chemotherapy induced DNA damage as manifested by histone H2AX phosphorylation in CD34+ leukemic blasts, with greater phosphorylation noted in responders. These investigators will be evaluating this therapeutic strategy in patients with newly diagnosed or relapsed refractory AML with an antecedent MPN in a Phase II trial (NCT03289910).
An ongoing clinical trial (NCT02878785) is evaluating the combination of PARPi with DNMTi is at the University of Maryland. In this multicenter phase 1/2 study, adults with untreated AML who are considered unfit for intensive chemotherapy or who have RR-AML are receiving decitabine intravenously daily for five days every 28 days together with escalating doses of talazoparib administered orally once daily on a continuous basis, commencing on cycle 1 day 1. The estimated study completion date of this study is December 2022.
7.2. Lymphomas and Chronic Lymphocytic Leukemia
Given the efficacy of olaparib in ATM deficient lymphoproliferative disorders, a multi-center phase I study was conducted of olaparib monotherapy in 15 patients with relapsed CLL (n-9), MCL (n=4), and T-prolymphocytic leukemia (n=2) (69). Median duration of therapy was 71 days (range 26–93 days). Olaparib was well tolerated with the most common adverse events (AEs) being anemia (66%), thrombocytopenia (53%), fatigue (53%), nausea (33%), and neutropenia (33%). AEs ≥ grade 3 were observed in 10 patients (66%), including anemia (33%), thrombocytopenia (33%), and neutropenia (20%). Median overall survival from initiation of therapy was 129 days with 9 deaths observed during the trial period.
8. Challenges and Future Directions
At the present time, many challenges remain before PARP inhibition can move into “prime time” consideration as a novel therapeutic approach for hematological malignancies. To date, all of the available PARPi appear to have limited single agent activity in leukemia and lymphomas. Of note, the greatest preclinical efficacy in tumors characterized by defective DDR. improved clinical efficacy will likely require identification of specific biological subtypes of AML (i.e. those expressing a “BRCAness” phenotype or cytogenetic and/or molecular aberrations affecting intrinsic tumor DNA damage repair). The most promising approach appears to be combining PARPi with agents that will either directly induce DNA damage to cancer cells (i.e. alkylating agents, topoisomerase inhibitors) or with other targeted agents capable of selectively impairing the cell’s ability to repair DNA. Agents directly altering epigenetic mechanisms, such as DNA methyltransferases and histone deacetylase inhibitors may be particularly suited for combinatorial approaches with PARPi, given their ability to alter chromatin structure and gene expression, and reduce the capacity of cells to undergo HR (30, 70, 71).
Combination regimens may also help in eradicating resistant disease and preventing the development of time-dependent PARP inhibitor resistant/refractory cells (72). Sullivan and colleagues recently identified the RAD52-RAD51 pathway as an escape route from PARP-mediated synthetic lethality in BRCA1/2 deficient cells. This group demonstrated that targeting PARP1 and RAD52 with small molecule inhibitors or dominant-negative mutants resulted in synergistic accumulation of DSBs and cell death of BRCA1/2 deficient leukemia cells, but not BRCA1/2 proficient cells. Simultaneous targeting of PARP1 and RAD52 also effectively reduced in vivo leukemia burden in preclinical models.
9. Conclusion
Poly (ADP-ribose) polymerase (PARP) inhibitors known to induce synthetic lethality in BRCA-mutant breast and ovarian cancers have now emerged as a novel therapeutic approach for hematological malignancies. This is based on accumulating laboratory evidence that select leukemia/lymphoma cells exhibit similar DNA repair deficiencies as BRCA mutant cells due to molecular and chromosomal events. To date, three PARP inhibitors (olaparib, velarinib, and talazoparib) have undergone clinical trial evaluation in hematological malignancies. Evidence to date supports some clinical benefit with excellent tolerability of PARPi. To further improve these results, PARPi are now being partnered with other therapies including cytotoxic agents, epigenetic modifiers, and small molecule inhibitors in early stage clinical trials in leukemia and lymphoma patients.
Figure 1.
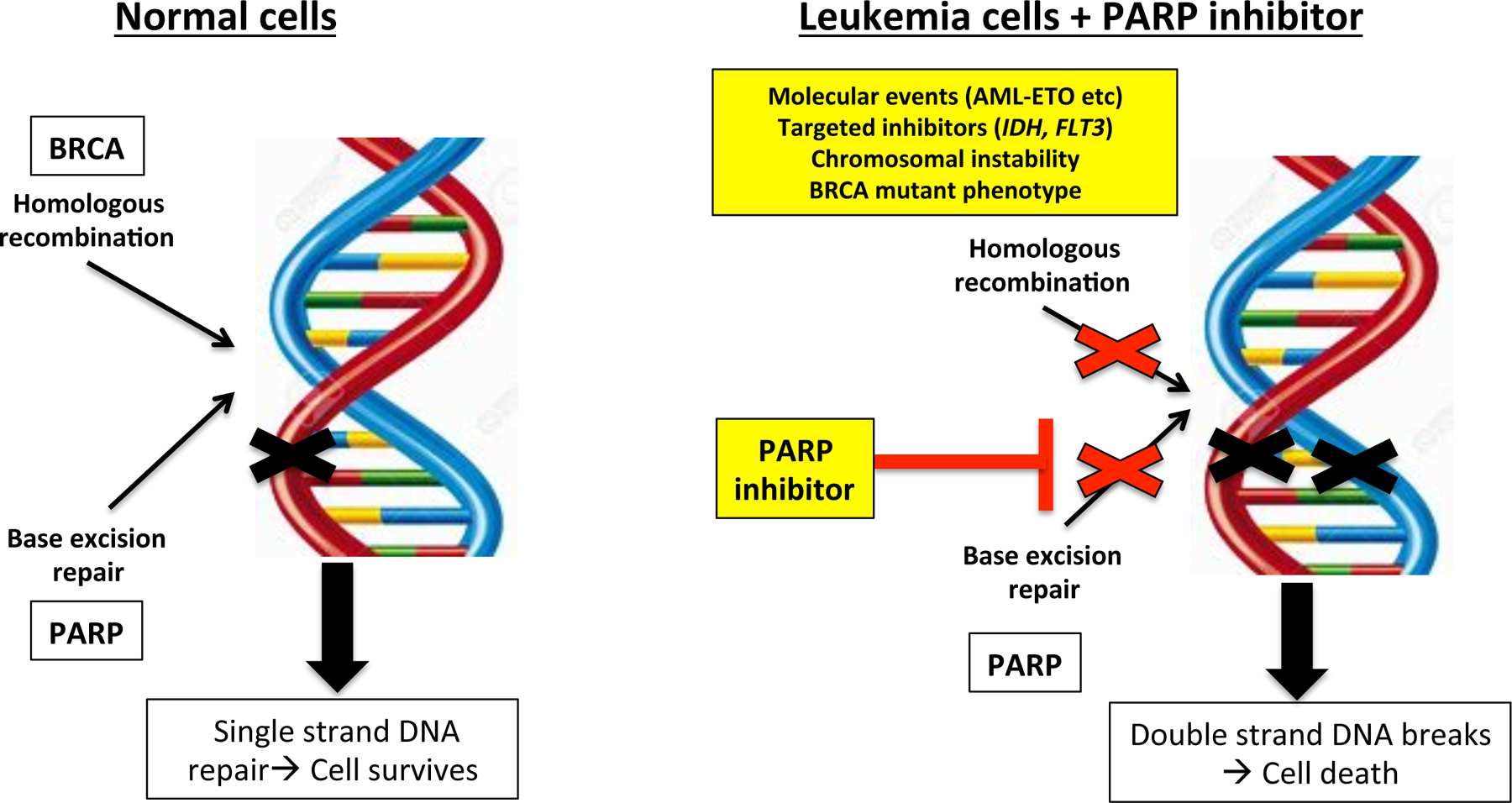
Mechanisms of sensitivity to PARP inhibition in hematological malignancies
Adapted from Iglehart JD et a. N Engl J Med 2009; 361:181–191
Table 2.
Clinical trials of PARP inhibition in hematological malignancies
| Treatment regimen | Phase trial | Disease | Patients | Outcomes | Adverse Events |
|---|---|---|---|---|---|
| Talazoparib (100– 2000 µg/day) | Phase 1 | Advanced heme cancers | 33 patients (21 AML, 4 MDS, 4 CLL, 4 MCL) | Stable disease in 12/24 pts, 1 MDS pt transfusion indpt | DLTs: febrile neutropenia and neutropenic sepsis at 2000 ug/day |
| Talazoparib daily plus decitabine 20 mg/m2 IV x 5 days per month | Phase 1 | AML | ND-AML unfit for intensive chemo or RR-AML | Ongoing | Ongoing |
| Veliparib (d1, d4–12 or d1–8) plus Temozolamide (d3–9, d1–5) | Phase 1 | ND-AML, RR-AML, CMML-2, sAML, tAML | 48 patients (AML) | CR (8/48)(16.6%) Median OS= 5.3 mos Median OS of responders = 20 mos | DLTs: Grade 3 mucositis/esophagitis; |
| Veliparib + carboplatin + topotecan | Phase 1 3+3 cohorts | RR-AML, MPN, CMML | 99 patients (34 RRAML, 35 sAML, 22 MPN, 4 ALL) | ORR= 33% (14 CR, 11 CRi, 8 PR) | |
| Veliparib 30 mg bid x 21d + carboplatin + topotecan (NCT03289910) | Phase 2 | AML | ND or RR-AML with antecedent MPN patients | Ongoing | Ongoing |
| Olaparib monotherapy | Phase 1 | Relapsed CLL, MCL and T-PLL | CLL (n-9), MCL (n=4), and T-PLL (n=2) | Median OS (all pts) = 129 days | AEs ≥ grade 3 in 10 pts (66%): anemia (33%), thrombocytopenia (33%), and neutropenia (20%) |
Practice Points:
PARP inhibitors have an emerging role for treatment of leukemia and lymphoma, even in the absence of known BRCA1/1 mutations.
Faulty DNA damage response occurring as a result of specific molecular and chromosomal aberrations in leukemia and lymphoma patients may be sufficient to render these patients sensitive to PARP inhibition.
To date, PARP inhibitors have exhibited limited single activity but excellent tolerability in a small number of clinical trials in hematological malignancies.
Research Agenda.
To determine the clinical efficacy of PARPi for patients with hematological malignancies (all comers vs. select molecular/cytogenetic disease categories)
Correlative testing should be performed on all individuals receiving PARPi on clinical trials to address whether specific molecular aberrations and/or defects in the underlying DNA repair processes can predict for response.
Acknowledgements/ Funding
This work was supported by a National Cancer Institute grant P30CA016506 to Roswell Park Comprehensive Cancer Center. EW and AP are also supported by the RPCI Alliance Foundation (Jacquie Hirsch Leukemia Research Fund and Leukemia Disease Site Research Group funding).
Funding:
This work was supported by a National Cancer Institute (NCI) grant to Roswell Park Comprehensive Cancer Center (P30CA016056). EW and AP are also supported by the Roswell Park Alliance Foundation (Jacquie Hirsch Leukemia Research Fund).
Footnotes
Conflicts of Interest
EW has served as a speaker for Novartis, Jazz, and Astellas. She has served in an advisory/consultant role for Pfizer, Amgen, Agios, Abbvie, Arog, Immunogen, Jazz, Daichi-Sanyo, and Ceylad. She previously received research funding from Immunogen. None of the other authors (CF, SP, AP) have any other conflicts of interest to declare.
References
- 1.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. The New England journal of medicine 2015;373(18):1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gafencu GA, Tomuleasa CI, Ghiaur G. PARP inhibitors in acute myeloid leukaemia therapy: How a synthetic lethality approach can be a valid therapeutic alternative. Medical hypotheses 2017;104:30–4. [DOI] [PubMed] [Google Scholar]
- 3.Zhao L, So CW. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Experimental hematology 2016;44(10):902–7. [DOI] [PubMed] [Google Scholar]
- 4.Gaymes TJ, Mufti GJ, Rassool FV. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer research 2002;62(10):2791–7. [PubMed] [Google Scholar]
- 5.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Molecular cell 2010;39(1):8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langelier MF, Planck JL, Roy S, Pascal JM. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. The Journal of biological chemistry 2011;286(12):10690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nature reviews Cancer 2010;10(4):293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Molecular aspects of medicine 2013;34(6):1217–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. The Journal of biological chemistry 1999;274(25):17860–8. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nature reviews Molecular cell biology 2006;7(7):517–28. [DOI] [PubMed] [Google Scholar]
- 11.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes & development 2012;26(5):417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maifrede SE, M., Nieborowski-Skorska M, Di Marcantonio D, Hulse M, Le BV, Zhao H, Piwocka K, Tempera I, Sykes SM, Skorski T MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Advances 2017;1:1467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 14.Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Molecular aspects of medicine 2013;34(6):1109–23. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine 2013;368(22):2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, et al. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Molecular aspects of medicine 2013;34(6):1066–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esposito MT, So CW. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014;123(6):545–61. [DOI] [PubMed] [Google Scholar]
- 18.Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014;514(7520):107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casorelli I, Tenedini E, Tagliafico E, Blasi MF, Giuliani A, Crescenzi M, et al. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: Focus on DNA repair genes. Leukemia 2006;20(11):1978–88. [DOI] [PubMed] [Google Scholar]
- 20.Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer cell 2016;30(4):637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y, Aoyagi-Scharber M, Wang B. Trapping Poly(ADP-Ribose) Polymerase. The Journal of pharmacology and experimental therapeutics 2015;353(3):446–57. [DOI] [PubMed] [Google Scholar]
- 22.Research CfDE. Approved Drug - Rucaparib 2017. [updated 5/182017. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm533891.htm.
- 23.Research CfDEa. Approved Drugs - Niraparib (ZEJULA) 2017. [Available from: http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm548487.htm.
- 24.Wang L, Cai W, Zhang W, Chen X, Dong W, Tang D, et al. Inhibition of poly(ADP-ribose) polymerase 1 protects against acute myeloid leukemia by suppressing the myeloproliferative leukemia virus oncogene. Oncotarget 2015;6(29):27490–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ES. Treating acute myeloid leukemia in older adults. Hematology American Society of Hematology Education Program 2014;2014(1):14–20. [DOI] [PubMed] [Google Scholar]
- 26.Faraoni I, Compagnone M, Lavorgna S, Angelini DF, Cencioni MT, Piras E, et al. BRCA1, PARP1 and gammaH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochimica et biophysica acta 2015;1852(3):462–72. [DOI] [PubMed] [Google Scholar]
- 27.Falzacappa MV, Ronchini C, Faretta M, Iacobucci I, Di Rora AG, Martinelli G, et al. The Combination of the PARP Inhibitor Rucaparib and 5FU Is an Effective Strategy for Treating Acute Leukemias. Molecular cancer therapeutics 2015;14(4):889–98. [DOI] [PubMed] [Google Scholar]
- 28.Robert C, Nagaria PK, Pawar N, Adewuyi A, Gojo I, Meyers DJ, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leukemia research 2016;45:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orta ML, Hoglund A, Calderon-Montano JM, Dominguez I, Burgos-Moron E, Visnes T, et al. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2’-deoxycytidine lesions. Nucleic acids research 2014;42(14):9108–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaymes TJ, Shall S, MacPherson LJ, Twine NA, Lea NC, Farzaneh F, et al. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009;94(5):638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamauchi T, Uzui K, Nishi R, Shigemi H, Ueda T. Gemtuzumab ozogamicin and olaparib exert synergistic cytotoxicity in CD33-positive HL-60 myeloid leukemia cells. Anticancer research 2014;34(10):5487–94. [PubMed] [Google Scholar]
- 32.Portwood SPRA, Walker RM, Wang ES Combining IMGN779, a Novel Anti-CD33 Antibody-Drug Conjugate (ADC), with the PARP Inhibitor, Olaparib, Results in Enhanced Anti-Tumor Activity in Preclinical Acute Myeloid Leukemia (AML) Models. Blood 2016;128:1645. [Google Scholar]
- 33.Yang A, Wilson NS, Ashkenazi A. Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Current opinion in cell biology 2010;22(6):837–44. [DOI] [PubMed] [Google Scholar]
- 34.Meng XW, Koh BD, Zhang JS, Flatten KS, Schneider PA, Billadeau DD, et al. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. The Journal of biological chemistry 2014;289(30):20543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, et al. A Small-Molecule Inhibitor of WEE1, AZD1775, Synergizes with Olaparib by Impairing Homologous Recombination and Enhancing DNA Damage and Apoptosis in Acute Leukemia. Molecular cancer therapeutics 2017. [DOI] [PMC free article] [PubMed]
- 36.Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Molecular cancer therapeutics 2009;8(8):2232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(8):1837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & development 2013;27(8):836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Science translational medicine 2017;9(375). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molenaar RJ, Radivoyevitch T, Nagata Y, Khurshed M, Przychodzen B, Makishima H, et al. IDH1/2 Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(7):1705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maifrede SN-SM, Sullival K, Dasgupta Y, Podszywalow-Bartnicka P, Le BV, Solecka M, Lian Z, Machnicki MM, Zhao H, Jelinek J, Piwocka K, Stoklosa T, Fischer T, Sykes SM, Koschmieder S, Bullinger L, Valent P, Huang J, Skorski T Tyrosine Kinase Inhibitor-Induced Defects in DNA Repair Sensitize FLT3(ITD)-Positive Acute Myeloid Leukemia Quiescent and Proliferative Cells to PARP Inhibitors. American Society of Hematology Annual Meeting; Atlanta, GA. Blood 2017. p. 2635. [Google Scholar]
- 42.Liu H, Cheng EH, Hsieh JJ. MLL fusions: pathways to leukemia. Cancer biology & therapy 2009;8(13):1204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nature medicine 2015;21(12):1481–90. [DOI] [PubMed] [Google Scholar]
- 44.Look AT. Oncogenic transcription factors in the human acute leukemias. Science 1997;278(5340):1059–64. [DOI] [PubMed] [Google Scholar]
- 45.Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. The Journal of clinical investigation 2003;112(11):1751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forster VJ, Nahari MH, Martinez-Soria N, Bradburn AK, Ptasinska A, Assi SA, et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia 2016;30(1):250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bamezai SHJ, Sahin D, Mohr F, Ciccarone F, Vegi N, Jose AP, Mulaw MA, Caiafa P, Dohner K, Dohner H, Feuring-Buske M, Buske C, Rawat VPS The PARP Inhibitor Olaparib Antagonizes Leukemic Growth Induced By TET1 Overexpression in AML1-ETO Positive Acute Myeloid Leukemia. Blood 2016;128:4063. [Google Scholar]
- 48.Zhou D OJ, Chen B. MiR-181a Down-Regulates PARP1 By Enhancing Its Acetylation in MLL -Rearranged AML. Blood 2017;130:1388.28912297 [Google Scholar]
- 49.Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer causes & control : CCC 2008;19(4):379–90. [DOI] [PubMed] [Google Scholar]
- 50.Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013;341(6144):395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO molecular medicine 2009;1(6–7):315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007;128(1):157–70. [DOI] [PubMed] [Google Scholar]
- 53.Kogan AAMLJ, Topper M, Muvarak N, Stojanovic L, Creed TM, Bentzen S, Civin CI, Baer MR, Kingsbury TJ, Baylin S, Rassool FV DNA Demethylating Agents Generate a Brcaness Effect in Multiple Sporadic Tumor Types: Prediction for Sensitivity to PARP Inhibitors in AML. Blood 2017;130:3347. [Google Scholar]
- 54.Shiloh Y The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006;31(7):402–10. [DOI] [PubMed] [Google Scholar]
- 55.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature reviews Molecular cell biology 2008;9(10):759–69. [DOI] [PubMed] [Google Scholar]
- 56.Williamson CT, Muzik H, Turhan AG, Zamo A, O’Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Molecular cancer therapeutics 2010;9(2):347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang P, Zhang W, Wang J, Liu Y, An R, Jing H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther 2018. [DOI] [PubMed]
- 58.Fang NY, Greiner TC, Weisenburger DD, Chan WC, Vose JM, Smith LM, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci U S A 2003;100(9):5372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Austen B, Powell JE, Alvi A, Edwards I, Hooper L, Starczynski J, et al. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 2005;106(9):3175–82. [DOI] [PubMed] [Google Scholar]
- 60.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010;116(22):4578–87. [DOI] [PubMed] [Google Scholar]
- 61.Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO molecular medicine 2012;4(6):515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maifrede S, Martin K, Podszywalow-Bartnicka P, Sullivan-Reed K, Langer SK, Nejati R, et al. IGH/MYC Translocation Associates with BRCA2 Deficiency and Synthetic Lethality to PARP1 Inhibitors. Molecular cancer research : MCR 2017;15(8):967–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valdez BC, Li Y, Murray D, Liu Y, Nieto Y, Champlin RE, et al. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2018;9(3):3908–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schaefer NG, James E, Wahl RL. Poly(ADP-ribose) polymerase inhibitors combined with external beam and radioimmunotherapy to treat aggressive lymphoma. Nucl Med Commun 2011;32(11):1046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Curtis A, Rueter J, Rajan S, Zhang R, Shopland L. Additive and synergistic inhibition of mantle cell lymphoma cell growth by combining olaparib with ibrutinib. J Cell Biochem 2018;119(7):5843–51. [DOI] [PubMed] [Google Scholar]
- 66.Mufti GJ EE, Popat R, Mattison R, Menne T, Azar J, Bloor A, Gaymes T, Khwaja A, Juckett M, Lennard A, Zhang C, Gallant GJA, Gopal AK Results of a Phase 1 Study of BMN 673, a Potent and Specific PARP-1/2 Inhibitor, in Patients with Advanced Hematological Malignancies. Haematologica 2014;99(s1):33–4. [Google Scholar]
- 67.Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, et al. A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(3):697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pratz KW, Rudek MA, Gojo I, Litzow MR, McDevitt MA, Ji J, et al. A Phase I Study of Topotecan, Carboplatin and the PARP Inhibitor Veliparib in Acute Leukemias, Aggressive Myeloproliferative Neoplasms, and Chronic Myelomonocytic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(4):899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pratt G, Yap C, Oldreive C, Slade D, Bishop R, Griffiths M, et al. A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br J Haematol 2017. [DOI] [PubMed]
- 70.Gaymes TJ, Padua RA, Pla M, Orr S, Omidvar N, Chomienne C, et al. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Molecular cancer research : MCR 2006;4(8):563–73. [DOI] [PubMed] [Google Scholar]
- 71.Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future oncology 2011;7(2):263–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. The Journal of clinical investigation 2017;127(6):2392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]