

Abstract

Activation of the IRE-1/XBP-1s pathway supports tumor progression. Here, we report a novel prodrug, TC-D-F07, in which a thiol-reactive dinitrobenzenesulfonyl (Dns) cage was installed onto the C8 hydroxyl of the covalent IRE-1 inhibitor D-F07. The electron-withdrawing Dns group in TC-D-F07 stabilizes the neighboring 1,3-dioxane acetal, allowing for stimulus-mediated control of its inhibitory activity. TC-D-F07 exhibits high sensitivity to intracellular thiols. Because tumor cells exhibit higher concentrations of glutathione and cysteine, treatment with TC-D-F07 results in more sustained levels of D-F07 in transformed versus normal cells. In addition, we show that a dinitrophenyl cysteine adduct resulting from cleavage of the Dns group induces endoplasmic reticulum (ER) stress, causing tumor cells to increase the expression of XBP-1s. The accumulated levels of D-F07 and its gradual decomposition into the active IRE-1 inhibitor eventually deprive tumor cells of XBP-1s, leading to more severe apoptosis than those treated with its uncaged analogue.
Keywords: cancer, IRE-1, PERK, ATF6, ATF4, endoplasmic reticulum stress, unfolded protein response
Introduction
Endoplasmic reticulum (ER) stress, resulting from the accumulation of misfolded protein, hypoxia, or calcium depletion in the ER, leads to activation of the ER stress response or unfolded protein response. Three major ER stress response pathways mediated by their respective ER-resident stress sensor proteins IRE-1, PERK, and ATF6 have been identified and characterized as critical mechanisms for normal cells to cope with ER stress. These pathways have also been shown to be critical for the survival of cancer.1 IRE-1 represents the most conserved ER stress response pathway and activates the production of the functional XBP-1s transcription factor through its ribonuclease (RNase) activity. Upregulation of XBP-1s results in the expression of specific chaperones and lipids and restoration of ER homeostasis. By genetic depletion of XBP-1s in chronic lymphocytic leukemia (CLL) and triple-negative breast cancer mouse models, we and others have shown that the IRE-1/XBP-1s pathway contributes to tumor progression.2,3 We have also developed tricyclic chromenone salicylaldehyde-based inhibitors such as B-I09 (Figure 1A) to inhibit the RNase activity of IRE-1, resulting in potent suppression of XBP-1s in cells and in vivo.3,4 With demonstrated pharmacokinetics to guide its dosing, B-I09 is effective in suppressing XBP-1s and improving diseased conditions in preclinical mouse models of CLL,3 Burkitt’s lymphoma,5 chronic graft-versus-host diseases (GVHD),6 and acute GVHD.7
Figure 1.
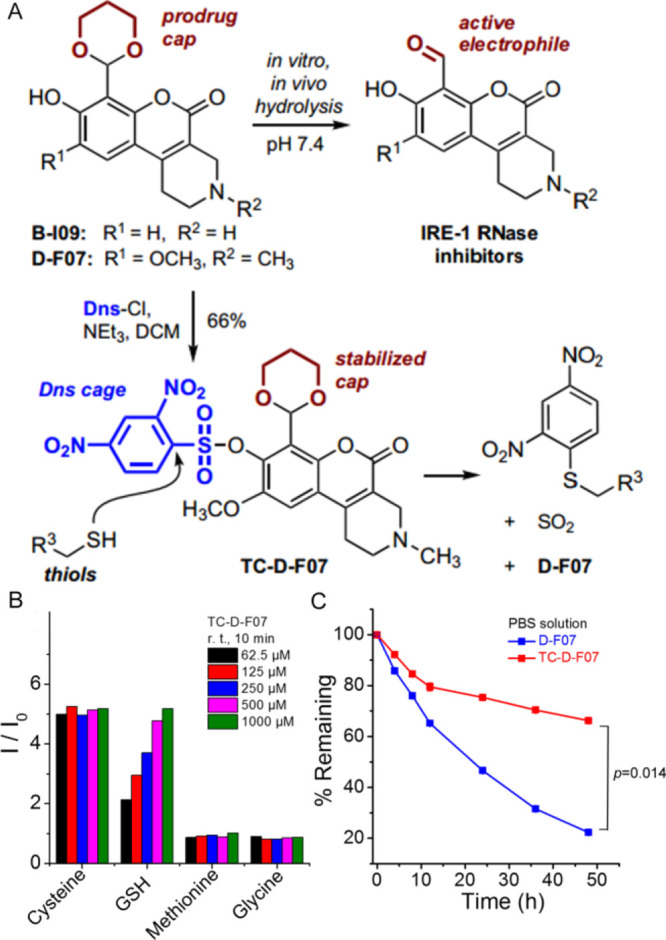
Installation of a thiol-reactive cage on the hydroxy group of D-F07 stabilizes the 1,3-dioxane acetal protecting group and allows for thiol-mediated release of fluorescent D-F07. (A) Structure of caged and prodrug derivatives of IRE-1 RNase inhibitors and mechanism of thiol-mediated uncaging. (B) Fluorescence readouts of D-F07 upon incubation of TC-D-F07 (2.5 μM in DMSO/PBS solution (v/v = 1:99), Ex = 360 nm) with increasing concentrations of cysteine, GSH, methionine, and glycine at room temperature for 10 min. I0 was the initial fluorescence intensity of TC-D-F07 at 2.5 μM. (C) Decomposition rates of D-F07 and TC-D-F07 were analyzed by HPLC and plotted as a function of time in PBS at 37 °C.
B-I09 was initially developed as a prodrug, in which we used a 1,3-dioxane acetal to protect the functional aldehyde group of the active IRE-1 inhibitor. Our recent studies have established that the installation of the 1,3-dioxane acetal results in sustained inhibition of XBP-1s in cells by B-I09 when compared with its aldehyde-exposed analogues. In addition, masking the aldehyde as 1,3-dioxane restores the inherent fluorescent properties of the coumarin structure within B-I09.8−10 Based on this discovery, we developed the fluorescent analogue D-F07 (Figure 1A), which exerts more potent and sustained inhibitory activity than B-I09.9 We demonstrated that chemical modification of the C8 phenol in D-F07 dramatically enhances the hydrolytic stability of the 1,3-dioxane acetal. This enabled the development of PC-D-F07 in which we modified the C8 hydroxyl motif with a photolabile cage group. The inhibitory activity of PC-D-F07 can thus be spatiotemporally controlled by UV irradiation,9 leading to a cascade mechanism of drug release.
Because IRE-1 RNase inhibitors trigger modest growth inhibition in multiple myeloma (MM) cell lines, studies utilizing these inhibitors for the treatment of MM have involved combination with bortezomib (Velcade)11 or other drugs.12 Bortezomib targets the ubiquitin-proteasome system by blocking proteasome activity and so leads to increased proteotoxic stress that can kill MM cells. However, treatment with bortezomib or MG132 also leads to the transient expression of XBP-1s,13 which may protect MM cells from death and lead to drug resistance. Thus, the combination of an IRE-1 inhibitor and an ER stress-inducing compound as an effective therapeutic strategy for MM or other types of B cell cancer warrants further investigation.
The effectiveness of a combination therapy can be influenced by the different membrane permeability of each drug, resulting in nonequimolar amounts of drugs inside the cells. Taking advantage of the fact that cancer cells often express higher levels of glutathione (GSH) and cysteine relative to normal cells,14,15 we chose to install a thiol-sensitive 2,4-dinitrobenzenesulfonyl (Dns) group16 onto the C8 hydroxy moiety of D-F07. The sulfonate cage can stabilize the 1,3-dioxane protecting moiety on D-F07 and quench the fluorescence of D-F07 via its strong electron-withdrawing property. The Dns moiety can also induce ER stress once it is cleaved, leading to the increased expression of XBP-1s and ATF4.17 Once liberated, fluorescent D-F07 can be visualized in cancer cells and potently inhibit XBP-1s expression. We have thus successfully designed and synthesized a caged prodrug, TC-D-F07, that demonstrates the possibility of delivering ER stress-inducing and XBP-1s-inhibiting activities in one entity for the treatment of cancer.
Experimental Section
General Synthesis Information
Unless stated otherwise, all reactions were carried out in flame-dried glassware under a positive pressure of nitrogen or argon gas using dry solvents. Reagents and solvents were obtained commercially and used without further purification except where noted. Toluene, dichloromethane (DCM), dimethylformamide, MeCN, and Et2O were used after passaging through the Pure Process Technologies (PPT) solvent purification system. Thin-layer chromatography (TLC) analysis was performed using silica gel precoated glass-backed plates (Merck 60 F254; 0.25 mm). Flash chromatography was conducted using silica gel cartridges (particle size: 40–65 μm). The progress of reactions was detected by TLC (single spot/two solvent systems) using a UV lamp, ninhydrin, ceric ammonium molybdate, or basic KMnO4 stain(s). NMR spectra were recorded using a 400 or 500 MHz spectrometer. Proton chemical shifts are reported as δ values relative to residual signals from deuterated solvents (CDCl3 or CD3CN). UV–vis absorbance and fluorescence spectra were observed and recorded using a BioTek Synergy NEO2 Multimode Reader. The purity (≥95%) of all synthesized compounds was confirmed by 1H NMR.
7-(1,3-Dioxan-2-Yl)-9-methoxy-3-methyl-5-oxo-1,3,4,5-tetrahydro-2H-chromeno[3,4-c]pyridin-8-Yl 2,4-Dinitrobenzenesulfonate (TC-D-F07)
2,4-Dinitrobenzenesulfonyl chloride (77 mg, 2900 μmol) was added to a mixture of D-F07(9) (50 mg, 1400 μmol) and triethylamine (29 mg, 2900 μmol) in DCM, and the reaction was stirred for 5 h at room temperature. The reaction was diluted with DCM, washed with H2O and brine, desiccated over anhydrous MgSO4, and concentrated. Further purification using silica gel flash chromatography (0–5% MeOH/CHCl3) yielded TC-D-F07 as an orange solid (55 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 8.70 (d, J = 2.2 Hz, 1H), 8.55 (dd, J = 8.7, 2.3 Hz, 1H), 8.37 (d, J = 8.7 Hz, 1H), 6.92 (s, 1H), 6.30 (s, 1H), 4.11 (dd, J = 11.3, 4.6 Hz, 2H), 3.98–3.83 (m, 2H), 3.52 (s, 3H), 3.41 (s, 2H), 2.83 (dt, J = 5.5, 3.2 Hz, 2H), 2.73 (t, J = 5.5 Hz, 2H), 2.48 (s, 3H), 2.26–2.09 (m, 1H), 1.32 (d, J = 13.4 Hz, 1H); HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H24N3O12S 578.1075, found 578.1084.
(S-2,4-Dinitrophenyl)cysteine (E-H01)
1-Chloro-2,4-dinitrobenzene (130 mg, 0.63 mmol) and l-cysteine (925 mg, 7.6 mmol) were dissolved in a 1:1 mixture of 2 M aq. NaOH and ethanol. After stirring for 24 h, the solution was neutralized with 1 M aq. HCl to generate a yellow precipitate which was further filtered and washed with H2O. The crude material was dissolved in a 1:1 mixture of MeCN/aq. PBS buffer and purified by RP-HPLC (C12 preparative column, 5–95% MeCN/H2O with 0.1% formic acid, linear gradient) to give E-H01 as a yellow powder after lyophilization (26 mg, 19%). 1H NMR (400 MHz, CD3CN) δ 9.12 (s, 1H), 8.99 (s, 1H), 8.26 (d, 1H), 7.05 (d, 1H), 4.84 (s, 1H), 3.15 (dd 2H); HRMS (ESI-TOF) m/z [M + H]+ calcd for 288.0285, found 288.0282.
Antibodies and Reagents
Antibodies against XBP-1s (Cell Signaling Technology), IRE-1 (Cell Signaling Technology), ATF6 (Proteintech), PERK (Cell Signaling Technology), p-eIF2α (Cell Signaling Technology), eIF2α (Cell Signaling Technology), ATF4 (Cell Signaling Technology), cleaved PARP (Cell Signaling Technology), and p97 (Fitzgerald) were obtained commercially. Cysteine, methionine, glycine, GSH, and N-methylmaleimide (NMM) were procured from Sigma-Aldrich.
Cell Culture
Cells were cultured in incubators containing 5% CO2 and maintained at 37 °C. RPMI-8226 and NCI-H929 human MM cell lines (purchased from ATCC), 5TGM1 mouse MM cell line (a gift from Dr. Lori A. Hazlehurst at the West Virginia University, Morgantown, WV), MEC2 and WaC3 human CLL cell lines (gifts from Dr. Javier A. Pinilla-Ibarz at the Moffitt Cancer Center, Tampa, FL), and primary B cells purified from spleens of mice were grown in RPMI 1640 media (Gibco) supplemented with heat-inactivated fetal bovine serum (FBS, 10%), l-glutamine (2 mM), sodium pyruvate (1 mM), nonessential amino acids (0.1 mM), β-mercaptoethanol (β-ME; 0.1 mM), penicillin G sodium (100 U/mL), and streptomycin sulfate (100 μg/mL). The J558 mouse myeloma cell line (purchased from ATCC) was cultured in DMEM media (Gibco) and 10% heat-inactivated horse serum together with the abovementioned supplemental nutrients. Human embryonic kidney 293 T cells (purchased from ATCC) and mouse hepatoma HEPA 1–6 cell line (purchased from ATCC) were cultured in DMEM media with 10% heat-inactivated FBS and the same supplemental nutrients.
Protein Isolation and Immunoblotting
Cells were lysed using RIPA buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and 1 mM EDTA) supplemented with protease inhibitors (Roche). BCA assays (Pierce) were used to determine protein concentrations. Samples were heated to 95 °C for 10 min in the SDS-PAGE sample buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, and 0.1% bromophenol blue) with β-ME before analysis by SDS-PAGE. Proteins were transferred to nitrocellulose membranes, which were blocked in 5% nonfat milk (wt/vol in PBS) and immunoblotted with indicated primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies (SouthernBiotech). Immunoblots were developed with Western Lightning Chemiluminescence Reagent (PerkinElmer).
Cell Proliferation Assays
Cells were suspended in phenol red-free media, seeded in 96-well cell culture plates, and treated with inhibitors. At indicated time points, cells were spun down and cell proliferation was assessed by XTT assays (Roche). We first combined 100 μL of phenol red-free RPMI media with 50 μL XTT labeling reagent and 1 μL electron-coupling reagent and then applied the reaction to each well of the 96-well plates. Cells were incubated in a 5% CO2 incubator at 37 °C for 4 h so that the yellow tetrazolium salt XTT was cleaved by mitochondrial dehydrogenases produced by metabolically active cells. The resulting orange formazan dye was quantified at 492 nm using a BioTek Synergy NEO2 Multimode Reader.
Compound Degradation Assay by HPLC
D-F07 and TC-D-F07 were dissolved in dimethyl sulfoxide (DMSO) to prepare for 20 mM stock solutions. D-F07 and TC-D-F07 were then diluted with PBS (pH 7.4) to 400 μM and incubated at 37 °C for different time periods. At each time point, compound aliquots were injected onto an analytical HPLC instrument and monitored by UV absorbance at 220 nm. The remaining intact compound relative to that found in the initial time point (t = 0) was plotted as a function of time. For compound degradation in lysates monitored by HPLC, HEPA 1–6 adherent cells (2.4 × 108 cells) were washed with cold PBS, harvested using trypsin, and spun down. Cell pellets were lysed with 300 μL of 3% NP-40 in cold PBS and spun down, and the supernatant was collected. Cell lysates were transferred to a clean tube, and proteins were precipitated from lysates using cold trichloroacetic acid (TCA). Briefly, 180 μL of ice cold 100% (w/v) TCA was combined with 900 μL lysates, incubated on ice for 10 min, and spun down, and the supernatant was collected. The supernatant was transferred to a clean tube and neutralized to pH 5.0 with 1 M NaHCO3. Sodium acetate buffer (0.2 M, pH 5.0) was used as the pH-controlled buffer. TC-D-F07 (20 mM in DMSO) was diluted to 500 μM in the deproteinized lysate or the pH-controlled buffer and incubated at 37 °C for different time periods. Compound aliquots from various time points were injected onto an analytical HPLC instrument and monitored by UV absorbance at 220 nm. The remaining intact compound relative to that found in the initial time point (t = 0) was plotted as a function of time.
Fluorescence Response of TC-D-F07 to Endogenous GSH
Primary B cells, 5TGM1, RPMI-8226, NCI-H929, J558, MEC2, or WaC3 cells (10 × 106 cells) were washed with cold PBS and spun down. 293 T and HEPA 1–6 adherent cells (0.5 × 106 cells) were washed with cold PBS, harvested using trypsin, and spun down. Cell pellets were lysed in 50 μL of 0.5% NP-40 in cold PBS and centrifuged for 15 min at 15,000 rpm at 4 °C. Cell lysates were transferred to a clean tube, and proteins were precipitated from lysates using cold TCA. Briefly, 8 μL of ice cold 100% (w/v) TCA was combined with 50 μL lysates, incubated on ice for 10 min, and centrifuged at 12,000×g for 5 min at 4 °C. The supernatant was transferred to a clean tube, neutralized to pH 6.0 with 1 M NaHCO3, and centrifuged again at 13,000×g for 15 min at 4 °C. The supernatant was transferred to a 96-well black plate, added with TC-D-F07 at the final concentration of 20 μM, and measured for the fluorescence intensity (Ex/Em: 350/454 nm)18 using a BioTek Synergy NEO2 Multimode Reader. Each fluorescent reading value was adjusted by subtracting the background measured from the supernatant alone.
GSH Concentration Assay
Primary B cells (25 × 106 cells), 5TGM1, NCI-H929, MEC2, or WaC3 cells (5 × 106 cells) were washed with cold PBS and spun down. Then, 293 T and HEPA 1–6 adherent cells (0.5 × 106 cells) were washed with cold PBS, harvested using trypsin, and spun down. GSH concentration was assayed using the Glutathione Fluorescence Detection Kit (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were lysed with 5% 5-sulfo-salicylic acid, incubated for 10 min at 4 °C, and spun down, and the supernatant was collected. Deproteinized lysate was diluted 1:5 with assay buffer followed by an additional dilution of 1:5 with a sample diluent for a final dilution of 1:25. Fifty microliters of diluted lysate and 25 μL detection reagent were added to a 96-well black plate, incubated for 15 min at room temperature, and measured for the fluorescence intensity (Ex/Em: 390/510) using a BioTek Cytation 5 Multimode Reader. Free GSH concentration was calculated by comparison to a standard curve and normalized by protein load.
Confocal Microscopy
First, 293 T or HEPA 1–6 cells were cultured for 12 h on a clean coverslip in each well of a 12-well plate before treatment. Live cells were incubated with DMSO (control group) or TC-D-F07 (20 μM) or pretreated with NMM (1 mM) for 1 h before incubation with TC-D-F07 (20 μM) for the indicated time. Cells were then washed three times with PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. In a different experiment, HEPA 1–6 cells were treated with DMSO, D-F07 (20 μM), or TC-D-F07 (20 μM) for the indicated time, washed three times with PBS, and fixed in 4% paraformaldehyde for 15 min at room temperature. Additionally, 5TGM1 or MEC2 cells were cultured for 12 h in a 12-well plate before treatment. Cells were then incubated with DMSO, D-F07 (20 μM), or TC-D-F07 (20 μM) for 3 h, washed three times with PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, spun down, and resuspended with 100 μL of PBS. Cells were subsequently seeded onto a clean coverslip, air-dried, and mounted on a glass slide. Confocal images were obtained using a Leica TCS SP5 II confocal microscope. D-F07, resulting from TC-D-F07, was excited at 488 nm to observe the fluorescence emission from 498 to 650 nm as green.
Mice
Wild-type mice were maintained in our animal facility strictly following the guidelines approved by the animal care and use committee at the Houston Methodist Research Institute.
Statistical Analysis
The t-test analysis was used to evaluate data. A P value of less than 0.05 was considered significant.
Results and Discussion
Installation of a Thiol Cage on the Phenol of D-F07 Stabilizes the 1,3-Dioxane Acetal Protecting Group
To develop an inhibitor that could target the expression of XBP-1s in tumor cells, we chose to install a thiol-reactive group onto the C8 hydroxy of D-F07 (Figure 1A). We modified the hydroxy group of D-F07 with a thiol-responsive Dns group, resulting in quenched fluorescence. TC-D-F07 initially emitted weak fluorescence in aqueous solution. A dose-dependent and time-dependent increase of fluorescence was observed upon incubating TC-D-F07 with GSH at room temperature (Figure 1B and S1). As expected, cysteine was also very efficient in cleaving the sulfonate of TC-D-F07 via a nucleophilic aromatic substitution to liberate D-F07. No change in fluorescence was observed when TC-D-F07 was incubated with increasing concentrations of methionine or glycine.
Consistent with our previous report showing that installation of a photolabile cage onto the C8 hydroxy group of D-F07 stabilizes the 1,3-dioxane acetal protecting group,9 we observed that installation of the thiol-reactive group had a similar effect, as evidenced by a slower decomposition rate of TC-D-F07 determined by HPLC (Figure 1C). More than 60% TC-D-F07 still remained intact after 48 h incubation at 37 °C in PBS solution (containing 1% DMSO), while only 22% D-F07 remained intact under the same condition. This indicates that installation of the thiol-reactive cage onto the hydroxy group of D-F07 not only quenches the fluorescence of D-F07 to achieve a fluorescence “Off–On” mode for tracing drug release, but also stabilizes the 1,3-dioxane acetal protecting group.
TC-D-F07 Is Effectively Cleaved To Liberate Fluorescent D-F07 in Tumor Cells That Express Higher Levels of GSH
To evaluate the selectivity of TC-D-F07 toward tumor cells as opposed to normal cells, we first treated SV40 large T antigen-transformed human embryonic kidney 293 T cells and mouse HEPA 1–6 hepatoma cells with TC-D-F07 at 20 μM for 30 or 60 min. We observed stronger fluorescence in TC-D-F07-treated HEPA 1–6 cells than in TC-D-F07-treated 293 T cells (Figure 2A). Both 293 T and HEPA 1–6 cells were also pretreated with NMM (a thiol scavenger) to confirm that the fluorescence in these cells was derived from thiol-mediated cleavage of TC-D-F07 to liberate the fluorescent D-F07 (Figure 2A). To quantify the fluorescence generated in cells, we incubated TC-D-F07 with the same volume of 293 T and HEPA 1–6 deproteinized lysates at room temperature for a course of 90 min and found continuous fluorescence increase in HEPA 1–6 lysate samples but not in 293 T samples (Figure 2B). This could be explained by the fact that HEPA 1–6 cells indeed produce significantly higher intracellular GSH than 293 T cells (Figure S2A). As an important control, we also detected by HPLC the rapid decomposition of TC-D-F07 into D-F07 in deproteinized HEPA 1–6 lysate but not in pH-controlled buffer (Figure 2C), confirming that fluorescence was resulted from the liberated D-F07. Because the IRE-1/XBP-1 pathway is hyperactivated in MM (5TGM1, RPMI-8226, NCI-H929, and J558) and CLL (WaC3 and MEC2) cell lines, we similarly examined whether TC-D-F07 could be more efficiently cleaved to liberate fluorescent D-F07 in these cell lines as opposed to in normal B cells. While a slight fluorescence increase was observed in the TC-D-F07 sample incubated with the deproteinized normal B cell lysate, significantly higher levels of fluorescence were detected in samples incubated with deproteinized lysates prepared from all other malignant B cell lines (Figure 2D). Together with our results showing that cancerous B cells produce significantly higher intracellular GSH than normal B cells (Figure S2B), we propose that TC-D-F07 is potentially a tumor-specific therapeutic agent.
Figure 2.

TC-D-F07 is more effectively uncaged in tumor cells versus normal cells. (A) 293 T or HEPA 1–6 cells were treated with DMSO (control group), TC-D-F07(20 μM), or pretreated with NMM (1 mM) for 1 h before incubation with TC-D-F07 (20 μM) for indicated times, washed three times with PBS, fixed, and analyzed by confocal microscopy. The fluorescence from D-F07 was recorded in the range of 498–650 nm. Scale bar = 10 μm. (B) Fluorescence response of TC-D-F07 to endogenous GSH produced by 293 T or HEPA 1–6 cells was investigated by incubating deproteinized lysates from both cell types with 20 μM TC-D-F07 and monitored at room temperature for 0 to 90 min. I0 was the initial fluorescence intensity of TC-D-F07 at 0 min. Results are representative of three independent experiments. (C) Decomposition rates of TC-D-F07 in the deproteinized HEPA 1–6 cell lysate and the pH-controlled buffer at 37 °C were analyzed by HPLC and plotted as a function of time. (D) Fluorescence response of TC-D-F07 to endogenous GSH produced by primary B cells, 5TGM1, RPMI-8226, NCI-H929, J558, MEC2, or WaC3 cells was investigated by incubating deproteinized lysates from these cell types with 20 μM TC-D-F07 and monitored at room temperature for 0 to 90 min. I0 was the initial fluorescence intensity of TC-D-F07 at 0 min. Results are representative of three independent experiments.
Cells Treated with TC-D-F07 Exhibit Higher Levels of Intracellular Fluorescent D-F07 Than Those Directly Treated with D-F07
We next compared the intracellular retention of TC-D-F07 with that of D-F07 by treating HEPA 1–6 cells with these compounds at 20 μM for a course of 6 h. The fluorescence of D-F07 in HEPA 1–6 cells was found to be highest at the 1 h time point (Figure 3A) and began to decay after that due to the hydrolysis of the 1,3-dioxane acetal in D-F07. TC-D-F07-treated HEPA 1–6 cells exhibited significantly higher fluorescence than D-F07-treated cells after 0.5 h (Figure 3A), and the fluorescence persisted for 6 h. To further evaluate the intracellular retention of both compounds, we treated HEPA 1–6 cells with TC-D-F07 or D-F07 at 20 μM for 1 h, washed cells with PBS, and re-cultured compound-treated cells in fresh media for 0, 1, and 3 h. TC-D-F07-treated cells exhibited consistently higher fluorescence than D-F07-treated cells after removal of compounds from the media for 3 h (Figure 3B,C). We further treated 5TGM1 MM and MEC2 CLL cells with TC-D-F07 and D-F07 at 20 μM for 3 h and compared intracellular fluorescence. Treatment with TC-D-F07 similarly resulted in significantly higher fluorescence than treatment with D-F07 (Figure 3D) in MEC2 (3.2-fold, Figure 3E) and 5TGM1 cells (2.6-fold, Figure 3E). TC-D-F07 also exhibited higher fluorescence in MEC2 cells than in 5TGM1 cells (Figure 3D,E), suggesting potentially different membrane permeabilities between these two cell lines.
Figure 3.

Cells treated with TC-D-F07 exhibit higher levels of intracellular fluorescent D-F07 than those directly treated with D-F07. (A) HEPA 1–6 cells were treated with DMSO, D-F07 (20 μM), or TC-D-F07 (20 μM) for 0, 0.5, 1, 3, or 6 h, washed three times with PBS, fixed, and analyzed by confocal microscopy. The fluorescence was recorded in the range of 498–650 nm. Scale bar = 10 μm. (B) HEPA 1–6 cells were first treated with DMSO, D-F07 (20 μM), or TC-D-F07 (20 μM) for 1 h, washed three times with PBS, re-cultured in fresh DMEM media for another 0, 1, or 3 h, fixed, and analyzed by confocal microscopy. The fluorescence was recorded in the range of 498–650 nm. Scale bar = 10 μm. (C) Within the linear range, the mean fluorescence intensity (I/I0; means ± SD) in the cytoplasm of DMSO-treated (69 cells), D-F07-treated (58 cells for 0 h, 66 cells for 1 h, and 68 cells for 3 h), or TC-D-F07-treated (48 cells for 0 h, 55 cells for 1 h, and 69 cells for 3 h) HEPA 1–6 cells was plotted as a function of time. I: the fluorescence intensity in cells treated with D-F07 or TC-D-F07 at indicated time points; and I0: the background fluorescence in cells treated with DMSO. (D) 5TGM1 or MEC2 cells were treated with DMSO, D-F07 (20 μM), or TC-D-F07 (20 μM) for 3 h, washed three times with PBS, fixed, and analyzed by confocal microscopy. The fluorescence was recorded in the range of 498–650 nm. Scale bar = 10 μm. (E) Within the linear range, the mean fluorescence intensity (I/I0) in the cytoplasm of 134 DMSO-treated, 138 D-F07-treated, or 198 TC-D-F07-treated 5TGM1 cells or 154 DMSO-treated, 101 D-F07-treated, or 112 TC-D-F07-treated MEC2 cells was plotted as means ± SD. I: the fluorescence intensity in cells treated with D-F07 or TC-D-F07; and I0: the background fluorescence in cells treated with DMSO.
TC-D-F07 Acts as an ER Stress-Inducing and XBP-1S-Inhibiting Compound To Kill MM and CLL Cells
To test whether TC-D-F07 is potent in suppressing XBP-1s expression in tumor cells, we treated 5TGM1 cells with TC-D-F07 at increasing concentrations for 3 h. We observed that TC-D-F07 at 2.5 μM could significantly suppress the expression of XBP-1s, indicating that free thiols in 5TGM1 cells rapidly reacted with the Dns group in TC-D-F07 to liberate D-F07 (Figure 4A). Intriguingly, the levels of XBP-1s together with those of the PERK pathway’s downstream effectors including phospho-eIF2α and ATF4 increased when 5TGM1 cells were treated with TC-D-F07 at 10 and 20 μM for 3 h (Figure 4A). We further performed time-course experiments to examine the levels of XBP-1s in 5TGM1 cells treated with TC-D-F07 at 2.5, 5, and 10 μM (Figure 4B). In 5TGM1 cells treated with TC-D-F07 at 2.5 μM for 12 h, the levels of XBP-1s began to recover similar to those treated with D-F07. When 5TGM1 cells were treated with TC-D-F07 at 10 μM, the levels of XBP-1s initially increased after 3 h treatment and subsequently decreased in response to continuous 12 h treatment, contributing to apoptosis as evidenced by the cleavage of PARP (Figure 4B). Because the increased XBP-1s expression only occurred in 5TGM1 cells treated with a high concentration of TC-D-F07 (10 or 20 μM) for a short time (Figure 4,BA), we hypothesized that TC-D-F07 might induce ER stress in 5TGM1 cells dose-dependently. Of note, the thiol-responsive Dns group has previously been shown to induce ROS-mediated stress through liberation of SO219 and inhibition of redox regulatory enzymes.20 However, we previously showed that treatment of MEC2 cells with H2O2 or a ROS scavenger did not significantly affect the levels of XBP-1s.12 To explore the possibility that a thiol adduct may be inducing XBP-1s expression, we synthesized E-H01, which is the product of cysteine-mediated Dns cleavage (Figure 4C). Like GSH, cysteine is present in high concentrations in tumor cells and is essential for their proliferation and survival.21 Our decaging studies also showed it to be more efficient than GSH in cleaving the Dns group (Figure 1B). When we treated 5TGM1 cells with TC-D-F07 and E-H01 at 10 μM for 24 h, we observed that indeed E-H01 could induce ER stress, as evidenced by the time-dependent increased expression of XBP-1s (Figure 4D). Such data suggest that TC-D-F07 induces XBP-1s through the production of E-H01 or similar thiol adducts, but that the resultant D-F07 subsequently decomposes to the active IRE-1 inhibitor to suppress XBP-1s.
Figure 4.

TC-D-F07 exerted enhanced cytotoxicity in killing MM cells. (A) 5TGM1 cells were treated with TC-D-F07 at indicated concentrations for 3 h, lysed, and analyzed for the expression of indicated proteins by immunoblots. (B) 5TGM1 cells were treated with D-F07 or TC-D-F07 at indicated concentrations for 0, 3, 6, or 12 h, lysed, and analyzed for the expression of indicated proteins by immunoblots. (C) Chemical structure of E-H01. (D) 5TGM1 cells were treated with TC-D-F07 or E-H01 at 10 μM for 0, 3, 6, 12, or 24 h, lysed, and analyzed for the expression of indicated proteins by immunoblots. (E–G) 5TGM1 cells were treated with DMSO, D-F07, or TC-D-F07 at 5 μM (E), 10 μM (F), or 20 μM (G) for 3, 6, 12, or 24 h and subjected to XTT assays. Percentages of growth were determined by comparing treated groups with control groups (DMSO). Each data point derived from four independent groups receiving the same treatment was plotted as means ± SD. Data were representative of three independent experiments. (H) 5TGM1 cells were treated with DMSO, E-H01, D-F07, E-H01 in combination with D-F07, or TC-D-F07 at 20 μM for 2 days, subjected to XTT assays, and similarly analyzed. (I) NCI-H929 cells were treated with DMSO, E-H01, D-F07, E-H01 in combination with D-F07, or TC-D-F07 at 20 μM for 2 days, subjected to XTT assays, and similarly analyzed.
To evaluate the cytotoxicity of TC-D-F07, we treated 5TGM1 cells with D-F07 or TC-D-F07 at 2.5, 5, 10, or 20 μM for 24 h (Figure 4E–G and S3A). Although both compounds at 2.5 or 5 μM showed no cytotoxicity (Figure 4E and S3A), clear cytotoxicity was observed in 5TGM1 cells treated with 10 or 20 μM TC-D-F07 (Figure 4F–G). The ER stress-inducing and XBP1-s-inhibiting activity of TC-D-F07 thus renders it more effective in killing MM cells relative to D-F07. Although our data showed that TC-D-F07 was more capable of retaining in 5TGM1 cells than D-F07 (Figure 3D,E), we sought to test whether the combination of D-F07 with E-H01 could exert cytotoxicity comparable to TC-D-F07. We further treated 5TGM1 cells with DMSO, E-H01, D-F07, E-H01 in combination with D-F07, or TC-D-F07 at 20 μM for 2 days (Figure 4H). Although E-H01 was not cytotoxic to 5TGM1 cells, it enhanced the cytotoxicity of D-F07. TC-D-F07 also exhibited stronger cytotoxicity than the combination of D-F07 and E-H01 in 5TGM1 cells. We next investigated human NCI-H929 cells using the same methods, and our results showed that treatment with TC-D-F07 at high concentration could similarly induce ER stress as evidenced by the expression of ER stress response markers and subsequently induce apoptosis (Figure S3B,C). TC-D-F07 indeed also exerted higher cytotoxicity than the combination of D-F07 and E-H01 (Figure 4I). To assure that our findings were not MM-specific, we similarly treated human MEC2 and WaC3 CLL cells with TC-D-F07. Our data suggested that higher concentrations of TC-D-F07 could also induce ER stress and potently inhibit cell growth by inducing apoptosis (Figure S4A–D). TC-D-F07 similarly exerted stronger cytotoxicity than the combination of D-F07 and E-H01 in both CLL cell lines (Figure S4E,F). These data highlight the advantage of combining ER stress-inducing and XBP-1s-inhibiting activities in one entity as in TC-D-F07 to achieve higher cytotoxicity for cancer therapy.
Conclusions
In summary, we have designed and synthesized a caged prodrug, TC-D-F07, by which ER stress-inducing and XBP-1s-inhibiting activities can be delivered into tumor cells simultaneously. The Dns cage installed onto the hydroxy group of TC-D-F07 not only stabilizes the 1,3-dioxane acetal protecting group for slow hydrolysis of D-F07 into the active drug, but also allows for stimulus-mediated release of D-F07. TC-D-F07 is more readily converted to prodrug D-F07 in tumor cells due to their overexpression of free thiol species. Upon decaging, a 1,3-dinitrophenyl cysteine adduct also acts as an ER stress inducer in tumor cells. A single agent that induces the ER stress and inhibits XBP-1s activity achieves higher cytotoxicity in tumor cells than an XBP-1s inhibitor alone or in combination with a separate ER stress-inducing compound. We believe that the higher cytotoxicity is due to the simultaneous entry of both the ER stress-inducing agent and the XBP-1s inhibitor at a 1:1 ratio, which is harder to achieve when two drugs potentially with different membrane permeabilities are administered. Different from TC-D-F07, D-F07 will begin to gradually decompose by losing the 1,3-dioxane acetal protecting group in media before it enters into the cells, compromising the cancer-killing activity in the combined treatment with E-H01. Additionally, the Dns cage group in TC-D-F07 may be replaced by other functional groups such as those that can induce ROS production to generate novel inhibitors for cancer therapy. We hypothesize that the Dns cage group may also be chemically linked to other inhibitors of ER stress response pathways for effective cancer therapy.
Acknowledgments
This study was partially supported by grants (K22CA248354, R01CA163910, and R01CA190860) from the NIH/NCI.
Glossary
Abbreviations
- XBP-1s
X-box binding protein 1 spliced form
- IRE-1
inositol-requiring enzyme 1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.1c00639.
NMR spectra of all newly synthesized compounds, absorbance curves, free GSH concentration, NCI-H929 cells, and MEC2 and WaC3 cells (PDF)
Author Present Address
§ School of Pharmaceutical Sciences, Jiangnan University, Wuxi, Jiangsu 214122, China
Author Contributions
A.S., J.R.D., and C.C.A.H. designed research; A.S., Q.X., C.W.K., C.F.C., A.C.L., C.H.A.T., J.R.D., and C.C.A.H. performed research; A.S., Q.X., C.W.K., C.F.C., A.C.L., C.H.A.T., J.R.D., and C.C.A.H. analyzed data; and A.S., J.R.D., and C.C.A.H. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Urra H.; Dufey E.; Avril T.; Chevet E.; Hetz C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. 10.1016/j.trecan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Chen X.; Iliopoulos D.; Zhang Q.; Tang Q. Z.; Greenblatt M. B.; Hatziapostolou M.; Lim E.; Tam W. L.; Ni M.; Chen Y. W.; Mai J. H.; Shen H. F.; Hu D. Z.; Adoro S.; Hu B.; Song M. Y.; Tan C.; Landis M. D.; Ferrari M.; Shin S. J.; Brown M.; Chang J. C.; Liu X. S.; Glimcher L. H. XBP1 promotes triple-negative breast cancer by controlling the HIF1 alpha pathway. Nature 2014, 508, 103–107. 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. H.; Ranatunga S.; Kriss C. L.; Cubitt C. L.; Tao J.; Pinilla-Ibarz J. A.; Del Valle J. R.; Hu C. C. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Invest. 2014, 124, 2585–2598. 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranatunga S.; Tang C. H.; Kang C. W.; Kriss C. L.; Kloppenburg B. J.; Hu C. C.; Del Valle J. R. Synthesis of novel tricyclic chromenone-based inhibitors of IRE-1 RNase activity. J. Med. Chem. 2014, 57, 4289–4301. 10.1021/jm5002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Tang C. H.; Song J. H.; Mancuso A.; Del Valle J. R.; Cao J.; Xiang Y.; Dang C. V.; Lan R.; Sanchez D. J.; Keith B.; Hu C. C.; Simon M. C. IRE1alpha RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Invest. 2018, 128, 1300–1316. 10.1172/JCI95864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutt S. D.; Wu Y.; Tang C. H.; Bastian D.; Nguyen H.; Sofi M. H.; Zhang M.; Liu C.; Helke K.; Wilson C.; Schnapp L. M.; Del Valle J. R.; Hu C. C.; Yu X. Z. Inhibition of the IRE-1alpha/XBP-1 pathway prevents chronic GVHD and preserves the GVL effect in mice. Blood Adv. 2018, 2, 414–427. 10.1182/bloodadvances.2017009068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts B. C.; Locke F. L.; Sagatys E. M.; Pidala J.; Walton K.; Menges M.; Reff J.; Saha A.; Djeu J. Y.; Kiluk J. V.; Lee M. C.; Kim J.; Kang C. W.; Tang C. H. A.; Frieling J.; Lynch C. C.; List A.; Rodriguez P. C.; Blazar B. R.; Conejo-Garcia J. R.; Del Valle J. R.; Hu C. C. A.; Anasetti C. Inhibition of Human Dendritic Cell ER Stress Response Reduces T Cell Alloreactivity Yet Spares Donor Anti-tumor Immunity. Front Immunol. 2018, 9, 2887. 10.3389/fimmu.2018.02887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G.; Zhang Z. X.; Li H. D.; Li D. S.; Wang X. R.; Qin C. G. Design, Synthesis and Application in Analytical Chemistry of Photo-Sensitive Probes Based on Coumarin. Crit. Rev. Anal. Chem. 2021, 51, 565–581. 10.1080/10408347.2020.1753163. [DOI] [PubMed] [Google Scholar]
- Shao A.; Kang C. W.; Tang C. H.; Cain C. F.; Xu Q.; Phoumyvong C. M.; Del Valle J. R.; Hu C. C. Structural Tailoring of a Novel Fluorescent IRE-1 RNase Inhibitor to Precisely Control Its Activity. J. Med. Chem. 2019, 62, 5404–5413. 10.1021/acs.jmedchem.9b00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D.; Liu Z.; Verwilst P.; Koo S.; Jangjili P.; Kim J. S.; Lin W. Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev. 2019, 119, 10403–10519. 10.1021/acs.chemrev.9b00145. [DOI] [PubMed] [Google Scholar]
- Mimura N.; Fulciniti M.; Gorgun G.; Tai Y.-T.; Cirstea D.; Santo L.; Hu Y.; Fabre C.; Minami J.; Ohguchi H.; Kiziltepe T.; Ikeda H.; Kawano Y.; French M.; Blumenthal M.; Tam V.; Kertesz N. L.; Malyankar U. M.; Hokenson M.; Pham T.; Zeng Q.; Patterson J. B.; Richardson P. G.; Munshi N. C.; Anderson K. C. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao A.; Xu Q.; Spalek W. T.; Cain C. F.; Kang C. W.; Tang C. A.; Del Valle J. R.; Hu C. A. Development of Tumor-Targeting IRE-1 Inhibitors for B-cell Cancer Therapy. Mol. Cancer Ther. 2020, 19, 2432–2444. 10.1158/1535-7163.MCT-20-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. H.; Chang S.; Paton A. W.; Paton J. C.; Gabrilovich D. I.; Ploegh H. L.; Del Valle J. R.; Hu C. C. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J. Cell Biol. 2018, 217, 1739–1755. 10.1083/jcb.201709137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Kwon Y.; Kim D.; Lee D.; Kim G.; Hu Y.; Ryu J. H.; Yoon J. Cyanine-Based Fluorescent Probe for Highly Selective Detection of Glutathione in Cell Cultures and Live Mouse Tissues. J. Am. Chem. Soc. 2014, 136, 5351–5358. 10.1021/ja412628z. [DOI] [PubMed] [Google Scholar]
- Wang Z. Q.; Wu H.; Liu P. L.; Zeng F.; Wu S. Z. A self-immolative prodrug nanosystem capable of releasing a drug and a NIR reporter for in vivo imaging and therapy. Biomaterials 2017, 139, 139–150. 10.1016/j.biomaterials.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Matsuno H.; Ushida M.; Katayama K.; Saeki K.; Itoh N. 2,4-Dinitrobenzenesulfonyl fluoresceins as fluorescent alternatives to Ellman’s reagent in thiol-quantification enzyme assays. Angew. Chem., Int. Ed. Engl. 2005, 44, 2922–2925. 10.1002/anie.200500114. [DOI] [PubMed] [Google Scholar]
- Holme J. A.; Nyvold H. E.; Tat V.; Arlt V. M.; Bhargava A.; Gutzkow K. B.; Solhaug A.; Lag M.; Becher R.; Schwarze P. E.; Ask K.; Ekeren L.; Ovrevik J. Mechanisms linked to differences in the mutagenic potential of 1,3-dinitropyrene and 1,8-dinitropyrene. Toxicol. Rep. 2014, 1, 459–473. 10.1016/j.toxrep.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. J.; Chai X. Z.; He X. P.; Kim H. J.; Yoon J.; Tian H. Fluorogenic probes for disease-relevant enzymes. Chem. Soc. Rev. 2019, 48, 683–722. 10.1039/C7CS00907K. [DOI] [PubMed] [Google Scholar]
- Malwal S. R.; Sriram D.; Yogeeswari P.; Konkimalla V. B.; Chakrapani H. Design, synthesis, and evaluation of thiol-activated sources of sulfur dioxide (SO(2)) as antimycobacterial agents. J. Med. Chem. 2012, 55, 553–557. 10.1021/jm201023g. [DOI] [PubMed] [Google Scholar]
- van Gisbergen M. W.; Cebula M.; Zhang J.; Ottosson-Wadlund A.; Dubois L.; Lambin P.; Tew K. D.; Townsend D. M.; Haenen G. R.; Drittij-Reijnders M. J.; Saneyoshi H.; Araki M.; Shishido Y.; Ito Y.; Arner E. S.; Abe H.; Morgenstern R.; Johansson K. Chemical Reactivity Window Determines Prodrug Efficiency toward Glutathione Transferase Overexpressing Cancer Cells. Mol. Pharmaceutics 2016, 13, 2010–2025. 10.1021/acs.molpharmaceut.6b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs J. A.; DeNicola G. M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. 10.3390/cancers11050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.