

Summary
Gut microbiota metabolism of dietary compounds generates a vast array of microbiome-dependent metabolites (MDMs), which are highly variable between individuals. The uremic MDMs (uMDMs) phenylacetylglutamine (PAG), p-cresol sulfate (PCS) and indoxyl sulfate (IS) accumulate during renal failure and are associated with poor outcomes. Targeted dietary interventions may reduce toxic MDMs generation; however, it is unclear if interindividual differences in diet or gut microbiome dominantly contribute to MDM variance. Here we use a 7-day homogeneous average American diet to standardize dietary precursor availability in 21 healthy individuals. During dietary homogeneity, the coefficient of variation in PAG, PCS, and IS (primary outcome), did not decrease, nor did interindividual variation in most identified metabolites; other microbiome metrics showed no or modest responses to the intervention. Host identity and age are dominant contributors to variability in MDMs. These results highlight the potential need to pair dietary modification with microbial therapies to control MDM profiles.
Graphical Abstract
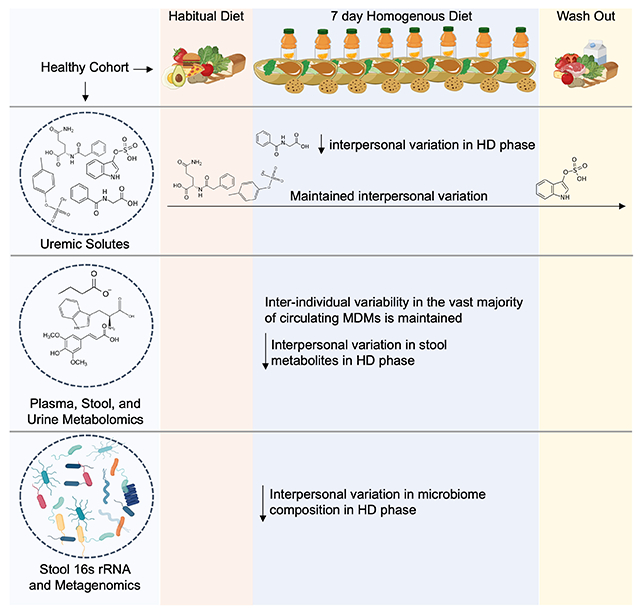
eTOC Blurb
Guthrie et al. determine the extent to which diet or the gut microbiome contribute to interpersonal variation in microbiome-dependent metabolites that are high priority targets of nutritional strategies to minimize levels. Diet homogeneity decreases inter-individual variability in hippurate, but not key uremic solutes phenylacetylglutamine, p-cresol sulfate and indoxyl sulfate.
Introduction
Dietary change is known to impact the human gut microbiome and physiological status including influencing metabolic and immune parameters (Martínez et al., 2013; Wu et al., 2016; Wastyk et al., 2021). Human diets are sources of chemically diverse substrates for gut microbiota metabolism, which generates over 30,000 microbiome-dependent metabolites (MDMs)(Glowacki and Martens, 2020). MDMs produced as a consequence of gut microbiota metabolism are associated with cardio-renal diseases(Tang and Hazen, 2014; Guldris, Parra and Amenós, 2017), obesity(Liu et al., 2017), metabolic syndrome(Pedersen et al., 2016) and cancer(O’Keefe, 2016), reach high concentrations (mM) in systemic circulation(Dobre, Meyer and Hostetter, 2020), and most are cleared by the kidneys and eliminated in urine. When kidney function declines, MDMs accumulate as some of the most abundant uremic solutes (uMDMs)(Mair et al., 2018; Dobre, Meyer and Hostetter, 2020), which have well defined mechanisms of toxicity(Koppe et al., 2013; Edamatsu et al., 2014; Xu et al., 2021), and are difficult to clear via dialysis alone(Dobre, Meyer and Hostetter, 2020). The levels of two uMDMs, p-cresol sulfate (PCS) and indoxyl sulfate (IS), are highly variable between individuals, but stable within an individual over time(Patel et al., 2012), indicating precision strategies to reduce their level of production could be clinically beneficial. While targeted dietary interventions are an attractive approach to modulate the accumulation of potentially toxic MDMs, the extent that diet can be used to modulate and control MDM production remains incompletely understood.
Several interventions have investigated the influence of diet modifiable factors on MDM levels. Protein restriction was shown in early studies to ameliorate uremic symptoms and also to reduce urinary excretion of compounds including PCS, consistent with many uMDMs being amino acid-derived (Evenepoel et al., 2009; Itoh et al., 2013). Increasing the intake of fiber, defined as carbohydrates that are not absorbed in the small intestine, has been hypothesized to program the microbiota to reduce degradation of amino acids(Sirich et al., 2014; Cases et al., 2019). Vegetarians produce less PCS and IS than omnivores, coincident with higher fiber and less protein intake(Patel et al., 2012). In hemodialysis patients, a randomized trial has shown that daily intake of 18 g of fiber in the form of resistant starch for 6 weeks reduced plasma levels of IS(Sirich et al., 2014).
We carried out the Microbiome Individuality and Stability Over Time (MISO) study to investigate interpersonal variation in diet derived MDM levels when diet is homogenized and highly standardized. MISO study participants’ urine, fecal and plasma metabolomes and stool metagenomes were profiled before, during, and after undergoing a 7-day standardized homogeneous diet. We designed the study to add a layer of stringency not previously achieved for microbiome focused studies, even in the most highly controlled in-patient diet intervention studies reported(Wu et al., 2011, 2016). Even when participants are receiving the same meals as one another, meal-to-meal variation (e.g., different content of breakfast vs. lunch) combined with variation in gut motility can result in differences in the chemical environment experienced by two participant’s respective microbiomes at a given sampling time point. Therefore, interindividual differences in, for example, serum metabolites, are difficult to attribute to temporal diet variation vs. individualized aspects of the microbiota. Our study, in providing a uniform chopped salad format for 7 days ensures that bite-after-bite is homogenized between participants; therefore, it is expected that dietary chemical flow through the colon should be as uniform as possible between participants over the course of sampling during the intervention period.
As factors that shape intestinal microbiota composition and its metabolic output are incompletely understood, the homogenous dietary intervention helped to identify both dietary and host factors impacting microbiota composition, function, and metabolic output. To address whether homogenization of diet between participants reduces interindividual variation in three key uMDMs associated with cardiovascular and renal toxicity(Ravid, Kamel and Chitalia, 2021), indoxyl sulphate (IS), p-cresol sulfate (PCS) and phenylacetylglutamine (PAG), a reduction in interindividual variation in the urine levels of these uMDMs served as the primary outcome. Beyond examining how diet impacts these three metabolites of interest, the study design enables us to examine how other aspects of microbiome composition, metabolic output, and function are affected when dietary variation between people and over time are eliminated. We used a microbiome-focused metabolomics pipeline to assess MDMs in fecal, urine, and plasma samples, and carried out shotgun metagenomic sequencing and 16S rRNA sequencing on fecal samples to assess microbiome functional potential and microbiome composition. Our approach identifies MDMs for which dietary strategies may be effective at impacting metabolite levels and identifies MDMs for which the microbiome or other individualized factors play a dominant role over this dietary intervention. Specifically, we identified host identity and age as dominant contributors to variability in microbiome-dependent metabolite levels broadly. The results suggest microbiome reprogramming or alternative dietary strategies may be required for changing much of the metabolic output of an individual’s gut microbial community.
Results
Overview of study design and dietary intervention
We performed a controlled feeding study to quantify the impact of a homogeneous diet on interpersonal variability in the excreted levels of MDMs in a healthy cohort (ClinicalTrials.gov Identifier: NCT04740684). Of the 61 individuals assessed for eligibility, 26 were enrolled and 21 participants completed the study and were used for the analysis (Figure 1A). The final cohort were adults (age 48 ± 14 y [mean ± SD]), with a mean body surface area (BSA) of 1.93 ± 0.27 m2 (Table 1). We enrolled healthy volunteers with diverse habitual diets, to understand the effects of homogenizing their diet, regardless of their baseline diet. We quantified and compared MDM levels in enrolled participants during three phases: the baseline diet (BD) phase (days 1-14), the homogeneous diet (HD) phase (days 15 - 21) and the washout (WO) phase (days 22-28) (Figure 1B). During the 14-day BD phase, study subjects were instructed to maintain their habitual diet and food logs were used to assess their dietary patterns. Participants showed diverse habitual diets, defined by a coefficient of variation CV greater than 25 % of macro nutrients (Figure S1). During the 7-day HD phase, study participants consumed single serve portions of a standardized diet ad libitum (Table S1) that was prepared in a commercial kitchen and packaged in 295 g portions.
Figure 1. MISO study enrollment, design, and data collection.
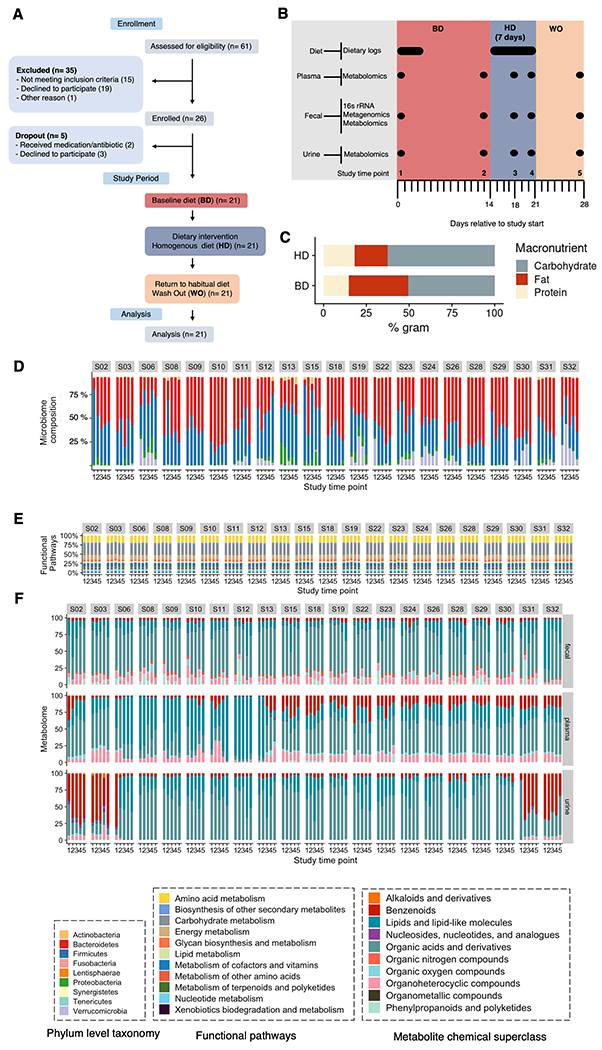
(A) CONSORT Flow diagram of participant enrollment and analysis in the MISO study.
(B) The 4-week study overview timeline, sample types collected, timepoints of sample collection, and corresponding experimental platforms.
(C) Average diet macronutrient composition during the baseline diet (BD) across the cohort and intervention homogenous diet (HD) study phases represented in percent grams. No baseline diet was available for participant 23.
Longitudinal microbiome (D) composition (phylum level), (E) functional category relative abundance, and (F) fecal, plasma and urine metabolomes categorized by chemical class for MISO study participants. Fecal metagenome functional profiles were derived from sequencing, mapping, and gene alignments. Urine and plasma metabolomes were quantified using a microbiome-focused metabolomics pipeline; metabolites were mapped to their structure-based chemical taxonomy (see Methods).
Table 1.
Participant characteristics
| subject | Sex | Age | BSA |
|---|---|---|---|
| S02 | F | 56 | 1.9 |
| S03 | F | 55 | 2 |
| S06 | M | 28 | 2.1 |
| S08 | F | 27 | 1.8 |
| S09 | M | 23 | 1.7 |
| S10 | M | 46 | 1.8 |
| S11 | F | 58 | 1.6 |
| S12 | F | 27 | 1.7 |
| S13 | M | 34 | 2.8 |
| S15 | F | 30 | 1.9 |
| S18 | M | 46 | 1.9 |
| S19 | M | 50 | 2 |
| S22 | F | 54 | 1.7 |
| S23 | F | 53 | 1.7 |
| S24 | F | 54 | 2 |
| S26 | F | 62 | 1.8 |
| S28 | F | 45 | 1.8 |
| S29 | M | 58 | 1.7 |
| S30 | M | 67 | 2.1 |
| S31 | M | 75 | 2.3 |
| S32 | M | 54 | 2.4 |
The HD was designed to recapitulate the diet quality, and specifically the fiber and macronutrient ranges(Grotto and Zied, 2010), common to adults in America based on the National Health and Nutrition Examination surveys and additional studies(Grotto and Zied, 2010). Although each participant consumed identical diets during HD (i.e., same composition and percentage of macronutrients), the amount of the HD consumed across participants varied as expected due to differences in individual caloric needs (we also did not want to confound the results with weight changes during this intervention) (Figure S1). During the 7-day WO phase study subjects resumed their habitual diet. In addition to collecting detailed participant food logs during the BD and HD study phases, we collected stool, blood, and urine samples at five different timepoints, inclusive of all three study phases, for microbiome and metabolome profiling (Figure 1B). The overall macronutrient composition of dietary protein, carbohydrates and fats were relatively stable across the BD and HD study phases (37.5 vs 35.1% for fat, 17.0 vs 14.9% for protein and 45.8 vs 51.3% for carbohydrates), amounting to a modest decline in total protein between the BD and HD (Figure 1C; Figure S1D, paired Wilcoxon signed rank test, p = 0.03) and significant decrease in overall fiber during the HD phase (Figure S1B, paired Wilcoxon signed rank test, p < 0.0001).
We wished to define how interpersonal and intrapersonal (i.e., temporal) variability in microbiome-dependent metabolites (MDMs), genes and species were affected by the homogeneous diet. Therefore, we performed shotgun metagenomic sequencing and 16S rRNA sequencing on fecal samples and targeted metabolomics of uremic solutes on urine samples; and we used a microbiome-focused metabolomics pipeline that we recently reported(Han et al., 2021) on fecal, plasma and urine samples from each participant. Participant microbiome composition profiles were varied, and as has been previously shown in studies of healthy individuals(‘The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease’, 2014), the most prevalent phyla were the Bacteroidetes and Firmicutes (Figure 1D). The relative abundance of the most prevalent microbiome functional pathways annotated in MISO participants remained stable throughout the study (Figure 1E). Our MDM-focused metabolomic profiling revealed extensive chemical diversity of the participants’ fecal, plasma and urine metabolomes; metabolites were mapped to their structure-based chemical ontology at the Superclass level (Figure 1F). MDM metabolome composition was more variable in some subjects across the study period and identity as a fecal, plasma, or urine MDM.
A 7-day homogeneous diet does not reduce interpersonal variation in three amino-acid derived uMDMs
To characterize the impact of homogenizing diet over a 7-day period on interpersonal variation in the output of uremic microbiota-dependent metabolites (uMDMs), we profiled uMDM levels in urine using targeted mass spectrometry during the BD, HD, and WO study phases. The primary outcome for this study was a 25% reduction of the coefficient of variation (CV) of 24 hour urinary excretion of at least 1 of 3 well-studied uMDMs that are derived from dietary amino acids: (p-cresol sulfate (PCS), indoxyl sulfate (IS), or phenylacetylglutamine (PAG)) measured and normalized to a body surface area of 1.73 m2 using the formula of Mosteller(Mosteller, 1987), during the HD phase as compared to the BD phase (Figure S2). The first (BD phase) and fourth (HD phase) time points were compared for change in CV calculations. Urine IS levels decreased by 2.8 mg/day/1.73 with a CV decrease of 1.7% (p = 0.86). Urine PCS levels increased by 3.9 mg/day/1.73, with a CV increase of 11.2% (p = 0.20). PAG levels increased by 7.7 mg/day/1.73 and had a CV reduction of 0.05 % (p = 0.49). Therefore, although all three uMDMs exhibited changes in CV during the 7-day homogeneous diet, they did not uniformly decrease, and did not meet the primary endpoint as each of the 3 metabolites had less than a 25% reduction in interpersonal variation as measured by %CV (Table 2) and the % CV between time points 1 and 4 did not uniformly decline (Figure 2A).
Table 2.
Primary and exploratory clinical trial outcomes.
Coefficient of variation at study time points 1 (BD phase), 4 (end of HD phase) and 5 (WO phase). A percent reduction in CV between time points 1 and 4 of 0.25 or greater meets the endpoint.
| Metabolite | Time point | Mean | SD | CV | *CV % | *p value |
|---|---|---|---|---|---|---|
| indoxyl sulfate | 1 | 23.8 | 11.9 | 0.49901 | −1.7% | 0.48 |
| indoxyl sulfate | 4 | 21.0 | 10.1 | 0.48164 | ||
| indoxyl sulfate | 5 | 28.5 | 15.4 | 0.54070 | ||
| p-cresol sulfate | 1 | 21.9 | 15.8 | 0.71993 | 11.2 % | 0.59 |
| p-cresol sulfate | 4 | 25.8 | 21.5 | 0.83168 | ||
| p-cresol sulfate | 5 | 29.6 | 20.2 | 0.68478 | ||
| phenylacetylglutamine | 1 | 84.0 | 55.0 | 0.65436 | −0.05 | 0.78 |
| phenylacetylglutamine | 4 | 91.7 | 59.9 | 0.65383 | ||
| phenylacetylglutamine | 5 | 107.0 | 74.7 | 0.69846 | ||
| Hippuric acid | 1 | 261.3 | 194.0 | 0.74231 | −27.9 % | p < .001 |
| Hippuric acid | 4 | 95.0 | 44.0 | 0.46304 | ||
| Hippuric acid | 5 | 277.4 | 212.6 | 0.76654 |
Time point 1 vs time point 4
Figure 2. HD diet results in reduction of interpersonal variation in hippuric acid but not other uMDMs.
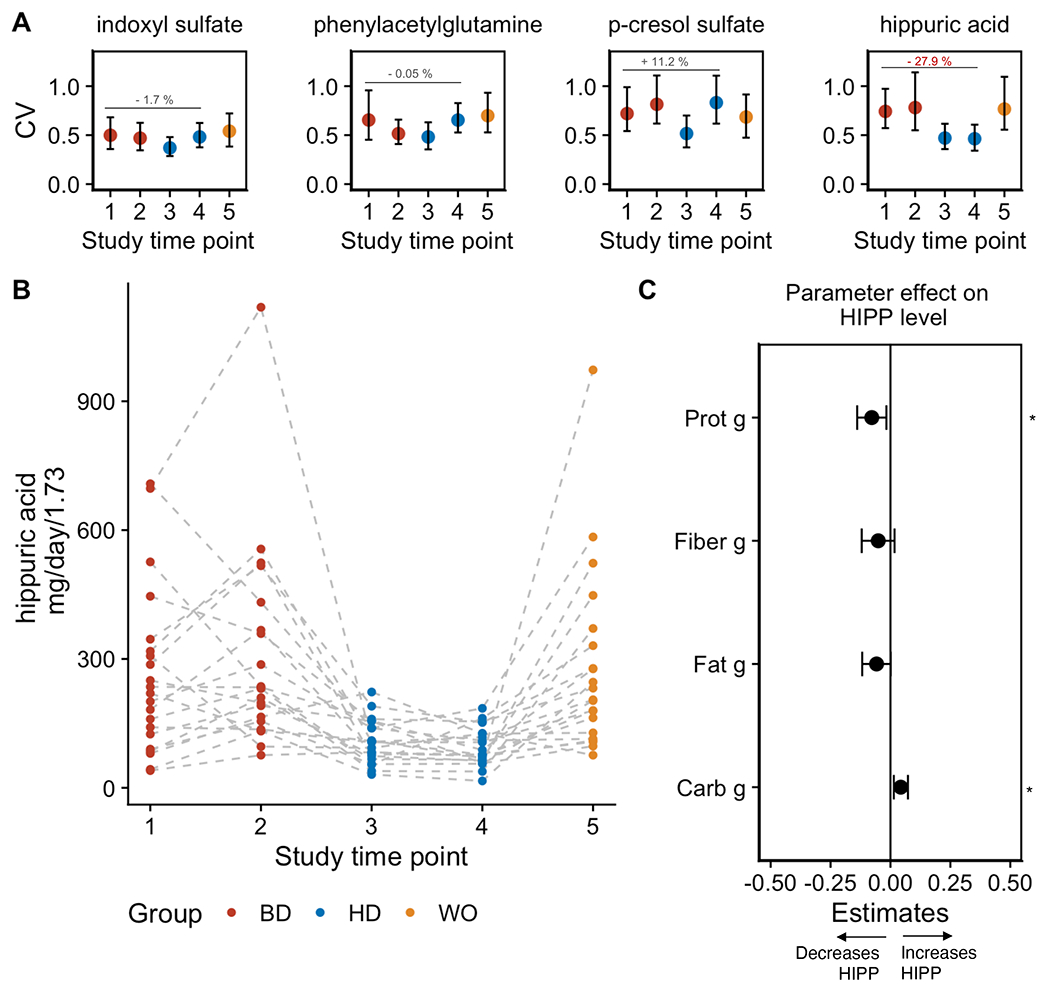
(A) Coefficient of variation (CV) with 95 % confidence interval in urine uremic solutes indoxyl sulfate (IS), phenylacetylglutamine (PAG), p-cresol sulfate (PCS) and hippuric acid (HIPP) at study time points across the BD, HD, and WO phases. Error bars represent 25th (bottom) and 75th (upper) quartiles.
(B) Urine levels of hippuric acid (HIPP) determined by targeted LC-MS during the baseline diet (BD), homogenous diet (HD) and wash out (WO) (****p < 0.0001; paired Wilcoxon signed rank test).
(C) Effect sizes of dietary macronutrients quantified by a mixed-effects model at time point 4 (along with a 95% CI) show that carbohydrates (p = 0.01) support higher levels of hippuric acid (HIPP), whereas total protein (p = 0.02) detracts from HIPP levels. Asterisks (*) indicate significant of specific parameters included in the model.
While we found no significant decline in CV for PCS, IS, or PAG, median macronutrient levels did decline between the BD and HD phases. Notably, urine urea nitrogen, an indicator of total protein breakdown, decreased during the intervention phase (Figure S1F, paired Wilcoxon signed rank test, p = 0.01 with a CV reduction of 12.7%; p = 0.0018), consistent with the decreased protein intake by participants during HD (Figure S1D). These data indicate that the reduction in total protein was not sufficient to reduce interpersonal variance in amino acid-derived metabolites, PCS, IS, and PAG. To determine the effects of any diet components on the levels of any of the 3 primary endpoint metabolites, we developed a mixed-effects model for each metabolite normalized measurements at time point 4, that considered biological sex. The effect sizes computed showed that carbohydrate consumption provided a significant increase in PAG levels, but no significant metabolite levels and diet for IS and PCS (Figure S2A). We also found no correspondence between Primary outcome uremic solute levels and the relative abundance of the major microbiome encoded gene responsible for their production (Figure S2C).
Dietary carbohydrates predict urine hippuric acid levels
Given that we saw a reduction in total consumption of fiber during the intervention HD phase (Figure S1B, paired Wilcoxon signed rank test, p < 0.0001), and a moderate decline in carbohydrates (Figure S1A), we wondered whether uMDMs derived from precursors found in carbohydrates and fiber were sensitive to the HD phase of the study. Specifically, we focused on hippuric acid, a uMDM that we profiled in urine using the same targeted mass spectrometry applied to IS, PCS and PAG. Host and microbial degradation of flavonoids like (−)-epigallocatechin result in the production of hippurate, which has urinary excretion values ranging from 700-1600 umol in healthy individuals(van der Hooft et al., 2012). Hippuric acid also accumulates at high levels in individuals with failing kidneys, but unlike IS, PCS, and PAG, there is no compelling evidence of adverse health associations, and therefore it was not included in our primary endpoint.
Hippuric acid (Figure 2B), is known to respond dynamically to dietary interventions(Martin et al., 2009; Lees et al., 2013), and can be derived from both plant polysaccharides (e.g., fiber) and protein sources. During the HD phase, the mean levels of hippuric acid decreased and the overall interpersonal variation decreased by 27.9 % difference in hippuric acid CV between time point 1 (BD) and time point 4 (HD) (Figure 2A, p = 0.011). During the WO phase, the CV returned to baseline levels, indicating that dietary heterogeneity between participants is a key determinant in hippuric acid production and variation. We next hypothesized that altered macronutrients and intrinsic microbiome capacity shaped hippuric acid levels during the HD phase. Using a mixed-effects model that considered dietary macronutrient intake parameters as covariates and controlling for host parameters (i.e., biological sex), we quantified the role of diet and bacterial metabolism (metagenomic data) on the levels of hippuric acid. Carbohydrate levels positively contribute to hippuric acid levels based on the computed effect sizes, and total protein negatively contributed to hippuric acid levels (Figure 2C, linear model, p = 0.04). We found no correspondence between hippuric acid levels and the relative abundance of the microbiome encoded enzyme responsible for producing a precursor, phenylalanine ammonia-lyase (PAL) (Figure S2C). These findings indicate that the observed reduction in the CV and urine levels for hippuric acid is dietary precursor dependent, consistent with the decrease in variability observed between study participants during the HD phase.
The HD intervention reduces inter- and intra- personal variation in microbiome composition, function, and metabolic output
We leveraged the study design and participant samples to examine how a homogeneous diet impacts a broader array of MDMs, genes, and microbial composition. We first investigated how the HD intervention shaped interpersonal variation in participant microbiomes.
Our microbiome focused metabolomics pipeline allowed us to broadly identify, beyond the IS,PCS,PAG, and hippuric acid, MDMs that decline in inter-individual variation due to the homogeneous average American diet consumed by participants in our study. We defined HD diet sensitive metabolites based on a 25% reduction of the coefficient of variation (CV) based on the first (BD phase) and fourth (HD phase) time points. Metabolomic data for urine, plasma, and fecal samples resulted in 168, 183, and 320 different metabolites, respectively, observed in 80% of participants for at least one time point. We evaluated the % change in CV between BD phase time point 1 and HD phase time point 4 for all fecal, urine, and plasma metabolites present in ≥80% of a given sample type (see Methods). We identified 45 fecal, 15 plasma and 16 urine metabolites with ≥25% reduction in interpersonal variation (CV) during the HD phase (Figure 3A). Although some of the metabolites identified by this pipeline are independent of microbiota metabolism (e.g., derived from host, diet, or medication), known urine MDMs, p-cresol glucuronide and indole-3-acetic acid (IAA) are colon-derived uremic solutes8, that share precursors with PCS and IS respectively, and show a 66.4 % and 37.9 % reduction in CV in urine levels during HD, respectively; IAA also shows a reduction in fecal CV (27.6 %). While variation in overall microbiome dependent metabolic output was largely independent of study phase (Figure 1), this analysis suggests that unifying diet may reduce interpersonal variation for a subset of uremic solutes.
Figure 3. Interpersonal variation in microbiome composition and fecal metabolic output declines during HD intervention.
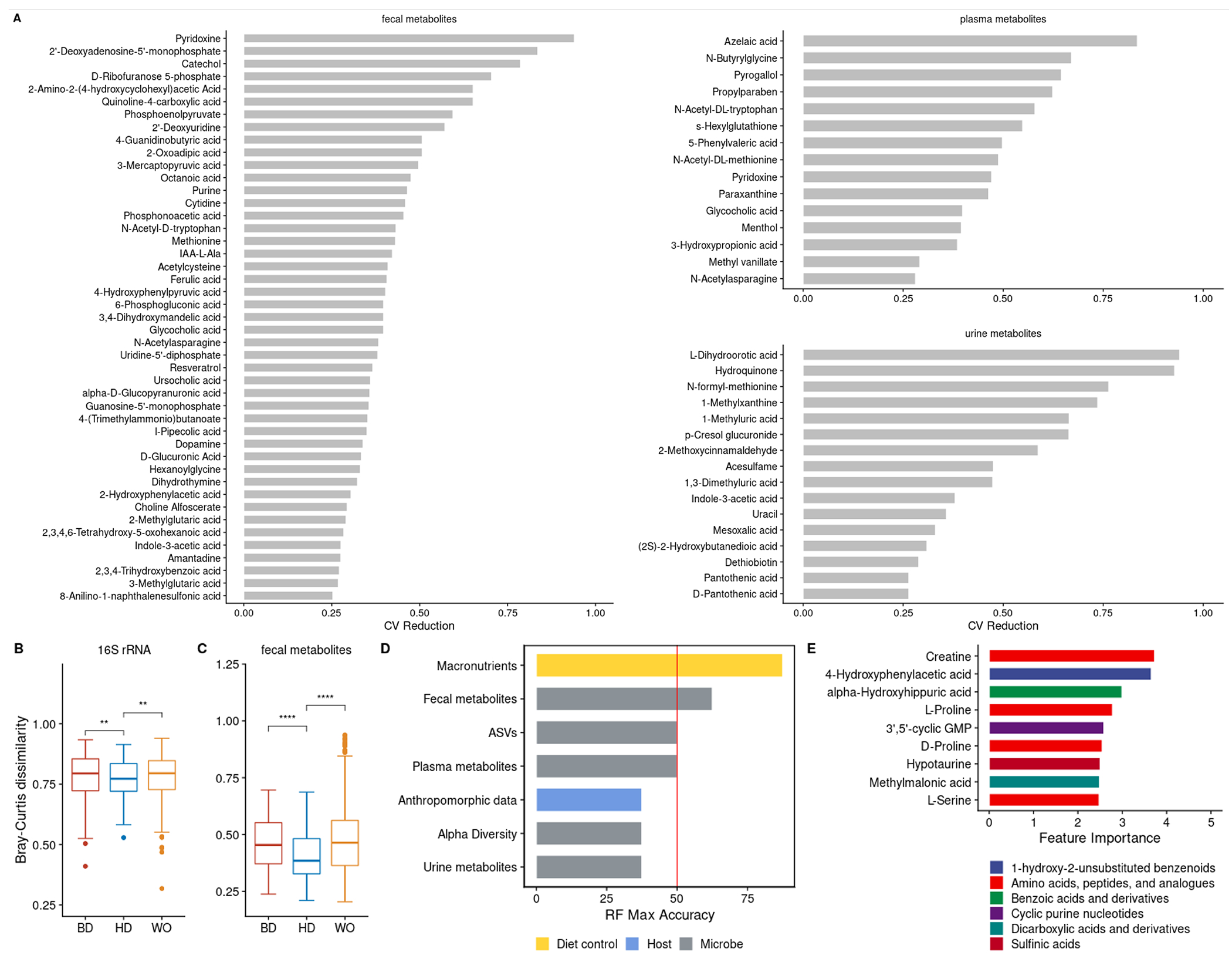
(A) Fecal, plasma and urine metabolites with ≥25% reduction in CV across the cohort comparing timepoint 1 (BD) with timepoint 4 (HD).
(B,C) Boxplots showing microbiome similarity measures for (B) ASV-level and (C) fecal metabolome based on Bray-Curtis dissimilarity metrics across study timepoints 1 (BD), 4 (HD) and 5 (WO). Boxplot error bars represent 25th (bottom) and 75th (upper) quartiles. At time point 1, the median Bray-Curtis value for ASVs and the fecal metabolome, are 0.79 (SD = 0.1 ) and 0.45 respectively (SD = 0.1). At time point 4, the median Bray-Curtis value for ASVs and the fecal metabolome, are 0.77 (SD = 0.08) and 0.38 (SD = 0.1) respectively. At time point 5, the median Bray-Curtis value for ASVs and the fecal metabolome, are 0.79 (SD = 0.1) and 0.46 (SD = 0.2) respectively. (D) Accuracy of leave-one-out cross-validation (LOOCV) of random forest models predicting study phase using different data types from study time points in BD (1 or 2) versus HD (3 or 4); diet macronutrient data (yellow), microbe-enriched data (grey) and anthropomorphic data from the host (blue).
(E) Percent importance of individual fecal metabolites contributing to the model. Metabolites are colored by their Subclass level chemical ontology.
To better understand the impact of the HD diet on inter-personal variation in all microbiome-dependent data types collected in the study, we calculated the Bray-Curtis dissimilarity (as a measure of uniqueness: the higher the value the more dissimilar from other samples) of participant microbiome composition (16S rRNA), fecal metagenomes, and metabolomes of urine, plasma, and stool. Data were collected at all study time points, reflecting all three phases of the study. 16S rRNA amplicon sequencing resulted in an average of 46,714 (range 17,989-70,638) reads per sample, an average of 252 ASVs per sample (range 77-331), and 534 ASVs that occurred in 80% of participants for at least one time point (a criteria set for further analysis; see STAR methods). We found that interpersonal variation in microbiome composition and fecal metabolomes declined in the HD phase relative to the BD phase and increased during the WO phase relative to HD phase (Figure 3B,C; paired Wilcoxon signed rank test, p = 0.002 and p < 0.003; respectively). These data illustrate that a small but measurable amount of variation between people in the compositional features and functional output of the gut microbiome are driven by differences in diet.
We next generated random forest models to determine which microbiome-associated data types were useful in differentiating participant samples into BD vs HD phases. We built independent models for ASV abundance, fecal metabolome, plasma metabolome, and urine metabolome, and assessed their ability to correctly classify study phase independently (Table S2, Figure S3). As a positive control, we developed a model based on participant macronutrient and fiber data, which at 87.5 % accuracy, had the highest accuracy for distinguishing BD (1 or 2) versus HD (3 or 4) study time points (Figure 3D). The best microbiome-associated data to classify participants by study phase were the fecal metabolomic profiles, with 62.5 % accuracy (Figure 3D). The fecal metabolite most explanatory for study phase was host derived creatine, followed by 4-hydroxyphenylacetic acid, an MDM of tyrosine (Figure 3E; time point 2 versus time point 3, LOOCV). Plasma and urine metabolomic profiles, and microbiome compositional features were no better than chance in classifying samples into the correct diet phase. Our results suggest that no individual metabolite had greater than 4 % explanatory power, however we note the limited generalizability of these findings given the small sample set, and the unique design of our intervention that lacks an appropriate external validation cohort.
Previous studies have demonstrated a dominant role of diet and environment in dictating features within the microbiome(David et al., 2013; Rothschild et al., 2018). We used permutational multivariate analysis of variance (PERMANOVA), to quantify the extent of variance in microbiome composition, function, and metabolic output explained by each host- and diet-associated variable that we measured. The majority of variation in microbiome features measured, which include species and genes found within the microbiome, and plasma, urine, and fecal metabolites, is due to host identity (Figure 4A), consistent with previous work demonstrating individuality of the microbiome in longitudinal dietary intervention studies(Zhu et al., 2015; Falony et al., 2016; Zmora et al., 2016; Zeevi et al., 2019). Age (15%), body surface area (8%), and biological sex (3%) also explain a significant portion of variance across all data types (p < 0.05). Host identity has a greater impact on plasma (47%) and urine (45%) derived host-microbe metabolomes compared to participant fecal metabolomes (23%). Age contributes to 11% of the plasma host-microbe metabolome, but not to the urine or fecal metabolomes, consistent with previous reports of age-related plasma metabolomic profiles(Oliphant and Allen-Vercoe, 2019; Zeevi et al., 2019; Asnicar et al., 2021). Overall, we found that the study time point explains 1-2% of the variance in microbiome composition, function, and metabolome.
Figure 4. Intrapersonal variation in microbiome composition, function, and fecal metabolic output shifts after the HD intervention, but host identity explains most variance.
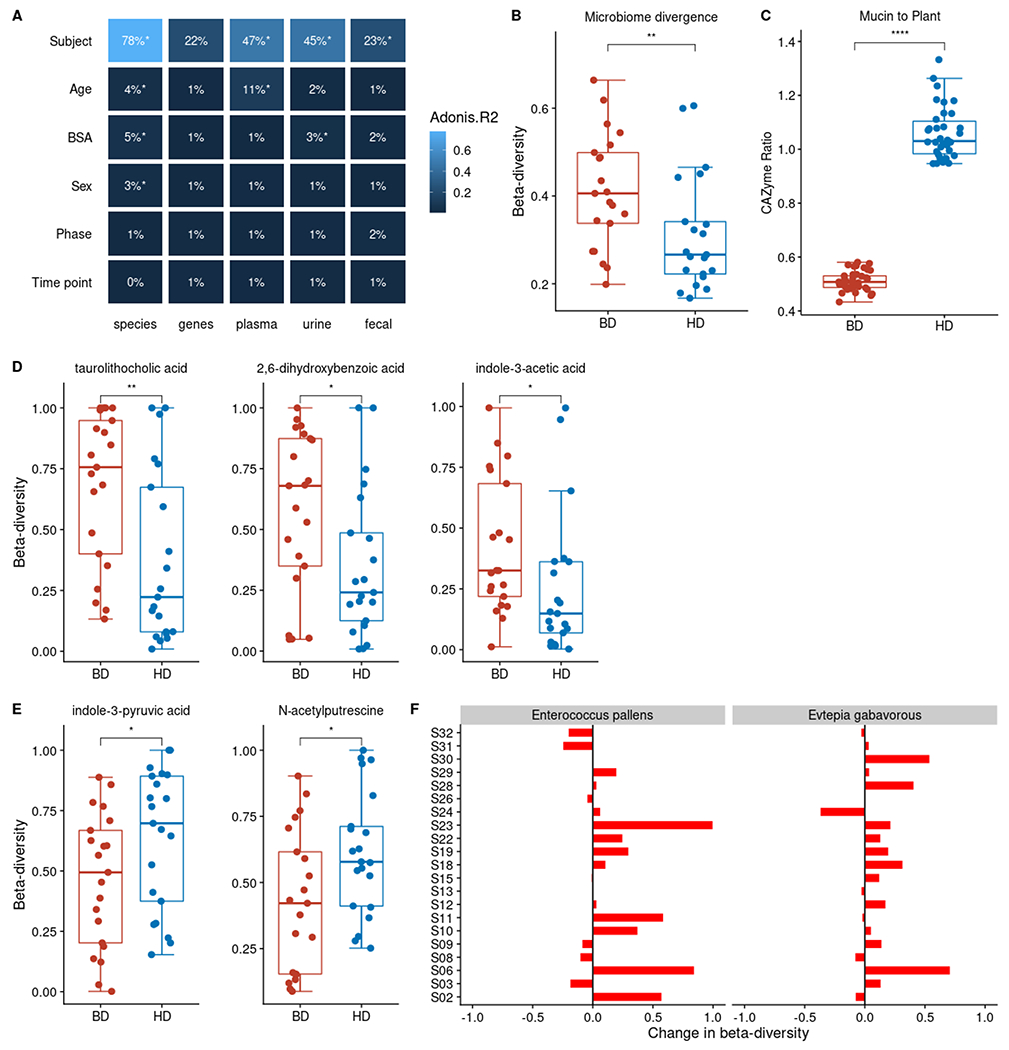
(A) Host identity followed by age are the largest factors contributing to variation in microbiome composition, function and metabolomes in the MISO Study based on PERMANOVA (*p<0.05). Additional significant study variables shown capture < 10% of variation. Variance explained (R2) is also indicated by blue color scale.
(B) Microbiome compositional intra-personal divergence collapses between the baseline (BD) and homogenous diet (HD) diet (paired Wilcoxon signed rank test,**p < 0.001;).
(C) Microbiome mucin-to-plant carbohydrate active enzyme ratios (CAZyme) shifts between the BD and SD diet (paired Wilcoxon signed rank test,****p < 0.0001).
(D) Levels of fecal metabolites, taurolithocholic acid, 2,6,-dihydroxybenzoic acid and indole-3-acetic acid levels are more similar during the HD (metabolite beta-diversity; Bray-Curtis distance) than the BD. Metabolite levels are based on 2 timepoints per study phase.
(E) Fecal indole-3-pyruvic acid and N-acetyl putrescine increased in intrapersonal divergence during the HD phase (metabolite beta-diversity; Bray-Curtis distance).
(F) Participant specific changes in beta-diversity for agmatinases from Enterococcus pallens (p < 0.05) and Evtepia gabavorous (p < 0.05) defined by the difference in beta-diversity between HD and BD phases. Specific microbial agmatinases involved in the production of N-acetyl putrescine increased in intrapersonal divergence during the HD phase (gene beta-diversity; Bray-Curtis distance) based on relative abundance counts of mapped agmatinase protein sequences against participant metagenomes.
Boxplot error bars represent 25th (bottom) and 75th (upper) quartiles.
We were intrigued by the limited impact of the HD intervention on interpersonal variation, and next sought to better understand how the HD diet impacted interpersonal variability in microbiome features when comparing the BD and HD phases. We hypothesized that, we would also observe that intrapersonal differences (i.e., temporal changes within study participants) would be greater in the BD phase, due to day-to-day variation in diet, in comparison to the HD phase. For each participant we calculated a Bray-Curtis dissimilarity matrix for the microbiome associated data types and computed the beta-diversity of each by comparing the difference in dissimilarity values for the two time points collected in the BD and HD phase. This approach allowed us to assess whether the homogeneous diet decreased overall interpersonal variation in these high-dimensional datasets. Participant microbiome 16S composition beta-diversity decreased significantly in the HD phase (Figure 4B, paired Wilcoxon signed rank test, p < 0.001). One caveat is that the two BD timepoints are separated by 14 days, whereas the HD timepoints are only 3 days apart. To assess the impact of the interval between time points on beta diversity, we compared additional timepoints where diet varies as controls and observed no relationship between time and b-diversity, supporting the impact of HD, rather than the time interval, on reduced b-diversity. Overall microbiota Shannon alpha diversity, a measure of species richness and diversity, did not decrease during the HD study phase or change cohort-wide over the study (Figure S4). These data are in keeping with the individuality and temporal stability of microbiome alpha and beta diversity of other short-term dietary interventions studies(David et al., 2013; Johnson et al., 2019).
Decreased dietary fiber consumption results in a shift in carbohydrate utilization within the microbiome towards mucin glycan degradation and away from plant carbohydrate degradation(Zhu et al., 2015; Smits et al., 2016, 2017; Sonnenburg and Bäckhed, 2016). Therefore, we hypothesized that the decrease in fiber during the HD phase would be reflected in an increased ratio of genes predicted to encode mucin- relative to plant-glycan-degrading enzymes in participants’ metagenomes. We assigned glycoside hydrolases and polysaccharide lyases (i.e., carbohydrate active enzymes, or CAZymes) represented within participant metagenomes to mucin or plant carbohydrate degradation. An increase in the ratio of mucin-to-plant CAZyme genes in the HD relative to the BD study phases is apparent, consistent with a shift towards mucin consumption in response to the low fiber HD diet (Figure 4C, paired Wilcoxon signed rank test, p < 0.0001). Similar differences in microbiome-encoded carbohydrate genes have been found in other diet studies (Kaoutari et al., 2013; Smits et al., 2017).
To further characterize the relationship between the dietary intervention and MDMs, we next investigated intrapersonal divergence in participant fecal, urine and plasma metabolomes. Only three fecal metabolites decreased between the BD and HD phase, taurolithocholic acid, 2,6-Dihydroxybenzoic acid and indole-3-acetic acid (Figure 4D). Interestingly, two additional fecal metabolites, indole-3-pyruvic acid, and N-acetyl putrescine, increased in intrapersonal variation despite identical dietary input during HD (Figure 4E), leading us to hypothesize that microbiome metabolism, rather than diet, plays a large role in their variation. Notably, all metabolites have consistent low levels of autocorrelation suggesting that the observed shifts were not due to time differences between sampling timepoints. To distinguish between the possibility that the increase was due to normal microbiome temporal dynamics or those induced by the shift in microbiome capacity on the HD, we investigated the intrapersonal divergence in the specific microbiome encoded genes responsible for production of these two metabolites. We mapped participant metagenomic profiles to a protein database of specific microbial genes encoding aromatic amino acid (AAA) aminotransaminases and agmatinases. These classes of enzymes include those that mediate the conversion of tryptophan into indole-3-pyruvic acid and arginine to putrescine, respectively (putrescine is converted into N-acetyl putrescine by host enzymes). Agmatinases that map to Enterococcus pattens and Evtepia gabavorous increase in overall intrapersonal divergence in the HD phase relative to the BD phase (Figure 4F) consistent with increased within-person variation in N-acetyl putrescine during HD. No similar pattern for aminotransaminase divergence was detected. Combined, these results suggest that homogenizing dietary input across healthy adults for 7 days contributes to some changes in microbiome composition, function, and specific MDMs, however, is insufficient to significantly reduce interpersonal and intrapersonal variation in many other facets of microbiome composition function and metabolic output.
Discussion
There is great interest in optimizing the production of microbiome-dependent metabolites relevant to human health, including minimizing those that contribute to uremic illness. For instance, microbiota generated products, p-cresol sulfate and indoxyl sulfate, derived from amino acids promote the progression and exacerbation of cardio-renal diseases(Koppe et al., 2013; Yang et al., 2017; Lim et al., 2021). The use of diet modification to alter specific aspects of microbiome functional output has shown some promise in previous studies(Suez et al., 2014; Salmean et al., 2015; Zeevi et al., 2015; Cho et al., 2017), but the extent to which inter-individual variation in MDM production is caused by a person’s microbiome vs. their habitual diet remains an open question. In this study, we employed a 7-day homogeneous diet (HD) modeled on an average diet in the U.S. to eliminate dietary variability between individuals while attempting to minimize perturbation when participants shifted to this dietary intervention. The homogeneous diet significantly reduced interpersonal variation in hippuric acid, and 76 additional fecal, plasma, and urine MDMs, but had no significant effect on other well studied uremic solutes including PAG, IS and PCS. The inability of HD to decrease the cohort-wide variability in these three uMDMs by 25% (the primary endpoint) provides strong evidence that diet-independent individualized features of the microbiome and host play a critical role in dictating individual-specific levels of PAG, IS, and PCS.
Interestingly, we found that the diet phase of the study contributed to less than 2% of the overall variance found between participants microbiome composition, functional profiles, fecal metabolomes, plasma metabolomes, and urine metabolomes. Additional studies will be required to assess whether a homogenous dietary intervention that is defined by a greater shift in diet composition from participant baseline diets, has a greater contribution to variance in microbiome composition and function. Diets limited in specific uMDM precursors may have the potential to reduce levels of specific solutes in individuals with failing kidneys, who notably are subjected to burdensome dietary restrictions(Patel et al., 2012). Of note, excretion rates of these MDMs remain widely variable in patients with chronic kidney disease as with individuals with functioning kidneys47–49. We hypothesize that given the interpersonal variability in the level of uMDMs such as PCS and IS in these individuals11, there is a potential for reduction given the appropriate diet.
Our finding that host identity and age, were dominant contributors to interpersonal variability, are consistent with other studies profiling healthy individuals(Human et al., 2012; ‘The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease’, 2014; Falony et al., 2016). While the overall impact of the HD intervention on interpersonal variability was limited, 16S-based composition analysis and fecal metabolite profiles show a small but significant decrease in interpersonal variation during HD consistent with diet contributing to some measurable extent of individuality. Our analysis also reveals that a homogenous diet reduces interpersonal variation in a subset of MDMs. These analyses broaden our understanding of MDMs that are sensitive and insensitive to dietary variation. Future studies expanding our understanding of metabolites that are sensitive to homogenous diets and modifiable by dietary strategies for clinical benefits are warranted.
Previously, studies of the gut microbiome in healthy cohorts have found that microbiome-food relationships are highly personalized(Johnson et al., 2019). Controlled feeding studies, which allow for the precise accounting of diet composition, can produce more reliable diet-microbiome interactions and associations(Wu et al., 2011, 2016; Pan et al., 2020); however, to date, controlled feeding studies have not specifically addressed the impact of homogenizing diet composition on interpersonal variation in microbiome features. Our data adds a stringent test to demonstrate that participants’ individualized responses to dietary interventions are likely governed in part by the individualized aspects of microbiome composition and function that are fairly recalcitrant to short-term change. The extent of dietary compliance depends upon self-reporting of our free-living participants, who were not monitored in a facility. To support dietary compliance, which was key to the study design, our study participants received prepared meals and had consistent interactions with a dietitian. A greater understanding of a participant’s habitual diet is critical to understanding how diet interacts with microbiome functionality to determine metabolite production, and the extent to which metabolite levels may respond to an intervention.
The design of our study allowed us to dissect the relative contributions of diet (i.e., macro nutrients) and microbiome functional potential (i.e., encoded genes) towards the interpersonal and intrapersonal variation in microbiome features (i.e., microbiome-dependent uremic solutes, 16S-based composition, metagenomic functions, and metabolome profiles). We found that the modest reduction in protein between the BD and HD phase (average 82 grams per day for BD, and 67 grams per day for HD) did not correspond to a reduction in the protein-derived metabolites and uremic solutes, PCS, IS and PAG. Previous studies quantifying IS and PCS, found that a very low protein diet (20 grams per day) was necessary to reduce their levels(Wishart DS, 2014; Thaiss et al., 2016; Wilson, Reedy and Krebs-Smith, 2016). We also found that tyrosine, phenylalanine, and tryptophan, which are precursors for p-cresol, phenylacetylglutamine and indoxyl sulfate; respectively, were stable across the BD and HD phase.
How changes in the chemical composition of diet can alter microbiome metabolite output needs to be tested under a variety of conditions (including extreme dietary control) and provision of specific chemical precursors) with an emphasis on understanding participants’ baseline dietary habits and microbiome functional capacity (metagenomes). Furthermore, future studies should focus on precision manipulation of the microbiome (e.g., microbial therapies) in combination with dietary interventions. To develop targeted approaches to modulate metabolite levels, factors contributing to individual MDM level variability should be understood. Thus, a detailed understanding of the relationship between dietary input and microbiota metabolism in shaping interpersonal variability provides a roadmap to develop diverse therapeutic approaches. Importantly, unifying how dietary intervention studies account for variation in participant habitual diets, record and characterize diet data, lifestyle factors, host genetics, as well as the range of metabolites measured(Hughes et al., no date; Leeming et al., 2021), will help fill gaps in understanding the utility of diet to modulate the production of specific solutes.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
All information and requests for further resources should be directed to and will be fulfilled by the Lead Contact, Justin Sonnenburg, jsonnenburg@stanford.edu.
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
All sequencing data has been deposition to the NCBI Sequence Read Archive under project PRJNA776530 and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table.
Key resources table
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Recruitment and selection of participants
Participants were recruited from the local community through online advertisement in different community groups as well as emails to past research participants that consented to being contacted for future studies. The current study assessed 61 participants for eligibility. They completed an online screening questionnaire and a clinic visit between September 2018 and January 2019. The primary inclusion criteria included age ≥ 18 y and general good health. Participants were excluded if they had a history of active uncontrolled inflammatory bowel disease (IBD) including ulcerative colitis, Crohn’s disease, or indeterminate colitis, irritable bowel syndrome (IBS) (moderate or severe), infectious gastroenteritis, colitis or gastritis, Clostridium difficile infection (recurrent) or Helicobacter pylori infection (untreated), malabsorptive intestinal disease (such as celiac disease), major surgery of the GI tract, with the exception of cholecystectomy and appendectomy, in the past five years, or any major bowel resection at any time. Other exclusion criteria included a BMI ≥ 40, diabetes, renal disease, significant liver enzyme abnormality, pregnancy or lactation, smoking, a history of CVD, inflammatory disease, or malignant neoplasm. CONSORT clinical trial flow diagram of participant recruitment shown in Figure 1A and demographics table shown in Table 1. 21 participants (11 female sex and gender identifying, 10 male sex and gender identifying) were used for full analysis with an average age of 48 +/− 14 years. All study participants provided written informed consent. The study was designed as an exploratory approach toward discovery of changes in the microbiota and the metabolome in response to a dietary intervention. The study was approved annually by the Stanford University Human Subjects Committee. Trial was registered at ClinicalTrials.gov, identifier: NCT04740684.
Specimen collection
Stool was collected at five study time points over a four-week period of the study and kept in participants’ home freezers (−20°C) wrapped in ice packs until they were transferred on ice to the research laboratory and stored at −80°C. 24 hour urine collection was obtained on Day 0 and 13 (while on BD), Day 17 (while on HD), and Day 28 (during WO phase). Plasma and spot urine samples were obtained during research clinic visits throughout the study at indicated time points.
METHOD DETAILS
Intervention
The study period lasted 28 days and consisted of a 14-day period while participants consumed their habitual baseline diet (BD), followed by 7 days of provided homogenous diet (HD), and concluding with a 7-day washout period (WO) of return to their prior habitual dietary pattern. They were asked to keep detailed food logs for the first 3 days upon initiation of the study. During the HD phase of study, participants were asked to record the quantity of packets consumed daily. Nutrient analyses were conducted with use of FOOD PROCESSOR (version 11.9.0; ESHA, Salem, OR).
Formulation of Homogenous Diet (HD)
The Homogenous Diet (HD) was provided for days 15-21 for the study period (7 days total). It was designed to recapitulate the diet quality, and specifically the fiber and macronutrient ranges(Grotto and Zied, 2010), common to adults in America based on the National Health and Nutrition Examination surveys and additional studies (Grotto and Zied, 2010). The food provided was a nutritionally adequate diet, designed by a registered dietitian. It was prepared by a professional chef in a commercial kitchen by mixing and cooking foods purchased from grocery stores. The food mixture was divided into 295 g portions that included a 210 g portion of the homogenous diet in addition to 59 g Orange Juice and 26 g of cookies to be consumed with each meal. The composition of the HD is described in Table S1.
Distribution of Food and Monitoring of Consumption
Food for the 7-Day homogenous diet was distributed after collecting baseline samples. Subjects were allowed to eat as much of the homogeneous diet as they wanted to meet their caloric needs. Subjects were asked not to eat anything other than the provided HD (no candy, snacks, etc.) and not to drink anything except water (no coffee, tea, sodas, or alcoholic drinks, etc.). Participants kept logs of time of day and number of pouches consumed during the 7 days of the controlled diet. These records were reviewed with the study dietitian to ensure completeness and accuracy. The ingestion of the HD study diet in each individual subject terminated with the urine, stool, and blood collection on the seventh day of eating the diet. After termination of the HD study diet, participants returned to their habitual diet.
Measurement of uMDMs
Solute excretion and nutrient consumption rates were corrected for body surface area calculated using the Mosteller formula(Mosteller, 1987).
16S amplicon sequencing
DNA was extracted from stool using the DNeasy PowerSoil HTP 96 kit according to the manufacturer protocol and amplified at the V4 region of the 16S ribosomal RNA (rRNA) subunit gene and 250 nucleotides (nt) Illumina sequencing reads were generated. 16S rRNA gene amplicon sequencing data from stool samples were demultiplexed using the idemp (https://github.com/yhwu/idemp). Amplicon sequence variants (ASVs) were identified with a learned sequencing error correction model (DADA2 method)(Callahan et al., 2016), using the dada2 package in R. ASVs were assigned taxonomy using the GreenGenes database (version 13.8)(DeSantis et al., 2006). ɑ-diversity was quantified as the number of observed ASVs, Shannon diversity, or PD whole tree, in a sample using the phyloseq package in R (version 4.0.2). There was an average of 46,714 reads per sample (range 17,989-70,638) recovered after filtering, denoising, and removing chimeras. A total 534 unique ASVs were observed ,with an average of 252 ASVs per sample (range 77-331), were used for further analysis.
Metagenomic Sequencing
DNA extraction for shotgun metagenome sequencing was done using the DNeasy PowerSoil HTP 96 kit as described in the 16S amplicon sequencing methods. For library preparation, the Nextera Flex kit was used with a minimum of 10ng of DNA as input and 6 or 8 PCR cycles depending on input concentration. A 12 base pair dual-unique-indexed barcode (CZ Biohub) was added to each sample and libraries concentration and size were quantified using an Agilent Fragment Analyzer. They were further size selected using AMPure XP beads (Beckman) targeted at a fragment length of 450bp (350bp size insert). DNA paired-end sequencing (2x146bp) was performed on a NovaSeq 6000 using S4 flow cells (CZ Biohub). The average target depth for each sample was 11.4 million paired-end reads with an average of 11,422,190 (range 2419034 -71084760) reads per sample. Data quality analysis was performed by demultiplexing raw sequencing reads and concatenating data for samples that required multiple sequencing runs for target depth before further analysis.
BBtools suite (https://sourceforge.net/projects/bbmap/) was used to process raw reads and mapped against the human genome (hg19) after trimming, with masks over regions broadly conserved in eukaryotes (http://seqanswers.com/forums/showthread.php?t=42552). Exact duplicate reads (subs=0) were marked using clumpify and adapters and low-quality bases were trimmed using bbduk (trimq=16, minlen=55).
Plasma, Urine and Stool metabolomics
Metabolites from three sample types (plasma, urine, and stool) metabolites were profiled using three complementary liquid chromatography- tandem mass spectrometry (LC–MS) methods designed to measure a broad range of microbial and microbe-host co-metabolites(Han et al., 2021). For all three sample types, samples were incubated for 5 minutes at room temperature and centrifugation at 5,000 x g for 10 minutes. Sample supernatants were then transferred, evaporated, and reconstituted in an internal standard mix in (50% Methanol). A Hydrophilic Interaction Liquid Chromatography (HILIC) method was used for the analysis of water-soluble polar metabolites in positive (HILIC-pos) ion mode, and two C18 column chromatography methods for measuring metabolites of intermediate polarity, in positive (C18-pos) or negative (C18-neg) ion mode. Raw data were processed, and compounds were annotated using MSDIAL software 3.988(Tsugawa et al., 2015). Analyses were conducted using the data obtained from all three LC-MS methods after removal of features observed in <80% of the samples and imputing missing values with half of the minimum observed measurement for each feature.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of Alpha and Beta Diversity Measures
Alpha and beta- diversity measures were calculated using the phyloseq package(McMurdie and Holmes, 2013) in R. Alpha diversity was calculated at the ASV level. For ASVs, metagenomes, plasma metabolomes, urine metabolomes and stool metabolomes, Bray-Curtis dissimilarity was computed at each timepoint and for each subject using the vegdist function in the R vegan package(Dixon, 2003). In order to investigate intra-individual divergence within a subject for single features, we computed a measure of divergence based on the difference in abundance values of each feature in the BD and HD phases, based on the two time points per phase.
Microbiome Functional Profiling
The assembled metagenomes were mapped against the following functional databases using USEARCH version 8 with an e-value cutoff of 1e-40 in order to ensure longer sequence hits for improved taxonomic and functional resolution: KEGG EC/KO (n = 2,000,708)(Kanehisa and Goto, 2000), Carbohydrate active enzyme (CAZyme) (n = 7215)(Cantarel et al., 2009), and a curated database of both cultured and metagenomically identified agmatinases and aminotransaminases developed in-house. The abundance of KEGG orthologous groups and modules were determined using the HUMAnN pipeline(Abubucker et al., 2012) with default parameters. CAZymes were classified as plant, animal, mucin, or fungal targeting based on the mapping file and approach described previously(Smits et al., 2017),which relies on a variety of sources for these categorizations(Cantarel, Lombard and Henrissat, 2012; Kaoutari et al., 2013).
Metabolite Chemical Class Mapping
All metabolites were mapped to their superclass, class and subclass using the ClassyFire automated chemical classification based on compound InChIKeys(Djoumbou Feunang et al., 2016).
Statistical Analysis
All statistical analyses were carried out in R. The coefficient of variation and 95% confidence interval for each uremic solute (IS, PCS, PAG and HIPP), at each study time point was determined using the R cvcqv package. Statistical difference in CV or beta-diversity between time points 1 and 4 and mean metabolite abundances between the BD and HD phases were determined using a paired Wilcoxon signed rank test. The autocorrelation decay was determined for each metabolite with significant changes in beta-diversity using the autocorrelation function in the timeseries R package.
Linear Mixed Effects Modeling
To study the effect of specific dietary parameters and the microbiome on the levels of hippuric acid, p-cresol sulfate, phenylacetylglutamine, and indoxyl sulfate during the HD phase at time point 4, when diet is homogenized, we used a linear mixed effects model. All variables included in the model were centered and scaled using the scale function in base R (scale function, center=TRUE, scale=TRUE). For each metabolite, metabolite levels were modeled as a function of the macronutrients, fiber, diet groupings, as well as microbiome encoded genes. A term for biological sex was included in the model as a covariate. The standardized regression coefficients, which reflect a standard deviation increase or decrease, were computed using the lme4(Bates et al., 2015) (1.1.26) package in R.
Recursive Feature Random Forest
Random forest regression models were built of the default set of 1000 trees, with the caret R package(Kuhn, 2008) to predict the study phase based on microbiome features. Training was achieved through 10-fold cross validation with ASV data, fecal metabolome, plasma metabolome, urine metabolome, dietary data, and anthropometry data. For each data type, one time point per participant for each phase (BD or HD) in our analysis (t1 vs t4, t1 vs t3, t2 vs t3, t2 vs t4) was selected to avoid accuracy inflation that could result from autocorrelation. The feature selection was performed by using the recursive feature elimination algorithm of the caret R package55. For the random forest model based on ASVs, 534 ASVs that occurred in 80% of participants for at least one time point that were used for further analysis. 320 fecal metabolites, 183 plasma metabolites and 168 urine metabolites were used to build separate models. The importance scores of features were determined based on the increase of prediction error when that feature was randomly permuted while all others were remained unchanged.
PERMANOVA
To calculate the variance explained by each of our collected study factors we performed an Adonis test implemented in the R vegan package (v2.5.7 using adonis) using 999 permutations, with random permutations constrained by using the “strata” option. We used Bray-Curtis dissimilarity as the distance measure for all five data types independently, which includes participants ASVs, metagenomes, plasma metabolomes, urine metabolomes and stool metabolomes. The total variance for each factor was determined independently.
Supplementary Material
Highlights.
Diet homogeneity did not decrease inter-individual variability in uremic MDMs
Homogenous diet results in reduction of interpersonal variation in hippuric acid
Host identity and age, but not diet, are dominant contributors to variability in MDMs
Acknowledgements
We wish to thank the participants for their engagement and effort to enable this study. Chef Laura Stec developed the recipe given many constraints and organized and led the cooking and packaging sessions. This work was funded through philanthropic support and the NIH/NIDDK 2R01DK101674 (J.L.S., M.A.F., and T.W.M.) P01 HL147823 (M.A.F.) and R01 3R01DK08502510 (J.L.S.); L.G. was supported by the NIH training grant T32AI007328 and the HHMI Hanna Gray Fellowship. S.P.S. was supported by the A.P. Giannini Foundation Research Fellowship and Leadership Award and the NIH Training Grant T32DK007056. S.H. was supported by Stanford Dean’s Postdoctoral Fellowship and NRSA F32AG062119. J.L.S. and M.A.F. are Chan Zuckerberg Biohub investigators.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors have no competing interests to disclose.
ADDITIONAL RESOURCES
Clinical trial registry #NCT04740684: https://clinicaltrials.gov/ct2/show/NCT04740684
References
- Abubucker S et al. (2012) ‘Metabolic reconstruction for metagenomic data and its application to the human microbiome.’, PLoS computational biology, 8(6), p. e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnicar F et al. (2021) ‘Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals’, Nature Medicine, 12(11), p. 13. doi: 10.1038/s41591-020-01183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D et al. (2015) ‘Fitting linear mixed-effects models using Ime4’, Journal of Statistical Software, 67(1). doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- Callahan BJ et al. (2016) ‘DADA2: High-resolution sample inference from Illumina amplicon data’, Nature Methods, 13(7), pp. 581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL et al. (2009) ‘The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics.’, Nucleic acids research, 37(Database issue), pp. D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Lombard V and Henrissat B (2012) ‘Complex Carbohydrate Utilization by the Healthy Human Microbiome’, PLoS ONE. Edited by V.D. Appanna, 7(6), p. e28742. doi: 10.1371/journal.pone.0028742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases A et al. (2019) ‘Vegetable-based diets for chronic kidney disease? It is time to reconsider’, Nutrients. MDPI AG. doi: 10.3390/nu11061263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CE et al. (2017) ‘Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial’, Molecular Nutrition and Food Research, 61(1). doi: 10.1002/mnfr.201600324. [DOI] [PubMed] [Google Scholar]
- David LA et al. (2013) ‘Diet rapidly and reproducibly alters the human gut microbiome’, Nature, 505(7484), pp. 559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ et al. (2006) ‘Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB’, Applied and Environmental Microbiology, 72(7), pp. 5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P (2003) ‘VEGAN, a package of R functions for community ecology’, Journal of Vegetation Science, doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- Djoumbou Feunang Y et al. (2016) ‘ClassyFire: automated chemical classification with a comprehensive, computable taxonomy’, Journal of Cheminformatics, 8(1), pp. 1–20. doi: 10.1186/s13321-016-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobre MA, Meyer TW and Hostetter TH (2020) ‘The Uremic Syndrome’, Chronic Renal Disease, pp. 199–210. doi: 10.1016/B978-0-12-815876-0.00014-0. [DOI] [Google Scholar]
- Edamatsu T et al. (2014) ‘Classification of Five Uremic Solutes according to Their Effects on Renal Tubular Cells’, International Journal of Nephrology, 2014, pp. 1–10. doi: 10.1155/2014/512178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evenepoel P et al. (2009) ‘Uremic toxins originating from colonic microbial metabolism’, Kidney International. Nature Publishing Group, doi: 10.1038/ki.2009.402. [DOI] [PubMed] [Google Scholar]
- Falony G et al. (2016) ‘Population-level analysis of gut microbiome variation.’, Science (New York, N.Y.), 352(6285), pp. 560–4. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- Glowacki RWP and Martens EC (2020) ‘In sickness and health: Effects of gut microbial metabolites on human physiology’, PLOS Pathogens. Edited by L.J. Knoll, 16(4), p. e1008370. doi: 10.1371/journal.ppat.1008370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotto D and Zied E (2010) ‘The Standard American Diet and Its Relationship to the Health Status of Americans’, Nutrition in Clinical Practice, 25(6), pp. 603–612. doi: 10.1177/0884533610386234. [DOI] [PubMed] [Google Scholar]
- Guldris SC, Parra EG and Amenós AC (2017) ‘Gut microbiota in chronic kidney disease’, Nefrología (English Edition), 37(1), pp. 9–19. doi: 10.1016/J.NEFROE.2017.01.017. [DOI] [PubMed] [Google Scholar]
- Han S et al. (2021) ‘A metabolomics pipeline for the mechanistic interrogation of the gut microbiome’, Nature, 595(7867), pp. 415–420. doi: 10.1038/s41586-021-03707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hooft JJJ et al. (2012) ‘Structural Elucidation and Quantification of Phenolic Conjugates Present in Human Urine after Tea Intake’, Analytical Chemistry, 84(16), pp. 7263–7271. doi: 10.1021/ac3017339. [DOI] [PubMed] [Google Scholar]
- Hughes RL et al. (no date) ‘The Role of the Gut Microbiome in Predicting Response to Diet and the Development of Precision Nutrition Models-Part I: Overview of Current Methods’. doi: 10.1093/advances/nmz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human T et al. (2012) ‘Structure, function and diversity of the healthy human microbiome.’, Nature, 486(7402), pp. 207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y et al. (2013) ‘Correlation between Serum Levels of Protein-Bound Uremic Toxins in Hemodialysis Patients Measured by LC/MS/MS.’, Mass spectrometry (Tokyo, Japan), 2(Spec Iss), p. S0017. doi: 10.5702/massspectrometry.S0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AJ et al. (2019) ‘Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans.’, Cell host & microbe, 25(6), pp. 789–802.e5. doi: 10.1016/j.chom.2019.05.005. [DOI] [PubMed] [Google Scholar]
- Kanehisa M and Goto S (2000) ‘KEGG: Kyoto Encyclopedia of Genes and Genomes’, Nucleic Acids Research, 28(1), pp. 27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaoutari A. El et al. (2013) ‘The abundance and variety of carbohydrate-active enzymes in the human gut microbiota’, Nature Publishing Group, 11(7), pp. 497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Koppe L et al. (2013) ‘p-Cresyl sulfate promotes insulin resistance associated with CKD.’, Journal of the American Society of Nephrology: JASN, 24(1), pp. 88–99. doi: 10.1681/ASN.2012050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M (2008) ‘Building predictive models in R using the caret package’, Journal of Statistical Software, 28(5). doi: 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- Leeming ER et al. (2021) ‘The complexities of the diet-microbiome relationship: advances and perspectives’, Genome Medicine. BioMed Central Ltd, pp. 1–14. doi: 10.1186/s13073-020-00813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees HJ et al. (2013) ‘Hippurate: The natural history of a mammalian-microbial cometabolite’, Journal of Proteome Research, 12(4), pp. 1527–1546. doi: 10.1021/pr300900b. [DOI] [PubMed] [Google Scholar]
- Lim YJ et al. (2021) ‘Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets’, Toxins, 13(2), p. 142. doi: 10.3390/toxins13020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R et al. (2017) ‘Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention’, Nature Medicine, 23(7), pp. 859–868. doi: 10.1038/nm.4358. [DOI] [PubMed] [Google Scholar]
- Mair RD et al. (2018) ‘Characteristics of Colon-Derived Uremic Solutes.’, Clinical journal of the American Society of Nephrology: CJASN, 13(9), pp. 1398–1404. doi: 10.2215/CJN.03150318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin FPJ et al. (2009) ‘Metabolic effects of dark chocolate consumption on energy, gut microbiota, and stress-related metabolism in free-living subjects’, Journal of Proteome Research, 8(12), pp. 5568–5579. doi: 10.1021/pr900607v. [DOI] [PubMed] [Google Scholar]
- Martínez I et al. (2013) ‘Gut microbiome composition is linked to whole grain-induced immunological improvements’, ISME Journal, 7(2). doi: 10.1038/ismej.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ and Holmes S (2013) ‘phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data’, PLoS ONE. Edited by M. Watson, 8(4), p. e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosteller RD (1987) ‘Simplified Calculation of Body-Surface Area’, New England Journal of Medicine, 317(17), pp. 1098–1098. doi: 10.1056/NEJM198710223171717. [DOI] [PubMed] [Google Scholar]
- O’Keefe SJD (2016) ‘Diet, microorganisms and their metabolites, and colon cancer’, Nature Reviews Gastroenterology and Hepatology. Nature Publishing Group, pp. 691–706. doi: 10.1038/nrgastro.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant K and Allen-Vercoe E (2019) ‘Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health’, Microbiome, 7(1), p. 91. doi: 10.1186/s40168-019-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S et al. (2020) ‘Gut microbial protein expression in response to dietary patterns in a controlled feeding study: A metaproteomic approach’, Microorganisms, 8(3), p. 379. doi: 10.3390/microorganisms8030379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KP et al. (2012) ‘The production of p-cresol sulfate and indoxyl sulfate in vegetarians versus omnivores.’, Clinical journal of the American Society of Nephrology: CJASN, 7(6), pp. 982–8. doi: 10.2215/CJN.12491211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HK et al. (2016) ‘Human gut microbes impact host serum metabolome and insulin sensitivity’, Nature, 535(7612), pp. 376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- Ravid JD, Kamel MH and Chitalia VC (2021) ‘Uraemic solutes as therapeutic targets in CKD-associated cardiovascular disease’, Nature Reviews Nephrology. Nature Research, pp. 402–416. doi: 10.1038/s41581-021-00408-4. [DOI] [PubMed] [Google Scholar]
- Rothschild D et al. (2018) ‘Environment dominates over host genetics in shaping human gut microbiota’, Nature, 555(7695), pp. 210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- Salmean YA et al. (2015) ‘Fiber supplementation lowers plasma p-cresol in chronic kidney disease patients’, Journal of Renal Nutrition, 25(3), pp. 316–320. doi: 10.1053/j.jrn.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirich TL et al. (2014) ‘Effect of increasing dietary fiber on plasma levels of colon-derived solutes in hemodialysis patients’, Clinical Journal of the American Society of Nephrology, 9(9), pp. 1603–1610. doi: 10.2215/CJN.00490114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SA et al. (2016) ‘Individualized Responses of Gut Microbiota to Dietary Intervention Modeled in Humanized Mice’, mSystems. Edited by P.C. Dorrestein, 1(5), pp. e00098–16. doi: 10.1128/mSystems.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SA et al. (2017) ‘Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania’, Science, 357(6353), pp. 802–805. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL and Bäckhed F (2016) ‘Diet—microbiota interactions as moderators of human metabolism’, Nature, 535(7610), pp. 56–64. doi: 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suez J et al. (2014) ‘Artificial sweeteners induce glucose intolerance by altering the gut microbiota’, Nature, 514(7521), pp. 181–186. doi: 10.1038/nature13793. [DOI] [PubMed] [Google Scholar]
- Tang WHW and Hazen SL (2014) ‘The contributory role of gut microbiota in cardiovascular disease.’, The Journal of clinical investigation, 124(10), pp. 4204–11. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss CA et al. (2016) ‘Persistent microbiome alterations modulate the rate of post-dieting weight regain’, Nature, 540(7634), pp. 544–551. doi: 10.1038/nature20796. [DOI] [PubMed] [Google Scholar]
- ‘The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease’ (2014) Cell Host & Microbe, 16(3), pp. 276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H et al. (2015) ‘MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis’, Nature Methods, 12(6), pp. 523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wastyk HC et al. (2021) ‘Gut-microbiota-targeted diets modulate human immune status’, Cell, 184(16), pp. 4137–4153.e14. doi: 10.1016/j.cell.2021.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MM, Reedy J and Krebs-Smith SM (2016) ‘American Diet Quality: Where It Is, Where It Is Heading, and What It Could Be’, Journal of the Academy of Nutrition and Dietetics, 116(2), pp. 302–310.e1. doi: 10.1016/j.jand.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS (2014) FooDB: the food database. Available at: http://foodb.ca/ (Accessed: 17 September 2018).
- Wu GD et al. (2011) ‘Linking long-term dietary patterns with gut microbial enterotypes’, Science, 334(6052), pp. 105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD et al. (2016) ‘Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production’, Gut, 65(1), pp. 63–72. doi: 10.1136/gutjnl-2014-308209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X et al. (2021) ‘The gut microbial metabolite phenylacetylglycine protects against cardiac injury caused by ischemia/reperfusion through activating β2AR’, Archives of Biochemistry and Biophysics, 697. doi: 10.1016/j.abb.2020.108720. [DOI] [PubMed] [Google Scholar]
- Yang K et al. (2017) ‘Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice.’, Blood, 129(19), pp. 2667–2679. doi: 10.1182/blood-2016-10-744060. [DOI] [PubMed] [Google Scholar]
- Zeevi D et al. (2015) ‘Personalized Nutrition by Prediction of Glycemic Responses’, Cell, 163(5), pp. 1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Zeevi D et al. (2019) ‘Structural variation in the gut microbiome associates with host health’, Nature, 568(7750), pp. 43–48. doi: 10.1038/s41586-019-1065-y. [DOI] [PubMed] [Google Scholar]
- Zhu A et al. (2015) ‘Inter-individual differences in the gene content of human gut bacterial species’, Genome Biology, 16(1), p. 82. doi: 10.1186/s13059-015-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmora N et al. (2016) ‘Taking it Personally: Personalized Utilization of the Human Microbiome in Health and Disease.’, Cell host & microbe, 19(1), pp. 12–20. doi: 10.1016/j.chom.2015.12.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data has been deposition to the NCBI Sequence Read Archive under project PRJNA776530 and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table.
Key resources table
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.