

Multisector analysis of malignant brain tumors highlights substantial differences in immunogenomic landscapes, with gliomas harboring greater spatial heterogeneity at a genomic, neoantigen, and T-cell repertoire level.
Abstract
Despite some success in secondary brain metastases, targeted or immune-based therapies have shown limited efficacy against primary brain malignancies such as glioblastoma (GBM). Although the intratumoral heterogeneity of GBM is implicated in treatment resistance, it remains unclear whether this diversity is observed within brain metastases and to what extent cancer cell–intrinsic heterogeneity sculpts the local immune microenvironment. Here, we profiled the immunogenomic state of 93 spatially distinct regions from 30 malignant brain tumors through whole-exome, RNA, and T-cell receptor sequencing. Our analyses identified differences between primary and secondary malignancies, with gliomas displaying more spatial heterogeneity at the genomic and neoantigen levels. In addition, this spatial diversity was recapitulated in the distribution of T-cell clones in which some gliomas harbored highly expanded but spatially restricted clonotypes. This study defines the immunogenomic landscape across a cohort of malignant brain tumors and contains implications for the design of targeted and immune-based therapies against intracranial malignancies.
Significance:
This study describes the impact of spatial heterogeneity on genomic and immunologic characteristics of gliomas and brain metastases. The results suggest that gliomas harbor significantly greater intratumoral heterogeneity of genomic alterations, neoantigens, and T-cell clones than brain metastases, indicating the importance of multisector analysis for clinical or translational studies.
This article is highlighted in the In This Issue feature, p. 1
Introduction
Malignant brain tumors consist of both primary tumors arising from within the central nervous system (CNS) and secondary metastases originating from extracranial sites. The most common malignant primary tumor of the CNS is glioblastoma (GBM), whereas secondary tumors typically develop from carcinomas of the lung, breast, or kidney or from melanoma (1, 2). Although historically both primary and metastatic malignancies carried poor prognoses, the use of checkpoint blockade immunotherapy has led to improved outcomes and robust intratumoral T-cell infiltration in a considerable subset of patients with brain metastases (BrMET; refs. 3, 4). However, these treatments and other immunotherapeutic approaches have not been effective in GBM. Indeed, anti–PD-1 monotherapy did not improve survival in patients with newly diagnosed or recurrent disease, and targeted vaccines and cell therapy approaches also have shown limited efficacy (5–9). Thus far, immunotherapy responses within GBM have been restricted to a selective group of patients with a hypermutated phenotype caused by germline deficiencies in DNA replication or repair (10, 11).
One cardinal feature of GBM that may be a particularly important contributor to therapy resistance is its extensive intratumoral molecular and cellular heterogeneity (12). Indeed, intratumoral genetic heterogeneity is found in many cancer types to varying degrees (13–16). The presence of a complex tumor subclonal genomic architecture likely plays a pivotal role in limiting the efficacy of both targeted therapies as well as immunotherapies. Specifically, studies in non–small cell lung cancer (NSCLC) and melanoma showed a strong association between checkpoint blockade immunotherapy response and the frequency of clonal nonsynonymous mutations, which likely serve as sources of spatially distributed neoantigen targets (17, 18). In GBM, extensive work has demonstrated that intratumoral heterogeneity of a range of tumor somatic changes, including mutations, copy-number alterations (CNA), and transcriptional signatures across spatially distinct tumor regions, is also a hallmark of this disease (19–21). In contrast, we have a more limited understanding of the extent of intratumoral heterogeneity in other intracranial malignancies such as BrMETs, as few corresponding analyses have been performed in these cancers (22). Moreover, further work is needed to understand the relationship between tumor genetic heterogeneity and other important features of the tumor ecosystem, including the immune microenvironment. Although numerous recent studies in other solid tumors, including NSCLC and ovarian cancer, have examined the relationship between tumor cell–intrinsic properties and immunologic parameters such as the T-cell receptor (TCR) repertoire (23–26), this analysis has not been extended to either primary or metastatic malignant brain tumors, in which the extent of heterogeneity in the immune microenvironment and its interplay with genomic and transcriptional diversity is unknown. In the setting of GBM, it is also unclear how the tumor genomic and immunologic landscapes evolve in the hypermutated state seen in close to 20% of recurrent disease (27).
To address these questions, we performed systematic and comprehensive multisector immunogenomic analyses on 93 samples from a cohort of 30 patients with primary or recurrent gliomas or metastatic brain tumors, representing the largest cohort of these brain cancers studied spatially to date. For each patient, we characterized multiple spatially distinct regions using whole-exome sequencing (WES), custom capture validation, RNA sequencing, and TCR sequencing. Our findings underscore the significant differences in clonal architecture between gliomas and metastatic brain tumors, which translates into distinct neoantigen landscapes and, in turn, tumor-infiltrating T-cell clonotypic diversity. These data therefore provide high-resolution insights into the immunogenomic landscapes within malignant brain tumors, which may inform tumor-specific therapeutic approaches.
Results
Genomic Features of Glioma and BrMET Cohorts
We obtained surgically resected tumor tissue and matched peripheral blood from a group of 30 patients with pathologically confirmed intracranial tumors (Supplementary Fig. S1A and S1B). Within this cohort, 14 tumors were primary GBM, 4 were recurrent GBM, 1 was an anaplastic oligodendroglioma, and 11 were BrMETs from lung, breast, or cutaneous malignancies. In total, 21 of these patients (15 primary gliomas and 6 BrMETs) were newly diagnosed and had received no prior therapy. The 4 patients with recurrent GBM had undergone standard-of-care chemoradiation therapy, whereas 5 of the patients with BrMET had received varying treatment regimens for their primary tumor (Supplementary Fig. S1B). Importantly, no patients had prior immunotherapy treatment, but all patients across the cohort were given preoperative steroids. Immediately following surgical resection via craniotomy, each sample was dissociated into multiple (2–4) spatially distinct tumor regions that each underwent comprehensive genomic and immunologic profiling including DNA WES, RNA sequencing, neoantigen prediction, and TCR sequencing (Fig. 1A). Using WES at an average coverage depth of 156×, we identified a total of 11,923 somatic variants (both single-nucleotide variants and indels) across the tumor cohort. Because of the subclonal nature of many variant calls derived from tumors with significant intratumoral heterogeneity, we developed a customized, targeted validation sequencing assay with a set of probes targeting all initially identified variants in addition to a select group of noncoding sites (e.g., TERT promoter mutation sites) to resequence all initially detected variants to confirm their presence. Using this approach, 87.6% of the cohort was characterized at a depth of at least 250× at >90% of positions captured by the custom reagent. This resulted in confirmation of 92% (10,254/11,181) of the original variants through validation sequencing after removal of those that could not be targeted (488 variants) or lacked sufficient minimum coverage (244 variants; Supplementary Table S1). These data allowed us to obtain high-precision estimates of the variant allele frequency (VAF) of each variant and provided greater confidence that variants were not missed owing to potential regional variability in neoplastic content or sequencing depth.
Figure 1.

Genomic landscape of brain tumor cohort. A, Overview of sample collection, sequencing, and data analysis. B, Variant counts per tumor pooled across samples. C, Summary of top 25 recurrently mutated genes (3 or more tumors) in 11 brain metastases. D, Summary of recurrently mutated genes (3 or more tumors) in 19 primary and recurrent gliomas. E, Cohort-level copy number variation in gliomas determined by the GISTIC algorithm. Dashed lines indicate significantly recurrent amplifications (red) and deletions (blue) at an FDR <0.1. F, NSCLC brain metastasis cohort GISTIC output. G, Breast cancer brain metastasis cohort GISTIC output. SCLC, small cell lung cancer.
As expected, the median number of aggregate somatic variants per tumor was higher in BrMETs (504 variants) than in either primary (93 variants) or recurrent glioma (141 variants; Fig. 1B). Within the glioma cohort, GBM065.Re represented an outlier with 5,750 somatic mutations identified across four distinct tumor regions. Most of the variants within this sample displayed the characteristic mutational signature associated with prior temozolomide treatment (Supplementary Fig. S2A and S2B), suggesting a treatment-induced hypermutated phenotype (28, 29). Within the BrMET specimens, recurrent mutations were identified in TP53 across all histologies (7/11 overall; 3/5 NSCLC, 2/4 breast carcinoma; Fig. 1C). KRAS alterations were identified within two of five NSCLC tumors, and PIK3CA mutations were present in two of four breast carcinomas consistent with their high incidence in studies of primary samples (30, 31). In addition, TERT promoter mutations were observed in both a melanoma and an NSCLC BrMET tumor. Across the GBM samples, we identified recurrent mutations in canonical GBM-associated genes such as the TERT promoter (14/18), TP53 (7/18), PTEN (4/18), NF1 (3/18), and EGFR (3/18; Fig. 1D). This high frequency of TERT promoter mutations mirrors earlier work estimating frequencies upward of 70% to 80% among GBM (32, 33). In addition, the frequency of TP53 alterations is consistent with prior studies observing mutations in 30% to 40% of GBM samples, whereas the frequency of EGFR mutations is slightly lower (21, 33). We next assessed the prevalence of CNAs within our cohort and identified classical GBM-associated changes such as frequent chromosome 7 amplification encompassing EGFR (17/18), chromosome 9 deletions of the CDKN2A locus (15/18), and chromosome 10 deletions of the PTEN locus (17/18; Fig. 1E; Supplementary Table S2). These amplifications and deletions have been detected at comparably high frequencies across several other studies (21, 33). The metastatic tumors displayed alterations similar to those previously defined for their corresponding primary tumor such as amplifications of KRAS (3/5) in NSCLC and ERBB2 (2/4) in breast carcinomas (Fig. 1F and G).
Intratumoral Genomic and Transcriptional Heterogeneity of Gliomas and BrMETs
Having characterized all variants in our glioma and BrMET cohorts, we next sought to characterize the extent of intratumoral genomic heterogeneity within all tumors. First, we categorized all variants in each sample as “clonal” (present in all sequenced regions of a tumor), “subclonal shared” (present in more than one region of a tumor but not all), or “subclonal private” (present in only one region of a tumor). We observed a striking difference between tumor types with a significantly (P < 0.001; unpaired t test) greater clonal variant fraction within the BrMETs (median = 0.88) than within gliomas (median = 0.41; Fig. 2A and B). Importantly, this observation was independent of the histology of the primary malignancy, as lung, breast, and melanoma metastases all harbored a high fraction of clonal variants. In contrast, gliomas (primary and recurrent samples pooled) contained a higher fraction of both subclonal shared (P < 0.05; unpaired t test) and subclonal private (P < 0.05; unpaired t test) variants than did the BrMETs (median subclonal private variant fraction 0.28 vs. 0.09; Fig. 2B). Overall, approximately 43% of mutations within the glioma cohort were categorized as clonal. Although this is slightly lower than two previous studies have reported, almost all of those tumors had only two spatially separate regions analyzed, in contrast to the three or four sectors sequenced in our study (21, 34). Focusing specifically on cancer driver genes, mutations in TP53, PIK3CA, and KRAS were clonal in all cases among BrMET samples. In contrast to the generalized heterogeneity of gliomas, most TERT promoter (13/14 clonal), TP53 (6/7), and EGFR (3/3) mutations were clonally distributed. However, most PTEN (1/4 clonal) and NF1 (0/3) alterations were not clonal within gliomas. These findings are largely consistent with a prior multisector sequencing study that noted significant clonality of TERT promoter and TP53 mutations (21). However, this same group did note that most identified EGFR mutations were subclonal private in contrast to our cohort (21).
Figure 2.

Intratumoral genomic heterogeneity of variants and CNAs. A, Variant clonality per tumor. B, Proportion of clonal, subclonal shared, and subclonal private variants in brain metastases and gliomas. Significance determined by unpaired t test. *, P < 0.05; ***, P < 0.001. C, Proportion of total identified variants that would have been captured through the sequencing of a random single site from within each tumor. Significance determined by unpaired t test. *, P < 0.05; **, P < 0.01. D, Total variants identified per tumor if one, two, or three samples were pooled for analysis. Significance determined by unpaired t test. *, P < 0.05. NS, not significant. E, CNV clonality per tumor in the gliomas (left), NSCLC brain metastases (middle), and breast cancer brain metastases (right) cohorts.
To explore the translational implications of this heterogeneity, we calculated the fraction of total tumor variants that would have been identified from sequencing a single glioma or BrMET tumor site, which simulates the information that would be obtained from a single-site biopsy at surgery. We observed that a higher fraction (P < 0.01; unpaired t test) of the total variants was identified within BrMETs (median = 0.92) compared with primary gliomas (median = 0.73) when sampling a single site (Fig. 2C). Given the limitations of single-site sampling to capture tumor-wide variant information in gliomas, we determined the extent to which multiregion sequencing could lead to the identification of additional tumor variants within our cohort. Among the BrMETs, sampling additional tumor regions did not identify significantly more variants. However, among glioma samples, sequencing three regions instead of one raised the median number of identified variants from 61 to 98 (Fig. 2D). Thus, multiregion sequencing in gliomas captures a more complete picture of the genomic landscape but provides only limited improvement in the characterization of BrMETs due to their increased comparative spatial genomic homogeneity.
Having characterized glioma and BrMET genomic architecture at the variant level, we next sought to characterize the intratumoral heterogeneity of CNAs to determine whether there is evidence for spatial architecture that is similar to that of the variants. Strikingly, the landscape of CNAs within gliomas was markedly more spatially heterogeneous than the pattern observed within BrMETs (Supplementary Fig. S3). When we classified each CNA event as clonal, subclonal shared, or subclonal private, we identified a significant fraction of clonal CNAs within BrMETs in contrast to the predominantly subclonal CNAs within gliomas (Fig. 2E). Thus, at both the variant and CNA levels, gliomas are significantly more spatially heterogeneous than BrMETs.
Finally, to characterize these samples at the transcriptional level, we made use of a previously defined gene expression–based molecular classification of GBM into proneural, neural, classical, and mesenchymal subtypes (35). We identified a distribution of transcriptional subtypes with 12 neural, 12 classical, 16 mesenchymal, and 6 proneural subtypes across the 46 primary or recurrent GBM tumor sectors. Intriguingly, in a majority (9/16) of the tumors with multiple regions analyzed, we observed intratumoral heterogeneity of these transcriptional subgroups (Supplementary Fig. S4), in line with prior reports (20).
Intratumoral Heterogeneity of Tumor Antigens in Gliomas and BrMETs
Numerous clinical trials developing personalized neoantigen vaccines for GBM and other brain cancers are ongoing (8, 9). To determine the consequences of tumor genetic heterogeneity on immunologic features of each tumor, we applied the pVacSeq neoantigen prediction pipeline (36, 37) to define the neoantigen landscape across all glioma and BrMET samples. When we aggregated the total number of predicted neoantigens for each tumor from all sampled regions, BrMETs harbored a higher number of HLA class I neoantigens per tumor (median = 186) than either primary (median = 39) or recurrent gliomas (median = 51; Supplementary Table S3). Moreover, BrMET class I neoantigens were significantly (P < 0.001; unpaired t test) more clonal than those identified in gliomas (Fig. 3A and B). Of note, all EGFR mutations did yield predicted clonal class I neoantigens. However, despite all carrying the IDH1 R132H mutation, only one of the four IDH-mutant patients within the cohort had a predicted class I neoantigen from this variant, displaying the HLA haplotype dependence of these results. BrMETs also exhibited a greater number of HLA class II neoantigens (300 antigens) than primary (60 antigens) or recurrent (81 antigens) gliomas, and these neoantigens were significantly (P < 0.001; unpaired t test) more clonal in their tumor distribution (Supplementary Fig. S5A and S5B; Supplementary Table S4). For most patients, the spatial distribution of both class I and class II neoantigens closely mirrored the underlying variant distribution (Supplementary Fig. S6A–S6C). Finally, whereas BrMET HLA class I neoantigens are mostly captured by single-site tissue sampling, additional glioma neoantigens continue to be identified as additional tissue sites are sampled (Fig. 3C). Taken together, these data demonstrate that neoantigens are distributed heterogeneously in gliomas compared with BrMETs.
Figure 3.

Intratumoral neoantigen and cancer/testis antigen heterogeneity. A, Class I neoantigen clonality per tumor. B, Proportion of clonal, subclonal shared, and subclonal class I neoantigens in brain metastases and gliomas. Significance determined by unpaired t test. **, P < 0.01; ***, P < 0.001. C, Impact of multiregion sequencing on total class I neoantigen load. Significance determined by unpaired t test. **, P < 0.01. D, Heat map of CT antigen scores for each sample calculated by normalizing tumor expression to normal “Brain-Cortex” expression (see Methods). E, Plot of the average intratumoral variation in CT scores between regions of the same tumor by the average cancer/testis antigen score for each gene among all brain metastases or gliomas.
In addition to neoantigens, cancer/testis (CT) antigens represent another group of tumor-specific antigens that can be recognized by the immune system. These antigens have highly restricted expression in normal tissue, can be expressed in reproductive cells, and are often upregulated in malignancies. Although CT antigens are targeted in a range of clinical trial efforts (38–41), their expression and distribution in brain cancers has not been described previously. We therefore sought to characterize CT antigen expression and spatial distribution within our tumor cohort. Because CT antigens are wild-type proteins, normal tissue expression affects both the degree of anticipated off-target effects from directed immunotherapeutic efforts as well as the extent of immunologic tolerance during development. Thus, we scored each candidate antigen from a curated list of CT antigens based on its log-transformed expression relative to normal brain tissue. Among gliomas, some of the highest scoring CT antigens were BIRC5 (Survivin), PMEL (gp100), and IL13RA2 (Fig. 3D). Although BIRC5 was also the highest-scoring antigen among the BrMET samples, it displayed relatively higher expression of many prominent CT antigens such as the MAGE family proteins, gp100, MART1, and HER2/neu compared with gliomas. One notable exception to this was IL13RA2, which was the lowest-scoring antigen among BrMETs but ranked third among gliomas. These findings were consistent regardless of whether brain or matched primary site tissue was used to generate BrMET CT antigen scores (Supplementary Fig. S7A). In contrast to the marked heterogeneity of tumor neoantigens, we observed no significant difference in the spatial distribution of CT antigen scores overall between gliomas and BrMETs as assessed by a cosine similarity index (Supplementary Fig. S7B). However, these characteristics do vary between CT antigens, with TERT, CTAG1B (NY-ESO-1), and the MAGE family trending to higher intratumoral variance (greater heterogeneity of expression) than the more clonal BIRC5 and gp100 (Fig. 3E).
Spatial Resolution of Immune Landscapes in Gliomas and BrMETs
We next sought to define the immune cell infiltration within each tumor and to describe the extent of intratumoral heterogeneity within the local immune microenvironments. We adopted previously published immune deconvolution methods that resolve immune cell populations from transcriptional data such as Danaher immune scores (42) and CIBERSORT (43) to characterize the 80 tumor regions for which there was sufficient RNA to generate RNA-sequencing data (Supplementary Table S5). We calculated Danaher immune scores for all regions and observed substantial intertumoral variation specifically among “CD8 T-cell” and “cytotoxic cell” scores (Fig. 4A). Surprisingly, we detected no significant difference in the aggregate immune scores between regions from glioma and BrMET samples.
Figure 4.

Spatial diversity of the immune microenvironment. A, Heat map of the immune cell scores for all samples as estimated by the method from Danaher and colleagues (42). Each column represents a tumor region and each row represents an immune population. Scores represent the average of the log-transformed expression of a collection of subset-specific genes. B, Difference in log-transformed expression of PD-L1 (left) and CXCL9 (right) between tumor types for all samples within cohort. Significance determined by t test with Benjamini–Hochberg multiple test correction. **, q < 0.01. C, Difference in macrophage polarization (left) or ontogeny (right) based on previously published gene sets (see Methods) between tumor types for all samples within the cohort. Significance determined by two-sided t test. *, P < 0.05; **, P < 0.01. D, Danaher scores for each sector from a tumor with three or more samples plotted in PC1–PC2 space following principal components analysis. DC, dendritic cell; NK, natural killer; Treg, regulatory T cell.
To probe potential differences in more detail, we next performed differential gene expression analysis on tumor regions from gliomas and BrMETs. We detected significantly higher levels of CD274 (PD-L1) and significantly lower levels of CXCL9 in gliomas compared with BrMETs (q < 0.01; t test with multiple comparison correction), consistent with the severe immunosuppression appreciated in the former (ref. 44; Fig. 4B). In addition, because there was a robust infiltration of macrophages in both gliomas and BrMETs as determined by CIBERSORT (Supplementary Fig. S8A), we explored two previously published gene sets (45, 46) to further characterize this population. Although we observed a slight polarization toward the immunosuppressive M2 phenotype characterized by higher expression of STAT3 and MRC1 (CD206) within gliomas, we identified a major distinction within macrophage ontogeny. Specifically, the BrMETs had a significant skewing toward higher expression of genes associated with monocyte-derived macrophages relative to gliomas, which were enriched for microglial-specific genes (Fig. 4C). This is consistent with recent work in the field using single-cell analyses (47, 48).
Finally, we assessed the degree of immunologic intratumoral heterogeneity as estimated by the Danaher scores. We performed principal components analysis (PCA) on the Danaher immune scores for all tumors with RNA from at least three sectors. Plotting each region in PCA space, we observed that most regions from the same tumor clustered together (Fig. 4D). To quantify the extent of heterogeneity, we calculated the area in PC1–PC2 space (termed Danaher intratumoral heterogeneity) of the triangle with vertices corresponding to each region of a tumor. In contrast to the substantial differences in variant and neoantigen heterogeneity, we detected no difference in the degree of intratumoral immune cell heterogeneity between the tumor types (Supplementary Fig. S8B). A similar result was obtained by calculating pairwise cosine similarity for Danaher immune scores between regions from the same tumor (Supplementary Fig. S8B). Overall, these data suggest that the intratumoral spatial variation in the immune microenvironment is similar between gliomas and BrMETs and generally is less than the intertumoral variation.
TCR Clonotypic Diversity and Heterogeneity
To evaluate the diversity, heterogeneity, and degree of clonal expansion of TCR clonotypes within the infiltrating T-cell populations of GBM and BrMET tumor samples, we performed TCR sequencing on 65 regions from 22 tumors in the cohort. The TCR β-chain complementarity-determining region 3 (CDR3) is highly diverse and plays a significant role in antigen recognition. Therefore, the TCR β-chain CDR3 sequences can function as unique barcodes of individual T-cell clones as they are activated and undergo clonal expansion within the tumor. We classified clonotypes within each tumor region as either the dominant clone (clone 1) or in predetermined clonotype groups based on frequency (i.e., clones 2–5, 6–20, 21–100, 101–1,000, or more than 1,000 clones; Fig. 5A; Supplementary Fig. S9A). Unexpectedly, we observed substantial clonal expansion within GBM. For example, each region of GBM055 harbored a dominant clone comprising >17% of the T-cell repertoire. In addition, regions from GBM047.Re, GBM056, GBM059, GBM065.Re, and GBM079 all contained dominant clones present at frequencies >12%. In contrast, of the sequenced BrMETs, only one region from BrMET025 (NSCLC) harbored a dominant T-cell clone present at a frequency >10%. Overall, the T-cell fraction among all cells (estimated through TCR sequencing; see Methods) was significantly (P < 0.05; unpaired t test) higher within the BrMETs, whereas the TCR repertoires within GBM were determined to have a higher degree of clonality (P < 0.05; unpaired t test; Fig. 5B).
Figure 5.

Intratumoral T-cell repertoire clonality and diversity. A, The proportion of each sample's T-cell repertoire consisting of clones from a given rank position when all TCR sequences are ordered by descending frequency in the sample. B, Comparison of T-cell fraction (left) and Simpson clonality (right) between regions from GBMs or BrMETs. T-cell fraction and Simpson clonality were calculated as described in the Methods. P values calculated by two-sided t test: *, P < 0.05. C, Heat maps showing the frequencies of the 10 most expanded intratumoral β-chain sequences per tumor in different regions. Each row represents one unique sequence and each column a tumor region. D, Quantification of TCR intratumoral heterogeneity by calculation of the Morisita overlap (see Methods) between pairs of distinct tumor regions in either GBM (left) or BrMETs (right). Values range from 0 (indicating no similarity) to 1 (identical TCR repertoires).
To investigate the degree of intratumoral heterogeneity of the T-cell repertoires, we next explored the distribution of the top 10 clones within each tumor. The distribution of the expanded TCRs within each tumor differed greatly between patients, with some exhibiting marked T-cell homogeneity among all examined regions, whereas others showed profound intratumoral diversity (Fig. 5C). Several of the GBMs were particularly heterogeneous, with locally expanded T-cell clones present at frequencies greater than 10% within one region of the tumor but significantly less than 1% in all other regions. We then quantified this diversity at a repertoire-wide level through pairwise comparisons using the Morisita overlap index (MOI), a measure of similarity between populations based on the number of shared sequences and their relative frequencies. As expected, the MOI values approached 0 (no similarity) for repertoires from different patients but were variable within patients (Fig. 5D). Comparing the tumor types, we detected a higher level of intratumoral repertoire similarity among BrMETs than among GBMs whether the MOI or a similar cosine similarity index was used (Supplementary Fig. S9B and S9C). Therefore, the increased spatial heterogeneity of variants and neoantigens within GBM is recapitulated at the TCR repertoire level.
Ultimately, to further validate that the expanded clones were specifically enriched within tumor tissue, we performed TCR sequencing on the peripheral blood from eight of these patients (five with GBM/three with BrMET). As expected, the T-cell repertoire from peripheral blood mononuclear cells (PBMC) trended toward lower clonality than the matched tumor-infiltrating lymphocytes (TIL; Supplementary Fig. S10A). Intriguingly, the degree of repertoire similarity between PBMCs and TILs appeared greater (P = 0.07) within BrMETs than within GBMs, perhaps suggestive of a stronger systemic immune response in patients with metastatic disease (Supplementary Fig. S10B). Tracking the most highly expanded intratumoral clones across each patient confirmed that most were detectable in peripheral blood but at substantially reduced frequencies (Supplementary Fig. S10C). In addition to this clonotypic diversity, we also observe differential V and J gene usage between peripheral blood and matched TILs, suggestive of a combination of both VJ-dependent and VJ-independent divergence as previously described (Supplementary Fig. S11; ref. 49).
Immunogenomics of a Patient with Hypermutated Recurrent GBM
Given the growing literature studying the genomics of hypermutated GBM and the ongoing assessment of whether tumors exhibiting this genotype may be more responsive to immunotherapeutic approaches (10, 11, 27), we characterized the immunogenomic landscape of a tumor with this phenotype. GBM065.Re presented with tumor progression of an IDH1-mutant anaplastic astrocytoma 5 years after initial resection. In the interim, the patient was treated with four cycles of vincristine, 1-(2-chloroethyl)-3-cyclohexyl-l-nitrosourea (CCNU), and procarbazine; proton radiotherapy; and six cycles of high-dose temozolomide (Fig. 6A). Using DNA WES of four tumor regions, we observed a substantial increase in the mutational burden across all regions of the recurrent tumor (mean 1,587 variants/sector) relative to the prior primary tumor (219 variants; Fig. 6B). Variant analysis revealed that two regions from the recurrent tumor contained the MSH6 T1219I variant previously identified in Lynch syndrome and known to act in a dominant-negative manner (refs. 50, 51; Supplementary Fig. S12A). The other two regions both contained unique MSH6 mutations not previously reported (G1148S and G1116D) but computationally predicted as “likely to impair molecular function” by a previously published tool for the prediction of MSH6 variant significance (52). Additional mutations in DNA mismatch repair genes such as MLH3 and POLD3 were detected in some but not all regions of the recurrent tumor. This heterogeneity of potentially pathogenic mismatch repair defects in hypermutated recurrent GBM was observed in one prior patient and suggests either the emergence of multiple unique routes to hypermutation occurring within the same tumor or a common alternative mechanism unrelated to these genes (34). An analysis of the mutational signatures within the recurrent tumor revealed a significant enrichment for signature 11, known to be associated with prior treatment with temozolomide (28, 29) and previously reported to be enriched in hypermutated recurrent gliomas (33).
Figure 6.

Immunogenomic profile of hypermutated recurrent GBM. A, Description of clinical course. RT, radiation therapy; PCV, procarbazine, CCNU, and vincristine; TMZ, temozolomide. B, Total variants identified from WES for primary tumor and each analyzed region of recurrence. C, UpSet plot of the distribution of all identified variants grouped by the set of tumor samples in which a variant is shared. D, Alluvial plot displaying the frequency of the five most expanded intratumoral TCR β-chain sequences from either region of recurrent tumor across peripheral blood and tumor samples. E, UMAP dimensionality reduction of the scRNA-seq data of T cells from GBM065.Re. The dashed outline delineates the general population each cell belongs to and the color indicates the degree of expansion for that T-cell clone (defined by α/β pair). F, Volcano plot of differentially expressed genes within hyperexpanded clones relative to rest of T cells.
We observed that most variants identified within each region of the recurrent tumor were subclonal private and not spatially distributed (Fig. 6C; Supplementary Fig. S12B). A small fraction (30 total) of variants were shared between all regions of the recurrent tumor, and an even smaller number (15 total) were shared by the primary and all sectors of the recurrence. Importantly, these included likely drivers of the tumor such as TP53 and IDH1 alterations. This remarkable heterogeneity is consistent with a prior report in which two regions of a hypermutated recurrent tumor were sequenced and shared less than 2% of all identified mutations (34). In contrast to the variant and neoantigen heterogeneity within this tumor, the TCR Vβ repertoires within two analyzed regions were more similar (MOI = 0.85) and had the highest clonality across all samples in the cohort. Remarkably, the top three T-cell clonotypes within GBM065.Re made up more than 37% of the intratumoral repertoire, suggesting a significant degree of clonal expansion. In addition, these dominant clones were all present at substantially reduced frequencies (<1%) within the patient's peripheral blood, confirming the specific intratumoral expansion of these cells (Fig. 6D).
Finally, to more deeply characterize the immunologic landscape of GBM065.Re, we performed single-cell RNA sequencing (scRNA-seq) with TCR enrichment on sorted CD45+ immune cells (n = 1,728) isolated from this tumor immediately following surgical resection. We found that most of the cells were of the lymphoid lineage and were predominantly cytotoxic CD8+ T cells (59% of sequenced cells) with smaller populations of naïve and CD4+ regulatory-like T cells (Fig. 6E; Supplementary Fig. S13). The highly expanded T-cell clones identified through bulk TCR Vβ sequencing also were represented in the scRNA-seq data but were found dispersed throughout the CD8+ T-cell clusters, suggesting heterogeneous gene expression patterns among these clonal populations. We performed differential expression analysis on the hyperexpanded clonotypes and found them enriched for markers of activation such as KLRC3, KLRC4, and GZMK together with MHC class II genes classically upregulated by activated T cells (Fig. 6F). Thus, despite substantial heterogeneity at the variant and neoantigen level, the hypermutated GBM065.Re contains clonally expanded T cells with an activated phenotype distributed throughout the tumor.
Discussion
Despite numerous advances in both targeted and immune-based therapies, the prognosis for patients with malignant brain tumors such as GBM or BrMETs remains poor. Within GBM, one potential rationale for the high rate of treatment failure is the extensive intratumoral cellular and molecular heterogeneity (12) owing to complex tumor clonal dynamics. However, the impact of this tumor cell diversity upon the tumor–immune microenvironment remains unclear. Furthermore, the spatial heterogeneity of both tumor and immune landscapes within BrMETs requires further study.
To address these knowledge gaps, we performed comprehensive immunogenomic profiling on multiple spatially distinct regions from a cohort of 30 patients with primary or secondary malignant brain tumors. We observed a striking distinction in the distribution of somatic variants, with most mutations usually shared by all analyzed regions within BrMETs, whereas gliomas were markedly heterogeneous and subclonal. This dichotomy extended to the distribution of candidate neoantigens within these tumors, whereas the intratumoral distribution of targetable CT antigens was more homogeneous. Furthermore, the intratumoral TCR repertoire was significantly more similar between spatially distinct regions of BrMETs than gliomas, which often harbored locally expanded T-cell clones.
Previous studies have reported on the significant genomic and transcriptional variability between spatially distinct regions of gliomas (19–21) and hypothesized the consequences of heterogeneity on treatment resistance. However, our results suggest that this spatial heterogeneity of genomic alterations is minimal within metastatic brain tumors, which instead display a markedly more clonal distribution. We envisage that this difference may be due to the separate evolutionary trajectories of these tumor types. Recent studies suggest that GBMs arise from the slow accumulation of somatic mutations in neural stem cells, during which time multiple subclones can develop before presenting as a clinically apparent tumor (53). However, secondary BrMETs likely develop quickly from the rapid growth of an already transformed subclone upon arrival into the CNS. This malignant clone will quickly develop into an apparent tumor, allowing less time for the development of genetically disparate subclones. Previous studies have shown that these metastatic clones can accumulate additional genomic alterations relative to the matched primary, thereby providing potentially unique therapeutic targets that are likely clonal within the metastatic tumor (22, 54). Future work should explore whether this tumor spatial homogeneity represents a specific feature of BrMETs or is a more general characteristic of secondary metastatic tumors.
Importantly, the comparative distinctions we observed extend to the distribution of candidate class I and class II neoantigens, as patients with metastatic tumors harbor a significantly higher proportion of clonal neoantigens. This dichotomy between BrMETs and gliomas could have profound implications on antitumor immunity, as studies have reported that T-cell immunoreactivity against clonal neoantigens drives sensitivity to checkpoint blockade treatment (17, 18). However, additional work is needed to characterize the degree of antitumor immunity within malignant brain tumors and determine the relative contributions of clonal and subclonal neoantigens in stimulating immune responses. In particular, detailed analysis of the antigenic targets of the T-cell clones within tumors will be critical to understanding how antigen clonality shapes immunogenicity.
Recent studies have broadened our understanding of the immune microenvironment within gliomas and BrMETs through methods such as mass cytometry, RNA sequencing, and immunofluorescence (47, 48). Our work builds on this by exploring the spatial heterogeneity of the immune response and performing a deeper analysis on the TIL populations. Through immune profiling from RNA-sequencing data, we did not detect significant intratumoral differences in either the immune infiltrate or activation state for either tumor type, in contrast to the significant heterogeneity of variants and neoantigens within gliomas. However, many of the immune profiling analyses performed are to some extent limited in their ability to resolve specific immune cell populations and activation states owing to their reliance on bulk RNA-sequencing data. Further spatial analysis with more sensitive metrics such as flow cytometry, immunofluorescence, scRNA-seq, and/or spatial transcriptomics would be needed to clarify whether this is true immune homogeneity or a limitation of bulk RNA-sequencing analysis.
In contrast, TCR repertoire sequencing did uncover substantial spatial heterogeneity. Although both GBMs and BrMETs demonstrated evidence of expanded intratumoral T cells, many of the dominant clones within GBM samples were highly spatially restricted. These data suggest that tumors harbor complex immune microenvironments in which a given tumor may contain pockets of clonally expanded T cells adjacent to regions with minimally expanded T cells. Whether this T-cell heterogeneity is due to the recognition of spatially diverse subclonal antigens or the result of extrinsic regional features such as inflammatory cytokines that promote clonal expansion requires further study.
Tumor mutational burden has been shown to correlate with checkpoint blockade response in some studies (18, 55), and hypermutated tumors resulting from germline or acquired DNA repair deficiencies have been shown to be uniquely responsive to immunotherapy across other tumor types (56). Within hypermutated GBM, which occurs in up to 20% of recurrent disease, the efficacy of checkpoint blockade and other immunotherapies remains an open question. Several reports have observed clinical responses from checkpoint blockade in patients with hypermutant GBM harboring germline DNA repair defects, but a recent retrospective analysis found no benefit in patients with mismatch repair–deficient GBM treated with the PD-1 blockade (10, 11, 27). Ongoing clinical trials are designed to address this question, such as the Alliance study A071702/NCT04145115 [a study testing the effect of immunotherapy (ipilimumab and nivolumab) in patients with recurrent glioblastoma with elevated mutational burden]. Although characterization of more patients is needed to generalize our findings, the immunogenomic profiling of GBM065.Re revealed potentially unique features of these tumors. First, most mutations and associated neoantigens within this hypermutant tumor were subclonal private. These data, together with our other findings and published work (21), indicate that single-site profiling analysis would be inadequate and dramatically underestimate both tumor complexity and neoantigen burden. Second, despite this dramatic heterogeneity of variants, we observed remarkably similar TCR repertoires with highly expanded CD8+ T-cell clones within different regions. scRNA-seq analysis on these expanded clones shows evidence of T-cell activation, suggesting potential tumor reactivity. Whether these clones react to the small subset of clonal neoantigens, the thousands of regional neoantigens, or overexpressed CT antigens or are responding in a nonspecific way to inflammatory stimuli will require further analysis. However, our data indicate that hypermutant tumors can contain highly activated and clonally expanded T cells while also representing an extreme of mutational and neoantigen heterogeneity. Ongoing work is directed at understanding how the immune system may direct responses to antigen targets in the context of this heterogeneity.
Taken together, these results carry immediate significance for the design and implementation of clinical studies in malignant brain tumors. The extensive intratumoral heterogeneity within gliomas indicates that single-site genomic analysis will not capture the totality of targetable mutations and neoantigens. This is of particular importance in the design of targeted therapy studies and/or neoantigen vaccines, as the analysis of multiple regions identifies a significantly higher number of targetable neoantigens than one site alone. We have already begun to implement this approach in a trial of a personalized neoantigen vaccine in patients with newly diagnosed GBM (NCT03422094). An additional benefit of this approach is increased confidence in the clonality of targeted antigens, as clonal neoantigens would presumably represent ideal targets. However, it remains to be seen whether sampling additional regions beyond the number in this study and the aforementioned clinical trial would shed further light on the molecular landscape. Furthermore, the complexity of the intratumoral T-cell repertoire argues that multiple sites should be analyzed for the potential expansion of neoantigen-reactive T cells or isolation of tumor-specific TCRs for therapy. On the other hand, the relative homogeneity of BrMETs suggests that a single region is sufficient for genomic and immunologic phenotyping.
Overall, this study provides an immunogenomic profiling of malignant brain tumors in a multisector approach showcasing substantial intratumoral heterogeneity within gliomas while highlighting surprising homogeneity among BrMETs. These distinctions hold for both cancer cell–intrinsic (genomic alterations) and cancer cell–extrinsic (TCR repertoire) features. A growing understanding of the tumor–immune microenvironment and an appreciation of its spatial complexity may improve the efficacy of immunotherapy in patients with malignant brain tumors.
Methods
Patient Recruitment
All study participants were neurosurgical patients at Barnes-Jewish Hospital with pathologically confirmed stage III/IV glioma or metastatic disease. Prior to surgery, we obtained written informed consent from the patients for a Washington University School of Medicine Institutional Review Board–approved protocol (#201111001) for the analysis of tumor tissue and peripheral blood and sharing of genomic data. All procedures and experiments were performed in accordance with the ethical standards of the 1964 Declaration of Helsinki. Clinical information related to each patient was collected at the time of surgery and is summarized in Supplementary Fig. S1.
Clinical Sample Processing and Nucleic Acid Extraction
Tumor samples were processed immediately following surgical resection under sterile conditions. Tissue was thoroughly washed in PBS to eliminate peripheral blood leukocyte contamination. For tumor samples that were resected en bloc, spatially distinct sectors were dissected out from the mass via scalpel. In other cases where multiple discrete regions demonstrated enhancement on MRI, tissue was separated at the time of surgery and a representative sample from each sector was chosen for analysis. In all cases, tissue was immediately flash-frozen and initially stored at −80°C or in liquid nitrogen until further use. Peripheral blood was separated through Ficoll-Paque PLUS density gradient (GE), and the buffy coat was collected and frozen for matched normal genomic DNA.
Total RNA and genomic DNA was extracted from peripheral blood mononuclear cells or frozen tissue using the Qiagen AllPrep DNA/RNA Kit (catalog no. 80204) according to the manufacturer's instructions (Qiagen). As melanin is a known inhibitor of enzymatic reactions and coprecipitates with RNA (57), further purification was performed for the melanoma sample as described (58), modified with an RNeasy (Qiagen) column-based cleanup to remove the additives used to bind the melanin. For GBM065.Re, regions 3 to 4 and the sample from the primary tumor came from formalin-fixed, paraffin-embedded tissue cores. In these cases, total RNA and genomic DNA were extracted using the Qiagen AllPrep DNA/RNA FFPE Kit (catalog no. 80234).
Sequencing and Somatic Variant Detection
All sequencing was performed on the Illumina NovaSeq (S4) platform. From each patient, a normal sample and two to four samples from a single tumor were subjected to WES. Eighty of 93 tumor samples had sufficient tissue to perform RNA sequencing. WES and RNA sequencing from 20 tumors were performed at the McDonnell Genome Institute, and 10 tumors underwent WES and RNA sequencing at the Institute for Genomic Medicine at Nationwide Children's Hospital. WES was performed on two additional recurrent GBM065 regions and a GBM065 primary tumor by Novogene. Read alignment, somatic variant calling, variant filtering, variant effect prediction, additional variant annotation, and RNA expression estimation were performed using a tumor analysis pipeline defined in the Genome Modeling System as previously described (59). Briefly, all fastq files were aligned to the human reference genome build GRCh38 with HISAT2(RRID:SCR_015530) for RNA (60) and BWA-MEM for DNA (61). Somatic variant calling was performed using Strelka (62), VarScan (63), Mutect (64), and Pindel (65). To remove any false-positive variants and discover TERT promoter mutations, custom capture validation sequencing was performed to an average depth of 582× for all unique variant sites using NimbleGen SeqCap EZ Prime Choice Probes (12,388 total probes created from 11,425 variants and 51 genes; 1.49 Mb of sequence targeted for capture). Tumor purity was estimated using TPES (66) for samples with sufficient variant count (>20), and for those with insufficient variants, the median variant allele frequency was used (2 × median VAF). SciClone (67) and ClonEvol (68) were used to assess the clonality of mutations and subclonal evolution of primary and recurrent GBM065 tumors.
CN Variation Detection
Matched tumor/normal WES data were used to predict CN variations (CNV) with CNVkit (69). Each tumor region sample was compared pairwise against the single matched normal data for the patient. CNVkit was run (via “cnvkit batch”) using default parameters. Regions of alignment were summarized to a resolution of 10 kb, and these data were subjected to segmentation using circular binary segmentation. The resulting segmentation files were divided into three cohorts: (i) primary and recurrent GBMs, (ii) breast cancer BrMETs, and (iii) NSCLC BrMETs. GISTIC2 (70) was run on the segments of each cohort with a q value cutoff of 0.1. Relative amplitude thresholds were used to create the heatmaps and clonality plots in Fig. 2.
GBM Molecular Subtyping
Single-sample gene set enrichment analysis scores (71) were calculated for each sample in the cohort using the previously defined gene sets (35) and the GSVA R package (72). The circlize (RRID:SCR_002141) R package was used for the visualization of these molecular subtypes (73).
Neoantigen Prediction
Clinical class I and class II HLA typing was performed on each normal sample by Histogenetics to two field resolution. For each tumor sample, a VCF file containing all passing somatic variants was annotated with Ensembl VEP (ref. 74; RRID: SCR_002344) using the parameters –everything, –flag_pick, and –plugin (Wildtype and Downstream). The VCF was further annotated with DNA and RNA read counts using VAtools. Using this VCF as input, a containerized version of pVACtools (ref. 37; DockerHub: griffithlab/pvactools:1.5.0) was used to predict and annotate likely neoantigens in each sample as previously described (75). Briefly, using the submodule “pvacseq run,” we performed peptide/MHC binding affinity predictions with eight class I and four class II algorithms (NNalign, NetMHC, NetMHCIIpan, NetMHCcons, NetMHCpan PickPocket, SMM, SMMPMBEC, SMMalign, MHCflurry, MHCnuggetsI, MHCnuggetsII). For this analysis, to be considered a neoantigen candidate, it must have arisen from a variant with tumor DNA VAF >5% and median binding affinity (across all algorithms) <500 nm. No sequences were excluded based on tumor RNA VAF, coverage, or gene expression values to ensure that no high-quality candidates were excluded based solely on RNA data. Neoantigens that pass this filter are designated “candidate neoantigens” in the article.
Genomic Alteration Distribution
All genomic alterations (variants and associated neoantigens, CNVs) were determined independently with each region as an individual sample. They were then aggregated together for the plots in Fig. 1 to provide an overall description of each tumor. We then determined the distribution of each alteration between regions from the same tumor. If the same change was observed in all regions of the tumor, it was defined as “clonal.” If it was identified in more than one but not all regions, it was defined as “subclonal shared.” Alterations observed in only one region of each patient sample were defined as “subclonal private.”
Cancer/Testis Antigen Analysis
A subset of cancer/testis antigens was chosen for analysis based on a combination of prior work targeting them in GBM (ref. 38; e.g., IL13Ra2) and/or previous studies in other malignancies (refs. 39, 41; e.g., NY-ESO-1, MAGE family proteins). Cancer/testis antigen scores were generated by performing a log2 transform on the expression (TPM) of each gene normalized to the median tissue expression for “Brain-Cortex” in the Genotype-Tissue Expression Project database. Cancer/testis antigen scores were generated for the metastatic samples through normalizing to “Brain-Cortex” expression or the associated matched primary tissue site (“Breast Mammary Tissue,” “Lung,” or “Skin Sun Exposed”). The intratumoral spread of cancer/testis antigen expression was assessed by calculating the variance of scores within the regions of a tumor or by calculating the cosine similarity between regions, where each antigen's score in a given region represents one component of the vector.
RNA-Sequencing Analysis and Immune Microenvironment Profiling
Gene and transcript quantification was performed using kallisto (76). Differential expression analysis between tumor types was then performed using sleuth (77). To quantify immune cell populations in the tumors, CIBERSORT (43) was run using the LM22 signature gene file and disabled quantile normalization for 100 permutations. Relative immune cell abundance was obtained using gene expression as previously described by Danaher and colleagues (42). PCA dimensionality reduction was implemented on Danaher scores for each tumor region in R version 3.6.2 using the ggbiplot package.
Intratumoral similarity was performed by representing Danaher scores as vectors and calculating the normalized dot product between pairs of regions from the same tumor. The immune intratumoral heterogeneity was defined for tumors with three distinct regions as the area in PC1–PC2 space of the triangle whose vertices were represented by each region's Danaher score.
The myeloid-specific analyses were performed through the generation of log-transformed scores, making use of gene sets defined in previous publications. A total of 11 and 10 genes were used to define the M1 and M2 scores, respectively, based on gene expression signatures of in vitro polarized M1 or M2 macrophages (45). A total of six distinct genes for each were used to define the scores for both microglial and monocyte-derived macrophages (46). These genes were selected based on differential expression in tumor-associated macrophages from distinct lineages in scRNA-seq data of human gliomas. To account for differential abundance of macrophages between samples as a reason for findings, the difference between these scores (corresponding to a ratio of gene expression) was taken to generate an “M2–M1 skew” or “MDM–microglial skew.”
TCR Repertoire Sequencing
Sequencing of the CDR3 regions of TCR β-chains was performed through the immunoSEQ Assay (Adaptive Biotechnologies) and used the same DNA extracted from tumor samples used for WES. Initial analysis was performed on the immunoSEQ ANALYZER 3.0. Additional analysis was performed using the immunarch package in R. Visualization of the V-J gene usage among T-cell repertoires was performed using the circlize (RRID:SCR_002141) package in R (73). The intratumoral T-cell fraction was calculated by dividing the number of productive TCR templates by the number of nucleated cells estimated by the amplification of reference genes. The Simpson clonality for each region was calculated by taking the square root of the Simpson diversity index for all productive rearrangements with possible values ranging from 0 (very polyclonal) to 1 (predominantly monoclonal or oligoclonal).
The similarity between TCR repertoires was assessed through either the normalized dot product (cosine similarity) or Morisita overlap between the vectors of TCR clonotype abundances. Both of these metrics are based on pairwise comparisons between two repertoires. The T-cell repertoires for two regions are represented by vectors with indices covering the union of TCRs observed in either of the corresponding regions. Each position of the vector then represents the abundance (as a count) of a given clonotype within that region. The cosine similarity of the T-cell repertoires in regions A and B can then be given by
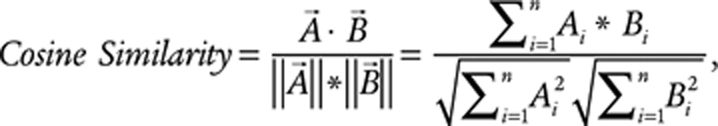 |
where Ai and Bi represent the counts of clonotype i within region A or B, respectively, and n represents the number of unique clonotypes observed in either A or B. Similarly, the Morisita overlap between the T-cell repertoires in regions A and B is defined as
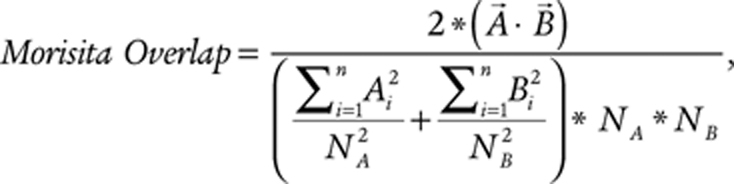 |
where Ai, Bi, and n represent the same values as before and NA and NB represent the total number of productive rearrangements observed in region A or B, respectively. Both of these metrics provide a value between 0 and 1, where 0 represents no similarity (orthogonal vectors or completely disparate populations) and 1 represents complete similarity (parallel vectors or completely identical populations). Comparisons between tumor types were performed by taking all intratumoral comparisons between GBMs and BrMETs and grouping them.
Sample Preparation for Single-Cell Sequencing
For GBM065.Re, the fresh surgical sample was rinsed with PBS to remove visual blood contaminant and manually dissociated using frosted microscope slides and gentle trituration. The resulting single-cell suspension was passed through 100-mm and 70-mm filters before undergoing Percoll (GE Healthcare Life Sciences) density gradient centrifugation to remove myelin contamination. Following this separation, the resulting cell pellet underwent RBC lysis with ACK Lysis Buffer (Lonza Biosciences) and was frozen in 90% FBS and 10% DMSO at −80°C and later stored in liquid nitrogen until further use.
The tumor sample was later thawed, and the single-cell suspension was stained with anti-CD45, anti-CD11b, anti-CD3, and Zombie NIR Viability Dye (BioLegend). Live CD45+ single cells were purified by FACS on a BD FACSAria II with an 85-mmol/L 45-psi nozzle into a buffer of PBS with 0.04% BSA. A total of 13,000 were submitted for analysis.
Single-Cell Library Preparation
cDNA was prepared after the Gel Beads in Emulsion (GEM) generation and barcoding, followed by the GEM-RT reaction and bead cleanup steps. Purified cDNA was amplified for 10 to 14 cycles before being cleaned up using SPRIselect beads. Samples were then run on a bioanalyzer to determine the cDNA concentration. TCR target enrichment was done on the full-length cDNA. GEX and enriched TCR libraries were prepared as recommended by the 10× Genomics Chromium Single Cell V(D)J Reagent Kits (v1 Chemistry) user guide with appropriate modifications to the PCR cycles based on the calculated cDNA concentration. For sample preparation on the 10× Genomics platform, the Chromium Single Cell 5′ Library and Gel Bead Kit (PN-1000006); Chromium Single Cell A Chip Kit (PN-1000152); Chromium Single Cell V(D)J Enrichment Kit, Human, T Cell (96rxns)(PN-1000005); and Chromium Single Index Kit T (PN-1000213) were used. The concentration of each library was accurately determined through qPCR using the KAPA library quantification kit according to the manufacturer's protocol (KAPA Biosystems/Roche) to produce cluster counts appropriate for the Illumina NovaSeq6000 instrument. Normalized libraries were sequenced on a NovaSeq6000 S4 Flow Cell using the XP workflow and a 151 × 8 × 151 sequencing recipe according to the manufacturer's protocol. A median sequencing depth of 50,000 reads/cell was targeted for each Gene Expression Library and 5,000 reads/cell for each V(D)J (T-cell) library.
Single-Cell Sequencing Analysis
Raw sequencing data were processed with Cell Ranger, version 3.0.1 (78), from 10× Genomics and mapped onto a human genome reference (GRCh38–2020-A). Downstream analysis was performed using the Seurat (RRID: SCR_007322) R package version 3.2.0 (79). In total, 2,147 cells passed the quality control steps performed by Cell Ranger. Low-quality cells and potential doublets were accounted for by removing cells that contained fewer than 500 expressed genes, a nCount value greater than the nCount value of the 93rd percentile of the total sample, or more than 10% mitochondrial transcripts. Following filtering, 1,728 cells remained within the final data set. Genes that were expressed in fewer than 100 cells were also removed. For each cell, expression of each gene was normalized to the sequencing depth of the cell, scaled to a constant depth (10,000), and log-transformed. Variable genes were selected with default settings, and PCA was performed on the variable genes. Dimensionality reduction and visualization were performed with the uniform manifold approximation and projection (UMAP) algorithm (Seurat implementation) using the first 15 PCA dimensions. Unsupervised graph-based clustering of cells was performed using the mentioned PCA dimensions with a resolution of 0.8.
Enriched gene expression levels in each cell cluster were identified by a Wilcoxon rank sum test-based function. These genes, along with common cell-type markers, were used to establish the cell identity of each cluster. Projection of average expression of marker genes into UMAP or violin plots was used for cell-type identification. Gene expression signatures used for definition of clusters were as follows: CD3E, CD3D, and CD3G (T cells); NKG7, PRF1, GZMH, and CD3− (natural killer cells); MS4A1, CD79B, and CD3− (B cells); and CD14, S100A8, S100A9, C1QC, CD68, CTSD, and HLA-DR+ (monocytes/macrophages).
Second-level clustering of T cells was performed by subsetting only T-cell clusters and rerunning scaling, log transformation, and variable gene selection. In addition, PCA was performed again on the new variable genes. Dimensionality reduction and visualization were performed with the UMAP algorithm using the first 10 PCA dimensions. Unsupervised graph-based clustering of cells was performed using the mentioned PCA dimensions with a resolution of 1.0. Gene expression signatures used for definition of clusters were as follows: CCR7, SELL, TCF7, and KLF2 (naive/central memory T cells); Il7R, KLRB1, and SELL− (effector memory T cells); TIGIT, CTLA4, and CD4 (CD4+ regulatory-like T cells); and NKG7, PRF1, CCL5, GZMH, CD3, CD8A, and CD8B (cytotoxic CD8+ T cells).
V(D)J libraries were processed with CellRanger V(D)J, version 2.0.0, from 10× Genomics and mapped onto a human VDJ reference (GRCh38–2.0.0). Clonotype analysis was performed with the scRepertoire (version 0.99.17) R package (80). Clonotypes were defined as the combination of the genes of the TCR A and B chains and nucleotide sequences as previously discussed (81).
Statistical Analysis
Data analysis and visualization in R was performed using the tidyverse package. Statistical significance for variant, neoantigen, CNV clonality estimates, heterogeneity estimates, T-cell fraction, and T-cell clonality was performed using an unpaired t test. Significance for differential expression was determined by a multiple t test with Benjamini–Hochberg adjustment with an FDR = 0.05.
Data Availability
Exome and RNA-sequencing read data have been made available in the controlled access repository, the database of Genotypes and Phenotypes (dbGaP), with accession number: phs002612.v1.p1. Original TCR sequencing data are available upon request.
Authors' Disclosures
Z.L. Skidmore reports grants from Washington University, St. Louis during the conduct of the study. A.H. Kim reports grants from Monteris Medical, Stryker, Collagen Matrix, and other support from Monteris Medical outside the submitted work. J.W. Osbun reports personal fees from Microvention, Inc., Medtronic, Inc., Terumo, Inc., and personal fees from InNeuroCo, Inc. outside the submitted work. E.C. Leuthardt reports personal fees and nonfinancial support from Monteris during the conduct of the study, other support from Neurolutions outside the submitted work, and stock ownership: Neurolutions, General Sensing, Osteovantage, Pear Therapeutics, Face to Face Biometrics, Immunovalent, Caeli Vascular, Acera, Sora Imaging Solutions, Inner Cosmos; consultant: Monteris Medical, E15, Acera, Alcyone, Intellectual Ventures, Medtronic, Inc., Neurolutions, Osteovantage, Pear Therapeutics, Inc., Sante Ventures, Microbot; SAB: Pear Therapeutics, Microbot; licensing from intellectual property: Neurolutions, Osteovantage, Caeli Vascular; licensing/product development agreements or royalties for inventions/IP: Cerovations, Intellectual Ventures. Washington University owns equity in Neurolutions. E.R. Mardis reports personal fees and other support from Qiagen N.V., personal fees and other support from PACT Pharma LLC, and personal fees and other support from Scorpion Therapeutics LLC outside the submitted work. G.P. Dunn reports personal fees from Ziopharm Oncology and personal fees from ImmunoGenesis outside the submitted work, and stock ownership from Immunovalent outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
M.O. Schaettler: Conceptualization, resources, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. M.M. Richters: Conceptualization, data curation, software, formal analysis, investigation, visualization, writing–review and editing. A.Z. Wang: Software, formal analysis, investigation, visualization. Z.L. Skidmore: Data curation, software, formal analysis, visualization. B. Fisk: Data curation, software, formal analysis, visualization. K.E. Miller: Data curation, software, formal analysis, writing–review and editing. T.L. Vickery: Resources. A.H. Kim: Resources. M.R. Chicoine: Resources. J.W. Osbun: Resources. E.C. Leuthardt: Resources. J.L. Dowling: Resources. G.J. Zipfel: Resources. R.G. Dacey: Resources. H.-C. Lu: Resources, formal analysis, investigation. T.M. Johanns: Conceptualization, resources, supervision, project administration, writing–review and editing. O.L. Griffith: Supervision, project administration, writing–review and editing. E.R. Mardis: Supervision, project administration, writing–review and editing. M. Griffith: Conceptualization, data curation, software, formal analysis, supervision, project administration. G.P. Dunn: Conceptualization, supervision, funding acquisition, methodology, project administration, writing–review and editing.
Supplementary Material
Acknowledgments
G.P. Dunn was supported by the Damon Runyon Cancer Research Foundation (grant #94-17). M. Griffith was supported by the National Human Genome Research Institute (NHGRI) of the NIH (award number: R00HG007940), the NIH National Cancer Institute (award number U01CA248235), and the V Foundation for Cancer Research (award number V2018–007). M. Schaettler was supported in part by the National Institutes of Health (grants T32 GM 7200–43 and T32 AI007163). M. Richters was supported in part by the NIH T32 Genome Analysis Training Program (2T32HG000045–21). E. Mardis and K. Miller were supported by the Nationwide Foundation Innovation Fund. We thank the patients for their participation in the research study and the physicians and staff at Barnes-Jewish Hospital and the Department of Neurosurgery at Washington University School of Medicine in St. Louis. We thank Dr. Katherine Schwetye from Saint Louis University School of Medicine for providing a tissue sample for analysis.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
References
- 1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol 2018;20:iv1–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schouten LJ, Rutten J, Huveneers HAM, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 2002;94:2698–705. [DOI] [PubMed] [Google Scholar]
- 3. Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol Met al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol 2016;17:976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kluger HM, Chiang V, Mahajan A, Zito CR, Sznol M, Tran Tet al. Long-term survival of patients with melanoma with active brain metastases treated with pembrolizumab on a phase II trial. J Clin Oncol 2019;37:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy Tet al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol 2018;20:674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte Het al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373–85. [DOI] [PubMed] [Google Scholar]
- 7. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJDet al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li Set al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas Cet al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019;565:240–5. [DOI] [PubMed] [Google Scholar]
- 10. Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry Aet al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 2016;6:1230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson Met al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016;34:2206–11. [DOI] [PubMed] [Google Scholar]
- 12. Aum DJ, Kim DH, Beaumont TL, Leuthardt EC, Dunn GP, Kim AH. Molecular and cellular heterogeneity: the hallmark of glioblastoma. Neurosurg Focus 2014;37:E11. [DOI] [PubMed] [Google Scholar]
- 13. Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch Cet al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016;22:105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang Jet al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014;346:256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah Set al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 16. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder Det al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SKet al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SMet al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 2018;50:1271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CYet al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014;343:189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JCet al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A 2013;110:4009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mahlokozera T, Vellimana AK, Li T, Mao DD, Zohny ZS, Kim DHet al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro Oncol 2018;20:472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RTet al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 2015;5:1164–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston Aet al. Publisher correction: spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med 2020;26:1148. [DOI] [PubMed] [Google Scholar]
- 24. Reuben A, Gittelman R, Gao J, Zhang J, Yusko EC, Wu C-Jet al. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov 2017;7:1088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia Q, Wu W, Wang Y, Alexander PB, Sun C, Gong Zet al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat Commun 2018;9:5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiménez-Sánchez A, Cybulska P, Mager KL, Koplev S, Cast O, Couturier D-Let al. Unraveling tumor–immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat Genet 2020;52:582–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CLet al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020;580:517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AVet al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi S, Yu Y, Grimmer MR, Wahl M, Chang SM, Costello JF. Temozolomide-associated hypermutation in gliomas. Neuro Oncol 2018;20:1300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis Ket al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pereira B, Chin S-F, Rueda OM, Vollan H-KM, Provenzano E, Bardwell HAet al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heidenreich B, Rachakonda PS, Hosen I, Volz F, Hemminki K, Weyerbrock Aet al. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget 2015;6:10617–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakthikumar S, Roy A, Haseeb L, Pettersson ME, Sundström E, Marinescu VDet al. Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biol 2020;21:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJet al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 2015;25:316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MDet al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ERet al. pVAC-Seq: a genome-guided in silico approach to identifying tumor neoantigens. Genome Med 2016;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hundal J, Kiwala S, McMichael J, Miller CA, Xia H, Wollam ATet al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol Res 2020;8:409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang W-Cet al. Bioactivity and safety of IL13R 2-redirected chimeric antigen receptor CD8 T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015;21:4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DEet al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II–restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol 2017;35:3322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach Met al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res 2014;20:2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster Eet al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020;367:eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Danaher P, Warren S, Dennis L, D'Amico L, White A, Disis MLet al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Yet al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SHet al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 2018;24:4175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SNet al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016;1:e85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri Aet al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017;18:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri Set al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020;181:1643–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJet al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020;181:1626–42. [DOI] [PubMed] [Google Scholar]
- 49. Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JLet al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci U S A 2016;113:E3529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berends MJW, Wu Y, Sijmons RH, Mensink RGJ, van der Sluis T, Hordijk-Hos JMet al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 2002;70:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin Met al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 2004;6:139–50. [DOI] [PubMed] [Google Scholar]
- 52. Terui H, Akagi K, Kawame H, Yura K. CoDP: predicting the impact of unclassified genetic variants in MSH6 by the combination of different properties of the protein. J Biomed Sci 2013;20:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJet al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018;560:243–7. [DOI] [PubMed] [Google Scholar]
- 54. Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti Eet al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet 2020;52:371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer Let al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring ADet al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eckhart L, Bach J, Ban J, Tschachler E. Melanin binds reversibly to thermostable DNA polymerase and inhibits its activity. Biochem Biophys Res Commun 2000;271:726–30. [DOI] [PubMed] [Google Scholar]
- 58. Lagonigro MS, Stefania Lagonigro M, De Cecco L, Carninci P, Di Stasi D, Ranzani Tet al. CTAB-urea method purifies RNA from melanin for cDNA microarray analysis. Pigment Cell Res 2004;17:312–5. [DOI] [PubMed] [Google Scholar]
- 59. Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AMet al. Genome modeling system: a knowledge management platform for genomics. PLoS Comput Biol 2015;11:e1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN] 2013. Available from: http://arxiv.org/abs/1303.3997. [Google Scholar]
- 62. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 63. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin Let al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez Cet al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Locallo A, Prandi D, Fedrizzi T, Demichelis F. TPES: tumor purity estimation from SNVs. Bioinformatics 2019;35:4433–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OLet al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 2014;10:e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dang HX, White BS, Foltz SM, Miller CA, Luo J, Fields RCet al. Clon-Evol: clonal ordering and visualization in cancer sequencing. Ann Oncol 2017;28:3076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016;12:e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IFet al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014;30:2811–2. [DOI] [PubMed] [Google Scholar]
- 74. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann Aet al. The ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Richters MM, Xia H, Campbell KM, Gillanders WE, Griffith OL, Griffith M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med 2019;11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bray N, Pimentel H, Melsted P, Pachter L. Near-optimal RNA-Seq quantification with kallisto. Nat Biotechnol 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 77. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 2017;14:687–90. [DOI] [PubMed] [Google Scholar]
- 78. Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson Ret al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM IIIet al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Borcherding N, Bormann NL, Kraus G. scRepertoire: an R-based toolkit for single-cell immune receptor analysis. F1000Res 2020;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Borcherding N, Crotts SB, Ortolan LS, Henderson N, Bormann NL, Jabbari A. A transcriptomic map of murine and human alopecia areata. JCI Insight 2020;5:e137424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Exome and RNA-sequencing read data have been made available in the controlled access repository, the database of Genotypes and Phenotypes (dbGaP), with accession number: phs002612.v1.p1. Original TCR sequencing data are available upon request.