

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer that has proven refractory to immunotherapy. Previously, treatment with the DNA hypomethylating drug decitabine (5aza-dC; DAC) extended survival in the KPC-Brca1 mouse model of PDAC. Here we investigated the effects of DAC in the original KPC model and tested combination therapy with DAC followed by immune checkpoint inhibitors (ICI). Four protocols were tested: PBS vehicle, DAC, ICI (anti-PD-1 or anti-VISTA), and DAC followed by ICI. For each single-agent and combination treatment, tumor growth was measured by serial ultrasound, tumor infiltrating lymphoid and myeloid cells were characterized, and overall survival was assessed. Single-agent DAC led to increased CD4+ and CD8+ tumor-infiltrating T cells (TILs), PD1 expression, and tumor necrosis while slowing tumor growth and modestly increasing mouse survival without systemic toxicity. RNA-seq of DAC-treated tumors revealed increased expression of Chi3l3 (Ym1), reflecting an increase in a subset of tumor-infiltrating M2-polarized macrophages. While ICI alone had modest effects, DAC followed by either of ICI therapy additively inhibited tumor growth and prolonged mouse survival. The best results were obtained using DAC followed by anti-PD-1, which extended mean survival from 26 to 54 days (p<0.0001). In summary, low-dose DAC inhibits tumor growth and increases both TIL and a subset of tumor-infiltrating M2-polarized macrophages in the KPC model of PDAC, and DAC followed by anti-PD-1 substantially prolongs survival. Since M2-polarized macrophages are predicted to antagonize anti-tumor effects, targeting these cells may be important to enhance the efficacy of combination therapy with DAC plus ICI.
Keywords: Pancreatic cancer, Immune checkpoint therapy, Epigenetic therapy, Tumor microenvironment
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy that has proven refractory to both conventional cytotoxic and targeted therapies. Notably, despite recent remarkable results with immunotherapy against other cancers, responses in PDAC have been elusive (1–3). Thus, developing more effective therapies using combinations of immune checkpoint inhibitors (ICIs) with other agents is being actively pursued (e.g. (4–8) and references therein). Epigenetic alterations are prominent in PDACs (9), and epigenetically-acting drugs are receiving increasing interest for possible utility against treatment refractory tumors. In a rapidly progressive mouse model of PDAC, KPC-Brca1, we previously found that treatment with decitabine (5aza-dC; DAC), a well-tolerated DNA hypomethylating drug, produced changes in DNA methylation in the epithelial and stromal cells of the tumors, inhibited tumor cell proliferation, and extended the survival of the animals (10). Given this background, we have now extended our approach to combination therapy, using the well characterized and highly chemoresistant KPC mouse model (KrasLSL.G12D/+; P53LSL.R172H/+; Pdx1Cretg/+), treated with single agent DAC and with monoclonal antibodies against two ICIs: PD-1 and V-domain Ig suppressor of T cell activation (VISTA; also known as PD-1H). The slower progressing KPC model is more widely used than KPC-Brca1, which facilitates comparisons of results across studies. Our results show that single-agent DAC induces tumor necrosis and inhibits tumor growth, without systemic toxicities, and that this hypomethylating agent alters specific sub-populations of tumor-infiltrating lymphoid and myeloid cells in the KPC model. We find that sequential combinations of DAC plus the ICIs are more effective than either type of agent alone in slowing tumor growth, and that DAC followed by anti-PD-1 substantially prolongs mouse survival.
Materials and Methods
Mouse strains
Procedures relating to the care and use of mice in this study were conducted in accordance with NIH guidelines, with the animals housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility. The KrasG12D/+;Trp53R172H/+;Pdx1-Cre (KPC) mice have been previously described (11). All animal studies were carried out in accordance with approved Institutional Care and Use Committee protocols for Columbia University Irving Medical Center (AC-AAAU2462).
U/S imaging of tumors
U/S imaging of normal and tumor-bearing mouse pancreata using the Vevo 770 System with a 35MHz RMV scanhead (Visual Sonics, Inc.) was performed in the Small Animal Imaging Shared Resource of the Herbert Irving Comprehensive Cancer Center (HICCC), and serial images at 0.25 mm intervals were collected as described (12). Tumor boundaries were marked on the U/S images and Vevo 770 software calculated a collective tumor volume based on close approximation of tumor topography. The tumor doubling time was calculated using the formula , where q1 is the tumor volume at treatment enrollment and q2 is the tumor volume at death or euthanasia, at their respective times, t1 and t2.
Treatment paradigms evaluating autochthonous tumors
We utilized three different types of treatment protocols (Supplementary Figure S1A–C). In Paradigm I (n=65), designed with a survival endpoint, we enrolled KPC mice after they developed 3-5 mm tumors, as measured by high-resolution U/S. Due to potential differences in mouse survival depending on the location of the tumors, in this treatment paradigm we only enrolled mice with tumors localized in the tail of the pancreas (TOP). Mice received 1μg/g body weight of DAC via IP injection every 72 hours for 3 doses, or PBS as vehicle control, then either PBS or 100μg anti-mouse PD-1H/VISTA (BioLegend) or anti-mouse PD-1 (Merck) IP every 72 hours for 3 doses. In the combination treatments (DAC + anti-VISTA or anti-PD-1), the mice received the first dose of the immune checkpoint inhibitor at 72 hours after the last DAC dose. Mice were sacrificed when moribund as defined by a scoring system of physical and behavioral symptoms. In Paradigm II (n=8) we assessed the effects of treatment with DAC on head-of-pancreas (HOP) tumors, using a timed endpoint. Mice received 1μg/g body weight of DAC, or PBS as control, via IP injection every 72 hours for 3 doses. Mice were euthanized and necropsied at 72 hours after completion of treatment. In Paradigm III (n=12) we assessed the effects of DAC when the drug was started earlier, when most mice have only pancreatic situ neoplasia (PanIN). Mice at 4 months of age, without U/S-detectable tumors, were given 1μg/g body weight of DAC IP, or PBS vehicle in the controls, every 72 hours for 5 doses, and were euthanized and necropsied at 72 hours after the last injection.
Tumor cell allografting
Tumor tissue was resected from KPC mice within our breeding colony and was trimmed and washed in sterile PBS and minced. Tumor fragments were incubated with collagenase (1mg/ml in DMEM) at 37°C for 45 min, filtered to remove non-dissociated fragments and debris, and the resulting single cell suspension plated and cultured in DMEM containing 10 percent fetal calf serum to obtain a purified tumor cell population. After no more than 10 passages, the tumor cells were injected subcutaneously at 2 × 106 cells in each flank of litter-mate recipient mice, which did not carry Pdx1-Cre and did not have autochthonous tumors. Starting at 6 days after the injection of the tumor cells, when small tumors were palpable, the mice received 1μg/g body weight of DAC or the equivalent volume of PBS I.P. every 72 hours for 3 doses and were sacrificed 48 hours after the last dose, to obtain the tumors for size measurements and histological studies.
In vitro DAC response
Primary KPC tumor cells, isolated as above, were seeded at 5×105 cells per 100 mm plate and cultured for 24 hours in DMEM containing 10 percent fetal calf serum. DAC was then added to achieve a range of final concentrations (see Results), and the medium, with the drug, replaced every 24h for 48h or 96h before harvesting the cells for DNA and RNA extraction.
Methylation-sensitive Pyrosequencing (MS-Pyroseq)
MS-Pyroseq for determining CpG methylation levels in B1 repetitive elements was performed at EpigenDx using genomic DNA samples from control and DAC-treated KPC cells maintained in tissue culture, and from KPC tumors obtained from the in vivo experiments.
IHC and IF
IHC and IF were performed on formalin-fixed paraffin-embedded (FFPE) tissue sections using primary antibodies against mouse CD4 (eBioscience EPR19514; 1:50), CD8 (Thermo Scientific; MA1-70041; 1:50), FoxP3 (Santa Cruz Biotechnology; sc-53876; 1:500), CD163 (Abcam; ab182422; 1:500), CD103 (BD Biosciences M290 FitC; 1:200), F4/80 (Ebioscience; 14-4801-85; 1:1000), Ki67 (Abcam; ab15580; 1:500), Arginase-1 (Santa Cruz Biotechnlogy; sc-18351; 1:200), p53 (Vector Labs; VP-P956; 1:200), PD-1H/VISTA (BioLegend; 143704; 1:100), Chi3l3/Chi3l4 (Abcam; ab192029; 1:500), p16 (Abcam; ab211542; 1:400), p21 (Abcam; ab188224; 1:400), PD1 (Abcam; ab214421; 1:400), dsRNA/9D5 (Absolute Antibody; Ab00458-23.0; 1:200), CD3 (Abcam; ab16669; 1:100), RELMα (Abcam; ab39626; 1:50), and CD11b (Thermo Fisher; 14-0112-81; 1:50). FITC anti-goat, FITC anti-mouse, FITC anti-rabbit, TRITC anti-goat, TRITC anti-mouse, TRITC anti-rat, TRITC anti-armenian hamster, and Alexa 647 anti-rabbit fluorescent (Jackson ImmunoResearch) secondary antibodies were used, all at 1:300 dilutions, for the IF imaging. Controls using secondary antibodies alone were negative, and all IHC and IF results were reproducible in at least two repeated experiments.
Flow cytometry
Cell suspensions from KPC tumors were prepared using the Mouse Tumor Dissociation Kit (Miltenyi) according to the manufacturer’s recommendations. Cells were Fc-blocked with purified rat anti-mouse CD16/CD32 (Clone: 2.4 G2, Becton Dickinson) for 30 minutes at 4°C. Dead cells were discriminated using the LIVE/DEAD (LD) Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) and samples were stained for the extracellular markers using directly labeled primary antibodies against Chi3l3/4 (Abcam; ab192029), CX3CR1 (Biolegend SA011F11 BV711), CD103 (BD Biosciences M290 FitC), CD45 (Biolegend 30-F11 BV605), CD11b (Biolegend M1/70 AF700), CD11c (Biolegend N418 BV650), and F4/80 (Biolegend BM8 PE/Cy7). Staining was visualized by fluorescence-activated cell sorting (FACS) analysis using a BD FACSCelesta Multicolor Cell Analyzer (BD Biosciences) and analyzed using FlowJo v10 software (FlowJo LLC).
Histological analysis and counting of tumor-infiltrating cells
We measured the areas of zonal necrosis in each tumor using low power (4X) images of two hematoxylin-eosin (H&E)-stained tissue sections of each tumor, with pixel counting using ImageJ. The two levels were chosen to be at least 6 sections or 30μm apart. Pixel counts were converted into mm2 by capturing a 1mm2 grid at 4X magnification.
For quantitating proliferating (Ki67) and tumor-infiltrating cells identified by IHC with cell type-specific antibodies, tumor area and cell counts were calculated over two entire tissue sections, obtained from the top and bottom of each paraffin-embedded tissue block (i.e. separated by a thickness of approximately 3 mm). Tumor area was calculated using a collection of 20X magnification images. Marker-positive cell counts were obtained from 10 random 40X fields per histological section using the Cell Counter function in NIH ImageJ (13) to tabulate manual counts. To minimize possible effects of an acute response to necrotic cell debris, cells immediately adjacent to necrotic areas (within 2mm) were not included in the final counts. However, recounts including these areas did not significantly alter the results. To count lymphocytes and macrophages around normal pancreatic ducts and PanIN lesions, we selected a 500 μm perimeter around the ducts and pre-malignant lesions in each field, counted the marker-positive cells in these areas, and averaged the results over the number of counted fields. All cell counts were visually quantified in a case-blinded fashion by two individuals. The assessment between the two readers showed high concordance in all cases and therefore they were averaged to produce the reported results.
RNA sequencing (RNA-seq), gene set enrichment and deconvolution analyses
RNA was isolated using Trizol (Invitrogen, MA, USA) and integrity was confirmed as RIN>7 on a Bio Analyzer (Agilent Technologies, CA, USA). Poly-A pull-down was used to enrich for mRNAs, and libraries were prepared using the Illumina TruSeq RNA kit. Libraries were sequenced on an Illumina HiSeq 4000 machine with 100 bp paired end reads, in the Genomics Shared Resource of the HICCC. After base calling and adaptor trimming, the reads were mapped to the mouse reference genome (UCSC/mm9) using Tophat, and the relative expression of genes was estimated by FPKM (Fragments Per Kilobase of transcript per Million mapped reads) using Cufflinks (14). Candidate in vivo genes were defined using Student’s t-test with unequal variance assumption and defined using nominal p-value<0.05, fold change >1.5 or <0.67 and FPKM>1 in at least one group. Candidate genes induced in KPC tumor cells by DAC exposure in tissue culture after 48h or 96h, using a range of DAC concentrations, were identified using linear regressions (Gene expression ~ DAC concentration) and defined using nominal p-value<0.05, R2>0.5, FPKM >1 in at least one DAC concentration, and the same dose effect trend for both time points (48h and 96h). All performed tests were two-sided. Multiple testing corrected p-values (FDRs) using the Benjamini-Hochberg method are reported in Supplementary Tables S1 and S2.
To test for enrichment of genes associated with immune and stromal cell lineages in our RNA-seq data, we downloaded the most recent expression data generated by the Immunological Genome Project (15) from primary myeloid, lymphoid and stromal cells from C57BL/6J mice using RNA-seq (NCBI GEO accession number GSE109125 ). Data were normalized using TMM method with edgeR R package. Using the ImmGen annotations, we categorized their samples according to cell type. In this dataset, we used expression in stromal cells as a baseline to identify genes that were relatively overexpressed in myeloid and lymphoid lineages (FDR<0.05 and fold change>2). Tests for enrichment of these myeloid and lymphoid signatures among the genes differentially expressed in our RNA-seq data from the KPC tumors were performed using logistic regression. Using statistical criteria (FDR<0.05 and fold change> 2), we identified genes specifically overexpressed in monocyte, macrophage and neutrophils, and tested for enrichment of each of these genes among the genes that we had found to be differentially expressed in our RNA-seq data from the KPC tumors.
Enrichment for biological pathways among our in vitro cultured KPC cell DAC-induced genes was performed using the Broad institute’s GSEA tool and the hallmark gene sets (MSigDB, Broad Institute), which contained expressed signatures derived by aggregating many MSigDB gene sets to represent well-defined biological pathways, including interferon (IFN) responses. For the deconvolution analysis, we used the current Immgen RNA-seq dataset (GEO accession GSE109125) and our tissue culture-based KPC tumor cell data to derive immune cell and cancer cell expression profiles, respectively. Since the Immgen RNA-seq data was aligned using STAR aligner with read counts, we realigned our data using the same pipeline. Read counts were normalized using the TMM method with edgeR R package (16) and data deconvolution was performed using the DeconRNASeq R package (17).
To assess the expression of ERV transcripts, we used two complementary approaches. First, we used the repeatmasker annotation of the mouse reference genome for ERV elements to calculate FPKM counts for known ERV elements, as described by Kazachenka, et al (18). Second, to be more comprehensive and assess ERV elements that were not present in the genome of the reference mouse, we generated a faux genome using 15 mouse provirus sequences, kindly provided by Dr. R. Gifford (references and NCBI accession numbers in Supplementary Table S4). RNA-seq data were then aligned against this genome to calculate FPKM of ERV transcripts. For repetitive elements, we focused on the SINE/LINE elements using the repeatmasker annotation to calculate FPKM. Lastly, to ask whether DAC-induced Irf7 expression might be ERV induction dependent, we tested the correlation of Irf7 expression with all significant ERVs using our in vitro cultured KPC tumor cell data and Spearman correlations (FDR<0.05).
Additional statistical methods
Statistical approaches for RNA-seq data are described above. The p-values for differences in cell counts by IHC are based on two-sided Student’s T-tests under the assumption of unequal variance. The statistical analyses of mouse survival and comparisons of survival curves were based on the Mantel-Cox (log-rank) test, performed using Graph Pad Prism 8 software. A value of P < 0.05 was considered as statistically significant for both methods of analysis.
Data repository URL and accession numbers
RNA-seq data have been deposited at NCBI GEO with accession GSE101336.
Results
Low dose DAC inhibits tumor growth and reduces global DNA methylation in the KPC model of PDAC
Cancer formation in KPC mice progresses from pre-invasive lesions, akin to human PanIN, to stroma-rich invasive adenocarcinomas, analogous to human PDACs. In this study we tested treatment with single-agent DAC and single-agent ICIs, and sequential combinations of DAC followed by ICIs, using survival and timed endpoint paradigms (Fig. 1A, and Supplementary Fig. S1). Paradigm I was a survival experiment in which we performed U/S imaging on the mice starting at 2 months of age and enrolled for treatment all mice with tumors that were 3-5 mm in diameter. Since our prior experience has suggested different survival times and differences in accuracy of tumor-detection by U/S for animals with tumors located in the head vs tail anatomical regions of the pancreas, we restricted this experiment to tail of pancreas (TOP) tumors (most reliable early tumor detection) and utilized the head of pancreas (HOP) tumors for biological assays but not survival, in a timed endpoint protocol that we refer to as Paradigm II. In Paradigm III, which we also used for biological assays but not survival data, we started treatment of the mice earlier, when the pancreata contained PanIN lesions but no overt tumors.
Figure 1. Effects of DAC on tumor growth, histology, and cell proliferation-related markers in KPC mice.

(A) Timing of treatment initiation and drug administration in treatment Paradigm I. (B) There is a significant increase in tumor doubling time in single-agent DAC treated mice compared to the mock-treated (PBS) control mice. (C) H&E stained tissue sections showing little or no necrosis in a tumor from a mock-treated control tumor, contrasting with large areas of zonal necrosis (asterisk) in a tumor from a DAC treated mouse. (D) Quantitative image analysis shows that DAC treatment significantly increases tumor necrosis. (E) IHC reveals a decrease in Ki67+ cells following DAC treatment. The fields are chosen to be representative. (F) Decreased numbers of Ki67+ cells per high power field (HPF) indicate a decrease in tumor cell proliferation with DAC treatment. (G) IHC reveals an increase in p21+ cells following DAC treatment. The fields are chosen to be representative. (H) The increased numbers of p21+ cells per high power field (HPF) are consistent with the decrease in tumor cell proliferation induced by DAC treatment. Numbers of cases for histology and cell counting reflect the number of histological blocks available. Original magnification, 20X; scale bars, 100 μm.
In treatment Paradigm I, we found a significant slowing of tumor growth (40 percent increase in tumor doubling time) in the mice treated with single-agent DAC, compared to mock-treatment with PBS vehicle (Fig. 1B). Importantly, consistent with our prior findings in the KPC-Brca1 model (10), with the low DAC dosage utilized in the current study (1μg/g body weight via IP injection every 72 hours; total of 3 doses) we found no evidence of systemic or hematologic toxicity (as evidenced by lack of anemia or neutropenia) during the period of drug. Circulating white blood cell counts were only mildly reduced in the DAC treated KPC mice compared to mock treated animals (Supplementary Table S3) and no other side effects were noted. In addition, we observed no increase in animal mortality during the first 3 weeks of treatment (when tumor sizes were small) in the DAC treatment arm compared to the PBS control arm, further indicating no systemic toxicity with this dosage of the drug.
To ask whether this low dosage of DAC led to measurable effects on global DNA methylation, we carried out MS-Pyroseq of B1 repetitive elements in tumor DNA samples, 72 hours after the last dose of DAC (Paradigm II), or at the survival endpoint (Paradigm I). The results from Paradigm II indicated that a modest (6 percent) but statistically significant drug-induced DNA global hypomethylation had occurred in vivo and persisted for at least 3 days after the last DAC dose (Supplementary Fig. S2). As would be expected, direct exposure of explanted KPC tumor cells to DAC in tissue culture produced greater reductions in B1 element methylation (Supplementary Fig. S2).
Treatment with DAC leads to increased tumor necrosis, decreased cell proliferation, and increased numbers of CD8-positive TILs
In tumors from Paradigm I the most striking finding in H&E-stained histological sections was an effect on zonal necrosis: measurements by digital image analysis revealed a significant (>3-fold) increase in the areas of necrosis in the tumors from the DAC treated compared to mock-treated mice (Fig. 1C, D). Further, IHC for Ki67, a marker for cells that are actively engaged in the cell cycle, revealed 2.5-fold fewer cycling tumor epithelial cells, at the survival endpoint, with DAC compared to PBS (Fig. 1E, F). Opposite to this reduction in Ki67 expressing cells, the DAC treatment produced a significant increase in the numbers of cells expressing cyclin-dependent kinase inhibitors p21 (Fig. 1G, H), and p16 (Supplementary Fig. S3), which inhibit cell cycle progression and are often reduced or silenced in the development of human pancreatic cancers (19).
Based on IHC followed by cell counting, we found a significant 5-fold increase in CD8+ TILs with single-agent DAC treatment. We observed these cells both diffusely infiltrating the tumors and concentrated in lymphoid aggregates. Both the total number of CD8+ cells per tumor area and the number of CD8+ cells per lymphoid aggregate were increased by DAC (Fig. 2A. B and Supplementary Fig. S4A), and we found a similar increase in these cells in tissue sections of the HOP tumors from the timed endpoint Paradigm II (Supplementary Fig. S4C). Numbers of CD4+ TILS were also increased by DAC (Fig. 2C, D). We found a significantly higher number of PD-1 expressing cells in DAC treated tumors (Fig. 2E, F), with most of the PD-1+ cells being CD8+ (Supplementary Fig. S4D). The number of FoxP3+ suppressor T cells was variable among the tumors but was not significantly altered by DAC, and the ratio of FoxP3+/CD8+ cells showed a non-significant decline (Supplementary Fig. S4B).
Figure 2. DAC treatment increases tumor-infiltrating CD8+ and CD4+ lymphocytes and PD-1 expressing cells.
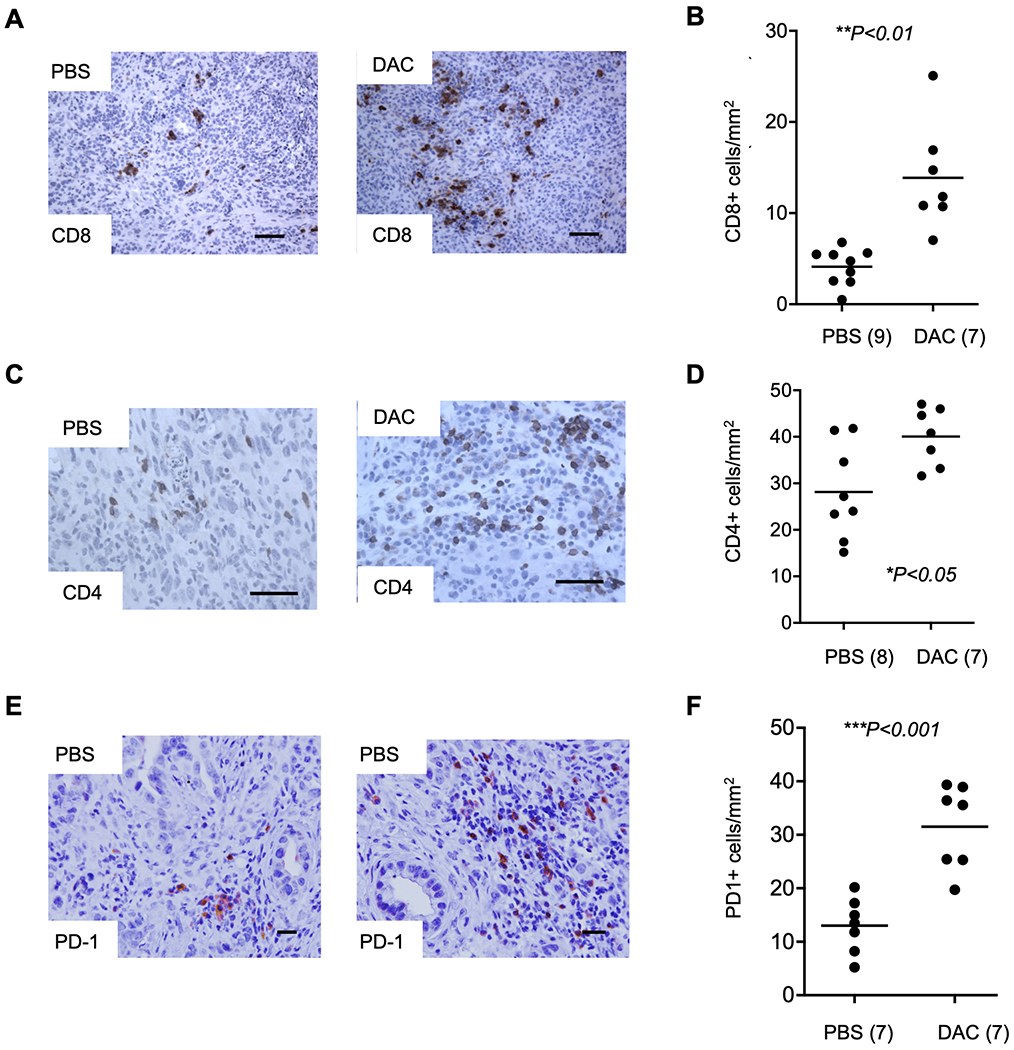
(A) IHC showing an increase in CD8+ TILs with DAC treatment compared to PBS vehicle. (B) Cell counting reveals a significant increase in CD8+ TILs following DAC treatment. (C) IHC images of CD4+ TILs with DAC compared to PBS. (D) Cell counting reveals an increase in the number of CD4+ cells between DAC and PBS treated tumors. (E) IHC shows that the number of PD-1 positive cells is increased by DAC. (F) The increase in PD-1+ cells is confirmed by cell counting. Data are from treatment Paradigm I. The number of cases for cell counting are indicated in parentheses and reflect the number of histological blocks available. Original magnification, 20X; scale bars, 100 μm.
In Paradigm III we tested the effects of DAC starting the treatment at 4 months of age, which is before the formation of tumors in most of the mice, but late enough to have allowed the formation of pre-invasive PanIN lesions. At three days after the last DAC dose, IHC of tumor sections revealed CD8+ lymphocytes within and adjacent to the PanIN lesions, with very few such cells around normal-appearing ducts in the same sections. However, despite this tropism of the CD8+ lymphocytes to the PanIN lesions, in this treatment paradigm we found no significant increase in the number of TILs with DAC compared to the PBS (Supplementary Fig. S5).
Gene expression profiling implicates effects of DAC on the cellular composition of the tumor microenvironment
To further understand the effects of DAC treatment on the tumor microenvironment, we profiled gene expression using RNA-seq in whole tumors from treatment Paradigm II, following three cycles of DAC treatment or PBS vehicle. The RNA-seq data revealed only small sets of treatment-responsive genes, with 195 genes upregulated and 20 downregulated by the in vivo DAC treatment (top-ranked over-expressed genes in Table 1; complete list in Supplementary Table S1). Among the strongly up-regulated mRNAs were transcripts from genes encoding a group of myeloid cell markers, including Chi3l3 (a.k.a. Ym1, Fig. 3A) and Chi3l4 (a.k.a. Ym2), Gpnmb, Ear6, Ear11, and Ms4a8a, several of which (Chi3l3, Chi3l4, Gpnmb, Ms4a8a) are associated with alternatively polarized “M2” macrophages (20–23).
Table 1. Gene expression profiling of DAC-treated tumors.
Genes with >2.5-fold increased expression in tumors from the DAC-treated mice. Expression values are in FPKM. Data are from treatment Paradigm II.
| Gene name | PBS | DAC | Fold increase | p-value |
|---|---|---|---|---|
| Chi3l3 | 44.1 | 754.8 | 17.1 | 0.032 |
| Ear6 | 0.5 | 4.8 | 9.0 | 0.029 |
| Chi3l4 | 1.3 | 9.6 | 7.6 | 0.041 |
| Ear11 | 1.3 | 8.2 | 6.3 | 0.018 |
| Zic3 | 0.3 | 1.6 | 5.4 | 0.006 |
| Spock3 | 1.1 | 6 | 5.2 | 0.037 |
| 2810047C21Rik1 | 0.5 | 2.5 | 4.6 | 0.011 |
| Wnt4 | 5.3 | 23.9 | 4.5 | 0.004 |
| Prss30 | 0.5 | 1.8 | 3.8 | 0.026 |
| Slc10a2 | 1.1 | 3.9 | 3.7 | 0.04 |
| Ms4a8a | 17.9 | 58.3 | 3.3 | 0.03 |
| Irx3 | 1.7 | 5.3 | 3.1 | 0.039 |
| Apol9a | 3.5 | 10.5 | 3.0 | 0.032 |
| Fam180a | 3.7 | 11 | 3.0 | 0.035 |
| Apol9b | 5.3 | 15.1 | 2.9 | 0.023 |
| Ephx4 | 1.4 | 4.2 | 2.9 | 0.009 |
| Hoxc6 | 0.6 | 1.8 | 2.9 | 0.031 |
| Atp6v0d2 | 5.2 | 14.3 | 2.7 | 0.003 |
| Muc6 | 8 | 21.6 | 2.7 | 0.039 |
| Gpnmb | 8.7 | 22.7 | 2.6 | 0.047 |
| Pamr1 | 10.3 | 26.8 | 2.6 | 0.038 |
| Sfi1 | 13 | 33.5 | 2.6 | 0.03 |
| Tnip3 | 2.5 | 6.5 | 2.6 | 0.013 |
| Wdr72 | 6.3 | 16.6 | 2.6 | 0.011 |
Figure 3. Gene expression profiling and data deconvolution confirms an abundant tumor stromal component and implicates an increase in a specific myeloid cell sub-population in the DAC-treated tumors.
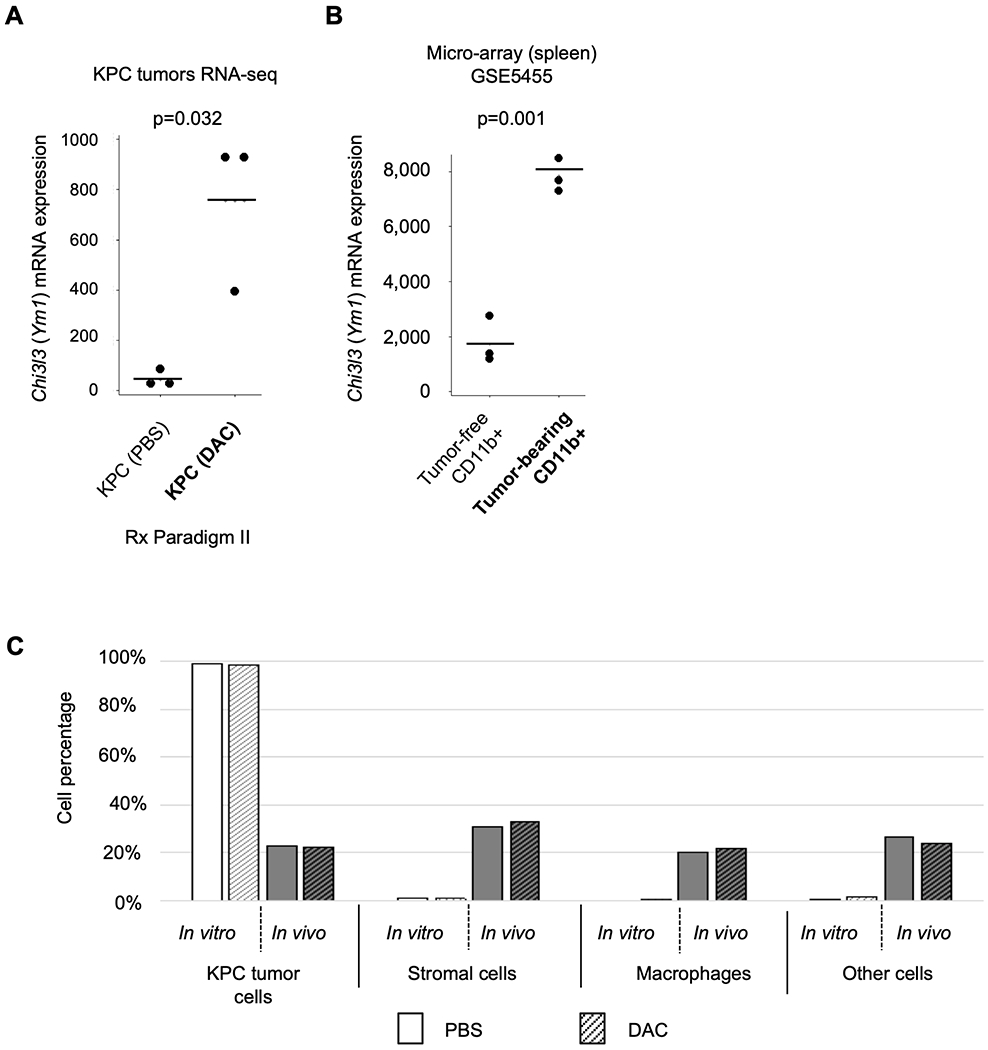
(A) Expression of Chi3l3 mRNA is significantly increased by DAC treatment in the KPC tumors in this study. (B) Chi3l3 mRNA is induced in CD11b+ splenic macrophages from mice hosting C26-GM colon cancer cells engineered to secrete GM-CSF (NCBI GSE5455), compared to CD11b+ cells isolated from tumor-free mice. (C) Results of deconvolution analysis for proportions of cell populations performed using the ImmGen RNA-seq reference dataset (see Methods). This analysis suggests that the tumors in vivo contain approximately equal (20 percent each) populations of malignant KPC cells, stromal cells, macrophage-lineage cells, and other cell types.
These findings led us to consider whether many of the gene expression changes induced by DAC in the KPC tumors might be due to changes in tumor-infiltrating myeloid lineage cells. To test this hypothesis, we adopted a targeted gene set enrichment approach, using an RNA-seq dataset (NCBI GEO accession GSE109125) from the ImmGen project (10,24) as a reference for expression levels in specific types of immune system cells and mesenchymal stromal cells. This analysis revealed an enrichment in myeloid lineage signature genes, with Odds Ratio (O.R.) = 1.9 and p = 8.6×10−5, with a 24% absolute overlap with the up-regulated genes in our dataset (including Chi3l3, Gpnmb, and Ear11). This enrichment was greater for signature genes of the subthreshold for monocytes (O.R.=1.9, p=0.06). The strongest expression of Chi3l3 in the ImmGen RNA-seq dataset is found in macrophages and granulocytes, and in several other publicly available expression datasets Chi3l3 mRNA is found to be inducible by tumor-derived cytokines or other stimuli to become highly expressed in CD11b+ macrophages (example in Fig. 3B). For completeness, it should be noted that Chi3l3 expression can be found either in CD11b+ macrophages or, in some studies alternatively in CD11b−/Cd11c+ myeloid dendritic cells, depending on tissue context, consistent with the known cytokine-dependent developmental fluidity of myeloid cells (25–28).
Using our RNA-seq data, with the ImmGen RNA-seq dataset as a reference, we performed a deconvolution analysis (Methods) to assess which cell types are predominantly represented in the expression profiling of the autochthonous KPC tumors. The results suggest that malignant KPC tumor cells represented 22%, cancer associated fibroblast-like stromal cells 30%, and macrophage/myeloid lineage cells 20% (Fig. 3C). This gross distribution of cell types did not change in response to DAC, suggesting that the significant shift in myeloid markers implied by our RNA-seq data might reflect a DAC induced increase in a specific sub-population of myeloid lineage cells – a presumption that is confirmed by immunostaining and flow cytometry data in the next section.
Overall, these results imply that many of the expression changes in the whole KPC tumors might be due to DAC-induced changes in a subpopulation of tumor-infiltrating cells, thus reflecting non-cell autonomous effects of the drug treatment. Nonetheless, we also sought to ask whether we could detect evidence for induction of classical DAC target genes in the treated tumors. We first performed a gene set enrichment analysis among upregulated genes that we observed using RNA-seq applied to KPC tumor cells in tissue culture exposed to a range of DAC concentrations and harvested at two time points (Supplementary Table S2). We found a significant induction of multiple immune pathways, and like our previously published observations in DAC treated KPC-Brca1 mouse tumors (10), a significant upregulation of interferon-responsive genes (Supplementary Fig. S6A). Induction of endogenous retroviruses (ERVs) and double-stranded RNA (dsRNA) has been shown to contribute to these pathways, and in the RNA-seq data from in vitro KPC cell DAC exposure we found ERV, SINE and LINE inductions (Supplementary Tables S4–S8). While the strongest findings came from the experiments using DAC exposure in vitro, one ERV transcript class was also significantly up-regulated in the autochthonous KPC tumors by DAC treatment in vivo (treatment Paradigm II; Supplementary Fig. 6B, C). Interestingly, among 59 ERV transcript classes induced by DAC treatment, 40 showed significant correlations with expression of the interferon responsive Irf7 gene (Spearman correlation FDR<0.05, Supplementary Table S9 and Supplementary Fig. S6D, E). In addition, IF with a monoclonal antibody against dsRNA revealed an increase in dsRNA in DAC treated tumors in vivo (Supplementary Fig. S6F).
DAC leads to increased tumor infiltrating Chi3l3/4+ macrophages
Two of the top-ranked over-expressed transcripts from expression profiling of the in vivo DAC treated tumors, Chi3l3 and Chi3l4, are from chitinase-like genes that are activated during myeloid-lineage cell differentiation in inflammatory conditions (20,29–32). These genes are known to be markers for “alternatively activated” or “M2-polarized” macrophages (20,29), although in some situations Chi3l3 is expressed by antigen-presenting dendritic cells (28). To ask whether the increased expression of these genes in the DAC-treated tumors reflected an increased number of tumor-infiltrating Chi3l3/4+ cells, we performed IHC on DAC-treated compared to PBS control tumors using a monoclonal antibody that recognizes both the Chi3l3 and Chi3l4 proteins. The results showed a highly significant 4-fold increase in the number of Chi3l3/4+ cells in the tumors from the DAC-treated mice in both the timed endpoint and survival paradigms (Fig. 4A–C), indicating a non-cell autonomous effect of DAC through recruitment and/or local expansion of this cell population. A combination of this strongly increased number of Chi3l3/4+ cells, presumably together with an increase in Chi3l3/4 mRNA expression per cell, likely accounts for the ~17-fold net over-expression of Chi3l3 mRNA that we observed by RNA-seq.
Figure 4. Increased Chi3l3/4-positive tumor-associated macrophages in KPC tumors following DAC and DAC+ICI treatment.
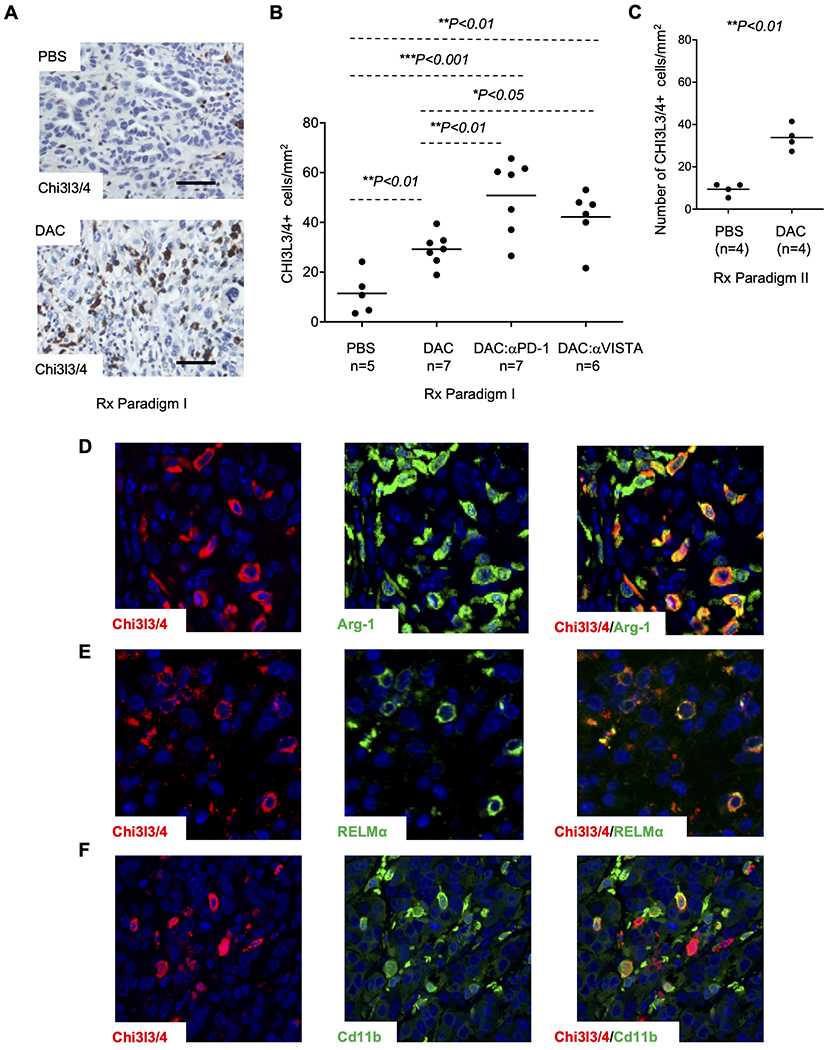
(A) IHC reveals a marked increase in tumor-infiltrating Chi3l3/4+ cells after DAC treatment. Fields are chosen to be representative. (B) Counting of Chi3l3/4+ cells per tumor area reveals a 3-fold increase in the numbers of these cells following DAC treatment, compared to PBS vehicle control, in treatment Paradigms I. In the combination treatment arms using DAC followed by ICIs there is a further increase in tumor-infiltrating Chi3l3/4+ cells (C) Counting of Chi3l3/4+ cells per tumor area reveals an a 3-fold increase in the number of these cells in DCA treated tumors compared to PBD in treatment Paradigm II. (D) Chi3l3/4 and Arginase-1 show substantial overlap, but there are also many Arginase-1+; Chi3l3/4− cells. (E) Double IF for Chi3l3/4 and RELMα shows substantial overlap of these two markers. (F) Double IF reveals partial overlap of Chi3l3/4 with CD11b. Original magnification, 20X; scale bars, 100 μm.
We next used a panel of markers in IHC/IF and flow cytometry to assign the tumor infiltrating Chi3l3/4+ cells to a specific cell type in the myeloid lineage. As expected, by two-color IF we found a lack of co-immunostaining with markers for malignant epithelial cells (mutant p53), stromal myofibroblasts (αSMA), and lymphocytes (CD3, CD103) (Supplementary Fig. S7A–D). Dual IF with known macrophage markers, showed that the majority of Chi3l3/4+ cells are also positive for the M2 macrophage markers Arginase-1 (Arg-1) and RELM-α, and a subset of these cells are positive for CD163, F4/80, and CD11b, with only a small percentage of the Chi3l3/4+ cells being positive for the dendritic cell marker CD103 (Fig. 4D–F and Supplementary Fig. S7C). The subset of Chi3l3/4+ cells that were positive for Cd11b had a variable and relatively low intensity of staining for this antigen; whereas those cells strongly positive for CD11b were mostly Chi3l3/4−. Although nearly all Chi3l3/4+ cells were positive for Arg-1, we also observed many Arg-1+;Chi3l3/4− and CD11b+;Chi3l3/4− cells, as well as some CD163+;Chi3l3/4− cells (Fig. 4D and Supplementary Fig. S7D). These results indicate the Chi3l3/4+ cells are a specific subset of M2 macrophages and are consistent with our finding that there was no difference in the mean number of Arg-1+ cells in the tumors from the PBS vs. DAC treatment arms (Fig. 5A). Lastly, relevant to a potential role in immune checkpoints, by two-color IF we also found that most Chi3l3/4+ cells also express PD-L1 (Fig. 5B).
Figure 5. The tumor-infiltrating Chi3l3-positive cells are a subset of Arginase-1 and DAC affects TILs.
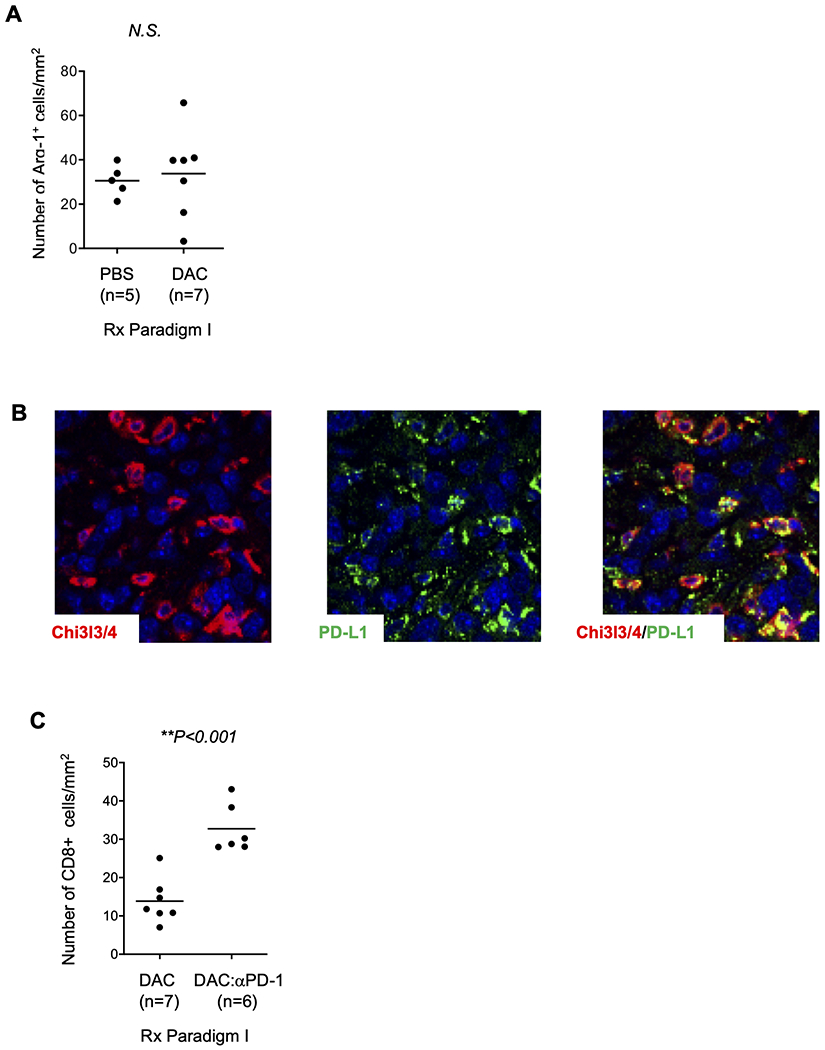
(A) The total number of Arg1+ cells is not increased by DAC treatment. (B) Double IF reveals partial overlap of Chi3l3/4 with PD-L1, although the two antigens have different subcellular distributions (punctate pattern for PD-L1). Data are from treatment Paradigm I. (C) Sequential combination therapy with DAC followed by anti-PD-1 leads to an increase in CD8+ TILs, compared to treatment with DAC alone. The number of cases for cell counting reflects the number of histological blocks available.
We supplemented these IHC/IF results with flow cytometry, which showed that most Chi3l3/4+ cells are CD11c− ;CD103−; CX3CR1+, indicative of macrophages, rather than dendritic cells. The majority of Chi3l3/4+ cells were positive for CD11b by flow cytometry, however the level of CD11b expression in most of these cells was low. Also, by flow cytometry, among CD11b+Chi3l3/4+ cells approximately 40% were F4/80+ (Supplementary Fig. S8A–C). Although a subset of Chi3l3/4-positive cells did not express high levels of the classical macrophage marker CD11b, these also did not express the dendritic cell marker CD103 (Supplementary Fig. S7C). Taken together, these results suggest that the majority of tumor-infiltrating Chi3l3/4+ cells are a unique subset of Arginase-1 and PD-L1-expressing M2-polarized macrophages that are recruited or polarized by paracrine signals.
When we examined tissue sections from treatment Paradigm III pancreata to assess PanIN lesions, we observed that Chi3l3/4+ macrophages are specifically present around these pre-malignant lesions, with very few such cells seen in the normal pancreatic parenchyma and around non-neoplastic ducts. Although these cells are clearly homing to the PanIN lesions, they were seen around PanIN both in the DAC-treated and in the PBS control mice, and we did not find a significant DAC-dependent increase in their numbers (Supplementary Fig. S9A, B). Similarly, we did not find a significant increase in Arg-1 or CD163-positive cells in DAC-treated mice with PanIN lesions (Supplementary Fig. S9C–E).
DAC effects can be studied in allograft tumors
While the autochthonous KPC tumors are the closest mimic of human PDACs, it is desirable to have alternative experimental systems that can give faster results without requiring serial U/S monitoring of large numbers of mice. To characterize DAC effects in an immunocompetent allograft system, we created syngeneic subcutaneous allografts using a KPC tumor cell explant line developed from a tumor occurring in our mouse breeding colony, with subcutaneous implantation of these cells into littermate host mice. Using an identical dose regimen to the in vivo experiments, we found a significant effect of DAC in reducing tumor size at a defined endpoint (similar to in vivo treatment Paradigm II), and we observed increased numbers of Chi3l3/4+ cells in the DAC treated allograft tumors (Supplementary Fig. S10A, B).
Combination therapy with DAC plus ICIs leads to further increases in tumor infiltrating CD8+ T cells and Chi3l3/4+ macrophages
Given this encouraging data from single-agent DAC, we next evaluated sequential DAC and immune checkpoint blockade, starting with an assessment of effects on tumor infiltrating lymphoid and myeloid cells. We chose to evaluate two ICIs, anti-PD-1 and anti-VISTA. VISTA is an immune checkpoint family member (33–35) that has been less studied as a therapeutic target than the related molecule PD-1. However, it is expressed in more broadly in lymphoid cells and has recently been shown to be one of the more abundant immune checkpoint mediators in PDAC (36,37). We observed a significant increase in VISTA+ TILs after DAC treatment (Supplementary Fig. S11A). By analyzing tumors from treatment Paradigm I, we found that both of the combination therapies (DAC followed by anti-PD-1 and DAC followed by anti-VISTA) led to a further increase in CD8+ TILs (Fig. 5C). Although this observation supports a potential benefit, we also noted that the putatively immunosuppressive Chi3l3/4+ myeloid cells were increased by the DAC plus ICI combinations (Fig. 4B and Supplementary Fig. S11B).
Combination therapy with DAC plus ICIs decreases tumor growth and increases overall survival
We next evaluated the effects of sequential DAC plus immune checkpoint blockade on tumor growth and mouse survival, using treatment Paradigm I. We observed a significant slowing of tumor growth (prolongation of doubling time) with single-agent DAC, anti-PD1, and anti-VISTA compared to PBS controls, with further tumor growth inhibition in the DAC + anti-VISTA and DAC + anti-PD-1 treatment arms (p<0.01 compared to PBS controls; Fig. 6A, B). As shown in Figure 6C, D, mouse survival was minimally prolonged by the single-agent ICIs, and DAC alone modestly prolonged median survival (32 vs 26 days; p<0.05). Both DAC + anti-VISTA and DAC + anti-PD-1 prolonged survival significantly compared to the PBS control arm (41 vs 26 days; p<0.0001 and 54 vs 26 days; p<0.0001, respectively) and there was a significant increase in survival in DAC + anti-PD-1 compared to DAC alone (54 vs 32 days; p<0.01) or anti-PD-1 alone (54 vs 30 days; p<0.01). Although the best combination of agents (DAC followed by anti-PD-1) doubled survival in the mice, the treatment was not curative, and few animals survived beyond 80 days (Figure 6D).
Figure 6. Effect on tumor growth and mouse survival of single-agent and combination therapy with DAC and immune checkpoint inhibitors.

(A) There is a significant increase in tumor doubling time (slowing of tumor growth) with single-agent DAC, single agent anti-VISTA, and sequential DAC followed by anti-VISTA, compared to PBS vehicle controls. The greatest inhibition of tumor growth is seen with the sequential combination therapy. Error bars indicated standard deviations. (B) Tumor doubling time is increased with single-agent DAC, and sequential DAC followed by anti-PD-1 treatment, compared to PBS vehicle controls. (C) Kaplan-Meier analyses show that combination therapy with DAC followed by anti-VISTA produces a modest but significant increase in mean survival compared to PBS (log rank ***p<0.0001), and that single-agent DAC confers a weaker benefit (log rank *p<.05). (D) Kaplan-Meier analyses show modest benefits with single agent DAC and single agent anti-PD-1 (both log rank *p<.05) and a major increase in mean survival with the DAC followed by anti-PD-1 combination treatment, (log rank ***p<0.0001) compared to PBS vehicle controls. The combination therapy was also significantly more effective than anti-PD-1 alone or DAC alone (log rank p<0.01 and log rank p<.05). Data are from treatment Paradigm I. #The PBS vehicle control arm includes 10 concurrent controls plus 3 historical controls from an identical tumor detection and mock treatment initiation procedure. The differences in mean survival between treatment arms remained statistically significant when only the concurrent controls were considered.
Discussion
Despite the efficacy of immune checkpoint inhibitors against some cancers, (38,39) such benefits have not yet been seen in human PDACs. Explanations could include high expression of immune checkpoint receptors, and a paucity of infiltrating cytotoxic T cells or antigen presenting cells (40), which may be due to low antigenicity of the cancer cells or immune suppressive properties of the tumor stroma (41–43). Strategies to overcome these roadblocks in preclinical models have included combinations of immune modulator agents and cytokines,(44,45) and combining immune modulators with epigenetically acting drugs (46–49). We previously found that low dose DAC inhibits tumor growth and prolongs survival in the rapid-onset KPC-Brca1 mouse model of PDAC (10). Here we have built on these findings to study the effects of DAC, and DAC plus immune modulating therapy, against PDACs in the more commonly used slower-onset KPC model, which is genetically similar to most human PDACs and which facilitates treatment protocols starting at an U/S-defined tumor size, which is more analogous to the human clinical situation.
Our most significant findings using single-agent DAC in this model were increased tumor necrosis, slowing of tumor growth, increased numbers of CD4+, CD8+, PD1+, and VISTA+ TILs, and an influx and/or intra-tumoral expansion of Chi3l3/4+ tumor-associated macrophages. While hypomethylating drugs are sometimes criticized for theoretically producing widespread changes in gene expression, in fact our RNA-seq data from the in vivo system revealed only small sets of differentially expressed genes in the whole KPC tumors, with the DAC-induced expansion and/or recruitment of the Chi3l3/4+ myeloid cell subpopulation reflected both in IHC findings and in the gene expression profiling, in which Chi3l3 was the most highly induced gene.
Our evidence based on IF and flow cytometry suggests that the tumor infiltrating Chi3l3/4+ cells are a sub-type of alternatively polarized (M2) macrophages. Prior studies have defined a population of M2-polarized macrophages that express Arginase-1, Chi3l3/4, and the resistin-like molecule (RELM)-alpha/FIZZ1 (50,51) – a marker profile that matches the cell population that expanded in response to DAC in our experiments. M2-polarized macrophages have been associated with immunosuppressive functions and unfavorable cancer outcomes (51–53). Among possible mediators, Arginase-1, which we have shown here is highly expressed by Chi3l3/4+ macrophages in the KPC tumors, has been proposed as a key regulator via its effects on reactive oxygen species (32,53,54). Recent data further suggest that myeloid cells in the pancreatic tumor stroma directly inhibit CD8 cell function (55).
Paracrine effects likely contribute to the recruitment or polarization of this myeloid cell population. Regarding possible direct effects of DAC on myeloid cells, the complex tumor microenvironment, as known from prior studies and highlighted here by our RNA-seq based deconvolution analysis, makes identification of methylation changes in subpopulations of cells difficult. Indeed, when we performed bisulfite sequencing on whole tumor DNAs we were unable to document detectable changes in Chi3l3 promoter methylation, despite the clear induction of this gene visualized at the level of protein products in subpopulations of cells. Nonetheless, we were able to identify direct effects of DAC on induction of ERVs and expression of dsRNA in tumor cells, which may have downstream consequences for immunogenicity of these cells.
Several different ICI molecules are expressed on the surface of T lymphocytes. We found that not only did the total number of effector T cells (CD8+ and CD4+ TILs) increase with DAC treatment, but that T cells expressing two checkpoint molecules, PD1 and VISTA, were also increased. VISTA/PD1-H is one of the more recently recognized family members, which has similarities with both PD-1 and PD-L1 (34,56). Antibodies against VISTA have not been used in humans, but genetic deletion of this molecule produced T cell activation and autoimmunity in mice (33,57), and in one study a greater accumulation of leukocytes was seen in the pancreas after inhibition of VISTA compared to PD-1 (35). Our results showing recruitment of these T cells and increased expression of both PD1 and VISTA provided a rationale for targeting both of these molecules in combination with DAC.
Based on these considerations, we tested the in vivo efficacy of sequential combination therapy using DAC followed by two different ICIs, anti-VISTA or anti-PD-1. We observed inhibition of tumor growth and a major extension of mean survival with DAC followed by anti-PD-1. Compared to DAC+anti-PD-1, there was slightly more inhibition of tumor growth with DAC followed by anti-VISTA, but this difference in growth inhibition was not a statistically significant, and we found less extension of survival with the DAC+anti-VISTA combination. Given the complicating considerations of tumor necrosis affecting the U/S interpretation of net tumor growth, and the role of locally invasive disease followed by sepsis, and potentially metastatic disease (11,58), as causes of death, a positive but somewhat variable correlation of tumor growth inhibition with extension of survival is not unexpected.
Overall, these findings have clear and promising implications for the design of future studies to improve the effectiveness of epigenetic modulation, with DAC or other DNA methyltransferase inhibitors, or with drugs targeting other epigenetically acting enzymes, in combination with immune checkpoint inhibition. Despite the clear beneficial effects of adding DAC to ICI therapy, our observations using both single agent DAC and its combination with the ICIs raise the possibility that the influx and/or local expansion of M2-polarized tumor-associated macrophages might be limiting the efficacy of this approach. Tumor promoting functions of M2-polarized TAMs are well documented both generally and in PDACs (53,59). Even without drug treatment, cancer-associated myofibroblasts (CAFs), which are abundant in KPC tumors, are known to produce cytokines that affect macrophages (60,61), and tumor epithelial cells in this model produce GM-CSF, which affects myeloid cell populations (62). In the current study, recruitment of the Chi3l3/Ym1-positive sub-class of M2 macrophages further increased when DAC was combined with ICIs. It is reasonable to suggest that the initial effect of DAC, perhaps in part driven by ERV induction and dsRNA expression, is beneficial and leads to accumulation of effector T cells. However, subsequently a more suppressive myeloid population accumulates in the tumor and this may restrict efficacy of the immune response. This observation suggests that targeting of myeloid cells might be a useful strategy to further enhance efficacy of epigenetic therapy. Encouragingly, approaches for such targeting in preclinical models are now becoming available (5,63). Lastly, given the low toxicity of DAC that we have observed both in the KPC model and previously in KPC-Brca1 mice, follow-up studies should also test more frequent DAC and anti-PD-1 dosing and longer treatment, as well as combining hypomethylating drugs with dual immune checkpoint inhibition. Encouragingly in this regard, the current dosing schedule for DAC was without systemic toxicities, and in two tumor-bearing KPC mice we have also tested a more aggressive 5-dose regimen of anti-PD1 given every 48 hours and found no evidence of adverse systemic effects during the period of ICI administration.
Supplementary Material
Significance.
In a pancreatic cancer model, a DNA hypomethylating drug increases tumor-infiltrating effector T cells, increases a subset of M2 macrophages, and significantly prolongs survival in combination with immune checkpoint inhibitors.
Acknowledgement
The authors thank Stephen Goff, PhD (Columbia University) and Robert Gifford, PhD (University of Glasgow) for their advice and assistance with retroviral analyses.
Grant support
Supported by grants from the National Institutes of Health U54 CA126513 (to T.C.W. and B.T), K08DK100544 (to T.G.), from the Merck Medical Innovation Science Program (to B.T. and T.G.), AACR-Landon Innovator Award for Cancer Prevention Research (to T.G.) and the Leokadia and Robert Lynn Research Fund (to T.G.).
Footnotes
Disclosure
The authors declare no potential conflicts of interest.
References
- 1.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. Journal of immunotherapy (Hagerstown, Md : 1997) 2010;33:828–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey P, Chang DK, Forget MA, Lucas FA, Alvarez HA, Haymaker C, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Scientific reports 2016;6:35848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan E, El-Rayes B. Pancreatic Cancer and Immunotherapy: Resistance Mechanisms and Proposed Solutions. J Gastrointest Cancer 2019;50:1–8 [DOI] [PubMed] [Google Scholar]
- 5.Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 2016;22:851–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lomberk G, Blum Y, Nicolle R, Nair A, Gaonkar KS, Marisa L, et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat Commun 2018;9:1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res 2013;73:885–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell 2005;7:469–83 [DOI] [PubMed] [Google Scholar]
- 12.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY) 2009;324:1457–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim CC, Lanier LL. Beyond the transcriptome: completion of act one of the Immunological Genome Project. Curr Opin Immunol 2013;25:593–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics 2013;29:1083–5 [DOI] [PubMed] [Google Scholar]
- 18.Kazachenka A, Bertozzi TM, Sjoberg-Herrera MK, Walker N, Gardner J, Gunning R, et al. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance. Cell 2018;175:1259–71 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G, Ji Y, Liu C, Li J, Zhou Y. Reduced levels of p15INK4b, p16INK4a, p21cip1 and p27kip1 in pancreatic carcinoma. Mol Med Rep 2012;5:1106–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 2014;123:e110–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmieder A, Schledzewski K, Michel J, Schonhaar K, Morias Y, Bosschaerts T, et al. The CD20 homolog Ms4a8a integrates pro- and anti-inflammatory signals in novel M2-like macrophages and is expressed in parasite infection. Eur J Immunol 2012;42:2971–82 [DOI] [PubMed] [Google Scholar]
- 22.Yu B, Sondag GR, Malcuit C, Kim MH, Safadi FF. Macrophage-Associated Osteoactivin/GPNMB Mediates Mesenchymal Stem Cell Survival, Proliferation, and Migration Via a CD44-Dependent Mechanism. J Cell Biochem 2016;117:1511–21 [DOI] [PubMed] [Google Scholar]
- 23.Ripoll VM, Irvine KM, Ravasi T, Sweet MJ, Hume DA. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J Immunol 2007;178:6557–66 [DOI] [PubMed] [Google Scholar]
- 24.Ericson JA, Duffau P, Yasuda K, Ortiz-Lopez A, Rothamel K, Rifkin IR, et al. Gene expression during the generation and activation of mouse neutrophils: implication of novel functional and regulatory pathways. PLoS One 2014;9:e108553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 2006;116:2777–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol 2012;22:289–97 [DOI] [PubMed] [Google Scholar]
- 27.Gordon S, Pluddemann A. Tissue macrophage heterogeneity: issues and prospects. Semin Immunopathol 2013;35:533–40 [DOI] [PubMed] [Google Scholar]
- 28.van Rijt LS, Vos N, Willart M, Muskens F, Tak PP, van der Horst C, et al. Persistent activation of dendritic cells after resolution of allergic airway inflammation breaks tolerance to inhaled allergens in mice. Am J Respir Crit Care Med 2011;184:303–11 [DOI] [PubMed] [Google Scholar]
- 29.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunology letters 2003;85:173–80 [DOI] [PubMed] [Google Scholar]
- 30.Welch JS, Escoubet-Lozach L, Sykes DB, Liddiard K, Greaves DR, Glass CK. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J Biol Chem 2002;277:42821–9 [DOI] [PubMed] [Google Scholar]
- 31.Webb DC, McKenzie AN, Foster PS. Expression of the Ym2 lectin-binding protein is dependent on interleukin (IL)-4 and IL-13 signal transduction: identification of a novel allergy-associated protein. J Biol Chem 2001;276:41969–76 [DOI] [PubMed] [Google Scholar]
- 32.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res 2001;61:1100–6 [PubMed] [Google Scholar]
- 33.Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci U S A 2014;111:14846–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med 2011;208:577–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A 2015;112:6682–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res 2017;23:3129–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A 2019;116:1692–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375:1823–33 [DOI] [PubMed] [Google Scholar]
- 39.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372:2521–32 [DOI] [PubMed] [Google Scholar]
- 40.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 2007;13:2151–7 [DOI] [PubMed] [Google Scholar]
- 41.Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res 2014;2:187–93 [DOI] [PubMed] [Google Scholar]
- 42.von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol 2017;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer cell 2017;32:654–68 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018;67:320–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dear AE. Epigenetic Modulators and the New Immunotherapies. N Engl J Med 2016;374:684–6 [DOI] [PubMed] [Google Scholar]
- 47.Hessmann E, Johnsen SA, Siveke JT, Ellenrieder V. Epigenetic treatment of pancreatic cancer: is there a therapeutic perspective on the horizon? Gut 2017;66:168–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017;171:1284–300.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A 2017;114:E10981–e90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nair MG, Du Y, Perrigoue JG, Zaph C, Taylor JJ, Goldschmidt M, et al. Alternatively activated macrophage-derived RELM-alpha is a negative regulator of type 2 inflammation in the lung. J Exp Med 2009;206:937–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016;150:1659–72 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wan G, Xiang L, Sun X, Wang X, Li H, Ge W, et al. Elevated YKL-40 expression is associated with a poor prognosis in breast cancer patients. Oncotarget 2017;8:5382–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Partecke LI, Gunther C, Hagemann S, Jacobi C, Merkel M, Sendler M, et al. Induction of M2-macrophages by tumour cells and tumour growth promotion by M2-macrophages: a quid pro quo in pancreatic cancer. Pancreatology 2013;13:508–16 [DOI] [PubMed] [Google Scholar]
- 54.Singer K, Gottfried E, Kreutz M, Mackensen A. Suppression of T-cell responses by tumor metabolites. Cancer Immunol Immunother 2011;60:425–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017;66:124–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol 2011;187:1537–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest 2014;124:1966–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer cell 2016;29:832–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Habtezion A, Edderkaoui M, Pandol SJ. Macrophages and pancreatic ductal adenocarcinoma. Cancer Lett 2016;381:211–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017;8:8633–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pitarresi JR, Liu X, Sharma SM, Cuitino MC, Kladney RD, Mace TA, et al. Stromal ETS2 Regulates Chemokine Production and Immune Cell Recruitment during Acinar-to-Ductal Metaplasia. Neoplasia 2016;18:541–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer cell 2012;21:822–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ekiz HA, Lai SA, Gundlapalli H, Haroun F, Williams MA, Welm AL. Inhibition of RON kinase potentiates anti-CTLA-4 immunotherapy to shrink breast tumors and prevent metastatic outgrowth. Oncoimmunology 2018;7:e1480286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.