

Abstract
The growth of cancer tissue is thought to be considered driven by a small subpopulation of cells, so-called cancer stem cells (CSCs). CSCs are located at the apex of a hierarchy in a cancer tissue with self-renewal, differentiation and tumorigenic potential that produce the progeny in the tissue. Although CSCs are generally believed to play a critical role in the growth, metastasis, and recurrence of cancers, the origin of CSCs remains to be reconsidered. We hypothesise that, chronic diseases, including obesity and diabetes, establish the cancer-inducing niche (CIN) that drives the undifferentiated/progenitor cells into CSCs, which then develop malignant tumours in vivo. In this context, a CIN could be traced to chronic inflammation that involves long-lasting tissue damage and repair after being exposed to factors such as cytokines and growth factors. This must be distinguished from the cancer microenvironment, which is responsible for cancer maintenance. The concept of a CIN is most important for cancer prevention as well as cancer therapy.
Subject terms: Cancer microenvironment, Cancer stem cells
Introduction
Cancer is a malignant tumour resulting from an uncontrolled division of cells. During the last century, scientists have looked into cancer from the viewpoint of a genetic disease. Boveri first hypothesised that cancer could be caused by specific combinations of chromosomal gains and losses [1]. In 1916, Tyzzer first applied the term “somatic mutation” to cancer and Bauer wrote that the mutations caused cancer [2]. Peyton Rous and Mackenzie identified those somatic changes, which involve DNA mutations and may be caused by viral or chemical carcinogens that finally develop tumour [3]. Several scientists not only follow and support the mutation theory but also develop the idea that cancer initiates and develops by the progressive accumulation of genetic mutations [4–15].
Sonnenschein and Soto proposed that the mutation theory should be abandoned because initial observations induced by carcinogens appeared to disrupt the normal interactions between the cells in an organ’s parenchyma and stroma [16]. Other scientists thought the disruption might be due to inflammation affecting the microenvironment, and looked at cancer from the point of inflammation and came to believe in the strong relationship between inflammation and cancer. Since then, the relationship between cancer and inflammation has persisted. However, the molecular mechanisms in this link are still unknown while this link has currently become widely accepted. Looking back to 1863, Virchow hypothesised that cancer initiated at the site of a chronic inflammation [17]. In part, some classes of irritants that cause tissue injury together with ensuing inflammation enhance cell proliferation based on his hypothesis [18]. Actually, this should be related to the result of inducing tumours on the ears of rabbits by painting them with a coal tar-benzene solution [19]. In inflammation, the homoeostasis of tissue is destabilised but will return to normal state after the inflammation ceases [20]. However, in chronic inflammation, normal cells are kept activated for tissue repair. Then they acquire the phenotype of the shifted homoeostasis and respond irreversibly to the continued inflammation, which causes the so-called malignancy.
More precisely, in a normal microenvironment tissues metabolically renew themselves by controlled cell division to keep a balance between growth, differentiation and death. In this condition of homoeostasis, the microenvironment plays a critical role in maintaining the usual tissue architecture by preventing the disorder leading to the malignant phenotype. The relationship between normal stem cells and inflammation in the surrounding microenvironment could occasional result in cancer initiation. Postulating the transformation of stem cells or progenitor cells into a cancer phenotype in inflammation, we may be able to uncover many missing links in carcinogenesis. Some scientists are now thinking about the cancer stem cell (CSC) theory as a suitable alternative for a mutation theory while others think it complementary.
CSC theory states that tumour growth is fuelled by a small subpopulation of cancer cells with a stem like character hidden in cancers. Tumours generated on the basis of CSCs are believed to follow a unidirectional hierarchy, in which only the CSC population is responsible for tumour growth [21, 22]. At the time of tumour initiation, it is suggested that CSCs divide asymmetrically to maintain the CSC pool [23]. Once CSCs appear, cells self-renew and differentiate into progeny through asymmetric division. If the self-renewing ratio is higher than the differentiation, cells will apparently undergo symmetric division resulting in proliferation. The CSCs have been demonstrated to possess the ability to differentiate into various cell types elucidating tumour heterogeneity [24–28].
Although researchers do not deny the presence of CSCs, few are investigating the origin of cancer, which is left in mystery; in other words, the point of appearance of CSCs at which normal cells acquire the malignancy is still not clear. In this context, a CSC could logically and simply be defined as a cell that has the capacity of self-renewal, differentiation and tumorigenicity. Essentially, the tumorigenic potential in vivo is important; otherwise, there is no difference between stem cells that develop an organ, a tissue or a body. According to this definition, several hypotheses have been proposed that the events in either stem and/or differentiated cells, such as genomic instability, inflammatory microenvironment, cell fusion, and lateral gene transfer, could be considered as the possible origin of CSCs (Fig. 1).
Fig. 1. An overview of the cancer initiation and development.

Both differentiated and undifferentiated (stem/progenitor state) normal cells could be the source of cancer initiation. Under chronic conditions, different cytokines and growth factors stimulate undifferentiated cells to become into cancer stem cells (CSCs). On the other hand, mutations by mutagens, viral infection and environmental stresses are traditionally believed to allow the cellular transformation. Cell fusion is also conceivable by the environmental stresses and infection of virus such as HVJ may induce cancer stem cells (CSCs) to originate from stem cells or differentiated cell affected by chronic inflammation, mutation, or viral infection. However, chronic conditions could be explained as a major part of the cause of cancer death, which is currently sporadic.
The breakthrough of induced pluripotent stem cells (iPSCs) has opened up unique prospects in scientific research societies, especially regeneration therapy. On the other hand, this progress in stem cell research opens the possibility of dropping the mutation theory by demonstrating the CSCs development. During a decade of research into CSCs development, Masaharu Seno’s group demonstrated that the inflammatory microenvironment could drive stem cells into CSCs that exhibited self-renewal, differentiation and tumorigenic potential [29–34]. All these studies contribute to the hypothesis of inducing CSCs from stem cells under cancerous microenvironments, the cancer-inducing niche (CIN). Aprecise definition of CIN would be a great help in preventing and defeating cancer from the outset.
Cancer-inducing niche concept
Carcinogenesis is the process of initiation of a cancer, during which normal cells are thought to acquire malignancy characterised by genetic and epigenetic levels alterations. Although the exact steps of carcinogenesis appear to be explained by somatic mutation theory, much evidence says that this theory is not always applicable. On the other hand, chronic inflammation is considered to be a potential risk factor for cancer development [35].
It is commonly accepted that more than 20% of human malignancies are related to chronic inflammation induced by viral and bacterial infection [36].
To understand the relationship between chronic inflammation and cancer initiation, it is necessary to consider in detail the process of inflammation, a physiologic process in response to tissue damage [37], that is usually caused by an immune response to a pathogen [38].
Inflammation is initiated by resident immune cells in tissues such as macrophages, histiocytes, Langerhans cells (LC), dendritic cells, (DCs) Kupffer cells, and mast cells [39–41]. These cells release chemical factors such as leukotrienes, histamines, and prostaglandins, which recruit immune cells and activate surrounding cells to express cytokines including interferons, interleukins, lymphokines, tumour necrosis factors and chemokines to establish an essential barrier against the pathogens [42, 43]. The activated immune cells produce reactive oxygen species (ROS), which may injure the tissues.
A variety of chemokines is induced by viral infection or bacterial invasion and determine the migration of inflammatory leucocytes into the damaged area. Typical chemokines are CXCL1, 2, 3, 5, 6, 7 and 8 for neutrophils [44] and CCL2, 3, 4, 5, 7 and 12 for monocytes/macrophages [45] while CCL1, 2, 17 and 22 recruit T-lymphocytes to the site of the wound or injury inducing inflammation. Then, wound healing is following by secreted growth factors such as epidermal growth factors (EGF) fibroblast growth factors (FGF), platelet derived growth factors (PDGF), vascular endothelial growth factors (VEGF) transforming growth factor betas (TGF-b), granulocyte macrophage colony stimulating factor (GM-CSF), and connective tissue growth factor (CTGF) [46]. This process is coordinated by several cell types (keratinocytes, fibroblasts, endothelial cells, macrophages, and platelets), that exhibit the migration, infiltration, proliferation, and differentiation. For the activation of gene expression, no mutations or genetic rearrangements are required but epigenetic effects, such as acetylation of histones and hypomethylation of CpG islands are required.
In the end, the site of the injury will recover over a short period while the time necessary for the completion of wound healing depends on the severity of the injury [47], and the epigenetic alterations will return to normal. However, repeated or continuous inflammation will maintain the epigenetic effects, thereby leading to chronic inflammation resulting in organ dysfunction such as fibrosis due to the continuous exposure to the cytokines, chemokines and especially growth factors, which in turn initiate cancer (Fig. 2). In fact, we can imagine that the continuously activated tyrosine kinases or serine/threonine kinases alter the character of cells just mutated transformed cells do in oncogenic tyrosine kinases or tumour suppressing phosphatases [35].
Fig. 2. Schematic drawing of the development of Cancer-inducing niche and stem cell alternation.

Against invaders, resident tissue macrophages start the attack while other immune cells infiltrate into the tissue from the blood vessels. These cells secrete cytokines and chemokines killing the invaders and damaging tissues in the microenvironment of inflammation, after which wound healing by activated cell growth occurs. Chronic inflammation results from a continuous immune response that keeps the microenvironment filled with pro-inflammatory mediators and growth factors, which in turn initiate cancer converting stem cells or progenitors into cancer stem cells.
Thus, the initial phase of tumour development could be considered to be the point of inflammation that affects epigenetic alterations from normal to pre-malignant lesions with enhanced proliferation and silenced tumour suppressor genes. This what, we define as the “cancer-inducing niche” (CIN), which convert stem cells into CSCss. We previously postulated that stem cells converted into CSCs if the CIN developed because of an irregular change in the microenvironment that affected on stem cell plasticity. Also, the mechanisms by which a CIN regulates or induces premalignancy are not yet established. Its mechanisms of normal to cancerous could be explained by continuous exposure to the components of chronic inflammation such as growth factors, cytokines, extracellular matrices and so on. The intracellular events should be an epigenetic modification that destabilises the genome to be transcribed coupled with the control of transcription factors by cytoplasmic signal transduction triggered by receptor ligand stimulation.
As for the changes in gene expression, genetic mutation or rearrangement are not always feasible in a chronic situation; however, epigenetic effects, such as nucleotide methylation, histone modification, chromatin remodelling, and non-coding RNA, such as microRNAs (miRNAs) are feasible. These mechanisms appear reversible and responsible for determining the landscape of cell fate [48], but the silencing and enhancing of gene expression over the long-term can reprogramme the cells with pluripotency, which is very important during normal mammalian development and ESCs differentiation [49] In this context, long-term effects such as chronic inflammation could determine the fate of cell plasticity by altering the intracellular signals that control self-renewal and differentiation, thereby converting stem cells into CSCs.
Several intracellular signalling cascades are triggered by growth factor receptor tyrosine kinases, which integrate a wide range of input signals. This leads to cell proliferation and survival as well as differentiation resulting in adhesion, migration and metabolism [50]. A number of chemokines including GROα/CXCL1, GROβ/CXCL2, GROγ/CXCL3 and IL-8/CXCL8 regulate neoplastic cell proliferation in melanoma [51].
Regarding intracellular signalling, the activation of the signal transducer and activator of the transcription (STAT) family, in particular STAT3, is broadly implicated in the tumorigenesis of multiple tissues, and is closely linked to inflammatory gastric, colon, liver, lung and pancreatic processes [52–54].
IL-6, produced by myeloid cells, activates STAT3, which promote the proliferation of pre-malignant cells and inhibits apoptosis in colitis-associated colorectal cancer [53, 55, 56]. The molecular function of STAT3 promoting cell proliferation is thought to upregulate the expression of cell-cycle regulators, such as cyclin D1, cyclin D2, and cyclin B, and the proto-oncogene MYC. It also promote cell survival by upregulating the expression of anti-apoptotic genes, such as BCL2 and BCL2-like1 (BCL2L1), which encodes BCL-XL [53, 55, 57]. Likewise, TNF was later shown to be a mediator of cancer initiation and progression by leucocytes infiltrating into the lamina propria and submucosa of the colon [58]). As a further response to inflammation, macrophages release cytokines (e.g., CCL3, IL-1RA, osteopontin, M-CSF1 and GDNF), which stimulate cell proliferation by activating ERK and Akt kinases [59]). Altogether, these data suggest that an inflammatory process accompanied by oncogenic activation may lead to the amplification of the NFκB/IL-6/STAT3 axis, which results in cancer initiation.
From another point of view, scientists are linking inflammation with mutation. Inflammation may be a factor causing neoplastic transformation by increasing the damage to DNA and impairing DNA repair mechanisms crucial for maintaining genomic integrity, thereby contributing to genomic instability. These mutations can either act directly or indirectly on epithelial cells, either through intrinsic or extrinsic action.
In an inflammatory microenvironment, highly reactive nitrogen and oxygen species released from inflammatory cells are thought to damage cells and the repeated regeneration of damaged tissue causes DNA mutations that result in permanent alterations such as point mutations, deletions, or rearrangements. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generally released by neutrophils and macrophages, or are intracellularly induced to cause DNA damage in pre-malignant cells [60–62].
TNF, IL-1, −4, -13, and TGFβ have all been shown to induce ectopic expression of activation-induced cytidine deaminase (AID), also called activation-induced cytosine deaminase (AICDA), a DNA and RNA cytosine deaminase family member which introduces mutations into crucial cancer-associated genes, which include TP53 (encoding p53) and MYC [63–65]. In fact, p53 mutations are observed at similar frequencies in tumours that arise in chronic inflammation associated with, for example, rheumatoid arthritis and inflammatory bowel disease [66].
All these data could uncover ways to define the CIN, which affects the first stage of CSC generation and be distinguished from “cancer niche”. These two niches are quite different because the “cancer-inducing niche” (CIN) is the microenvironment that supports the appearance of cancer-related cells and can differ from organ to organ; while “cancer niche” is the microenvironment provided by the cancer cells themselves to maintain or survive. This difference in CIN from organ to organ, will develop different conversion stages, which depend on the content of cytokines, chemokines, and other inflammatory factors which eventually lead to developing different CSCs. The tissue-specific phenotype of the CSCs could be determined by the environment specific to each organ during the conversion of stem cells or progenitor cells. We describe the cases of the liver, mammary gland and pancreas in the following sections.
Liver cancer-inducing niche
Many studies have demonstrated that inflammation is closely related to cancer initiation and development. Most liver cancer arise from chronic inflammation, which results from hepatitis induced by viral infection from HBV and HCV, hemochromatosis and chronic alcohol abuse [67–69]. Other cases are irregular conditions in the liver affected by diabetes and obesity, nonalcoholic fatty liver disease and nonalcoholic steatohepatitis (NASH).
Inflammation is a fundamental immune response that follows injury and wound healing. During this process, several molecules growth factors, cytokines, and chemokines are involved which are released by damaged tissues and activated immune cells. If successful, the wounds will be cured in a short period, but if not, inflammation will last a long time and become chronic.
Typically, HBV and HCV infections are considered as the main risk factors for liver cancer [70]. After infection, the human hepatocyte starts to express the viral antigen to activate CD4+T– and CD8 + T-cells, which help clear the virus by cytolytic and noncytolytic effector mechanisms.
In response to the stimulation, the CD4 + T cells differentiate into Th1 to secrete IFN-γ, IL-2, TNF-α, and IFN to activate CD8 + T cells [71, 72] or into Th2 cells to secrete IL-4, -5, -9, -10, -13 and -25 to suppress Th1 cells and Th17 cells (Fig. 3), also induced from CD4 + T cells, and secrete specific inflammatory cytokines such as IL-17, -21, -22 and IFN-γ, which suppress the function of Treg cells [72–77]. The CD8 + T cells inhibit viral replication mediated by cytokines such as TNF-α, IFN-γ, IL-4, -5, -9, -10, -13, -17 and -21, and directly kill infected hepatocytes [71, 78–80].
Fig. 3. Schematic drawing of inflammatory microenvironment in the liver depicting the immune responses of different cells.

Different immune cells participate in the inflammatory microenvironment to stop or eliminate the invaders while damaged tissues are being repaired. When the immune response and wound healing repeatedly continues, the microenvironment becomes a cancer-inducing niche leading to the conversion of stem cells or progenitors into CSCs.
Cytokines activate different signalling pathways of which enhancement leads to cancer initiation. The mechanism could be similar to that observed in hepatocellular carcinoma (HCC) models induced with diethylnitrosamine, which mimics the inflammation induced by obesity- activating STAT3 pathway from IL-6 and TNF [81]. Similarly, IL-22 produced by infiltrating leucocytes initiates HCC by activating STAT3 and its downstream survival genes [82]. Thus, CIN could be explained by the role of the IL-6/STAT3 axis.
It could also be explained by the expression of the genes activated by NF-κB in inflammation resulting in cellular proliferation and survival [83–85]. NF-κB was the first molecular link established between inflammation and liver cancer [86].
Collectively, the liver CIN could be proposed as a microenvironment consisting of a wide range of cytokines and growth factors secreted by inflammatory responses of the immune system.
Pancreatic cancer-inducing niche
In the case of the pancreas, stromal desmoplasia and sustained activation of the immune system are also hallmarks of cancer initiation and development. Inflammation, in which activated immune cells secrete various cytokines and chemokines, has been identified as one of the cofactors in pancreatic carcinogenesis [87].
The ceaseless and continuous crosstalk between the progenitor cells and the innate and adaptive immune cells could promote cancer initiation by converting normal progenitors to cancer stem cells, thereby inducing malignancy such as angiogenesis. During crosstalk, interleukins (e.g., IL-1, −4, -8, -10), and chemokines (e.g., CXC-3, -4, -5, -8, -12, -13) regulate cellular migration, proliferation, and survival (Fig. 4), factors that are believed to be involved in tumorigenicity and characteristics of cancer cells. Pancreatic intra-epithelial neoplasia (PanIN), which is the precursor to invasive ductal adenocarcinoma, is thought to develop through different stages to pancreatic ductal adenocarcinoma (PDAC), which forms approximately 90% of pancreatic neoplasms [88, 89].
Fig. 4. Scheme of cancer initiation from inflammation in pancreas.

Generally, pancreatic duct cells contain progenitors differentiating into acinar and endocrine cells. In response to pancreatitis, immune system is activated and form the microenvironment of inflammation with cytokines and growth factors. Once the inflammation becomes chronic, the microenvironment turns to CIN converting the progenitors into CSCs, which in turn lead to tumour development.
The PanIN lesion microenvironment includes macrophages, neutrophils and fibroblasts. These cells are enriched around developing PanIN lesions and change the composition of the acinar cell microenvironment during PanIN progress [90]. Secreting CXCL17, macrophages and neutrophils recruit immature dendritic cells during PanIN progress. Therefore, downregulation of CXCL17 develops immune tolerance toward tumour cells. Once they become immunotolerant, some types of immune cells such as T helper type 2 cells (Th2) and T regulatory cells are attracted to the site of cancer initiation where pro-tumorigenic cells increase [91]. Th2 secretes IL-10 preventing the activation of dendritic cells and attracting different immune cells to PanIN lesions, the sites of cancer initiation [92]. IL-6 is essential for the maintenance and the progression of PanIN lesions. In the absence of IL-6, PanIN cells re-differentiated into normal acini cells and PanIN lesions gradually disappear. IL-6 was shown to activate MAPK and PI3K/AKT pathways in PanIN lesions with activated KRAS; hence, KRAS mutations are considered to be a link to PanIN development. It is conceivable that the progression of PanIN depends on the inflammatory microenvironment to allow the crosstalk between the lesion and the surrounding cells such as macrophages and fibroblasts [93].
Thus, the inflammatory factors in the microenvironment appear to support the expansion of PanIN lesions by establishing immunosuppressive and tumour-promoting conditions that mediate cancer initiation. Acinar cells with mutated KRAS were shown to stimulate inflammation probably because the mutant gene derived from a viral oncogene that had been expressed by the viral infection, which recruited immune cells (e.g., macrophages), resulting in the induction of inflammatory factors such as chemokine ligand 5 (CCL5) and tumour necrosis factor (TNF). This would lead to establishment of a chronic environment contributing to the formation of pre-neoplastic pancreatic lesions. The macrophages and fibroblasts in the inflammation help remodel the damaged acinar tissue by a wound-healing mechanism that inhibits metalloproteinases, which results in the induction of fibrosis [90, 94].
Swidnicka-Siergiejko et al. [95] recently reported that chronic inflammation induces pancreatic cancer in the absence of p53. They showed that upregulated expression of either IĸB kinase-2 (IKK2) or cyclooxygenase-2 (COX-2) led to chronic inflammation-induced DNA mutations such as on KRAS gene in the absence of p53 and to pancreatic cancers through various signalling pathways. The increase of acinar cells and infiltrated immune cells into pancreatic tissue were associated with the sustained expression of COX-2/IKK2 together with the mutation.
Kong et al. [96] independently defined the steps involved in pancreatic duct adenocarcinoma (PDAC) resulting from pancreatitis. They revealed that three phases inflammation, regeneration and refinement were responsible for carcinogenesis. In chronic pancreatitis, the infiltration of immune cells led to apoptosis and vacuolisation resulting in acinar-to-ductal metaplasia, which was sustained in KRAS mutant mice without regeneration but followed by fibrosis and the development of pancreatic intra-epithelial neoplastic lesions (PanINs). The loss of proliferation of suitable cells after inflammation due to changes of a complex molecular network were found to be responsible for early-stage carcinogenesis in KRAS mutant cells. Another relation between inflammation and oncogenic KRAS was reported by Lee et al. [97]. In that study, pancreatic ductal cell senescence caused by inflammation was found to be suppressed by expression of the oncogenic KRAS through transcription factor Twist, which was found to suppress p16INK4A. This mechanism is also thought to contribute to the initiation of PDAC. Consistent with these reports, Li et al. [98] found that inflammation accelerated tumorigenesis in mice with mutant KRAS allowing the appearance of proto-oncogenic progenitor cells. The cooperation of pancreatic lineage transcription factors and KRAS-activated transcription factors Fosl1 and Junb enhanced the oncogenic transformation of progenitor cells with the KRAS mutation.
Collectively, chronic inflammatory condition could cause PDAC by triggering CIN as one of the probable causes. The secreted factors will keep stimulating progenitors to promote the stages of cancer initiation. The KRAS mutations are not enough by themselves but repeated activation and crosstalk among different signalling pathways appear to be responsible for the proliferation and migration of the cells, which amplifies the effects of cellular interactions in the chronic microenvironments resulting in the initiation of PanIN lesions.
Ovarian cancer-inducing niche
Ovaries are oval, thumb-sized organs located on both sides of the uterus. They release mature ovules periodically until the end of menses. Notwithstanding important mutations in genes such as BRCA1 and BRCA2 [99–101], menopause is thought to be the risk factor of ovarian cancer. For postmenopausal women, this is the most common cancer and is usually found in the advanced stages, causes death in more than the half of all patients [99, 100].
At menopause, which is biologically significant and inevitable event for a female, the ovary loses all follicles and stops responding to both the follicle-stimulating and luteinizing hormones. As a results, the ovary will cease to produce progesterone or oestrogen. Progesterone induces immune tolerance and suppresses T cells [102], and oestrogen has various effects on immune cells, including B cell activation and T cell suppression [103]. In the menopausal and postmenopausal environment, pro-inflammatory serum markers such as IL-1, IL-6 and TNF-α will increase. Responding to these cytokines, immune blood cells increase, B and CD4+ T lymphocytes decrease, and the cytotoxic activity of NK cells is suppressed [104]. Hence, the immune system will become relatively downregulated resulting in the incomplete recovery of inflammation, leading to the chronic inflammation that works as the CIN.
Pelvic inflammatory disease (PID) is induced by infection of the female reproductive organs and is well known to cause ovarian cancer [105–107]. PID most often occurs by sexually transmitted microbials such as Chlamydia trachomatis or Neisseria gonorrhoeae, which spread from the vagina to the uterus, fallopian tubes or ovaries [106, 108, 109]. These can be treated with antibiotics, but the patients will not notice the disease until it gets worse if therapy is not completed. In the meantime, inflammation will be kept in the upper reproductive tract, including the ovaries, thereby starting the infection [110–112]. During infection, gene expressions of TNF, IL-8, CXCL1, CXCL12, GM-CSF, IL-6, and IL-1α are upregulated and secretion of these factors is also increased (Fig. 5). These chemokines attract immune cells and initiate and enhance inflammation [113]. Thus, the inflammation will become chronic and act as the CIN. Other studies showed that the microbials stimulate the secretion of IL-6, TNF-α, CXCL1, MIP-1α, and RANTES from macrophages more than from endothelial cells [114, 115].
Fig. 5. Ovarian cancer-inducing niches.
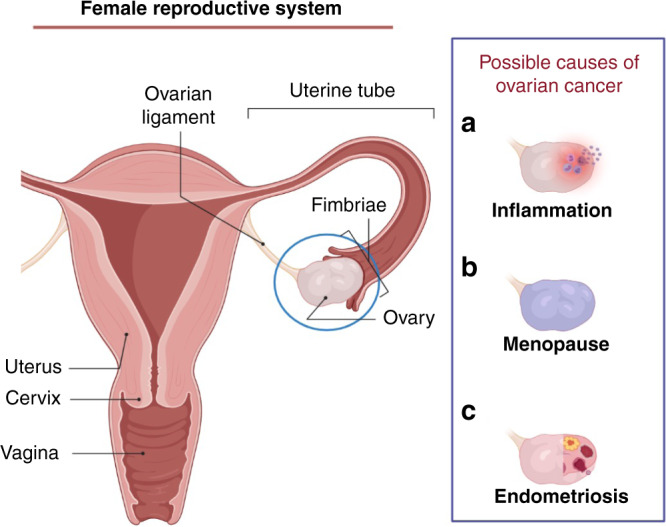
a Microbial infection sometimes occurs in the upper part of female reproductive system, which will cause inflammation in ovary in the end. During the inflammation, noted cytokines will be secreted and the niche can induce cancer. b At time of menopause, hormone secretions from the ovary will stop. This change will cause significant changes to the body including immune regulation, which can allow cancer development. c In chronic endometriosis cysts, some cytokines will attack their progenitor cells and cause cancer in the niche. IL, interleukin; TNF, tumour necrosis factor; CXCL, chemokine (C-X-C motif) ligand; CX3CL, chemokine (C-X3-C motif) ligand.
Moreover, cells similar to those in the endometrium, the layer of tissue that normally covers the inside of the uterus, are known to grow outside the uterus as endometriosis, which is still controlled by oestrogen. The progenitor population is self-renewing and differentiating and thought to be included in the endometrium as well as in a normal state [116]. Most often endometriosis occurs on the ovaries, fallopian tubes, and tissue around the uterus and ovaries [117, 118]. Although the cause is not entirely clear [119], the areas of endometriosis bleed during each menstrual period, resulting in inflammation, wound and growth [120]. Among the inflammatory factors, the CXC chemokine family, fractalkine, prostaglandin E2, IL-1β and IL-6 were also found, and the chronic inflammatory environment may act as the CIN that leads to the development of ovarian cancers [121, 122].
Endometrial-associated ovarian cancer might be induced by additional pathways related to oxidative stress [123] and hyperestrogenism [124], which are caused by either long-term infection or stimulation of the normal progenitors in the endometrium by endometriosis.
Although inflammatory cytokines have a significant importance in CIN as described above, their production in phagocytic cells, such as neutrophils, macrophages and oligodendrocytes, is generally triggered by innate immune system recognition of pathogens with molecular patterns derived from various microbes. Typically, toll like receptors (TLRs) are currently well characterised and those bound to ligands are thought to transduce signals through four adaptor proteins: MyD88, TIRAP, TRIF and TRAM. These proteins are involved in signal transduction to IRAK/TRAF and finally the activation of transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulatory factor (IRF), which are responsible for transcribing the genes for inflammatory cytokines and interferon, respectively [125]. Accordingly, chronic infection will help form CIN to maintain continuous production of inflammatory cytokines that trigger proliferation, differentiation, growth, and migration of progenitor/undifferentiated cells. Moreover, IL-6 expression was reported to be upregulated in human ovarian cancer cell lines by NF-κB, which was activated through the lysophophatidic acid-induced Gi/PI3K/AKT pathway independent of TLRs [126]. This indicated that the PI3K pathway could work in parallell with the MyD88 pathway in the ovarian CIN.
Although finding the origin of ovarian cancers is currently still challenging, as their symptoms are not valid in their early development, these insights surely indicate the presence of ovarian CIN.
Conclusion and future research
As reviewed here, there is growing evidence supporting the role of cytokines and growth factors in cancer initiation. The CIN is the chronic inflammation explained by cytokines and growth factors playing their roles in the microenvironment where the various factors secreted keep stimulating the normal progenitors of undifferentiated cells over the long-term, thereby inducing CSCs. This should be critically distinguished from the cancer niche, which is the microenvironment provided after the cancer is initiated. The exact definition of the cancer-inducing niche (CIN) for each type of cancer will be crucial for preventing and defeating cancer.
This perspective showed greater detail of the involvement of key upstream mediators of inflammatory responses and inducers such as TNF, IL-1β, -4, -6, -13, -22, TGFβ, in addition to multiple other cytokines/chemokines associated with cancer initiation. It is also becoming clear that network-based crosstalk among intracellular signalling pathways stimulated by these mediators and the heterogeneous cell-to-cell interactions in the microenvironment are responsible for cancer initiation. Therefore, targeting these molecules within the CIN should be effective for improving cancer prevention and therapy. Although there are many studies targeting these molecules for cancer therapy, treating chronic inflammation is another way to prevent cancer by targeting these molecules.
Author contributions
MS and SMA developed the concept and performed the literature. SMA wrote the manuscript. GH and AS assisted in collecting the draft. MS and SMA revised the final manuscript. All authors read and approved the final manuscript.
Data availability
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Said M. Afify, Email: saidafify@s.okayama-u.ac.jp
Masaharu Seno, Email: mseno@okayama-u.ac.jp.
References
- 1.Boveri T. Zur Frage der Entstehung maligner Tumoren. Jena, Germany: Verlag von Gustav Fischer; 1914. p. 29–32.
- 2.Bauer KH. Mutationstheorie der Geschwulst-Entstehung. Berlin: Julius Springer Verlag; 1928.
- 3.Mackenzie I, Rous P. The experimental disclosure of latent neoplastic changes in tarred skin. J Exp Med. 1941;73:391–416. doi: 10.1084/jem.73.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller HJ. Detection of mutations in the second chromosome by use of the “sifter” stock. Dros Inf Serv. 1951;25:117–8. [Google Scholar]
- 5.Nordling CO. A new theory on the cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burdette WJ. The significance of mutation in relation to the origin of tumors: a review. Cancer Res. 1955;15:201–26. [PubMed] [Google Scholar]
- 7.Fisher J. Multiple-mutation theory of carcinogenesis. Nature. 1958;181:651–52. doi: 10.1038/181651b0. [DOI] [PubMed] [Google Scholar]
- 8.Burch PR. Mutation, autoimmunity, and ageing. Lancet. 1963;2:299–300. doi: 10.1016/S0140-6736(63)90200-9. [DOI] [PubMed] [Google Scholar]
- 9.Burch PR. Genetic carrier frequency for lung cancer. Nature. 1964;202:711–2. doi: 10.1038/202711a0. [DOI] [PubMed] [Google Scholar]
- 10.Ashley DJB. The two “hit” and multiple “hit” theories of carcinogenesis. Br J Cancer. 1969;23:313–28. doi: 10.1038/bjc.1969.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foulds L. Neoplastic development. New York: Academic; 1969 p. 72–74.
- 12.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genome. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. doi: 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays. 2004;26:1097–107. doi: 10.1002/bies.20087. [DOI] [PubMed] [Google Scholar]
- 17.Virchow R, Rudolf V. Die krankhaften Geschwülste: dreissig Vorlesungen, gehalten während des Wintersemesters 1862-1863 an der Universität zu Berlin. Vol 1–3. Berlin: Verlag von August Hirschwald; 1863.
- 18.Balkwill F, Mantovani A. Inflammation, and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 19.Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of cancer. J Cancer Res. 1918;3:1–29. doi: 10.3322/canjclin.27.3.174. [DOI] [PubMed] [Google Scholar]
- 20.Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue Homeostasis, and Inflammation. Annu Rev Immunol. 2021;39:557–81. doi: 10.1146/annurev-immunol-061020-053734. [DOI] [PubMed] [Google Scholar]
- 21.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 22.Afify SM, Seno M. Conversion of stem cells to cancer stem cells: undercurrent of cancer initiation. Cancers (Basel) 2019;11(Mar):345. doi: 10.3390/cancers11030345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bu P, Chen K-Y, Lipkin SM, Shen X. Asymmetric division: a marker for cancer stem cells? Oncotarget. 2013;4:950–1. doi: 10.18632/oncotarget.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan T, Mizutani A, Chen L, Takaki M, Hiramoto Y, Matsuda S, et al. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J Cancer. 2014;5(Jul):572–84. doi: 10.7150/jca.8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osman A, Oze M, Afify SM, Hassan G, El-Ghlban S, Nawara HM, et al. Tumor-associated macrophages derived from cancer stem cells. Acta Histochem. 2020;122:151628. doi: 10.1016/j.acthis.2020.151628. [DOI] [PubMed] [Google Scholar]
- 26.Osman A, Afify SM, Hassan G, Fu X, Seno A, Seno M. Revisiting cancer stem cells as the origin of cancer-associated cells in the tumor microenvironment: a hypothetical view from the potential of iPSCs. Cancers (Basel) 2020;12:879. doi: 10.3390/cancers12040879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumon K, Afify SM, Hassan G, Ueno S, Monzur S, Nawara HM, et al. Differentiation of cancer stem cells into erythroblasts in the presence of CoCl2. Sci Rep. 2021;11:23977. doi: 10.1038/s41598-021-03298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan G, Afify SM, Nair N, Kumon K, Osman A, Du J, et al. Hematopoietic cells derived from cancer stem cells generated from mouse induced pluripotent stem cells. Cancers (Basel) 2019;12:82. doi: 10.3390/cancers12010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L, Kasai T, Li Y, Sugii Y, Jin G, Okada M, et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS ONE. 2012;7:e33544. doi: 10.1371/journal.pone.0033544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calle AS, Nair N, Oo AK, Prieto-Vila M, Koga M, Khayrani AC, et al. A new PDAC mouse model originated from iPSCs-converted pancreatic cancer stem cells (CSCcm) Am J Cancer Res. 2016;6(Dec):2799–815. [PMC free article] [PubMed] [Google Scholar]
- 31.Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci Rep. 2017;7(Jul):6838. doi: 10.1038/s41598-017-07144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Afify SM, Sanchez Calle A, Hassan G, Kumon K, Nawara HM, Zahra MH, et al. A novel model of liver cancer stem cells developed from induced pluripotent stem cells. Br J Cancer. 2020;122(Apr):1378–90. doi: 10.1038/s41416-020-0792-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du J, Xu Y, Sasada S, Oo AKK, Hassan G, Mahmud H, et al. Signaling inhibitors accelerate the conversion of mouse iPS cells into cancer stem cells in the tumor microenvironment. Sci Rep. 2020;10(Jun):9955. doi: 10.1038/s41598-020-66471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassan G, Ohara T, Afify SM, Kumon K, Zahra MH, Fu X, et al. Different pancreatic cancer microenvironments convert iPSCs into cancer stem cells exhibiting distinct plasticity with altered gene expression of metabolic pathways. J Exp Clin Cancer Res. 2022;41(Jan):29. doi: 10.1186/s13046-021-02167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(Nov):759–71. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 36.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(Dec):2373–80. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 37.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9:7204–18. doi: 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu W, Pasare C. Location, location, location: tissue-specific regulation of immune responses. J Leukoc Biol. 2013;94:409–21. doi: 10.1189/jlb.0413207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivera A, Siracusa MC, Yap GS, Gause WC. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol. 2016;17(Apr):356–63. doi: 10.1038/ni.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129:2619–28. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulfone-Paus S, Nilsson G, Draber P, Blank U, Levi-Schaffer F. Positive and negative signals in mast cell activation. Trends Immunol. 2017;38(Sep):657–67. doi: 10.1016/j.it.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(Jul):263. doi: 10.1038/s41392-021-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palomino DC, Marti LC. Chemokines and immunity. Einstein (Sao Paulo) 2015;13:469–73. doi: 10.1590/S1679-45082015RB3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(Jan):a016303. doi: 10.1101/cshperspect.a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(Sep-Oct):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 47.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Y, Yan F, Ying L, Xu D. Emerging roles for epigenetic programming in the control of inflammatory signaling integration in heath and disease. Adv Exp Med Biol. 2017;1024:63–90. doi: 10.1007/978-981-10-5987-2_3. [DOI] [PubMed] [Google Scholar]
- 49.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 50.Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer. 2012;12(May):387–400. doi: 10.1038/nrc3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richmond A, Thomas HG. Purification of melanoma growth stimulatory activity. J Cell Physiol. 1986;129(Dec):375–84. doi: 10.1002/jcp.1041290316. [DOI] [PubMed] [Google Scholar]
- 52.Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(Apr):441–55. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(Feb):103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(Apr):456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 55.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(Feb):91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 56.Waldner MJ, Foersch S, Neurath MF. Interleukin-6–a key regulator of colorectal cancer development. Int J Biol Sci. 2012;8:1248–53. doi: 10.7150/ijbs.4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(Nov):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118(Feb):560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dang T, Liou GY. Macrophage cytokines enhance cell proliferation of normal prostate epithelial cells through activation of ERK and Akt. Sci Rep. 2018;8(May):7718. doi: 10.1038/s41598-018-26143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(Jul):436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 61.Campregher C, Luciani MG, Gasche C. Activated neutrophils induce an hMSH2-dependent G2/M checkpoint arrest and replication errors at a (CA)13-repeat in colon epithelial cells. Gut. 2008;57(Jun):780–7. doi: 10.1136/gut.2007.141556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev. 2003;194(Aug):77–95. doi: 10.1034/j.1600-065X.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 63.Takai A, Toyoshima T, Uemura M, Kitawaki Y, Marusawa H, Hiai H, et al. A novel mouse model of hepatocarcinogenesis triggered by AID causing deleterious p53 mutations. Oncogene. 2009;28(Jan):469–78. doi: 10.1038/onc.2008.415. [DOI] [PubMed] [Google Scholar]
- 64.Okazaki IM, Kotani A, Honjo T. Role of AID in tumorigenesis. Adv Immunol. 2007;94:245–73. doi: 10.1016/S0065-2776(06)94008-5. [DOI] [PubMed] [Google Scholar]
- 65.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135(Sep):889–98. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 66.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002;99(Jul):10025–30. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bishayee A. The role of inflammation and liver cancer. Adv Exp Med Biol. 2014;816:401–35. doi: 10.1007/978-3-0348-0837-8_16. [DOI] [PubMed] [Google Scholar]
- 68.Yang YM, Kim SY, Seki E. Inflammation and liver cancer: molecular mechanisms and therapeutic targets. Semin Liver Dis. 2019;39:26–42. doi: 10.1055/s-0038-1676806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sällberg M, Pasetto A. Liver, tumor and viral hepatitis: key players in the complex balance between tolerance and immune activation. Front Immunol. 2020;11(Mar):552. doi: 10.3389/fimmu.2020.00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gehring AJ, Sun D, Kennedy PT, Nolte-‘t Hoen E, Lim SG, Wasser S, et al. The level of viral antigen presented by hepatocytes influences CD8 T-cell function. J Virol. 2007;81:2940–49. doi: 10.1128/JVI.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–56. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Cobleigh MA, Lian JQ, Huang CX, Booth CJ, Bai XF, et al. A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology. 2011;141:1897–906. doi: 10.1053/j.gastro.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kared H, Fabre T, Bedard N, Bruneau J, Shoukry NH. Galectin-9 and IL-21 mediate cross-regulation between Th17 and Treg cells during acute hepatitis C. PLoS Pathog. 2013;9:e1003422. doi: 10.1371/journal.ppat.1003422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS ONE. 2011;6:e18909. doi: 10.1371/journal.pone.0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 77.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–38. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 78.Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150:353–60. [PubMed] [Google Scholar]
- 79.Mittrucker HW, Visekruna A, Huber M. Heterogeneity in the differentiation and function of CD8(+) T cells. Arch Immunol Ther Exp (Warsz) 2014;62:449–58. doi: 10.1007/s00005-014-0293-y. [DOI] [PubMed] [Google Scholar]
- 80.Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114:596–99. doi: 10.1182/blood-2009-02-203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(Jan):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54(Sep):900–9. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 83.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(Oct):749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 85.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12(Jul):715–23. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 86.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(Sep):461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 87.Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2009;7(Nov):S44–7. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lunardi S, Jamieson NB, Lim SY, Griffiths KL, Carvalho-Gaspar M, Al-Assar O, et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget. 2014;5(Nov):11064–80. doi: 10.18632/oncotarget.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liou GY. Inflammatory cytokine signaling during development of pancreatic and prostate cancers. J Immunol Res. 2017;2017:7979637. doi: 10.1155/2017/7979637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Storz P, Crawford HC. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology. 2020;158(Jun):2072–81. doi: 10.1053/j.gastro.2020.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Steele CW, Jamieson NB, Evans TR, McKay CJ, Sansom OJ, Morton JP, et al. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J Cancer. 2013;108(Mar):997–1003. doi: 10.1038/bjc.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Med (Baltim) 2016;95(Dec):e5541. doi: 10.1097/MD.0000000000005541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73(Oct):6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Inman KS, Francis AA, Murray NR. Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol. 2014;20(Aug):11160–81. doi: 10.3748/wjg.v20.i32.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Swidnicka-Siergiejko AK, Gomez-Chou SB, Cruz-Monserrate Z, Deng D, Liu Y, Huang H, et al. Chronic inflammation initiates multiple forms of K-Ras-independent mouse pancreatic cancer in the absence of TP53. Oncogene. 2017;36(Jun):3149–58. doi: 10.1038/onc.2016.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kong B, Bruns P, Behler NA, Chang L, Schlitter AM, Cao J, et al. Dynamic landscape of pancreatic carcinogenesis reveals early molecular networks of malignancy. Gut. 2018;67(Jan):146–56. doi: 10.1136/gutjnl-2015-310913. [DOI] [PubMed] [Google Scholar]
- 97.Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 2010;18(Nov):448–58. doi: 10.1016/j.ccr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y, He Y, Peng J, Su Z, Li Z, Zhang B, et al. Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nat Cancer. 2021;2:49–65. doi: 10.1038/s43018-020-00134-z. [DOI] [PubMed] [Google Scholar]
- 99.Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69(Jul):280–304. doi: 10.3322/caac.21559. [DOI] [PubMed] [Google Scholar]
- 100.Prat J. New insights into ovarian cancer pathology. Ann Oncol. 2012;23(Sep):x111–7. doi: 10.1093/annonc/mds300. [DOI] [PubMed] [Google Scholar]
- 101.Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N, Sessa C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 S6(Oct):24–32. doi: 10.1093/annonc/mdt333. [DOI] [PubMed] [Google Scholar]
- 102.Shah NM, Lai PF, Imami N, Johnson MR. Progesterone-related immune modulation of pregnancy and labor. Front Endocrinol (Lausanne) 2019;10(Mar):198. doi: 10.3389/fendo.2019.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28(Aug):521–74. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 104.Finn OJ. Cancer immunology. N. Engl J Med. 2008;358(Jun):2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 105.Lin HW, Tu YY, Lin SY, Su WJ, Lin WL, Lin WZ, et al. Risk of ovarian cancer in women with pelvic inflammatory disease: a population-based study. Lancet Oncol. 2011;12(Sep):900–4. doi: 10.1016/S1470-2045(11)70165-6. [DOI] [PubMed] [Google Scholar]
- 106.Rasmussen CB, Jensen A, Albieri V, Andersen KK, Kjaer SK. Is Pelvic inflammatory disease a risk factor for ovarian cancer? Cancer Epidemiol Biomark Prev. 2017;26(Jan):104–9. doi: 10.1158/1055-9965.EPI-16-0459. [DOI] [PubMed] [Google Scholar]
- 107.Zhou Z, Zeng F, Yuan J, Tang J, Colditz GA, Tworoger SS, et al. Pelvic inflammatory disease and the risk of ovarian cancer: a meta-analysis. Cancer Causes Control. 2017;28(May):415–28. doi: 10.1007/s10552-017-0873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ingerslev K, Hogdall E, Schnack TH, Skovrider-Ruminski W, Hogdall C, Blaakaer J. The potential role of infectious agents and pelvic inflammatory disease in ovarian carcinogenesis. Infect Agent Cancer. 2017;12(May):25. doi: 10.1186/s13027-017-0134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rasmussen CB, Jensen A, Albieri V, Andersen KK, Kjaer SK. Increased risk of borderline ovarian tumors in women with a history of pelvic inflammatory disease: a nationwide population-based cohort study. Gynecol Oncol. 2016;143(Nov):346–51. doi: 10.1016/j.ygyno.2016.08.318. [DOI] [PubMed] [Google Scholar]
- 110.Brunham RC, Gottlieb SL, Paavonen J. Pelvic inflammatory disease. N. Engl J Med. 2015;372(May):2039–48. doi: 10.1056/NEJMra1411426. [DOI] [PubMed] [Google Scholar]
- 111.Curry A, Williams T, Penny ML. Pelvic inflammatory disease: diagnosis, management, and prevention. Am Fam Physician. 2019;100(Sep):357–64. [PubMed] [Google Scholar]
- 112.Gradison M. Pelvic inflammatory disease. Am Fam Physician. 2012;85(Apr):791–6. [PubMed] [Google Scholar]
- 113.Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, et al. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest. 1997;99(Jan):77–87. doi: 10.1172/JCI119136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Makepeace BL, Watt PJ, Heckels JE, Christodoulides M. Interactions of Neisseria gonorrhoeae with mature human macrophage opacity proteins influence production of proinflammatory cytokines. Infect Immun. 2001;69(Mar):1909–13. doi: 10.1128/IAI.69.3.1909-1913.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramsey KH, Schneider H, Cross AS, Boslego JW, Hoover DL, Staley TL, et al. Inflammatory cytokines produced in response to experimental human gonorrhea. J Infect Dis. 1995;172(Jul):186–91. doi: 10.1093/infdis/172.1.186. [DOI] [PubMed] [Google Scholar]
- 116.Cousins FL, Dorien OF, Gargett CE. Endometrial stem/progenitor cells and their role in the pathogenesis of endometriosis. Best Pr Res Clin Obstet Gynaecol. 2018;50(Jul):27–38. doi: 10.1016/j.bpobgyn.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 117.Anglesio MS, Yong PJ. Endometriosis-associated ovarian cancers. Clin Obstet Gynecol. 2017;60(Dec):711–27. doi: 10.1097/GRF.0000000000000320. [DOI] [PubMed] [Google Scholar]
- 118.Worley MJ, Welch WR, Berkowitz RS, Ng SW. Endometriosis-associated ovarian cancer: a review of pathogenesis. Int J Mol Sci. 2013;14(Mar):5367–79. doi: 10.3390/ijms14035367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zondervan KT, Becker CM, Missmer SA. Endometriosis. N. Engl J Med. 2020;382(Mar):1244–56. doi: 10.1056/NEJMra1810764. [DOI] [PubMed] [Google Scholar]
- 120.Bulletti C, Coccia ME, Battistoni S, Borini A. Endometriosis and infertility. J Assist Reprod Genet. 2010;27(Aug):441–7. doi: 10.1007/s10815-010-9436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Melin A, Sparén P, Persson I, Bergqvist A. Endometriosis and the risk of cancer with special emphasis on ovarian cancer. Hum Reprod. 2006;21(May):1237–42. doi: 10.1093/humrep/dei462. [DOI] [PubMed] [Google Scholar]
- 122.Wei JJ, William J, Bulun S. Endometriosis and ovarian cancer: a review of clinical, pathologic, and molecular aspects. Int J Gynecol Pathol. 2011;30(Nov):553–68. doi: 10.1097/PGP.0b013e31821f4b85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamaguchi K, Mandai M, Toyokuni S, Hamanishi J, Higuchi T, Takakura K, et al. Contents of endometriotic cysts, especially the high concentration of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin Cancer Res. 2008;14(Jan):32–40. doi: 10.1158/1078-0432.CCR-07-1614. [DOI] [PubMed] [Google Scholar]
- 124.Zanetta GM, Webb MJ, Li H, Keeney GL. Hyperestrogenism: a relevant risk factor for the development of cancer from endometriosis. Gynecol Oncol. 2000;79:18–22. doi: 10.1006/gyno.2000.5905. [DOI] [PubMed] [Google Scholar]
- 125.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;25:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, et al. Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-kappaB pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis. 2005;26:45–52. doi: 10.1093/carcin/bgh301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.