

Abstract
Aims
Transgender women (TGW) have been underrepresented in trials and use gender‐affirming hormonal therapies (GAHT) that may alter renal function by significantly increasing creatinine clearance. Population pharmacokinetic (popPK) models and simulations would aid in understanding potential differences in emtricitabine/tenofovir disproxil fumarate (F/TDF) parent–metabolite concentrations in TGW on GAHT when compared to cisgender men (CGM) not exposed to GAHT.
Methods
Pharmacokinetic (PK) data from a Phase 1, open‐label clinical trial with directly observed therapy of daily F/TDF consisting of 8 TGW and 8 CGM was utilized for model building. PopPK analysis was performed using nonlinear mixed effects modelling (NONMEM 7.5.0). Covariates of body weight, creatinine clearance, and gender were evaluated. Final models were subjected to Monte Carlo simulations to compare drug exposure following once daily and on‐demand (IPERGAY 2 + 1 + 1) dosing of F/TDF.
Results
Tenofovir (TFV) and emtricitabine PK were best described by a 2‐compartment model, first‐order absorption/elimination with absorption lag time. Parent models were linked to their metabolites by first order formation and elimination. Creatinine clearance was a significant covariate influencing clearance in both models. Simulations demonstrated that at least 2, weekly 2 + 1 + 1 cycles of on‐demand dosing in TGW on GAHT is necessary for TFV‐diphosphate to reach similar exposure after the initial week of on‐demand dosing in CGM not on GAHT.
Conclusion
PopPK models of TFV, emtricitabine and intracellular metabolites in TGW were established. Dose simulations revealed that TGW should be treated for at least 2 weeks to have comparable exposures to CGM.
Keywords: HIV; popPK model; population pharmacokinetics; pre‐exposure prophylaxis; tenofovir, transgender women
What is already known about this subject
Transgender women (TGW) remain at high‐risk for HIV infection.
The relationship between emtricitabine/tenofovir disoproxil fumarate (F/TDF) and gender‐affirming hormonal therapy (GAHT) is not completely understood.
Studies have shown lower plasma emtricitabine and tenofovir concentrations in TGW when compared to cisgender men (CGM).
What this study adds
A joint F/TDF parent–metabolite population pharmacokinetic model in TGW on GAHT and CGM demonstrate that drug concentrations are influenced by renal function.
Simulations indicate that on‐demand F/TDF requires 2 weekly 2 + 1 + 1 cycles in TGW on GAHT to achieve comparable tenofovir‐metabolite concentrations observed during a single 2 + 1 + 1 cycle in CGM.
1. INTRODUCTION
Transgender women (TGW) remain at high‐risk for HIV infection, with an estimated global prevalence of 19%; they are 49‐times more likely to acquire HIV than cisgender women of reproductive age. 1 TGW are a key population that would benefit from pre‐exposure prophylaxis (PrEP) for HIV prevention. However, there are barriers to PrEP uptake, including access to care, social stigma and patient concerns regarding the impact of PrEP on gender‐affirming hormonal therapies (GAHT). 2 , 3 , 4 TGW frequently access oestrogen‐based GAHT to induce physical changes and promote congruence with their gender identity; these therapies may also include anti‐androgenic agents. 5 , 6 The World Professional Association for Transgender Health has published transgender‐focused recommendations to promote feminization and reduce gender dysphoria in TGW. 7 There is also heterogeneity in dosages and formulations of GAHT accessed by TGW. Given the variety in GAHT options available to TGW, in conjunction with existing concerns regarding unfavourable interactions between GAHT and PrEP regimens, more research is required in this area.
Daily oral tenofovir disoproxil fumarate (TDF) 300 mg/emtricitabine (FTC) 200 mg (F/TDF) was the first drug regimen approved by the US Food and Drug Administration for PrEP. Daily product use was shown to be highly effective in reducing HIV acquisition; subsequent studies showed that an alternative on‐demand (IPERGAY 2 + 1 + 1) PrEP regimen in men who have sex with men is associated with an 86% relative risk reduction in HIV infection. 8 , 9 , 10 Earlier trials evaluating the efficacy of F/TDF demonstrated decreased PrEP efficacy and lower adherence in TGW in comparison to cisgender men (CGM) who have sex with men. 9 However, the relationship between F/TDF, and their active metabolites, tenofovir‐diphosphate (TFV‐DP) and emtricitabine‐triphosphate (FTC‐TP), and GAHT, is not completely understood. Two studies reported 24–32% lower TFV and FTC plasma concentrations in TGW when compared to contemporaneously studied CGM. 11 , 12 However, Shieh et al. did not identify statistically significant differences in TFV‐DP concentrations in peripheral blood mononuclear cells (PBMC) between populations 12 ; a similar finding was observed in a more recent study, which showed comparable intraerythrocytic TFV‐DP concentrations collected as dried blood spots between CGM and TGW on stable GAHT. 13 Further, neither study found an impact of PrEP on GAHT. 12 However, due to the variety and variability of analytical methods, as well as heterogeneity of GAHT regimens accessed among these studies, additional efforts to comprehensively assess the impact of GAHT on oral F/TDF for PrEP are warranted.
Population pharmacokinetic (popPK) modelling is used to describe sources of PK parameter variability as well as their covariate relationships. PopPK models can facilitate clinical trial design, dose selection, and dose timing optimization. Numerous popPK studies describe the relationship between plasma TFV or FTC and PBMC TFV‐DP or FTC‐TP concentrations. 14 , 15 , 16 , 17 , 18 , 19 However, none of these studies include TGW on GAHT. In this report, we developed a joint parent–metabolite model to characterize the interrelationship of plasma TFV/FTC with PBMC TFV‐DP/FTC‐TP in non‐HIV‐infected TGW and CGM based on data first reported in a descriptive noncompartmental analysis from our group. 12 We then used the model to simulate a comparison of daily and on‐demand F/TDF PrEP in TGW on GAHT.
2. METHODS
2.1. Trial design
The clinical study generating the data utilized in the popPK models and assay performance characteristics are described elsewhere. 12 This analysis was approved by the North Texas Regional Institutional Review Board and the Johns Hopkins Medicine Institutional Review Board. Study data were collected between April 2016 and April 2018, and all participants provided informed consent to participate in the clinical study. 12 Briefly, 8 TGW and 8 CGM were enrolled in a phase 1, open‐label PK study of once daily TDF 300 mg/FTC 200 mg under direct observation in a research clinic for 8 days. Study participants (CGM and TGW) were required to have a baseline creatinine clearance (CrCl) ≥70 mL/min, using the Cockcroft–Gault formula for inclusion in the clinical trial. Additional inclusion criteria for the protocol required TGW to be on oestrogen‐based GAHT with serum total oestradiol concentration >100 pg/mL. GAHT consisting of oestrogen ± progesterone or ± antiandrogen medication were used by TGW in this cohort. After 7 days of directly observed dosing, blood was collected predose, and 1, 2, 4, 6, 8 and 24 hours postdose, and PBMC were collected predose, and 2, 8 and 24 hours postdose.
All doses of TDF and FTC and drug analyte concentrations were converted to micromolar units for comodelling. The intracellular metabolite concentrations were converted to micromolar units using 282 fL/cell as cellular volume. 20 The molecular weights used for conversion were: TDF, 635.52 g/mol; TFV, 287.216 g/mol; and FTC, 247.248 g/mol.
2.2. PopPK model development
PopPK analysis was performed using NONMEM (version 7.5.0, ICON Development Solution, Ellicott City, MD, USA) with Perl speaks NONMEM (PsN version 4.9.0) as an interface. R software (version 4.0.3) was used for dataset preparation and graphics. PopPK data analyses and subsequent simulations were performed between June 2020 and April 2021.
Parent and metabolites were modelled simultaneously, in which plasma drug disposition was evaluated first, then the PBMC compartment was added to describe metabolite concentrations. One‐ and 2‐ compartment models with first‐order absorption, with and without a lag time, were tested for structural models. Metabolite compartments were linked to a first‐order formation rate constant once a reasonable parent model was established. An exponential error model was used to describe interindividual variability, which was assumed to be normally distributed with mean of zero and variance of ω2. Additional information on the popPK models may be found in Supplemental File 1.
For each analyte, additive, proportional, and combined additive and proportional residual variability models were assessed separately, and were assumed to be normally distributed with mean of zero and variance of 2.
Model selection was driven by a global evaluation based on NONMEM objective function value (OFV), the Akaike information criteria (AIC), successful convergence, goodness‐of‐fit (GOF) plots, and precision and biological plausibility of parameter estimates. A decrease in the objective function by 3.84 units (P < .05, 1 degree of freedom) for hierarchical models was considered to be significant. For non‐nested model comparisons, the AIC was used for model selection using the following formula: AIC = NONMEM OFV + 2*number of parameters in the model. A drop in AIC of 2 or more was a threshold for considering 1 model over another. 21
For covariate screening, we used biological rationale, parameter vs. covariate scatterplots for continuous variables, and boxplots for categorical variables. Body weight, CrCl (estimated by Cockcroft–Gault equation using the female term for TGW), and self‐identified gender were evaluated for the impact on PK parameters. Continuous covariates were modeled using a median‐normalized power model. Categorical covariate was modeled using a proportional model. We used forward inclusion and backward elimination procedures along with the likelihood ratio test to evaluate the effect of each covariate on model parameters. Covariates were considered significant and included in the basic model if the decrease in OFV was >3.84 units (P < .05, 1 degree of freedom) for forward inclusion, and the increase in OFV was >6.64 units (P < .01, 1 degree of freedom) for backward elimination. 22
2.3. Model evaluation
GOF plots, visual predictive checks (VPCs), 23 and nonparametric bootstrap analysis were utilized to evaluate the final models and parameter estimates. 24 GOF plots include individual (IPRED) and population (PRED) predictions vs. observed concentrations, and conditional weighted residuals (CWRES) vs. time after dose and PRED. 25 VPCs were conducted using PsN and R. A total of 500 replicates of the simulated dataset were produced from the final models and used for the determination of prediction intervals. The 5th, 50th and 95th percentiles of the simulated concentrations were plotted against the time after dose and compared with observed TFV, FTC, TFV‐DP and FTC‐TP. The nonparametric bootstrap method was performed to evaluate reliability and stability of the final models. Nonparametric bootstraps of 500 iterations were conducted to estimate 95% confidence intervals (CIs) for the population parameters.
2.4. Monte Carlo simulations to compare on‐demand vs. QD dosing regimen
Monte Carlo simulations of plasma (TFV and FTC) and PBMC (TFV‐DP and FTC‐TP) were performed for 500 virtual subjects with gender specific CrCl using the final population PK models. For gender‐specific CrCl, random samples (n = 500) of CrCl were generated from mean and standard deviation from the trial data using R. Four different dosing scenarios were simulated with respect to a sexual activity (potential exposure of virus) as a reference time point:
Scenario 1: Subjects were on conventional daily dosing (F/TDF) before sexual exposure and at steady state, consistent with Food and Drug Administration label dosing
Scenario 2: Subjects initiate conventional daily dosing on the day of sexual exposure to represent initial phase of conventional daily dosing
Scenario 3: On‐demand dosing in which subjects took a double dose 2 hours before exposure followed by a single dose 24 hours after the first dose, and a single dose 24 hours after the second dose (1 cycle), consistent with the on‐demand IPERGAY 2 + 1 + 1 regimen. On‐demand dosing is interchangeably referred to as 2 + 1 + 1 regimen in the remaining text.
Scenario 4: Same as Scenario 3 but repeat the regimen for a total of 2 weeks (2 cycles)
For each simulated scenario, a mean concentration–time profile and boxplot of C24 (trough) concentrations for TFV/FTC/TFV‐DP/FTC‐TP after the exposure were compared.
3. RESULTS
3.1. Data summary
Demographic summary of the participants is shown in Table 1. A total of 179 concentration values from 16 subjects were available for the model building exercise. Most participants were Black (75%). TGW showed no differences in age or weight compared to CGM (P > .05). There were statistically significant differences in serum creatinine and CrCl between genders (P < .05), with TGW showing 24% lower creatinine and 61% higher CrCl when compared to CGM (Figure S1). These observations were not accounted for by a statistically nonsignificant 18% weight difference (P > .05). No analyte concentrations were below the limit of quantification.
TABLE 1.
Demographics of the participants
| TGW (n = 8) | CGM (n = 8) | P value | |
|---|---|---|---|
| Age, y a | 29 (27–38) | 46 (38–51) | .195 b |
| Weight, kg a | 98 (85–113) | 83 (73–89) | .130 |
| CrCl (mL/min) a | 174 (117–184) | 108 (85–121) | .038 |
| SCr (mg/dL) a | 0.80 (0.80–0.83) | 1.05 (1.00–1.12) | .005 |
| Race | >.999 c | ||
| African ancestry | 6 | 6 | |
| European ancestry | 1 | 2 | |
| Asian ancestry | 1 | 0 |
Data expressed as median (interquartile range).
Exact 2‐sided P value, Wilcoxon rank sum test.
Fisher's exact test.
Abbreviations: CGM, cisgender men; CrCl, creatinine clearance; SCr, serum creatinine; TGW, transgender women.
3.2. PopPK model of FTC and FTC‐TP
Comparison of 1‐ and 2‐compartment models for plasma FTC using AIC and OFV confirmed that a 2‐compartment model provided a better fit and significantly smaller objective function (ΔOFV of −107.8 and ΔAIC of −101.8 for FTC). With statistical criteria as well as GOF plots, FTC was best described by a 2‐compartment model with first‐order absorption and elimination. The addition of absorption lag time further reduced the objective function (ΔOFV of 33.0 for FTC) and improved the fit of the observations in the absorption phase. It was found that parent FTC was best linked with intracellular FTC‐TP by first‐order formation and elimination. Residual error was modelled using a proportional error model. CrCl calculated using female correction factor for TGW was shown to be a significant covariate on clearance (ΔOFV of −11.7). Gender and CrCl were colinear. Based on biological plausibility, CrCl was included in the model. In a sequential step, gender was tested as a covariate in addition to CrCl, but was not a significant covariate. Inclusion of CrCl as a covariate reduced the between‐subject variability (BSV; %CV) on CL to 7.5% from 12.9% in the base model. A schematic of the model is shown in Figure 1. The intracellular half‐life for FTC‐TP was calculated as 15 hours.
FIGURE 1.
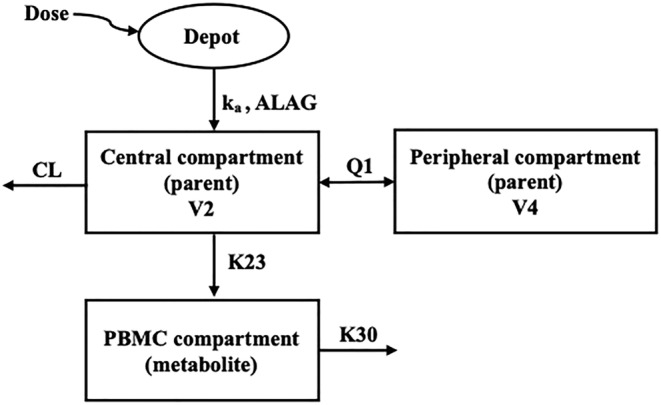
Model schematic for tenofovir disoproxil fumarate and emtricitabine disposition. ka, absorption rate constant; ALAG, lag time; CL, clearance from the central compartment; V2, central compartment volume of distribution; V4, peripheral compartment volume of distribution; Q1, intercompartmental clearance; K23, formation rate constant of metabolite stimulated by plasma drug; K30, elimination rate constant of intracellular metabolite
The percent relative standard error (%RSE) of the PK parameter estimates was acceptable with <30% for fixed effect parameters and residual error, and <65% for random effects. The shrinkage was <30% for all random effects parameters that were estimated on absorption rate constant, except BSV (37% shrinkage). GOF plots of the final model are shown in Figure S2. These plots showed no major bias and were considered acceptable. The VPC plot represented in Figure S3 indicated a good predictive performance of the model showing an agreement between the distribution of observed and predicted data. Final parameters along with bootstrap 95% confidence intervals (CIs) are provided in Table 2.
TABLE 2.
Emtricitabine and emtricitabine triphosphate population pharmacokinetic parameter estimates for the final models and bootstrap results for the final model
| Parameter (unit) | Final model | Bootstrap |
|---|---|---|
| Estimate (% RSE) | Median (95% CI) | |
| Fixed effects | ||
| ka (1/h) | 3.10 (20) | 2.95 (1.09–32.32) |
| V2 (L) | 76.1 (6) | 71.7 (51.7–83.4) |
| V4 (L) | 77.9 (13) | 70.5 (55.6–108.1) |
| Q1 (L/h) | 7.89 (12) | 9.07 (5.95–15.22) |
| CL (L/h) | 19.8 (3) | 19.2 (16.7–21.4) |
| ALAG (h) | 0.522 (26) | 0.464 (0.005–0.979) |
| K30 (1/h) | 4.69E−2 (13) | 4.78E−2 (1.82E−2–8.50E−2) |
| K23 (1/h) | 1.08E−2 (15) | 1.16E−2 (4.20E−3–2.96E−2) |
| Covariate effect | ||
| CrCl on CL* | 2.96E−2 (18) | 3.12E−1 (1.76E−1–4.63E−1) |
| Interindividual variability (%CV) | ||
| ka | 139.6 (33) | 112.1 (36.5–235.6) |
| ALAG | 65.3 (65) | 78.0 (0.7–361.8) |
| Q1 | 42.3 (53) | 27.3 (0.4–68.9) |
| CL | 7.5 (51) | 4.5 (0.1–12.4) |
| K30 | 28.5 (34) | 25.8 (7.3–36.0) |
| Residual variability (% CV) | ||
| Proportional error on parent | 18.3 (18) | 17.9 (13.5–22.7) |
| Proportional error on metabolite | 43.4 (29) | 42.6 (30.3–56.7) |
Abbreviations: ALAG, lag time; CL, clearance; CrCl, creatinine clearance; K23, formation rate of constant of metabolite stimulated by plasma drug; K30, elimination rate constant of intracellular metabolite; ka, absorption rate constant; Q1, intercompartmental clearance; V2, central compartment volume of distribution; V4, peripheral compartment volume of distribution.
3.3. PopPK model of TFV and TFV‐DP
A decrease in objective function and AIC, and an improvement in GOF plots were observed in plasma TFV when comparing 1‐ and 2‐compartment models (ΔOFV of −133.9 and ΔAIC of −127.9 for TFV). A 2‐compartment model with first‐order absorption best described plasma TFV concentration. After TFV‐DP was linked to the plasma compartment with the first‐order formation by the simultaneous estimation approach, higher interindividual variability, RSE and correlation of the parameters (K23 ‐ K32 and K30 ‐ ALAG with 0.95 and 0.90, respectively) were observed. The shrinkage was <30% for all random effects parameters that were estimated. The implementation of nonlinear metabolite formation using the indirect response model approach 16 to describe plasma TFV and PBMC TFV‐DP concentrations failed to converge. The sequential estimation approach showed similar PK parameter estimates when compared to the simultaneous approach. Gender and CrCl were significant covariates on CL when testing with a forward inclusion step (ΔOFV of −5.2 for gender and ΔOFV of −10.2 for CrCl). Following backward elimination, CrCl was a significant covariate of CL. Inclusion of CrCl as a covariate reduced the BSV (%CV) on CL to 18.6% from 31.2% in the base model. Residual error was modeled using a proportional error model. Final parameters along with 95% bootstrap CIs are provided in Table 3. A schematic of the model is shown in Figure 1. The intracellular half‐life for TFV‐DP was estimated as 173 hours.
TABLE 3.
Tenofovir and tenofovir diphosphate population pharmacokinetic parameter estimates for the final models and bootstrap results for the final model
| Parameter (unit) | Final model | Bootstrap |
|---|---|---|
| Estimate (% RSE) | Median (95% CI) | |
| Fixed effects | ||
| ka (1/h) | 3 FIXED | 3 FIXED |
| V2 (L) | 234 (38) | 234 (74–332) |
| V4 (L) | 657 (92) | 656 (489–848) |
| Q1 (L/h) | 227 (27) | 227 (136–298) |
| CL (L/h) | 50 (13) | 50 (43–54) |
| ALAG (h) | 0.290 (123) | 0.289 (0.003–0.403) |
| K30 (1/h) | 9.14E−3 (303) | 8.94E−3 (9.14E−5–1.17E−2) |
| K23 (1/h) | 2.03E−5 (309) | 2.03E−5 (2.00E−7–4.52E−5) |
| Covariate effect | ||
| CrCl on CL* | 5.97E−1 (72) | 5.89E−1 (1.50E−1–8.74E−1) |
| Interindividual variability (%CV) | ||
| ka | 130.8 (789) | 130.5 (39.9–167.7) |
| CL | 18.6 (142) | 18.6 (10.5–23.1) |
| V2 | 22.6 (238) | 22.1 (0.5–31.6) |
| V4 | 42.5 (155) | 41.5 (3.7–47.0) |
| ALAG | 114.0 (522) | 113.8 (77.2–307.3) |
| Residual variability (% CV) | ||
| Proportional error on parent | 15.7 (27) | 15.7 (11.7–18.3) |
| Proportional error on metabolite | 37.1 (70) | 37.1 (26.6–45.3) |
Abbreviations: ALAG, lag time; CL, clearance; CrCl, creatinine clearance; K23, formation rate of constant of metabolite stimulated by plasma drug; K30, elimination rate constant of intracellular metabolite; ka, absorption rate constant; Q1, intercompartmental clearance; V2, central compartment volume of distribution; V4, peripheral compartment volume of distribution.
GOF plots of the final model are shown in Figure S4. These plots showed no major bias. The VPC plot represented in Figure S5 indicated a good predictive performance of the model showing an agreement between the distribution of observed and predicted data. Final parameters along with bootstrap 95% confidence intervals are provided in Table 3.
3.4. Simulations of the conventional daily dose and on‐demand dosing
Simulated continuous concentration–time profiles (Figure 2) and discrete Cmax and Cmin concentrations on day 3 and 10 (Table S1) for TFV, TFV‐DP, FTC, and FTC‐TP contrasted differences between daily and on‐demand PrEP, as well as differences between TGW and CGM. At all times over 2 weeks, PBMC TFV‐DP concentrations after on‐demand dosing were below median steady‐state concentrations with daily dosing consistent with expectations for 4 vs. 7 doses per week (18.8 vs. 36.3 fmol/106 cells); this was also observed for PBMC FTC‐TP, although with far smaller differences (4 doses: 0.6 pmol/106 cells; 7 doses: 2.2 pmol/106 cells). The highest trough (Cmin) and peak (Cmax) PBMC TFV‐DP concentrations during on‐demand dosing were 44 and 52% of daily dosing concentrations in week 1 (occurring on day 3) and increased to 63 and 67% of daily dosing concentrations in week 2 (occurring on day 10) and thereafter, with pseudo‐steady‐state achieved in week 2. Contrasting TGW and CGM with on demand dosing, the highest trough and peak PBMC TFV‐DP concentrations in TGW were 83 and 84% of CGM during weeks 1 and 2. In TGW on an on‐demand regimen, the highest trough and peak PBMC TFV‐DP concentrations were 38 and 43% of daily dosing steady‐state concentrations in CGM during the first week (day 3), and improved to 53 and 55% in week 2 (day 10) and thereafter. With a far shorter half‐life, the highest trough concentrations of PBMC FTC‐TP in all weeks of on‐demand dosing were similar to steady‐state daily dosing; on‐demand dosing in TGW yielded PBMC FTC‐TP concentrations only 8% below CGM on the same on‐demand regimen.
FIGURE 2.
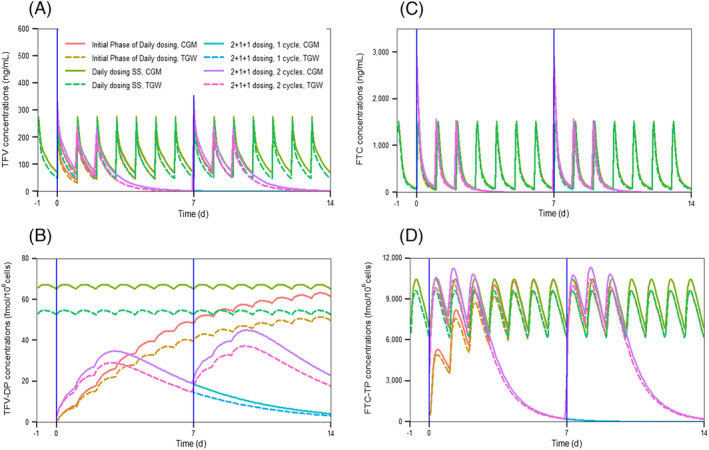
Simulated mean concentration–time profiles of (a) tenofovir (TFV), (b) TFV‐diphosphate (TFV‐DP), (c) emtricitabine (FTC) and (d) FTC‐triphosphate (FTC‐TP) on conventional daily dose and 2 + 1 + 1 dosing. Blue line represents exposure point. CGM, cisgender men; TGW, transgender women
4. DISCUSSION
This report describes the generation of an F/TDF popPK model that specifically focuses on TGW and the application of drug exposure simulations that take into consideration different populations and dosing regimens. Further, these simulations reflect current, on‐demand modalities for F/TDF PrEP use. This work focuses on an at‐risk population and aims to determine optimal dosing strategies in TGW to ensure drug exposures consistent with protection.
We developed a joint parent–metabolite popPK model describing plasma TFV/FTC and intracellular TFV‐DP/FTC‐TP based on both TGW on GAHT and CGM, which demonstrated the substantial influence of CrCl variability on drug clearance for both TFV and FTC. Our parameter estimate for the CrCl effect on drug clearance is generally in agreement with previous studies. 14 , 16 , 26 , 27 , 28 Because gender was associated with CrCl, and based on biological rationale of the influence of hormones on renal function, we included only CrCl in the final model.
The use of the 0.85 correction factor recommended for females in the Cockcroft–Gault CrCl calculation results in a 15% decrease in calculated CrCl in TGW when compared to CGM. In the phase 1 clinical trial used for model generation, median CrCl was 174 mL/min and 108 mL/min in TGW and CGM, respectively. This represents a 61% increase in CrCl in TGW, which is not accounted for by the Cockroft–Gault female correction factor, or the statistically insignificant 15% higher median body weight that can also influence CrCl. As CrCl was calculated using the 0.85 multiplier for TGW, it is expected that the model parameters estimates would need to be re‐estimated if any other multiplier is used. However, we anticipate that the conclusions of the simulations would remain similar. More direct measurement of glomerular filtration rate using other methods, such as iohexol clearance, would provide a more accurate covariate of TFV and FTC renal clearance.
A 2‐compartment model with first order absorption and elimination best described TFV plasma PK. Final parameters are in general agreement with previous reports for TFV. 14 , 15 , 16 , 17 , 18 , 19 , 27 Metabolite formation rate constant (K23) is in reasonable agreement with our prior report, once we take into consideration the difference in units for metabolite compartment. 14 The elimination half‐life for the metabolite estimate (calculated as 0.693/K30) from this report is higher than Burns et al., 14 but consistent with prior publications, which ranged from 150 to 180 hours. 29 , 30 , 31 Potential sources of such variability across studies may include sampling time differences and limits of quantification used in metabolite measurements.
Plasma FTC was best described as a 2‐compartment model with first order absorption and elimination. PK parameter estimates are in general agreement with previous reports. 17 , 18 , 19 , 28 , 32 Saturable functions for metabolite formation were not used, given only 1 dose level. The previous elimination half‐life of FTC‐TP was 17–36 hours 33 , 34 ; however, a slightly shorter half‐life of FTC‐TP of 15 hours was observed in this study.
Several studies have shown that the use of F/TDF for PrEP with GAHT is associated with decreased plasma TFV/FTC concentrations. 11 , 12 , 35 Hiransuthikul et al. reported 12 and 18% reductions in AUC and trough concentration of plasma TFV in TGW using GAHT; however, the study did not evaluate plasma FTC or intracellular metabolite concentrations. 11 Cirrincione et al. reported a reduction of 24 and 14% AUC in plasma TFV and FTC, respectively, in TGW compared to historical controls; however, they reported significantly higher (>2‐fold higher) PBMC concentrations of metabolites in TGW. 35 Shieh et al. evaluated both plasma TFV/FTC along with their intracellular components. They reported a reduction of 27 and 24% in AUC for plasma TFV and FTC concentrations in TGW compared to CGM, respectively. A reduction of 24 and 12% in AUC for TFV‐DP and FTC‐TP, respectively, was reported in this study, although the observation was not statistically significant. 12 The clinical data described by Shieh and colleagues were used to generate the models described in this manuscript.
We conducted simulations to compare daily dosing and on‐demand dosing strategies as well as TGW and CGM in terms of TFV, FTC, TFV‐DP, and FTC‐TP exposure. As expected, the combination of fewer doses per week and the days long PBMC TFV‐DP half‐life resulted in lower concentrations with on‐demand dosing throughout the simulated dosing periods. Comparing the highest PBMC TFV‐DP concentrations during the initial (week 1) 4‐dose on‐demand F/TDF regimen, TGW and CGM achieved only 45 and 53% of the lowest steady‐state daily dosing concentrations in CGM. In the second week and thereafter (at pseudo‐steady‐state), peak on‐demand dosing TFV‐DP concentrations increased to 57 and 69%; notably, PBMC TFV‐DP concentration in TGW during the second week of a weekly on‐demand regimen catches up with CGM concentrations achieved in the first on‐demand dosing week. The IPERGAY and PREVENIR trials studied on‐demand 2 + 1 + 1 F/TDF dosing with most participants taking the regimen weekly. Both studies demonstrated low to nonexistent instances of PrEP failure.8 In the original IPERGAY study, only a small number of participants reported taking the on‐demand regimen less than weekly, but there was no apparent difference in PrEP efficacy in this group. 8 In addition, because there are no studies of an on demand 2 + 1 + 1 regimen with lower F/TDF doses than those used in the largely CGM populations in IPERGAY and PREVENIR, we previously raised concern that the 24%–32% reduction in plasma TFV concentrations 11 , 12 could reduce the efficacy of on‐demand regimens, though have no negative impact on daily regimens. Combined with the simulations performed here, this provides some level of reassurance that, at least in the second week of on‐demand dosing, F/TDF protection in TGW is likely to be as high as in the initial on‐demand week in CGM, despite the observed impact of GAHT on TFV and TFV‐DP concentrations. 12 This is further reinforced by the simulations indicating minor differences in FTC‐TP across gender and dosing conditions. An essential caveat, however, is that neither the relative importance of peaks or troughs on any given day of the on‐demand regimen, nor the relative contribution of TFV‐DP and FTC‐TP with the F/TDF fixed dose combination, are established.
Our models have several limitations. First, poor precision was observed in parameter estimates due to the small dataset used for model building. Second, our models assume linear PK and simulations conducted in terms of accumulation of exposure. Third, the sampling times for TFV‐DP in the study do not ensure true steady‐state conditions; therefore, this limits the accumulation ratio estimate; however, we do not believe this limitation impacts the overall findings of the described work. Extrapolation of models beyond this PrEP formulation is not ideal and should be carefully considered. This present study serves as an important starting point to explore an alternative regimen for TGW to help clinical study designs. Further validation of models is required before clinical implementation for alternative dosing decisions. Lastly, TFV alafenamide, which has very different blood and tissue TFV‐DP PK when compared to TDF, requires its own models and simulations.
5. CONCLUSIONS
Joint popPK models for describing TFV/FTC and TFV‐DP/FTC‐TP concentrations after orally administered F/TDF in TGW have been developed and described in this report. Simulations indicate the on‐demand regimen requires 2 weekly 2 + 1 + 1 cycles in TGW on GAHT to ensure achievement of similar PBMC TFV‐DP concentrations observed during the initial 2 + 1 + 1 regimen in CGM. Further, the 2 + 1 + 1 regimen in CGM, proven highly effective as PrEP, protects with only 1/2 to 2/3 of the TFV‐DP concentrations associated with daily dosing.
COMPETING INTEREST
M.A.M., E.J.F. and C.W.H. receive funded research support from Gilead Sciences, Merck and ViiV Healthcare. C.W.H. sits on a scientific advisory board for Merck. E.J.F. and C.W.H. are founders and officers of Prionde Biopharma. The other authors note no conflicts of interest.
CONTRIBUTORS
C.W.H., E.J.G., E.S., R.P.B. and M.A.M. performed the original research study. A.T., A.C., C.W.H. and M.A.M. designed this research study. A.T. and A.C. developed the models and performed simulations. A.T., A.C., C.W.H. and M.A.M. drafted and edited manuscript. All authors read successive versions of the manuscript and approved the final manuscript.
STUDY PI DECLARATION
The authors confirm that the PI for this paper is Mark Marzinke, PhD. The authors also claim that the PI for the clinical study in which these data are based upon is Craig Hendrix, MD, and that he had direct clinical responsibility for patients.
Supporting information
FIGURE S1 Comparison of gender vs. CrCl.
FIGURE S2 Goodness‐of‐fit plots of the final plasma FTC and PBMC FTC‐TP models. (a, e) Individual‐predicted concentrations (IPRED) against observed concentrations (DV), (b, f) Population‐predicted concentrations (PRED) against DV, (c, g) Time after dose against conditional weighted residuals (CWRES), and (d, h) PRED against CWRES.
FIGURE S3 Visual predictive check (VPC) for (a) plasma FTC and (b) PBMC FTC concentrations vs. time. The dashed black and solid black lines depict the 5th, 50th and 95th percentiles of the observed and simulated concentrations, respectively. The shaded area represents the 95% confidence interval around the simulated percentiles.
FIGURE S4 Goodness‐of‐fit plots of the final plasma TFV and PBMC TFV‐DP models. (a, e) Individual‐predicted concentrations (IPRED) against observed concentrations (DV), (b, f) Population‐predicted concentrations (PRED) against DV, (c, g) Time after dose against conditional weighted residuals (CWRES), and (d, h) PRED against CWRES.
FIGURE S5 VPC for (a) plasma TFV and (b) PBMC TFV concentrations vs. time. The dashed black and solid black lines depict the 5th, 50th, and 95th percentiles of the observed and simulated concentrations, respectively. The shaded area represents the 95% confidence interval around the simulated percentiles.
TABLE S1 Simulated concentrations of intracellular metabolites for various dosing regimen of TDF/FTC
ACKNOWLEDGEMENTS
We thank the study participants for their essential contributions to the clinical research that formed this basis of the described model. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins CFAR or NIH.
This work was supported by the and National Institutes of Health, National Institute of Allergy and Infectious Diseases under award number R01 AI145675 (Principal Investigator: Marzinke). In addition, data generated during the phase 1 study were supported, in part, by the Johns Hopkins Center for AIDS Research through a developmental research grant (P30 AI042855; Principal Investigator: Hendrix) and the Clinical Pharmacology Training Program grant (Dr. Shieh, T32 GM066691).
Tanaudommongkon A, Chaturvedula A, Hendrix CW, et al. Population pharmacokinetics of tenofovir, emtricitabine and intracellular metabolites in transgender women. Br J Clin Pharmacol. 2022;88(8):3674‐3682. doi: 10.1111/bcp.15310
Funding information Clinical Pharmacology Training Grant, Grant/Award Number: T32 GM066691; Division of Intramural Research, National Institute of Allergy and Infectious Diseases, Grant/Award Number: R01 AI145675; Johns Hopkins Center for AIDS Research, Grant/Award Number: P30 AI042855
DATA AVAILABILITY STATEMENT
The data that support the findings of this study may be available from the corresponding author upon reasonable request and in compliance with relevant institutional policies.
REFERENCES
- 1. Baral SD, Poteat T, Strömdahl S, Wirtz AL, Guadamuz TE, Beyrer C. Worldwide burden of HIV in transgender women: a systematic review and meta‐analysis. Lancet Infect Dis. 2013;13(3):214‐222. doi: 10.1016/S1473-3099(12)70315-8 [DOI] [PubMed] [Google Scholar]
- 2. Wang Z, Lau JTF, Yang X, et al. Acceptability of Daily Use of Free Oral Pre‐exposure Prophylaxis (PrEP) Among Transgender Women Sex Workers in Shenyang, China AIDS Behav. 2017;21(12):3287‐3298. doi: 10.1007/s10461-017-1869-4 [DOI] [PubMed] [Google Scholar]
- 3. Sevelius JM, Deutsch MB, Grant R. The future of PrEP among transgender women: the critical role of gender affirmation in research and clinical practices. J Int AIDS Soc. 2016;19(7[Suppl 6]):21105. doi: 10.7448/IAS.19.7.21105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomlins L. Prescribing for transgender patients. Aust Prescr. 2019;42(1):10‐13. 10.18773/austprescr.2019.003. Epub 2019 Feb 1. Erratum in: Aust Prescr. 2019 Aug;42(4):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamidi O, Davidge‐Pitts CJ. Transfeminine Hormone Therapy. Endocrinol Metab Clin North Am. 2019;48(2):341‐355. doi: 10.1016/j.ecl.2019.02.001 [DOI] [PubMed] [Google Scholar]
- 6. Unger CA. Hormone therapy for transgender patients. Transl Androl Urol. 2016;5(6):877‐884. doi: 10.21037/tau.2016.09.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Professional Association for Transgender Health Standards of Care for the Health of Transsexual Transgender, and Gender Nonconforming People, Version 7.; 2010.
- 8. Molina JM, Capitant C, Spire B, et al. On‐Demand Preexposure Prophylaxis in Men at High Risk for HIV‐1 Infection. N Engl J Med. 2015;373(23):2237‐2246. doi: 10.1056/NEJMoa1506273 [DOI] [PubMed] [Google Scholar]
- 9. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587‐2599. doi: 10.1056/NEJMoa1011205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367(5):399‐410. doi: 10.1056/NEJMoa1108524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hiransuthikul A, Janamnuaysook R, Himmad K, et al. Drug‐drug interactions between feminizing hormone therapy and pre‐exposure prophylaxis among transgender women: the iFACT study. J Int AIDS Soc. 2019;22(7):e25338. doi: 10.1002/jia2.25338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shieh E, Marzinke MA, Fuchs EJ, et al. Transgender women on oral HIV pre‐exposure prophylaxis have significantly lower tenofovir and emtricitabine concentrations when also taking oestrogen when compared to cisgender men. J Int AIDS Soc. 2019;22(11):e25405. doi: 10.1002/jia2.25405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grant RM, Pellegrini M, Defechereux PA, et al. Sex Hormone Therapy and Tenofovir Diphosphate Concentration in Dried Blood Spots: Primary Results of the iBrEATHe Study. Clin Infect Dis. 2020:ciaa1160, 73, 7, e2117, e2123. doi: 10.1093/cid/ciaa1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burns RN, Hendrix CW, Chaturvedula A. Population pharmacokinetics of tenofovir and tenofovir‐diphosphate in healthy women. J Clin Pharmacol. 2015;55(6):629‐638. doi: 10.1002/jcph.461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duwal S, Schütte C, von Kleist M. Pharmacokinetics and pharmacodynamics of the reverse transcriptase inhibitor tenofovir and prophylactic efficacy against HIV‐1 infection. PLoS ONE. 2012;7(7):e40382. doi: 10.1371/journal.pone.0040382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baheti G, Kiser JJ, Havens PL, Fletcher CV. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV‐1‐infected patients. Antimicrob Agents Chemother. 2011;55(11):5294‐5299. doi: 10.1128/AAC.05317-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen X, Seifert SM, Castillo‐Mancilla JR, et al. Model Linking Plasma and Intracellular Tenofovir/Emtricitabine with Deoxynucleoside Triphosphates. PLoS ONE 2016;11(11):e0165505. doi: 10.1371/journal.pone.0165505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garrett KL, Chen J, Maas BM, et al. A Pharmacokinetic/Pharmacodynamic Model to Predict Effective HIV Prophylaxis Dosing Strategies for People Who Inject Drugs. J Pharmacol Exp Ther. 2018;367(2):245‐251. doi: 10.1124/jpet.118.251009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dumond JB, Collins JW, Cottrell ML, et al. p16INK4a, a Senescence Marker, Influences Tenofovir/Emtricitabine Metabolite Disposition in HIV‐Infected Subjects. CPT Pharmacometrics Syst Pharmacol. 2017;6(2):120‐127. doi: 10.1002/psp4.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simiele M, D'Avolio A, Baietto L, et al. Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother. 2011;55(6):2976‐2978. doi: 10.1128/AAC.01236-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2(4):e38. doi: 10.1038/psp.2013.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15(9):1463‐1468. doi: 10.1023/A:1011970125687 [DOI] [PubMed] [Google Scholar]
- 23. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. doi: 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ette EI, Onyiah LC. Estimating inestimable standard errors in population pharmacokinetic studies: the bootstrap with Winsorization. Eur J Drug Metab Pharmacokinet. 2002;27(3):213–24, 224. doi: 10.1007/BF03190460 [DOI] [PubMed] [Google Scholar]
- 25. Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187‐2197. doi: 10.1007/s11095-007-9361-x [DOI] [PubMed] [Google Scholar]
- 26. Castillo‐Mancilla JR, Zheng JH, Rower JE, et al. Tenofovir, emtricitabine, and tenofovir diphosphate in dried blood spots for determining recent and cumulative drug exposure. AIDS Res Hum Retroviruses. 2013;29(2):384‐390. doi: 10.1089/aid.2012.0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu Y, Goti V, Chaturvedula A, et al. Population Pharmacokinetics of Tenofovir in HIV‐1‐Uninfected Members of Serodiscordant Couples and Effect of Dose Reporting Methods. Antimicrob Agents Chemother. 2016;60(9):5379‐5386. doi: 10.1128/AAC.00559-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dickinson L, Yapa HM, Jackson A, et al. Plasma Tenofovir, Emtricitabine, and Rilpivirine and Intracellular Tenofovir Diphosphate and Emtricitabine Triphosphate Pharmacokinetics following Drug Intake Cessation. Antimicrob Agents Chemother. 2015;59(10):6080‐6086. doi: 10.1128/AAC.01441-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pruvost A, Negredo E, Benech H, et al. Measurement of intracellular didanosine and tenofovir phosphorylated metabolites and possible interaction of the two drugs in human immunodeficiency virus‐infected patients. Antimicrob Agents Chemother. 2005;49(5):1907‐1914. doi: 10.1128/AAC.49.5.1907-1914.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pruvost A, Negredo E, Théodoro F́́, et al. Pilot pharmacokinetic study of human immunodeficiency virus‐infected patients receiving tenofovir disoproxil fumarate (TDF): investigation of systemic and intracellular interactions between TDF and abacavir, lamivudine, or lopinavir‐ritonavir. Antimicrob Agents Chemother. 2009;53(5):1937‐1943. doi: 10.1128/AAC.01064-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hawkins T, Veikley W, St Claire RL 3rd, Guyer B, Clark N, Kearney BP. Intracellular pharmacokinetics of tenofovir diphosphate, carbovir triphosphate, and lamivudine triphosphate in patients receiving triple‐nucleoside regimens. J Acquir Immune Defic Syndr. 2005;39(4):406‐411. doi: 10.1097/01.qai.0000167155.44980.e8 [DOI] [PubMed] [Google Scholar]
- 32. Greene SA, Chen J, Prince HMA, et al. Population Modeling Highlights Drug Disposition Differences Between Tenofovir Alafenamide and Tenofovir Disoproxil Fumarate in the Blood and Semen. Clin Pharmacol Ther. 2019;106(4):821‐830. doi: 10.1002/cpt.1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anderson PL, Kiser JJ, Gardner EM, Rower JE, Meditz A, Grant RM. Pharmacological considerations for tenofovir and emtricitabine to prevent HIV infection. J Antimicrob Chemother. 2011;66(2):240‐250. doi: 10.1093/jac/dkq447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seifert SM, Chen X, Meditz AL, et al. Intracellular Tenofovir and Emtricitabine Anabolites in Genital, Rectal, and Blood Compartments from First Dose to Steady State. AIDS Res Hum Retroviruses. 2016;32(10–11):981‐991. doi: 10.1089/aid.2016.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cirrincione LR, Podany AT, Havens JP, et al. Plasma and intracellular pharmacokinetics of tenofovir disoproxil fumarate and emtricitabine in transgender women receiving feminizing hormone therapy. J Antimicrob Chemother. 2020;75(5):1242‐1249. doi: 10.1093/jac/dkaa016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Comparison of gender vs. CrCl.
FIGURE S2 Goodness‐of‐fit plots of the final plasma FTC and PBMC FTC‐TP models. (a, e) Individual‐predicted concentrations (IPRED) against observed concentrations (DV), (b, f) Population‐predicted concentrations (PRED) against DV, (c, g) Time after dose against conditional weighted residuals (CWRES), and (d, h) PRED against CWRES.
FIGURE S3 Visual predictive check (VPC) for (a) plasma FTC and (b) PBMC FTC concentrations vs. time. The dashed black and solid black lines depict the 5th, 50th and 95th percentiles of the observed and simulated concentrations, respectively. The shaded area represents the 95% confidence interval around the simulated percentiles.
FIGURE S4 Goodness‐of‐fit plots of the final plasma TFV and PBMC TFV‐DP models. (a, e) Individual‐predicted concentrations (IPRED) against observed concentrations (DV), (b, f) Population‐predicted concentrations (PRED) against DV, (c, g) Time after dose against conditional weighted residuals (CWRES), and (d, h) PRED against CWRES.
FIGURE S5 VPC for (a) plasma TFV and (b) PBMC TFV concentrations vs. time. The dashed black and solid black lines depict the 5th, 50th, and 95th percentiles of the observed and simulated concentrations, respectively. The shaded area represents the 95% confidence interval around the simulated percentiles.
TABLE S1 Simulated concentrations of intracellular metabolites for various dosing regimen of TDF/FTC
Data Availability Statement
The data that support the findings of this study may be available from the corresponding author upon reasonable request and in compliance with relevant institutional policies.