

Abstract
The immune system employs recognition tools to communicate with its microbial evolutionary partner. Among all the methods of microbial perception, T cells enable the widest spectrum of microbial recognition resolution, ranging from the crudest detection of whole groups of microbes to the finest detection of specific antigens. The application of this recognition capability to the crucial task of combatting infections has been the focus of classical immunology. We now appreciate that the coevolution of the immune system and the microbiota has led to development of a lush immunological decision tree downstream of microbial recognition, of which an inflammatory response is but one branch. In this review we discuss known T cell–microbe interactions in the gut and place them in the context of an algorithmic framework of recognition, context-dependent interpretation, and response circuits across multiple levels of microbial recognition resolution. The malleability of T cells in response to the microbiota presents an opportunity to edit immune response cellularity, identity, and functionality by utilizing microbiota-controlled pathways to promote human health.
Keywords: intestinal microbiota, Th17 cells, Tregs, γδ T cells, MAIT cells, segmented filamentous bacteria, dendritic cells, enteric nervous system
INTRODUCTION
A profound realization in the last decade has been that human health relies significantly on the composition and function of the commensal microbiota. Indeed, the microbiota promotes homeostasis in multiple tissues, either directly (e.g., in skin, gastrointestinal, respiratory, and urogenital tracts) or indirectly (e.g., in the central and peripheral nervous system, liver, adipose tissue, skeletal muscles, and cardiovascular system). In turn, the host provides the microbiota with nutrients, supports its diversity and stability, and protects it from domination by pathogens. The evolutionary positive feedback loop of host–microbiota quid pro quo has led to the development of microbial perception tools by the immune system. Such recognition was likely critical to the establishment of mutually beneficial host–microbe interactions, and its application to the crucial task of combatting infections has been the focus of classical immunology. We now appreciate that invasive pathogens represent only a small fraction of the microbial world with which the immune system interacts around the clock and that, concordantly, an inflammatory response is but one branch of a lush decision tree downstream of a microbial recognition event.
The immune system’s perception of microbes involves three stages: recognition, interpretation, and response (Figure 1). The immune microbial perception circuits can be hierarchically organized according to detection resolution from low to high. The basic level of recognition is the metabolic activity of microbes. Microbial groups possessing specific classes of enzymatic pathways can be recognized via detection of metabolic derivatives, such as short-chain fatty acids (SCFAs), secondary bile acids, and tryptophan derivatives. One resolution level higher is identification of groups of similar bacteria and their geographical distribution via common structural patterns, for example via pattern recognition receptors. At the metabolic and structural pattern resolution levels, bacteria are recognized in large groups on the basis of an evolutionarily relevant common denominator, keeping the energy cost of recognition per bacterial species low. One order higher in terms of resolution is a filter for coevolutionary partners. Examples of such selection include species within the classes Clostridia and Bacteroidia that induce homeostatic T cell activation. The highest level of resolution is antigen-specific recognition, which enables identification of bacteria at the species or even strain level. To achieve such specificity, signal amplification processes are required. Therefore, the energy cost per bacterium detected is high and its targets tend to be of great evolutionary importance, either as mutualistic partners or as life-threatening infectious agents.
Figure 1.
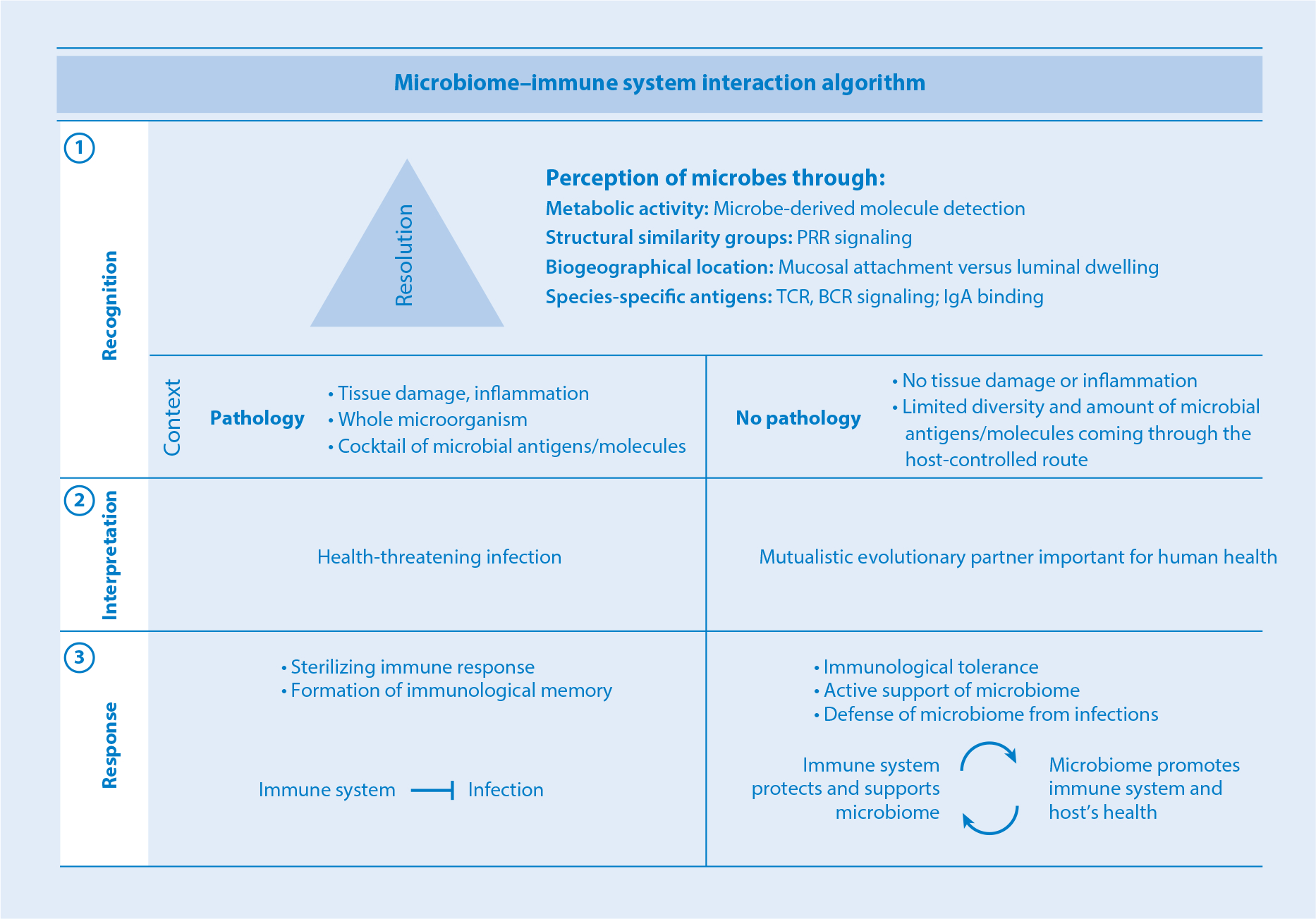
Microbiota–immune system interaction algorithm. Immune microbial perception involves three steps: (①) recognition, (②) context-dependent interpretation, and (③) response. Depending on the function of the recognition circuit, different resolution may be required. At the low end of the resolution spectrum, groups of microbes are recognized via detection of microbial metabolites. The next level up involves detection of common structural patterns, for instance, via PRR signaling. Higher still are circuits involving detection of species-specific coevolutionary adaptations, such as the ability to colonize epithelium. The highest resolution characterizes antigen-specific recognition circuits. Following detection, the microbial signature is classified either as pathological or as commensal/mutualistic according to context, including the presence or absence of inflammation, tissue damage, and mode of antigen acquisition. Signatures determined to be infectious are sterilized, while those perceived as mutualistic are tolerated and receive support and protection. Abbreviations: BCR, B cell receptor; IgA, immunoglobulin A; PRR, pattern recognition receptor; TCR, T cell receptor.
Downstream of detection is the interpretation stage, which includes signal processing and classification of the microbial agent. Even normally harmless commensals may become pathogenic in specific circumstances, thus precluding universal classification. As with any recognition algorithm that classifies a microbial signal, the immune system must also evaluate its context. In the context of tissue invasion or damage, a microbe will be identified as pathogenic and targeted for sterilization. Such recognition usually occurs in the context of the whole microorganism and entails a unique cocktail of microbial antigens and microbe-associated molecular patterns. These microbial signals activate a set of antigen-presenting cells (APCs) and innate immune cells in a specific manner, eventually inducing an inflammatory response as an output. In contrast, under homeostatic conditions, commensal microorganisms are noninvasive and sequestered from direct contact with immune cells. This is achieved through several physical and immunological barriers, such as epithelial and mucus layers, antimicrobial peptides, and secretory IgA. As a result, the diversity and number of commensal-derived products that reach APCs and other innate immune cells are controlled and limited. Thus, in the absence of tissue damage and in the context of legitimate biogeography (e.g., Peyer’s patches), and/or in combination with positive signals (e.g., SCFAs), the microbial signature will be judged as commensal/mutualistic and will result in an output entailing immunological tolerance or nutritional support. If commensals deviate from normal routes of entry, for example, due to breach of the intestinal barrier during injury or acute inflammatory processes, then they will activate a proinflammatory eradicative immune response.
T cells are unique in that they are capable of microbial recognition at all levels of resolution and in the context of both pathology and mutualistic coevolution. The characteristics of a successful T cell response to a given microbe (whether pathogenic or commensal) are antigen specificity and appropriate functional differentiation, e.g., T regulatory cells (Tregs); T follicular helper (Tfh) cells; and T helper 1, 2, and 17 (Th1, Th2, and Th17) cells. Whereas proinflammatory T cell responses are aimed at clearing invaders, T cell responses to commensals aim to survey and control our autochthonous ecosystem. Accordingly, T cell responses (unconventional T cell responses in particular) to commensal microbes are often associated with promiscuous specificity. Moreover, the unique mechanisms of sampling commensals and the nature of commensal-derived molecules promote engagement with tolerogenic innate immune cells or APCs, which leads to either outright induction of Tregs or induction of effector T cells with reduced inflammatory functions. Commensal-induced T cells can also actively participate in decreasing tissue damage as well as in promoting tissue repair and tolerance (Figure 1).
The functional objectives of T cell responses to the microbiota are not completely understood and are likely context dependent. It is safe to assume that they promote mutualism by directly and indirectly regulating the numbers, composition, and distribution of commensal microbes. However, characterizing the mechanistic details of these functions will be the next exciting area of research in this field. In this review, we discuss known T cell–microbe interactions across multiple levels of microbiota recognition resolution, with a focus on the intestine. Furthermore, we discuss how the malleability of T cells in response to microbiota composition presents an opportunity to edit T cellularity, identity, and functionality by utilizing microbiota-driven molecular pathways in order to promote human health.
T CELL ALGORITHMS OF MICROBIAL RECOGNITION
Among the immunological programs of microbial perception, T cell recognition circuits are arguably the most versatile, covering the full spectrum of microbial recognition resolution with outputs ranging from promoting tolerance and tissue repair to sterilizing inflammatory responses. Unconventional T cells such as γδ T cells, mucosal-associated invariant T (MAIT) cells, and natural killer T (NKT) cells recognize groups of microbes on the basis of microbe-derived metabolites. Following microbial detection, activation of unconventional T cell circuits leads to fortification of the mucosal barrier and protection against infection. This likely reflects an early stage of host–microbiota coevolution. Conventional CD4 and CD8 T cells enable microbial recognition down to specific microbial antigens and generate non-zero-sum outputs, benefiting both the host and the microbiota (e.g., immunological tolerance) and representing the evolutionary state of the art in host–microbiota adaptation.
Antigen-Specific Recognition Circuits
Conventional CD4 and CD8 T cells in the intestine are constitutively exposed to antigen. T cells can be primed by a diverse array of antigens, including those derived from the microbiota, together with their unique contexts. Activated conventional T cells differentiate into a wide spectrum of effector cells with functions ranging from regulatory to proinflammatory. The full range of participating T cell functionalities and microbiota species and antigens is an exciting area of intense research. Here we discuss current examples of how specific microbiota species/strains induce T cell subsets with unique functions.
Th17 cells.
Th17 cells provide one of the first- and best-studied examples of antigen-specific recognition circuits resulting from mutualistic coevolution of a commensal and its host. Segmented filamentous bacteria (SFB) are beneficial commensals and part of the healthy microbiota in many animal species that colonize the small intestine, particularly the terminal ileum (1, 2). SFB carry mucolytic genes and can cross the mucus barrier and attach to the epithelial lining (3, 4); however, unlike pathogens, SFB do not penetrate or invade the epithelial layer. Importantly, only SFB strains indigenous to the host species are capable of attaching to the epithelium interface; for instance, rat SFB inoculated into mice remain restricted to the lumen and vice versa (4). Therefore, at steady state, SFB are recognized and classified as evolutionary partners. SFB induce Th17 cells that recognize antigens encoded by SFB, which are predicted to be secreted or expressed on the bacterial surface (5, 6). Mechanistically, upon attachment, SFB antigens can be sampled by intestinal epithelial cells (IECs) via microbial adhesion–triggered endocytosis (MATE) (7). MATE is a clathrin-independent, dynamin-dependent, and cell division control protein 42 (CDC42)-mediated endocytosis mechanism in IECs elicited by SFB adhesion. SFB adhesion is also associated with activation of a unique IEC gene expression program that includes upregulation of serum amyloid A proteins (SAAs) (1, 4, 8, 9). SAAs activate lamina propria CD11c+ cells to produce interleukin (IL)-1ββ, which in turn stimulates IECs to secrete more SAAs (4). This feedback loop functions as a signal amplifier, likely increasing recognition circuit sensitivity. Its importance is highlighted by the existence of a redundant signal amplification loop, in which SFB activate production of IL-22 by group 3 innate lymphoid cells (ILC3s), and IL-22 enhances the expression of SAAs in IECs (4, 8). Downstream of SFB attachment to the epithelium, an amplified SAA signal is converted into the circuit’s output, including Th17 cell differentiation (4, 8, 9). Counterbalancing the output of the SFB recognition circuit, the number and function of induced Th17 cells are kept in check by peripherally induced Treg (pTreg) differentiation controlled by another recognition circuit (10, 11), discussed in more detail in the section in the section titled T regulatory cells. Commensal-induced Th17 cells reinforce the epithelial barrier through upregulation of Nox1 (an apical NADPH oxidase), α-defensins, and polymeric immunoglobulin receptor required for the transcytosis of secretory IgA, ultimately bolstering the host’s immunity against pathogens (12–14) (Figure 2).
Figure 2.
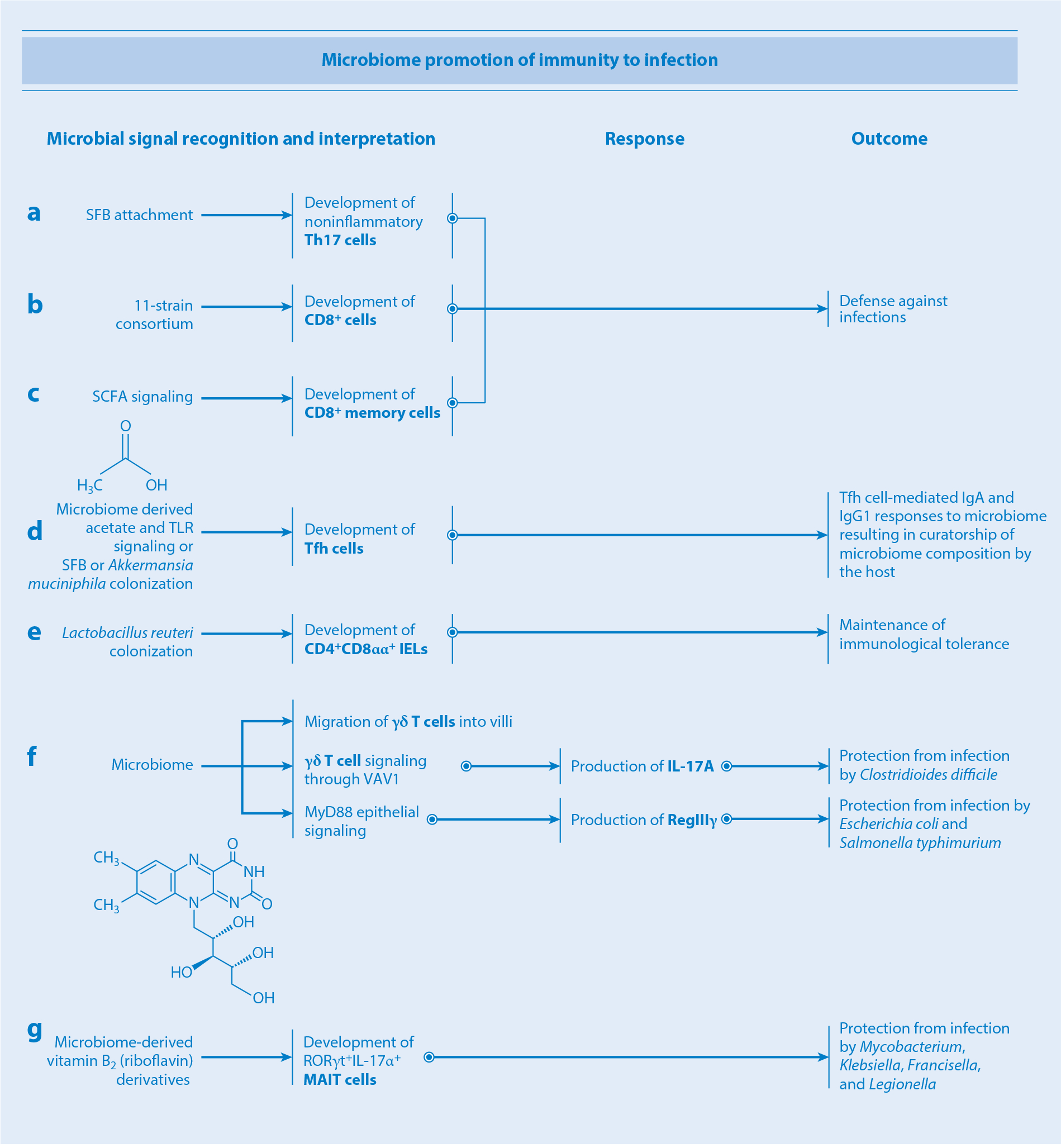
How the microbiota promotes immunity to infection. (a–c) Attachment of SFB to the intestinal epithelium, colonization by an 11-strain consortium, and SCFA signaling promote differentiation of noninflammatory Th17, CD8+, and memory CD8+ T cells, respectively, ultimately bolstering the host’s and microbiota’s protection from infections. (d) Microbiome-derived acetate and TLR signaling or colonization with SFB or Akkermansia muciniphila promote differentiation of Tfh cells, which in turn supports the development of IgA and IgG1 responses. (e) Colonization with Lactobacillus reuteri promotes differentiation of regulatory CD4+CD8αα+ IELs, thus enhancing intestinal immunological tolerance. (f) Microbial signals shape the biogeographical distribution of γδ T cells within the epithelial layer. Microbial signaling through guanine nucleotide exchange factor VAV1 on γδ T cells stimulates production of IL-17A, bolstering protection against Clostridioides difficile infection. MyD88 epithelial signaling triggers secretion by γδ T cells of the antimicrobial peptide RegIIIγ, conferring protection against Escherichia coli and Salmonella typhimurium infections. (g) Microbiota-derived vitamin B2 derivatives promote development of MAIT cells, conferring protection against infection by Mycobacterium, Klebsiella, Francisella, and Legionella. Abbreviations: IEL, intraepithelial lymphocyte; Ig, immunoglobulin; IL, interleukin; MAIT cell, mucosal-associated invariant T cell; SCFA, short-chain fatty acid; SFB, segmented filamentous bacteria; Tfh cell, T follicular helper cell; Th17, T helper 17; TLR, Toll-like receptor.
In addition to SFB, Bifidobacterium adolescentis also induces intestinal Th17 cells through an as-yet-undefined mechanism (15). Ketone bodies accumulated by consumption of a very low-carbohydrate and high-fat ketogenic diet inhibit the growth of B. adolescentis and reduce Th17 cell frequency (16). In contrast, a high-salt diet enhances Th17 cell differentiation (17–19). This effect is due to a reduction in Lactobacillus species and a consequent reduction in the levels of Th17-inhibitory tryptophan metabolites, such as indole-3-lactic acid and indole-3-acetic acid, in the intestine (17). High-salt conditions also directly activate nuclear factor of activated T cell 5 and serum glucocorticoid-regulated kinase 1 in T cells to promote Th17 cell differentiation (18, 19). An accumulation of Th17 cells mediated by a high-salt diet can lead to hypertension and cognitive impairment (17, 20). Therefore, Th17 induction by the microbiota is connected across diverse host physiological processes. It is also possible that microbe-induced Th17 differentiation helps improve their competitive fitness for their ecological niches.
T regulatory cells.
One of the clearest examples of a microbial recognition circuit that benefits both the host and the microbiota is that responsible for maintaining the population of commensal Tregs. FOXP3+ pTregs positive for RORγt and c-Maf and negative for Helios and Neuropilin 1 are capable of inhibiting antigen-specific as well as bystander T cells via constitutive expression of cytotoxic T lymphocyte–associated protein 4 (CTLA4) and inducible expression of T cell costimulator, IL-10, IL-35, and transforming growth factor β (TGF-β) (21–23). In contrast, a significant proportion of colonic Foxp3+ Tregs express GATA-binding protein 3 (GATA3) and the IL-33 receptor ST2 (24, 25). Most GATA3+ Tregs are positive for Helios and are unaffected by the absence of the gut microbiota, suggesting that they are derived mostly from thymic Tregs. The pTreg population is induced as a result of detection and classification of specific groups of bacteria as commensals, such as spore-forming Clostridia clusters XIVa and IV, which are drastically reduced in germ-free and antibiotic-treated mice (26–28). A group of 46 murine-derived (28) and 17 human-derived (27) Clostridia strains induce the differentiation of a Clostridia-specific population of pTregs in germ-free mice. Among Clostridia, Clostridium ramosum alone is sufficient to induce colonic RORγt+ Tregs (29). Helicobacter hepaticus colonization also induces antigen-specific RORγt+ pTregs in the large intestine during homeostasis (11, 30). These reports suggest that specific members provide the rest of the microbiota with, to borrow an economics term, a nonexcludable public good, in the form of broad immunological tolerance that results from recognition of a small number of species but benefits all of the microbiota. The host, in turn, benefits as Treg circuits provide a counterbalance to potentially proinflammatory self-amplifying circuits, such as the Th17 circuits described above. Moreover, the host may also benefit from establishment of immunologically tolerated, and therefore functional, microbiota. It follows that antibiotics targeting Clostridia species should be deployed with caution; conversely, knowledge of the workings of Treg circuitry opens an opportunity for development of Clostridia-based biotherapeutic interventions.
T follicular helper cells.
Tfh cells are responsible for the differentiation of affinity-matured antigen-specific B/plasma cells. Recognition of Akkermansia muciniphila or SFB directs the differentiation of Tfh cells (31, 32), thereby supporting IgA- and IgG1-mediated curatorship of microbiota composition that, in turn, promotes the overall diversity and stability of the microbiota (Figure 2). Mechanistically, colonization with A. muciniphila induces microbial antigen-specific Tfh cells in Peyer’s patches and mesenteric lymph nodes (MLNs). The microbial antigen-specific Tfh cell differentiation were clearly demonstrated when T cells from Akkermansia antigen-specific T cell receptor (TCR) transgenic mice were transferred into gnotobiotic mice colonized with A. muciniphila but not with altered Schaedler flora (31). Interestingly, Akkermansia-specific CD4 T cells adopt various fates including Th1 and Th17 cells in the intestinal lamina propria, depending on the composition of the surrounding microbiota (31). SFB induce Tfh cell differentiation in Peyer’s patches by inhibiting the IL-2 signaling pathway in CD4 T cells (32). In contrast to A. muciniphila antigen-specific Tfh cell differentiation, cognate recognition of SFB antigens is not required for SFB-mediated Tfh cell induction (32).
Th1 cells.
Recognition of bacterial species belonging to the genus Klebsiella, such as K. aeromobilis and K. pneumoniae, drives Th1 cell responses in the intestine (33). The Klebsiella-mediated Th1 cell induction is dependent on both basic leucine zipper ATF-like transcription factor 3 (Batf3)-dependent CD11b−CD103+ dendritic cells (DCs) and MyD88-dependent signaling pathways [including Toll-like receptor (TLR) and IL-18 signaling]. Many induced Th1 cells recognize Klebsiella antigens, including OmpX (outer membrane protein X) (33), although it remains unclear how the bacterial antigens reach lamina propria DCs. Although it induces Th1 cells, Klebsiella colonization does not necessarily cause inflammatory changes in the intestine. Instead, it leads to upregulation of induced interferon (IFN)-inducible genes in IECs to enhance intestinal barrier integrity. However, Klebsiella-specific Th1 cells can elicit severe gut inflammation in the absence of homeostatic control, for instance, in Il10−/− mice (33). Similarly to Klebsiella, an Escherichia coli strain isolated from the small intestine of a patient with Crohn’s disease possesses a potent ability to induce Th1 cells in germ-free mice (34). While these Klebsiella and E. coli species are minor components of the healthy human gut microbiota, they expand and induce Th1 cells following disruption of colonization resistance mechanisms, such as dysbiosis. Indeed, Klebsiella and E. coli are significantly enriched in the fecal microbiota of patients with inflammatory bowel disease (IBD) in comparison to healthy controls (35), suggesting that they play a role in perpetuating intestinal inflammation.
CD8 T cells.
Recognition of a specific consortium of 11 bacterial strains isolated from the microbiota of a healthy donor leads to differentiation of intestinal IFN-γ+ CD8 T cells (36) (Figure 2). Concordantly, these cells are present in the intestinal lamina propria in normal mice but are less abundant in germ-free mice (36). Mechanistically, the 11 strain–driven T cell induction is dependent on CD103+ DCs via major histocompatibility (MHC) class Ia (conventional MHC class)-mediated commensal antigen presentation (36). The 11 strains promote IFN-γ+ CD8 T cell accumulation in subcutaneously engrafted tumors, enhancing the efficacy of anti-PD1 (programmed cell death protein 1) or anti-CTLA4 checkpoint blockade cancer therapy (36). In addition to having an obvious benefit for the host, the CD8 circuit may benefit the 11 strains by protecting their ecological niches from pathogens. Indeed, immunity stimulated by the 11 strains helps repel an orally administered Listeria monocytogenes infection. Microbe-derived metabolites can support CD8 T cell accumulation. Mevalonate and dimethylglycine derived from these 11 strains, for example, are associated with IFN-γ+ CD8 T cell induction (36). Similarly, gut microbiota–derived SCFAs promote the transition of antigen-activated CD8 T cells into memory cells by fueling oxidative phosphorylation (37). Therefore, specific bacterial consortia–derived metabolites enter circulation and may be involved in the systemic activation of CD8 T cells.
A series of studies on the skin microbiota has revealed that colonization with Staphylococcus epidermidis promotes noninflammatory accumulation of IFN-γ+ and IL-17+ CD8 T cells in the skin (38–41). These S. epidermidis–driven immune responses help protect the host from infection by diverse pathogens, including Leishmania major and Candida albicans, and promote tissue repair after injury (38–40). S. epidermidis–driven CD8 T cell induction is mediated through the activation of TLRs (TLR2 in particular) and Dectin 1 (41), and by presentation of N-formyl methionine–containing peptides by nonclassical MHC class I (H2M3) on DCs (40).
CD4+CD8αα+ intraepithelial lymphocytes.
Along with the Clostridia-mediated Treg-inducing circuit, recognition of Lactobacillus reuteri generates regulatory CD4+CD8αα+ double-positive intraepithelial lymphocytes (IELs) in the small intestine of mice (42) (Figure 2). Mechanistically, L. reuteri metabolizes tryptophan to indole-3-lactic acid, a ligand for the transcription factor aryl hydrocarbon receptor (AhR). Indole-3-lactic acid–bound AhR acts in concert with TGF-β to downregulate the Cd4 gene regulator ThPOK (T helper–inducing POZ/Krüppel-like factor) in CD4+ T cells (42, 43). This process leads to RUNX3 and T-bet upregulation, which in turn results in CD8αα expression. Therefore, the presence of Lactobacillus plus a tryptophan-rich diet promotes differentiation of CD4+CD8αα+ IELs and consequently protects the host from food allergy and celiac disease (44, 45).
Microbial Metabolite Recognition Circuits
Complementary to state-of-the-art but energy-demanding antigen-specific recognition enabled by conventional T cell circuits, unconventional T cells recognize whole groups of microbes simultaneously via detection of microbe-derived lipid and glycolipid antigens as well as microbial metabolic intermediates via monomorphic antigen-presenting molecules, such as MHC class I–related protein 1 (MR1) and CD1d. Such recognition is characterized by lower-resolution offset by lower energy demand per microbe species detected, but greater speed, in comparison to antigen-specific recognition. These trade-offs make unconventional T cell–mediated recognition circuits ideal for immune applications that do not require an antigen-specific level of resolution, such as reinforcement of the intestinal barrier, wound repair, and augmentation of conventional T cell–mediated responses.
γδ T cells.
γδ T cells express semi-invariant TCRs composed of a γ and a δ chain and have been implicated in a wide range of homeostatic processes ranging from immune surveillance and wound repair (46) to regulation of thermogenesis (47) and central nervous system (CNS) plasticity (48). They represent a minor component of the circulating T cell population, but are much more abundant as tissue-resident IELs at sites including the skin and gastrointestinal tract (49). The two major γδ T cell subsets in humans are Vγ9Vδ2+ and Vδ1+, which recognize lipids and phosphoantigens (50). In particular, Vγ9Vδ2+ cells recognize microbial metabolic pathway intermediates including (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP), a microbial precursor of isopentenyl pyrophosphate (IPP) (51). HMB-PP is produced by most gram-negative bacteria as well as by pathogens including Mycobacterium tuberculosis, Clostridioides difficile, and L. monocytogenes. In contrast, humans do not produce HMB-PP, instead generating IPP via the mevalonate pathway. The microbiota modulates γδ IEL cytokine production, dynamics, and epigenetics. For example, colonization with species able to penetrate the intestinal mucosal barrier (e.g., E. coli and Salmonella typhimurium) but not others (e.g., Bacteroides thetaiotaomicron and SFB) induces γδ IEL expression of the antimicrobial peptide RegIIIγ. This induction occurs in an epithelial cell–intrinsic, MyD88-dependent manner (52), suggesting that invasive microbes trigger a signaling cascade in epithelial cells that, in turn, communicate with γδ IELs.
γδ T cells function as early responders in several models of infectious disease and are often responsible for the initial production of cytokines such as IL-17 and IFN-γ (46, 53) (Figure 2). While the gut microbiota does not seem to affect the absolute number of intestinal γδ IELs (52, 54), certain commensals are able to increase the frequency of IL-17+IL-1R1+ γδ T cells via signaling through the guanine nucleotide exchange factor VAV1, ostensibly protecting against disease (55). In fact, IL-17A production by γδ T cells protects mouse models against C. difficile infection (56). Additionally, γδ IEL positioning and motility within the intestinal epithelial compartment are microbiota dependent: γδ IELs are physically shifted downward along the crypts in germ-free versus specific-pathogen-free mice (57). The commensal-regulated localization and motility of γδ IELs along the epithelial layer are required for efficient surveillance of the entire epithelial surface. Mobilization patterns also differ in the context of enteropathogenic infection, mediated in part by a γδ IEL metabolic switch and epithelial cell–intrinsic MyD88 signaling (57). The gut microbiota also affects chromatin accessibility of enhancers in both γδ and αβ T cells, thereby modulating myriad pathways and potentially accounting for some of the observed differences in T cell function between mice with disparate microbiota compositions (58).
Mucosal-associated invariant T cells.
MAIT cells are innate-like lymphocytes expressing predominantly CD8α, with a smaller subset lacking both CD4 and CD8. MAIT cells express an invariant TCR α chain (Vα19 Jα33 in mice and Vα7.2 Jα33 in humans) paired with one of several Vβ chains, and they recognize antigens presented by the MHC class Ib protein MR1 (59). Several MR1-bound antigens have been described; these include a range of microbe-derived vitamin B2 (riboflavin) derivatives, such as the ribityl-lumazines 7-hydoxy-6-methyl-8-d-ribityllumazine, 6,7-dimethyl-8-d-ribityllumazine, and 5-(2-oxopropylideneamino)-6-d-ribitylaminouracil (5-OP-RU) (60–62). The microbiota plays a critical role in the production of riboflavin derivatives, and numbers of MAIT cells are severely decreased in germ-free animals (63, 64). Carriage of the genes necessary for 5-OP-RU production (namely ribA, ribD, and pyrp2) is widespread in both gut and skin commensals, and monocolonization of germ-free mice with microbes competent in this pathway (including Proteus mirabilis or Enterococcus hirae) is sufficient to induce thymic MAIT cells to levels observed in specific-pathogen-free mice (63, 64). Interestingly, the induction of tissue-resident, but not thymic, MAIT cells specifically requires early-life exposure to MR1-bound antigens such as 5-OP-RU—the absence of exposure during this critical developmental period results in permanently impaired tissue-resident MAIT cell frequencies (63). The various roles these cells play in host health and disease remain to be fully elucidated, though in mice both the thymic and skin-resident microbiota-induced MAIT cells tend to be RORγt+, typically adopting a type 17 effector phenotype (63, 64). In particular, skin MAIT cells can secrete IL-17A upon local sensing of commensals in an IL-1- and IL-18-dependent manner (63). They can also promote wound healing (63), much like other, nonclassical, MHC-restricted commensal-specific T cells. MAIT cells confer protection against infection with pathogens including Mycobacterium, Klebsiella, Francisella, and Legionella species, among others (59) (Figure 2). MAIT cells also recognize acute viral infections via sensing of IL-18, IL-15, and type I IFNs and play important roles in subsequent boosting of the adaptive immune response, as exemplified by the observation that MAIT cells are required for the generation of antigen-specific CD8+ T cells by adenovirus vector–based vaccines (65). Therefore, MAIT cells act as a link between the innate antimicrobial immune response and the generation of adaptive immunity for pathogens and vaccines alike.
Invariant natural killer T cells.
Invariant natural killer T (iNKT) cells recognize microbial and host-derived glycolipids presented by the MHC class Ib molecule CD1d, and they express an invariant α chain paired with a limited selection of β chains (50). iNKT cells rapidly produce cytokines upon activation, and subsets analogous to Th1, Th2, and Th17 cells have been identified in mice, though these distinctions are not as clear in humans (66). Factors contributing to iNKT cell development, particularly in the periphery, remain poorly understood, though it is becoming increasingly appreciated that reciprocal cross talk exists between these cells and the microbiota. Germ-free mice exhibit increased relative and absolute numbers of iNKT cells in the colonic lamina propria and lung, secondary to Cxcl16 hypermethylation and subsequent increased mucosal recruitment (67). Interestingly, conventionalization of neonatal but not adult mice normalizes iNKT cell levels and protects against oxazolone-induced colitis and models of asthma (67). Monocolonizing neonatal mice with Bacteroides fragilis produces a similar effect, albeit by a distinct mechanism. B. fragilis synthesizes a bacterial sphingolipid called GSL-Bf717, which competitively inhibits CD1d and thus suppresses iNKT cell proliferation in the gut (68). Nevertheless, other B. fragilis–derived sphingolipids weakly stimulate iNKT cell expansion, suggesting that these cells are likely controlled by the net effect of an individual’s overall gut sphingolipid profile (68, 69). iNKT cell–microbiota cross talk is bidirectional, as mice lacking iNKT cells are more prone to small-intestinal bacterial overgrowth and translocation, potentially due to impaired stimulation and degranulation of Paneth cells (70).
MICROBIOTA-MEDIATED PRIMING AND SPECIFICATION OF T CELLS
As described above, multiple examples of commensal effects on T cell responses have been characterized. In contrast, mechanistic knowledge about how the differentiation and function of microbiota-induced T cells are controlled is still in its infancy. The issue is further complicated by the potential heterogeneity of mechanisms utilized by different microbes. The sheer number and diversity of microbes and microbiota-derived antigens in the adult intestine suggest that a large proportion of homeostatic intestinal T cells are microbiota specific. Although commensal-specific homeostatic T cells are indeed present in the intestine in both mice and humans (1, 5, 6, 11, 27, 31, 71), fundamental questions about the prevalence and heterogeneity of such responses remain unanswered. For example, whether homeostatic CD4 T cells are induced by many, or only a few, commensal microbes is unclear. In mice, intestinal T cells generally do not respond to purified microbial lysates, and only a few species have been demonstrated to induce such responses in monocolonized mice (1, 5, 6, 15, 29). These observations have led to the concept that a few “keystone” species are responsible for the homeostatic T cell response in the gut (72). However, these experiments investigated relatively small numbers of selected microbes, and no studies to date have examined this topic systematically. Interestingly, the few known examples of such species show remarkable diversity in terms of antigen acquisition and innate immune cell involvement.
Pathways for Acquisition of Microbiota-Derived Antigens
Host–microbiota coevolution has led to the development of dedicated communication interfaces. In the intestine, antigen sampling can occur through several mechanisms. The relative contribution of these mechanisms to overall mucosal homeostasis and even to the sampling of individual commensal species remains elusive. Individual microbes are likely detected in several ways, depending on the size or type of specific antigens (e.g., secreted, membrane bound, or intracellular). Indeed, the subcellular localization of bacterial antigen dictates the T cell response to S. epidermidis. Cell wall component antigens preferentially stimulate CD8 T cells, whereas secretory antigens predominantly induce CD4 T cell responses (73). Therefore, the mode of antigen acquisition is crucial for determining the nature of the resulting T cell response.
Acquisition of antigens in the intestine occurs either through the specialized follicle-associated epithelium (FAE) or through the villous epithelium (74, 75). The FAE is present on the luminal surface of Peyer’s patches and isolated lymphoid follicles. It is anatomically designed to facilitate sampling of luminal contents with the clear goal of engaging mucosal adaptive immunity. FAE epithelial cells lack microvilli and mucus- or antimicrobial peptide–secreting capabilities, facilitating access by noninvasive commensals (75). The sampling workhorse of FAE are the M cells, which can internalize particulate material through endocytosis or macropinocytosis and release their cargo within intraepithelial pockets (76, 77). Antigen uptake is mediated by receptors on the surface of M cells, such as GP2 (77, 78). M cells can also deliver secretory IgA-coated commensal bacteria into Peyer’s patches (79, 80). The basolateral pocket of M cells and the surrounding subepithelial dome area of the Peyer’s patches contain specialized subsets of DCs, which take up M cell–trafficked antigens to prime T and B cells and imprint them with gut-homing capabilities. Conditional deletion of tumor necrosis factor receptor–associated factor 6 (TRAF6) in IECs causes loss of M cells, which leads to significant impairment of commensal-specific IgA responses (81), underscoring their importance for induction of adaptive immunity.
Multiple mechanisms exist for antigen sampling through the villous epithelium. These include epithelium transcellular and paracellular mechanisms (82). Transcellular mechanisms in enterocytes include endocytosis and transcytosis of luminal antigens, neonatal Fc receptor–mediated sampling of IgG-bound luminal contents (83), and release of MHC class II–containing exosomes (82, 84). Engulfment of apoptotic IECs by lamina propria macrophages or DCs may also play a role (85). In addition to enterocytes, villous M cells have been described (86), though their functionality and contribution to commensal sampling are unclear. Luminal antigens can also be delivered through goblet cell–associated passages (87), a process that provides commensal T cell antigens during early life (88). Paracellular mechanisms include sampling of luminal contents by intestinal APCs through extension of transepithelial dendrites (89–91). Although transepithelial APC dendrites respond to microbial stimuli and are capable of sampling luminal contents (89–91), whether this process samples commensal microbes and modulates T cell responses is not known. Enterocytes can also receive antigens from attached bacteria under physiological conditions through MATE for induction of SFB-specific Th17 cells (7).
Microbial Signal Processing
Following acquisition, antigens are delivered to resident APCs. Antigen-capturing APCs migrate to inductive sites such as MLNs and T cell zones of Peyer’s patches, where they prime and expand naïve T cells. Importantly, APCs initiate the T cell differentiation program and imprint the activated T cells for homing back to their mucosal tissue of origin. In the mucosal tissue of origin, activated antigen-specific T cells complete their differentiation program into terminal effectors under the control of tissue-resident signals.
To produce a certain output, the microbial signature needs to pass through appropriate processing by APCs as well as by innate immune cells, epithelial cells, and enteric neurons (Figure 3). In steady state, sampling of microbiota-derived molecules occurs in the absence of tissue damage or overt inflammation (i.e., so-called signal 3 in the classical model of T cell activation). In this context, sampling of commensal microbes leads to focused activation of epithelial and resident innate immune cells, which ensures that microbiota antigens are delivered to appropriate, in most cases tolerogenic, APCs to prime T cells at the correct location, as well as to jump-start the T cell differentiation program.
Figure 3.
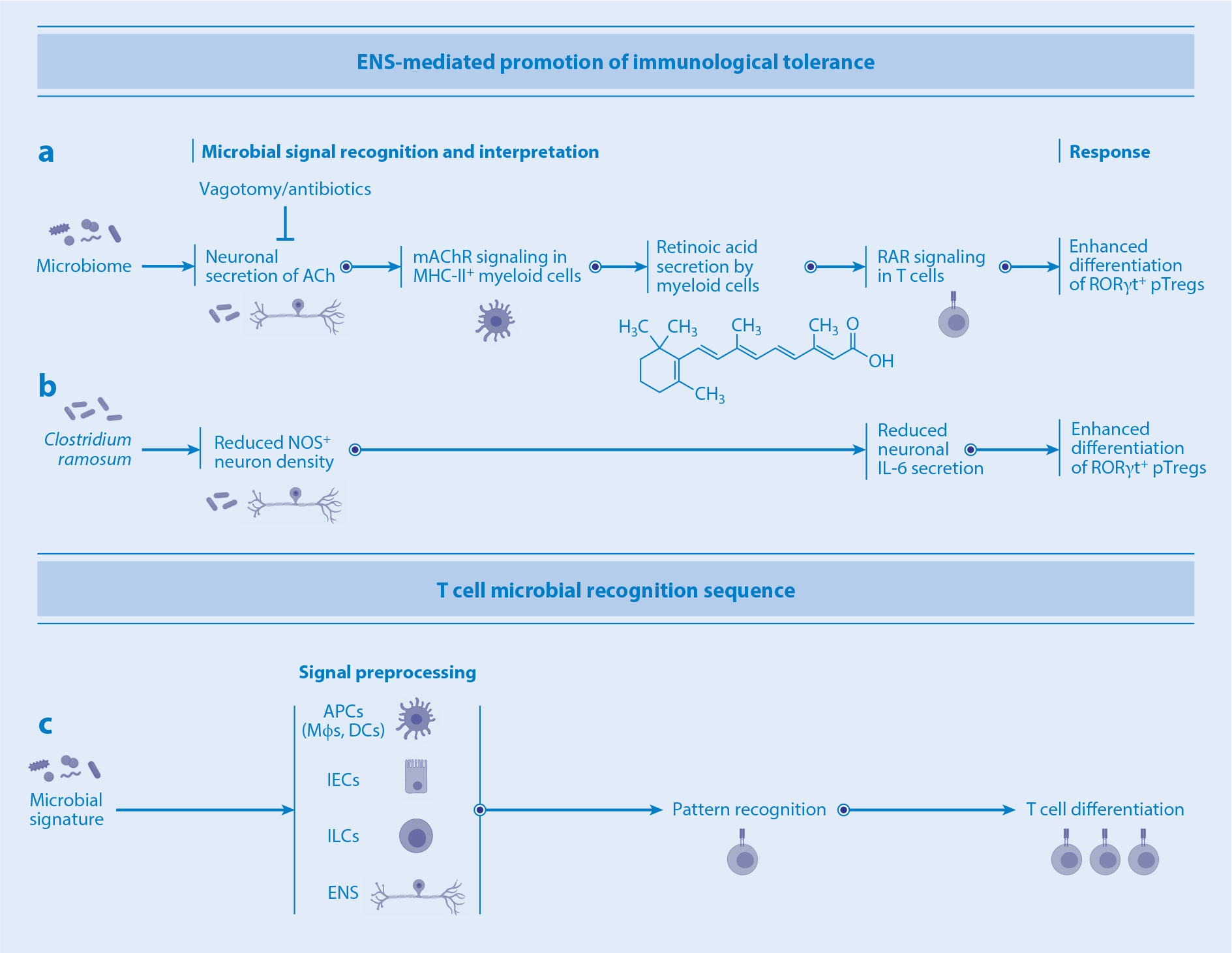
Innate immune cells and enteric neuronal modulation of T cell differentiation. (a) Microbial signals stimulate enteric neurons to secrete ACh, leading to the production of retinoic acid by myeloid cells. Retinoic acid in turn promotes pTreg differentiation via RAR signaling. (b) Clostridium ramosum reduces NOS+ IL-6-secreting neurons, promoting pTreg differentiation. (c) The T cell microbial recognition sequence involves preprocessing of microbial signals by APCs and/or IECs, ILCs, or the ENS prior to the recognition and differentiation stages. Abbreviations: ACh, acetylcholine; APC, antigen-presenting cell; DC, dendritic cell; ENS, enteric nervous system; IEC, intestinal epithelial cell; IL, interleukin; ILC, innate lymphoid cell; mAChR, muscarinic acetylcholine receptor; MHC, major histocompatibility complex; Mφ, macrophage; NOS, nitric oxide synthase; pTreg, peripherally induced T regulatory cell; RAR, retinoic acid receptor.
Dendritic cells.
To prime commensal-specific T cell responses in the gut, antigen-loaded APCs migrate from the lamina propria to draining MLNs. MLNs are particularly important for priming tolerogenic intestinal T cell responses (92–94). It is generally agreed that migratory DCs control induction of intestinal pTregs to commensal bacteria or to establish oral tolerance to dietary antigens (92–95). CD103+ DCs are particularly tolerogenic, due to their ability to induce intestinal Tregs (96–98). In the intestinal lamina propria, CD103+ DCs come in two main types: classical CD11b− cDC1s, which require BATF3 and IFN regulatory factor 8 (IRF8), and CD11b+ cDC2s, which require IRF4 and Notch2 (99–102). CD11b+CD103+ cDC2s are unique to the intestine and have important homeostatic functions, including inducing pTregs and noninflammatory mucosal Th17 cells (102–104). However, whether CD103+ DCs are necessary for such responses remains unclear. For example, a recent study found that CD103+ DCs were dispensable for induction of Helicobacter-specific pTregs (95). Interestingly, similar results were obtained for SFB-induced Th17 cell differentiation, where both cDC1 and cDC2 CD103+ DCs were dispensable (105). It has been suggested that intestinal DC subsets may share responsibility for controlling homeostatic commensal T cell responses (95). However, a drawback of studies on the functional roles of individual DC subsets is that they rely almost exclusively on genetic models of depletion. The deletion of DC subsets may provide an opportunity for alternative subsets to fill the void in antigen presentation and perform a semi-redundant function. Therefore, the absence of phenotype in depletion models may not always exclude a major role for the deleted subset under physiological conditions. An alternative strategy is conditional deletion of MHC class II molecules on individual APC subsets, which thus far has not been applied to commensal-specific T cell responses.
Macrophages.
In addition to DCs, intestinal macrophages, IECs, and ILCs can all express MHC class II and can potentially participate in T cell priming and specification. Intestinal CX3CR1+ cells express high levels of MHC class II and were initially classified as DCs due to their unique morphology, including transepithelial dendrites (90, 106). However, they were later shown to undergo macrophage developmental and phenotypic programs (107). Intestinal macrophages are constantly replenished from bone marrow-derived CCR2+ monocytes, although a subset of these cells have fetal origin (108, 109). Intestinal macrophages have multiple mechanisms by which to acquire commensal and other luminal antigens. Depletion of intestinal macrophages abolishes SFB-specific Th17 cell differentiation (105). Similarly, deletion of colonic CX3CR1+ macrophages impedes induction of Candida albicans–specific Th17 responses (110). However, whether these cells actually present such antigens to CD4 T cells and are capable of priming commensal-specific T cell responses in vivo has not been unequivocally demonstrated. As lung-resident CCR2+ monocytes are known to initiate CD4 T cell responses by delivering fungal antigens to draining lymph nodes for presentation by DCs (111), intestinal macrophages may acquire and deliver, or in some other way make available, commensal antigens for T cell priming. In support of this idea, colonic macrophages can promote T cell–dependent secretory IgA responses against Salmonella infection in an MHC class II–dependent manner (112). An argument against the ability of macrophages to prime commensal T cell responses is based on the observation that they reside in the lamina propria and do not normally migrate to MLNs. However, not all lamina propria CD4 T cell responses require Peyer’s patches and MLNs. For example, macrophage migration to MLNs is not required for induction of Salmonella adaptive immunity, which instead occurs in tertiary lymphoid structures in the lamina propria (112). Similarly, MLNs and Peyer’s patches are not required for generation of commensal-specific Th17 cells (5, 113). Therefore, macrophages may affect the pool of commensal-specific CD4 T cells through various mechanisms.
Epithelial cells.
Villous IECs can also play a role in induction and modulation of homeostatic T cell responses. As discussed above, IECs have multiple mechanisms for acquisition of luminal antigens. In addition, IECs can serve as APCs in vitro (114). Intestinal stem cells and enterocytes express not only MHC class II but also the full machinery for antigen presentation, including unconventional costimulatory molecules (115). Recent studies have demonstrated a role for IEC-mediated antigen presentation via MHC class II in the regulation of intestinal CD4 T cell and CD4+CD8αα+ IEL development (115–117). However, whether IECs can serve as nonconventional APCs in vivo remains to be demonstrated. Interestingly, epithelial MHC class II expression is induced only by a subset of commensal microbes (5, 118) in a circadian manner (119). Therefore, the role of epithelial MHC class II needs to be examined in the context of individual commensal antigens and appropriate host T cell responses.
Innate lymphoid cells.
ILC3s express MHC class II and affect CD4 T cell responses (5, 120). ILC3s can directly engage CD4 T cells in MLNs and mediate MHC class II–dependent deletion of commensal-reactive proinflammatory CD4 T cells in a unique process of extrathymic negative selection (120). Therefore, ILC3s can contribute to the overall T cell unresponsiveness to commensal antigens.
ILC2s also express MHC class II and costimulatory receptors (CD80 and CD86). ILC2s can acquire and process antigens and can induce antigen-specific activation and proliferation of T cells (121). Activated T cells in turn produce IL-2 and promote production of type 2 cytokines from ILC2s. In this manner, cross talk between ILC2s and T cells contributes to the development of efficient type 2 responses to the parasitic worm Nippostrongylus brasiliensis (121).
Following priming, commensal-induced T cells home to mucosal sites, where they complete their terminal differentiation program. This process is largely controlled by microbiota-mediated activation and cytokine production by local innate immune subsets. For example, intestinal pTreg induction and maintenance are regulated by CSF2 (granulocyte-macrophage colony-stimulating factor) production by ILC3s. Microbiota-driven IL-1β production by macrophages promotes the release of CSF2 by ILC3s, which in turn controls DCs and macrophages to maintain colonic Treg homeostasis (122). SFB-primed CD4 T cells in MLNs complete their Th17 differentiation, including IL-17 production, in the terminal ileum. This process is controlled by SFB-dependent activation of ILC3s and IECs (4, 8). In contrast, T cells are critical for preventing excessive activation of ILC3s and IECs induced by the microbiota (123). In the absence of CD4 T cells, an extensive and persistent phosphorylation of STAT3 is induced in ILC3s and IECs by SFB in an IL-22- and IL-23-dependent manner. Both Tregs and Th17 cells contribute to preventing ILC3 activation: Tregs do so by decreasing IL-23 production from myeloid cells, whereas Th17 cells regulate the bacterial burden (123). In the absence of adaptive immunity, most bacteria, including SFB, are increased, while SFB abundance is further increased in the absence of innate immunity, such as in Il23a−/−Rag1−/− mice (123), indicating that cross talk between innate and adaptive immune cells shapes the commensal microbiota and maintains tissue homeostasis.
Enteric nervous system.
The enteric nervous system (ENS) is composed of the myenteric plexus (also termed Auerbach’s plexus), located within the muscularis layer between the longitudinal and the circular muscles, and the submucosal plexus, located between the mucosa and the circular layer of muscles (also termed Meissner’s plexus). The ENS innervates all layers of the intestinal tissue, from the epithelial layer through the lamina propria, submucosa, muscularis, serosa, and mesentery. In addition to gut intrinsic neurons, the intestine is innervated by different classes of extrinsic neurons, which detect and convey information within the gut to extrinsic ganglia and the CNS. Extrinsic ganglia, such as the sensory nodose ganglion and dorsal root ganglion, as well as the sympathetic celiac-superior mesenteric ganglia, project neurons to the gut (124). Several recent reports have demonstrated cross talk among the microbiota, ENS, and innate/adaptive immune cells in the gut wall. For instance, MHC class II+ myeloid cells and Tregs reside close to projections of neurons in the colonic lamina propria (125–127). Colonic myeloid cells express muscarinic acetylcholine (ACh) receptor (mAChR) at high levels and upregulate retinoic acid–synthesizing enzymes upon ACh stimulation. Interfering with mAChR signaling by vagotomy results in a significant reduction in retinoic acid production by myeloid cells, accompanied by a reduction in the number of RORγt+ pTregs in the colon (127) (Figure 3). Consequently, vagotomized mice exhibit increased susceptibility to dextran sulfate sodium–induced colitis and decreased establishment of oral tolerance, suggesting that the cholinergic pathway plays a crucial role in regulation of Tregs (127, 128). Tonic microbial signals are required for neuronal modulation of Treg differentiation, as antibiotic-treated mice are refractory to vagotomy (127). At the same time, at least a part of the ENS functions to negatively regulate Treg differentiation. Indeed, colonization of germ-free mice with commensals, such as Clostridium ramosum, leads to a reduction in the number of neuronal cell bodies in the myenteric plexus and in the density of nerve projections to the lamina propria, thereby inducing accumulation of Tregs in the colon (125). Under certain conditions, enteric nitric oxide synthase–positive neurons negatively regulate pTreg differentiation via IL-6 production (126). Therefore, the ENS is composed of neurons with different functions and regulates Tregs both positively and negatively, perhaps in a context-dependent manner. It is interesting to consider the functional differences between Tregs induced by pathways modulated by the ENS with input from outside the gut and the ones simply induced by intestinal APCs. One major difference might be in the spatial organization of resulting Treg distribution.
MICROBIOTA-MEDIATED MODIFICATION OF T CELLS
Modulation of T Cell Function by Microbiota-Derived Molecules
Both circulating and tissue-resident populations of T cells are influenced by intestinal microbiota–derived metabolites, exemplified by the observation that a significant proportion of intestinal Tregs depends on the metabolic activity of specific gut microbiota members that ferment dietary fiber into SCFAs (23, 27, 129–131) (Figure 4). SCFAs, particularly butyrate, act as histone deacetylase inhibitors and epigenetically enhance expression of Foxp3 by promoting histone acetylation at the promoter and at the conserved noncoding sequence 1 (CNS1) of the Foxp3 gene locus. SCFAs also signal through G protein–coupled receptors, such as GPR43 and GPR109a, on T cells and myeloid cells to promote Foxp3+ Treg development and help protect the host from colitis (132, 133).
Figure 4.
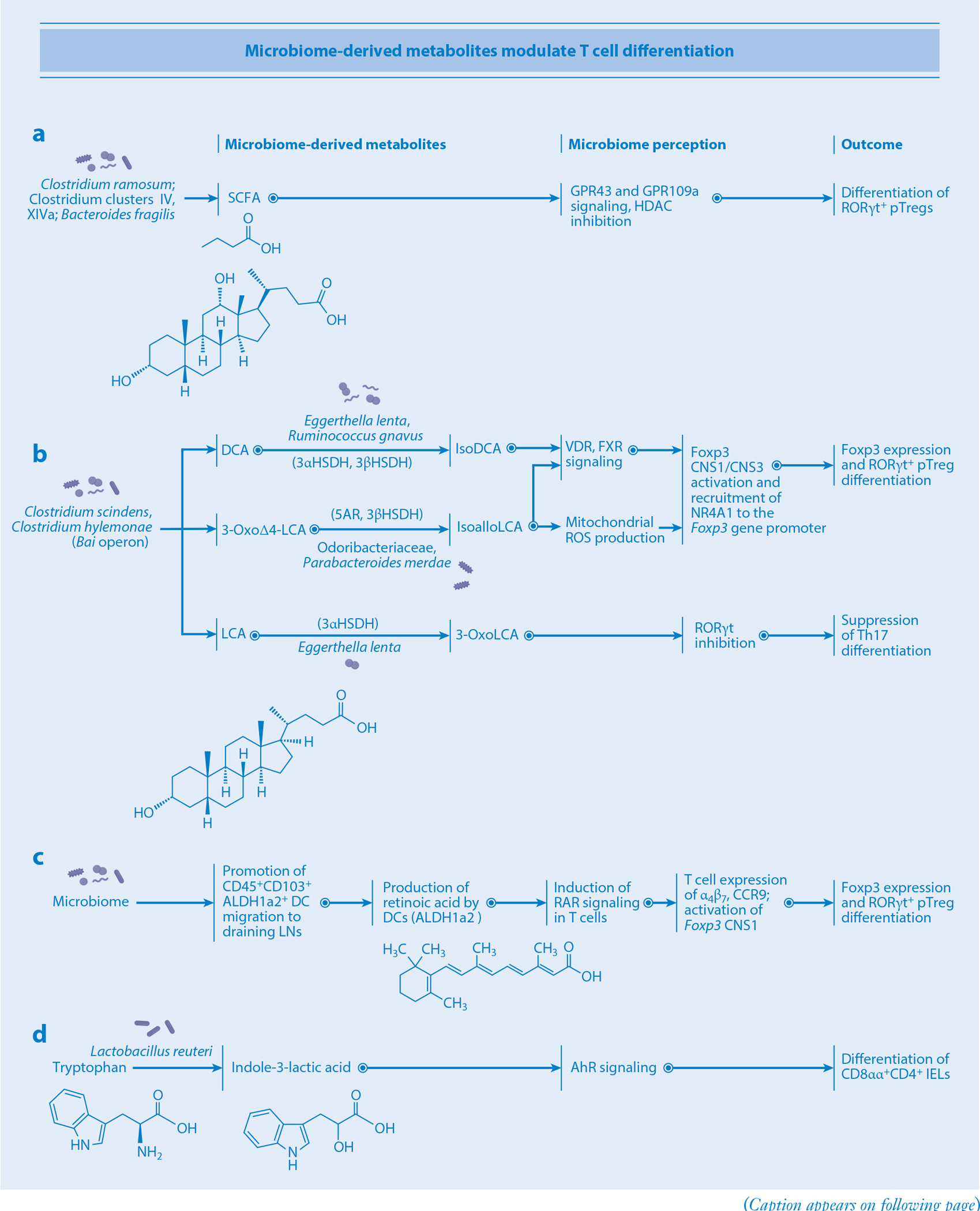
Microbiota-derived metabolites modulate T cell differentiation. (a) SCFAs derived from Clostridium ramosum and Clostridia clusters IV and XIVa signal through GPR43 and GPR109a and inhibit HDAC in T cells, thus promoting pTreg differentiation. (b) Bai operon–carrying bacteria such as Clostridium scindens and Clostridium hylemonae produce DCA, 3-oxoΔ4-LCA, and LCA. DCA is subsequently converted to isoDCA by 3αHSDH- and 3βHSDH-carrying bacteria such as Eggerthella lenta and Ruminococcus gnavus. IsoDCA signals through the T cellular receptors VDR and FXR, triggering activation of CNS1/CNS3 regions of the Foxp3 gene, a process that ultimately leads to pTreg differentiation. 3-OxoΔ4-LCA is converted to isoalloLCA by the 5AR- and 3βHSDH-carrying bacteria Odoribacteraceae and Parabacteroides merdae. IsoalloLCA triggers mitochondrial ROS production and VDR, FXR, and NR4A1 signaling in T cells, promoting their differentiation into pTregs via CNS1/CNS3 activation. LCA is converted into 3-oxoLCA by 3αHSDH-carrying E. lenta. 3-OxoLCA then suppresses RORγt, inhibiting the Th17 route of differentiation. (c) Microbial signals promote the migration of CD45+CD103+ DCs to gLNs as well as the production of retinoic acid by DCs utilizing the ALDH1a2 enzyme. Retinoic acid receptor signaling on T cells leads to activation of Foxp3 CNS1 and expression of integrins α4β7 and CCR9, ultimately leading to pTreg differentiation. (d) Lactobacillus reuteri converts dietary tryptophan into indole-3-lactic acid, stimulating AhR signaling and resulting in CD8αα+CD4+ IEL differentiation. Abbreviations: AhR, aryl hydrocarbon receptor; CNS1/3, conserved noncoding sequence 1/3; DC, dendritic cell; DCA, deoxycholic acid; FXR, farnesoid X receptor; gLN, lymph node; HDAC, histone deacetylase; HSDH, hydroxysteroid dehydrogenase; IEL, intraepithelial lymphocyte; LCA, lithocholic acid; NR4A1, nuclear receptor subfamily 4 group A member 1; pTreg, peripherally induced T regulatory cell; RAR, retinoic acid receptor; ROS, reactive oxygen species; SCFA, short-chain fatty acid; Th17, T helper 17; 5AR, 5α-reductase.
Microbiota-derived specific secondary bile acids help modulate the function and differentiation of T cells. Biosynthesis of secondary bile acids is a multistep process involving deconjugation and 7α-dehydroxylation reactions by the microbiota. A small fraction of intestinal microbes, such as Clostridium scindens and Clostridium hylemonae, possess the complete 7α-dehydroxylation pathway gene (Bai operon) and produce deoxycholic acid (DCA) and lithocholic acid (LCA) from cholic acid and chenodeoxycholic acid, respectively. In addition, several microbes (regardless of carriage of Bai operon) can mediate oxidation and epimerization reactions to produce oxo, iso, and allo forms of bile acids (134). For example, Eggerthella lenta can generate 3-oxoDCA and 3-oxoLCA from DCA and LCA, respectively, by the action of 3α-hydroxysteroid dehydrogenase (3αHSDH) (135–137). E. lenta can also transform 3-oxoDCA and 3-oxoLCA into isoDCA and isoLCA, respectively, by the action of 3βHSDH (135, 136). Bacteroidetes species such as Odoribacteraceae spp. and Parabacteroides merdae synthesize isoalloLCA from 3-oxoΔ4-LCA by 5α-reductase and 3βHSDH (137, 138). In addition, the microbiota can promote amino acid conjugations of host bile acids to produce phenylalanocholic acid (Phe-chol), tyrosocholic acid (Tyr-chol), and leucocholic acid (Leu-chol) (139). Clostridium bolteae can synthesize both Phe-chol and Tyr-chol, which activate farnesoid X receptor (FXR) (139). Feeding mice with mixtures of bile acids, such as cholic acid/ursodeoxycholic acid or 3-oxoLCA/LCA, results in activation of vitamin D receptor and FXR signaling in T cells and consequent induction of pTregs in the intestine (140) (Figure 4). IsoDCA also enhances the differentiation of naïve T cells into Tregs via FXR-mediated CNS1 activation (141). IsoalloLCA has a different mode of action and promotes Foxp3 expression via CNS3 through production of mitochondrial reactive oxygen species (ROS) (142). Concurrently, isoalloLCA induces an open chromatin region at the Foxp3 gene promoter, promoting binding of the nuclear hormone receptor NR4A1 and activation of its promoter (138). Bile acids also regulate Th17 cell differentiation. In particular, 3-oxoLCA and isoLCA inhibit differentiation of Th17 cells via direct binding to RORγt (136, 142).
Retinoic acid is another metabolite that affects the mucosal T cell differentiation program. Retinoic acid is produced from dietary retinol (vitamin A) by sequential activities of retinol dehydrogenases and retinaldehyde dehydrogenases (ALDHs) (143). Retinoic acid functions as a ligand for DNA-binding retinoic acid receptors (RARs) that directly regulate transcription of specific target genes. Retinol is packaged in chylomicrons and is transferred from IECs into the lymphatics with a gradient from the upper to the lower intestine. This retinol gradient results in region-specific retinoic acid concentration and, consequently, differential function of gut-draining lymph nodes (94). Retinol and retinoic acid upregulate ALDH1a2 expression in CD103+ DCs and promote the accumulation of retinoic acid in the microenvironment. The retinoic acid and RAR complex imprints T cells with a gut tropism by enhancing the expression of α4β7 integrins and CC–chemokine receptor 9 (CCR9) (Figure 4). Moreover, RAR acts together with Smads activated by TGF-β signaling to induce pTregs through activation of the CNS1 of the Foxp3 gene. Additionally, retinoic acid supports proinflammatory CD4+ T cell responses, and depletion of retinol reduces the proportion of Th17 cells in the small intestine (144), suggesting that retinoic acid plays critical roles of in T cell activation and regulation.
AhR is expressed at high levels in the skin, lung, and gut and recognizes dietary tryptophan derivatives produced by the microbiota and the host. In host cells, indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan hydroxylase 1 are involved in the production of AhR ligands. IELs are one of the prominent T cell populations that respond to AhR ligands. Lactobacillus reuteri catabolizes dietary tryptophan into the AhR agonist indole-3-lactic acid, thereby inducing differentiation of CD8αα+CD4+ IELs (42) (Figure 4). In addition to facilitating IEL development, AhR signaling also regulates the function and differentiation of CD4 T cells and ILC3s. AhR regulates the expression of several genes such as Cyp1a1, Il22, and Il17 in T cells. Cyp1a1 metabolizes AhR ligands and thus plays a negative feedback role in AhR signaling. CD4 T cells from mice that constitutively express Cyp1a1 under control of the Rosa26 promoter (R26Cyp1a1 mice) are less capable of developing IL-22-producing Th17 cells in response to AhR ligands (145). Furthermore, R26Cyp1a1 mice have decreased numbers of ILC3s in the colon and small intestine. The decreased AhR-dependent Th17 and ILC3 responses result in increased susceptibility to enteric pathogen infections. AhR signaling also regulates the function of iNKT cells. Bacterial products that are structurally related to oxazolone, such as E. coli–derived microcin B17 and heterocyclic compounds like oxazoles and thiazoles, can promote IEC IDO1 activity, thereby catabolizing tryptophan into AhR ligands like kynurenine and 3-hydroxy-kynurenic acid (146). Subsequent signaling through the AhR modulates CD1d-mediated lipid antigen presentation to iNKT cells, triggering them to release the proinflammatory cytokines IL-13 and IFN-γ.
The polyamines (putrescine, spermine, and spermidine) are abundant within the gastrointestinal tract and have been implicated in T cell immunity. In the biosynthesis of polyamines, ornithine decarboxylase (ODC) acts as the rate-limiting enzyme and catalyzes the conversion of ornithine derived from arginine into putrescine (147). Putrescine is then converted by spermidine synthase into spermidine. The polyamine spermidine is used to hypusinate (i.e., modify a specific lysine residue to hypusine) the translation factor eukaryotic initiation factor 5A (eIF5A) via the enzymatic activities of deoxyhypusine synthase (DHPS) and deoxyhypusine hydroxylase (DOHH). Hypusinated eIF5A promotes the expression of mitochondrial molecules to enhance the tricarboxylic acid (TCA) cycle–dependent cellular metabolism, resulting in acetyl-CoA accumulation and histone acetylation. Upon activation, naïve CD4 T cells upregulate expression of ODC to generate high levels of polyamines (147). In the absence of Odc, CD4 T cells show dysregulated expression of transcription factors and cytokines, such as high levels of T-bet and IFN-γ. Moreover, similar phenotypes are observed in Dhps- and Dohh-deficient T cells, and these mutant mice develop colitis (147), indicating that polyamines and hypusinated eIF5A have a critical role in correct specification of the CD4 T cell differentiation program.
In summary, numerous microbial molecules activate receptors on and in T cells to induce specific signaling. Several other molecules are capable of entering cells to affect transcription factor activity and chromatin structure. In addition, as exemplified by the critical contribution of polyamines, microbial metabolites help promote proper allocation of metabolic resources to ensure optimal induction of energetically costly T cell responses.
Influence of the Microbiota on T Cell Metabolism
The field of immunometabolism is fast-developing, prompted by the realization that T cell activation and differentiation induce metabolic reprogramming that supports proliferation and cytokine production (148–150). Naïve T cells are metabolically quiescent and utilize primarily oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) pathways to meet their limited anabolic needs. Upon antigen recognition, T cells shift their metabolism toward aerobic glycolysis and glutaminolysis at both the transcriptional [via c-Myc and hypoxia-inducible factor (HIF)-1α] and the posttranscriptional (via the PI3K/Akt/mTOR pathway) levels (150–152), thereby promoting generation of substrates necessary for nucleotide, fatty acid, and effector cytokine synthesis (152, 153). As effector T cells differentiate into memory cells during the contraction phase of the immune response, they revert to a metabolic state reminiscent of naïve T cells (via TRAF6-dependent signaling in the case of CD8 T cells), characterized by OXPHOS and FAO (154). These memory T cells also exhibit an increased mitochondrial reserve capacity secondary to enhanced mitochondrial biogenesis, thus supporting their long-term survival (148, 154).
It is becoming increasingly apparent, however, that metabolism is not merely a passive process but rather can directly affect immune cell fate and effector function. For example, blocking glycolytic flux via mTOR inhibition or HIF-1α knockout dampens Th17 cell development and promotes a reciprocal increase in Tregs (155, 156). Conversely, microbially derived lipopolysaccharide induces succinate accumulation in macrophages, which in turn stabilizes HIF-1α and promotes inflammation by enhancing both glycolysis and IL-1β production (157, 158). In another example, Tfh cells possess unique lipid metabolic programs and concentrate phosphatidylethanolamine in the outer layer of the plasma membrane, where CXCR5 is localized and prevented from internalization and degradation (159).
The gut microbiota plays a critical role in modulating T cell metabolism and, by extension, T cell function and differentiation. For example, fermentation products like SCFAs enhance the CD8 T cell cytotoxic response by boosting both glycolysis and mitochondrial respiration (160). These effects have been replicated in vivo in mice fed a high-fiber diet, which skews gut microbiota composition toward SCFA-producing species and ultimately accelerates clearance of influenza virus by promoting CD8 T cell activation (160). Microbiota-derived butyrate enhances the differentiation of effector into memory CD8 T cells by uncoupling the TCA from glycolysis, thereby promoting the use of glutamine and fatty acids to fuel OXPHOS (37). SCFAs can also directly promote OXPHOS by serving as substrates for FAO (161, 162). Secondary bile acids represent another class of gut commensal-derived molecules that regulate immunometabolism. As described above, isoalloLCA in particular enhances oxidative phosphorylation and mitochondrial ROS generation, which support Treg differentiation (142). Bifidobacterium species also enhance Treg mitochondrial activity via an incompletely understood mechanism, promoting IL-10 production and conferring protection against immune checkpoint blockade–induced colitis (163).
These multifaceted mechanisms by which metabolism dictates T cell function and fate represent a prime target for therapeutic intervention in inflammatory diseases. Indeed, pathogenic Th17 cells in a mouse model of experimental autoimmune encephalomyelitis express higher levels of glycolysis pathway genes such as glucose phosphate isomerase 1 (Gpi1) than do commensal-induced homeostatic Th17 cells (164). Inhibition of the glycolysis pathway by Gpi1 deletion leads to increased mitochondrial respiration in homeostatic Th17 cells, but not in pathogenic Th17 cells. Accordingly, inactivation of Gpi1 preferentially depletes pathogenic Th17 cells by subjecting them to metabolic stress (164). The high metabolic demands of pathogenic T cells can be exploited in a similar manner in IBD models. CD4 T cells that recognize microbial flagellin have been implicated in IBD, and stimulating these TCRs while suppressing glycolysis via mTOR inhibition induces metabolic stress that results in pathogenic T cell depletion in transgenic mouse models (165). Overall, as the immunometabolic pathways modulated by gut commensals become better understood, therapies may be designed and tailored to manipulate specific aspects of T cell function, thereby combating inflammatory disease.
ROLE OF GUT-TRAINED T CELLS
T cell responses in the gut are generated through unique mechanisms of antigen acquisition and engagement of specific cells in the mucosa (such as epithelial cells) and in the ENS and innate immune cells. Gut-trained T cells exert multiple functions, one of which is curation of microbiota composition with the goal of sustaining maximal diversity of commensals while preventing infection by pathogens. T cell–dependent IgA induction is an important mechanism of shaping the microbiota. Microbial exposure at the intestinal mucosa generates IgA+ plasma cells that are polyreactive and predominantly specific to bacterial surface antigens; in contrast, systemic exposure to microbes elicits a diverse IgG repertoire specific to both microbial cytoplasmic and cell surface antigens (166). IgA-secreting plasma cells in the intestine are highly mutated. T cells mediate somatic hypermutation primarily in germinal centers in Peyer’s patches, which is critical to establish a fully functional secretory IgA system (167). The T cell–dependent IgA response requires T cell–intrinsic innate immune signals from the microbiota (168). In particular, TLR stimulation together with acetate supplementation facilitates differentiation of CD4 T cells into IgA-inducing Tfh cells (169). In contrast, IgA-producing cells can be induced in a T cell–independent manner as well, and selection of microbiota-reactive and polyreactive IgAs can occur independently of germinal centers. As IgA-producing cells in tissues other than the small intestine are severely reduced in the absence of microbiota and T cells, T cells might be more crucial for generation of extraintestinal IgA-producing cells (170).
Whether induced by T cell–dependent or –independent pathways, secretory IgA antibodies bind a broad, but distinct, subset of microbiota in vivo. In particular, Enterobacteriaceae as well as mucus-resident bacteria are heavily coated with IgA (169, 171). IgA induces agglutination, enchained growth, neutralization, and immune exclusion of microbes. IgA can also bind to commensal species and promote their growth (172). Although it remains unclear what factors determine whether IgA coating enhances or diminishes the fitness of microbes in the intestine, accumulating evidence indicates that IgA coating leads to alterations in microbial gene expression, motility, and/or spatial localization (173, 174). In addition to IgA B cell responses, IgG1 responses specific to commensal species, such as A. muciniphila, can also be elicited in the intestine (31). While microbiota exposure is required for induction of IgA and IgG1, it appears that the opposite is true for IgE. IgE is elevated in the sera of germ-free mice and decreases upon colonization with commensal microbes (175). Germ-free mice begin to produce IgE shortly after weaning age to high levels, and they maintain these levels unless colonized with microbes within the first week of life. As antigen-free mice (fed a diet devoid of macromolecules) show suppressed levels of IgE even under germ-free conditions, food antigens appear to be the major drivers of spontaneous IgE elevation (176).
Another task fulfilled by mucosal T cells is maintenance of mucosal barrier integrity. Th17 cell cytokines, such as IL-17A, IL-17F, and IL-22, stimulate IECs to promote antimicrobial protein production, tight junction formation, and IgA transport. Consequently, Th17 cells have an indispensable role in prevention of infection by several extracellular mucosal pathogens. Indeed, genetic defects in the IL-17/IL-17R axis and RORγt in humans have been linked to susceptibility to chronic mucocutaneous candidiasis (13, 14), and the deficiency of both Il17a and Il17f genes in mice results in opportunistic mucocutaneous infection by Staphylococcus aureus (177). Commensal-specific T cells can develop as long-lived tissue-resident memory (TRM) T cells. A significant fraction of long-lived CD4 TRM cells in the mucosa, such as in the lungs, are derived from effector Th17 cells (ex-Th17 cells) (178). Ex-Th17 TRM cells are maintained by IL-7 from lymphatic endothelial cells. IL-17-experienced cells play an essential role in the protective immune response against secondary infection by pathogens such as Klebsiella (178). Moreover, IL-17A+ T cells educated by commensals have the potential to participate in type 2 immune response against tissue injury and other challenges (179).
Although homeostatic Th17 cells help maintain epithelial barrier integrity and afford protection against pathogens, Th17 responses are also implicated in IBD and other autoimmune disorders, including psoriasis, multiple sclerosis, and rheumatoid arthritis. Therefore, Th17-inducing microbiota may have proinflammatory effects. Twenty epithelium-adhesive bacterial strains isolated from feces of a patient with ulcerative colitis induce Th17 cells in a gnotobiotic setting (4). These strains are capable of promoting maternal inflammation and contribute to autism phenotype development in progeny (180). Another example involves mice colonized with H. hepaticus that normally induces RORγt+ pTregs (11). In an inflammatory setting, such as in Il10-deficient mice, H. hepaticus drives development of proinflammatory IFN-γ+ Th17 cells instead (9, 11). The induction program of proinflammatory Th17 cells is driven by SAAs derived from epithelial cells or by a glycolysis-promoting hypoxic inflammatory environment (164, 181). These reports add further support for the idea of context-dependent induction, specification, and output of T cells by the microbiota.
IMPLICATIONS AND PERSPECTIVES
While an individual’s genome remains stable throughout their life, the microbiota is malleable and editable. Intestinal microbiota affects the health of virtually all organ systems and is subject to T cell–mediated curatorship. In reverse, T cells in the gut and throughout the body serve as conduits of microbial influence on the homeostasis of remote organs. The T cell–microbiota entanglement opens promising avenues for future research. Carefully designed consortia of microbes or microbiota-derived molecules can be used to shape T cell identity and cellularity locally in the gut, as well as in remote organs. Studies of T cell–mediated curatorship of microbiota will help define what constitutes healthy human microbiota or what are the functional entities needed for one. As one of the long-term goals of microbiota research is to construct a predictive model of healthy human microbiota, T cell recognition circuits that shape the microbiota should not only be studied on a qualitative level but also be parameterized. Conceptually, similarly to how the visual cortex interprets visual information, T cell recognition circuits may be viewed as processors of microbial signals that provide a view of the environment and appropriately modify the behavior of the immune system. This perspective on host–microbiota interaction opens the field to the use of tools developed in the framework of information theory and methods of artificial pattern recognition and, therefore, might help pave the way toward defining healthy human microbiota.
ACKNOWLEDGMENTS
K.H. is funded by the AMED Project Focused on Developing Key Technology for Discovering and Manufacturing Drugs for Next-Generation Treatment and Diagnosis, “The next-generation drug discovery and development technology on regulating intestinal microbiome (NeDD Trim)” (grant JP21ae0121041); a Grant-in-Aid for Specially Promoted Research from the Japan Society for the Promotion of Science (20H05627); the Naito Foundation; and the Takeda Science Foundation. I.I.I. is funded by grants from the National Institutes of Health (R01 DK098378, R01 AI144808, U01 AI163069, and R21 AI146817), a Burroughs Wellcome Fund PATH award (1019125), and a Pew Charitable Trusts Innovator Award (PEW00031379). I.I.I. is a Pew Charitable Trusts Scholar in Biomedical Sciences and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease. A.N.S. is supported by the National Institutes of Health Medical Scientist Training Program (T32 GM07170).
Footnotes
DISCLOSURE STATEMENT
K.H. is a scientific advisory board member of Vedanta Biosciences and 4BIO CAPITAL. The other authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, et al. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139(3):485–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, et al. 2009. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31(4):677–89 [DOI] [PubMed] [Google Scholar]
- 3.Sczesnak A, Segata N, Qin X, Gevers D, Petrosino JF, et al. 2011. The genome of Th17 cell–inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe 10(3):260–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, et al. 2015. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163(2):367–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goto Y, Panea C, Nakato G, Cebula A, Lee C, et al. 2014. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40(4):594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, et al. 2014. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510(7503):152–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ladinsky MS, Araujo LP, Zhang X, Veltri J, Galan-Diez M, et al. 2019. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 363(6431):eaat4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sano T, Huang W, Hall JA, Yang Y, Chen A, et al. 2015. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163(2):381–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J-Y, Hall JA, Kroehling L, Wu L, Najar T, et al. 2020. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell 180(1):79–91.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, et al. 2011. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell–mediated inflammation. Immunity 34(4):566–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Pokrovskii M, Ding Y, Yi R, Au C, et al. 2018. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554(7692):373–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, et al. 2016. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity 44(3):659–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, et al. 2015. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349(6248):606–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Casanova J-L, Puel A. 2018. Mucocutaneous IL-17 immunity in mice and humans: host defense versus excessive inflammation. Mucosal Immunol. 11(3):581–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, et al. 2016. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. PNAS 113(50):E8141–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ang QY, Alexander M, Newman JC, Tian Y, Cai J, et al. 2020. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell 181(6):1263–75.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, et al. 2017. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551(7682):585–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, et al. 2013. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496(7446):518–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, et al. 2013. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496(7446):513–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benakis C, Brea D, Caballero S, Faraco G, Moore J, et al. 2016. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med 22(5):516–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. 2020. Regulatory T cells and human disease. Annu. Rev. Immunol 38:541–66 [DOI] [PubMed] [Google Scholar]
- 22.Niec RE, Rudensky AY, Fuchs E. 2021. Inflammatory adaptation in barrier tissues. Cell 184(13):3361–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanoue T, Atarashi K, Honda K. 2016. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol 16(5):295–309 [DOI] [PubMed] [Google Scholar]
- 24.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, et al. 2011. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J. Clin. Investig 121(11):4503–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, et al. 2014. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513(7519):564–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, et al. 2014. Commensal bacteria protect against food allergen sensitization. PNAS 111(36):13145–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, et al. 2013. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500(7461):232–36 [DOI] [PubMed] [Google Scholar]
- 28.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, et al. 2011. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331(6015):337–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, et al. 2015. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349(6251):993–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, et al. 2017. Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci. Immunol 2(13):eaal5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansaldo E, Slayden LC, Ching KL, Koch MA, Wolf NK, et al. 2019. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364(6446):1179–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, et al. 2016. Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer’s patch T follicular helper cells. Immunity 44(4):875–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, et al. 2017. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 358(6361):359–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagayama M, Yano T, Atarashi K, Tanoue T, Sekiya M, et al. 2020. TH1 cell–inducing Escherichia coli strain identified from the small intestinal mucosa of patients with Crohn’s disease. Gut Microbes 12(1):1788898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, et al. 2019. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569(7758):655–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, et al. 2019. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565(7741):600–5 [DOI] [PubMed] [Google Scholar]
- 37.Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, et al. 2019. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity 51(2):285–97.e5 [DOI] [PubMed] [Google Scholar]
- 38.Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, et al. 2012. Compartmentalized control of skin immunity by resident commensals. Science 337(6098):1115–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naik S, Bouladoux N, Linehan JL, Han S-J, Harrison OJ, et al. 2015. Commensal–dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520(7545):104–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linehan JL, Harrison OJ, Han S-J, Byrd AL, Vujkovic-Cvijin I, et al. 2018. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172:784–96.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YE, Bouladoux N, Hurabielle C, Mattke AM, Belkaid Y, Fischbach MA. 2019. Decoding commensal–host communication through genetic engineering of Staphylococcus epidermidis. bioRxiv 664656. 10.1101/664656v1 [DOI] [Google Scholar]
- 42.Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, et al. 2017. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science 357(6353):806–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. 2013. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nat. Immunol 14(3):271–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, et al. 2016. Tissue adaptation of regulatory and intraepithelial CD4+ T cells controls gut inflammation. Science 352(6293):1581–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDonald BD, Jabri B, Bendelac A. 2018. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol 18(8):514–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribot JC, Lopes N, Silva-Santos B. 2021. γδ T cells in tissue physiology and surveillance. Nat. Rev. Immunol 21(4):221–32 [DOI] [PubMed] [Google Scholar]
- 47.Hu B, Jin C, Zeng X, Resch JM, Jedrychowski MP, et al. 2020. γδ T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature 578(7796):610–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, et al. 2019. Meningeal γδ T cell–derived IL-17 controls synaptic plasticity and short-term memory. Sci. Immunol 4(40):eaay5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen MM, Witherden DA, Havran WL. 2017. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol 17(12):733–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pellicci DG, Koay HF, Berzins SP. 2020. Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nat. Rev. Immunol 20(12):756–70 [DOI] [PubMed] [Google Scholar]
- 51.Eberl M 2020. Antigen recognition by human γδ T cells: one step closer to knowing. Immunol. Cell Biol 98(5):351–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ismail AS, Severson KM, Vaishnava S, Behrendt CL, Yu X, et al. 2011. γδ intraepithelial lymphocytes are essential mediators of host–microbial homeostasis at the intestinal mucosal surface. PNAS 108(21):8743–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chien YH, Meyer C, Bonneville M. 2014. γδ T cells: first line of defense and beyond. Annu. Rev. Immunol 32:121–55 [DOI] [PubMed] [Google Scholar]
- 54.Bandeira A, Mota-Santos T, Itohara S, Degermann S, Heusser C, et al. 1990. Localization of γ/δ T cells to the intestinal epithelium is independent of normal microbial colonization. J. Exp. Med 172(1):239–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duan J, Chung H, Troy E, Kasper DL. 2010. Microbial colonization drives expansion of IL-1 receptor 1–expressing and IL-17-producing γ/δ T cells. Cell Host Microbe 7(2):140–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen YS, Chen IB, Pham G, Shao TY, Bangar H, et al. 2020. IL-17-producing γδ T cells protect against Clostridium difficile infection. J. Clin. Investig 130(5):2377–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoytema van Konijnenburg DP, Reis BS, Pedicord VA, Farache J, Victora GD, Mucida D. 2017. Intestinal epithelial and intraepithelial T cell crosstalk mediates a dynamic response to infection. Cell 171:783–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Semenkovich NP, Planer JD, Ahern PP, Griffin NW, Lin CY, Gordon JI. 2016. Impact of the gut microbiota on enhancer accessibility in gut intraepithelial lymphocytes. PNAS 113(51):14805–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Godfrey DI, Koay HF, McCluskey J, Gherardin NA. 2019. The biology and functional importance of MAIT cells. Nat. Immunol 20(9):1110–28 [DOI] [PubMed] [Google Scholar]
- 60.Koay H-F, Gherardin NA, Xu C, Seneviratna R, Zhao Z, et al. 2019. Diverse MR1-restricted T cells in mice and humans. Nat. Commun 10:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, et al. 2012. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491(7426):717–23 [DOI] [PubMed] [Google Scholar]
- 62.Corbett AJ, Eckle SBG, Birkinshaw RW, Liu L, Patel O, et al. 2014. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509(7500):361–65 [DOI] [PubMed] [Google Scholar]
- 63.Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, et al. 2019. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 366(6464):eaax6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Legoux F, Bellet D, Daviaud C, El Morr Y, Darbois A, et al. 2019. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 366(6464):494–99 [DOI] [PubMed] [Google Scholar]
- 65.Provine NM, Amini A, Garner LC, Spencer AJ, Dold C, et al. 2021. MAIT cell activation augments adenovirus vector vaccine immunogenicity. Science 371(6528):521–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crosby CM, Kronenberg M. 2018. Tissue-specific functions of invariant natural killer T cells. Nat. Rev. Immunol 18(9):559–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olszak T, An D, Zeissig S, Vera MP, Richter J, et al. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336(6080):489–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.An D, Oh SF, Olszak T, Neves JF, Avci FY, et al. 2014. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156(1/2):123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, et al. 2013. Production of α-galactosylceramide by a prominent member of the human gut microbiota. PLOS Biol. 11(7):e1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nieuwenhuis EES, Matsumoto T, Lindenbergh D, Willemsen R, Kaser A, et al. 2009. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J. Clin. Investig 119(5):1241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, et al. 2017. Circulating and tissue-resident CD4+ T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology 153(5):1320–37.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157(1):121–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen YE, Atabakhsh K, Dimas A, Nagashima K, Fischbach MA. 2021. Eliciting a potent antitumor immune response by expressing tumor antigens in a skin commensal. bioRxiv 2021.02.17.431662. 10.1101/2021.02.17.431662 [DOI] [Google Scholar]
- 74.Allaire JM, Crowley SM, Law HT, Chang S-Y, Ko H-J, Vallance BA. 2018. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. 39(9):677–96 [DOI] [PubMed] [Google Scholar]
- 75.Schulz O, Pabst O. 2013. Antigen sampling in the small intestine. Trends Immunol. 34(4):155–61 [DOI] [PubMed] [Google Scholar]
- 76.Neutra MR, Frey A, Kraehenbuhl J-P. 1996. Epithelial M cells: gateways for mucosal infection and immunization. Cell 86(3):345–48 [DOI] [PubMed] [Google Scholar]
- 77.Kanaya T, Williams IR, Ohno H. 2020. Intestinal M cells: tireless samplers of enteric microbiota. Traffic 21(1):34–44 [DOI] [PubMed] [Google Scholar]
- 78.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, et al. 2009. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune response. Nature 462(7270):226–30 [DOI] [PubMed] [Google Scholar]
- 79.Rochereau N, Drocourt D, Perouzel E, Pavot V, Redelinghuys P, et al. 2013. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA–antigen complexes by intestinal M cells. PLOS Biol. 11(9):e1001658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mantis NJ, Rol N, Corthésy B. 2011. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 4(6):603–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kanaya T, Sakakibara S, Jinnohara T, Hachisuka M, Tachibana N, et al. 2018. Development of intestinal M cells and follicle-associated epithelium is regulated by TRAF6-mediated NF-κB signaling. J. Exp. Med 215(2):501–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ménard S, Cerf-Bensussan N, Heyman M. 2010. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 3(3):247–59 [DOI] [PubMed] [Google Scholar]
- 83.Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, et al. 2006. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Investig 116(8):2142–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Niel G, Mallegol J, Bevilacqua C, Candalh C, Brugière S, et al. 2003. Intestinal epithelial exosomes carry MHC class II/peptides able to inform the immune system in mice. Gut 52(12):1690–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cummings RJ, Barbet G, Bongers G, Hartmann BM, Gettler K, et al. 2016. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539(7630):565–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jang MH, Kweon M-N, Iwatani K, Yamamoto M, Terahara K, et al. 2004. Intestinal villous M cells: an antigen entry site in the mucosal epithelium. PNAS 101(16):6110–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, et al. 2012. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483(7389):345–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, et al. 2017. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci. Immunol 2(18):eaao1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, et al. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol 2(4):361–67 [DOI] [PubMed] [Google Scholar]
- 90.Niess JH, Brand S, Gu X, Landsman L, Jung S, et al. 2005. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307(5707):254–58 [DOI] [PubMed] [Google Scholar]
- 91.Chieppa M, Rescigno M, Huang AYC, Germain RN. 2006. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med 203(13):2841–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, et al. 2011. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34(2):237–46 [DOI] [PubMed] [Google Scholar]
- 93.Esterházy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D. 2016. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat. Immunol 17(5):545–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, et al. 2019. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 569(7754):126–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Russler-Germain EV, Yi J, Young S, Nutsch K, Wong HS, et al. 2021. Gut Helicobacter presentation by multiple dendritic cell subsets enables context-specific regulatory T cell generation. eLife 10:e54792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coombes JL, Siddiqui KRR, Arancibia-Caŕcamo CV, Hall J, Sun C-M, et al. 2007. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β- and retinoic acid–dependent mechanism. J. Exp. Med 204(8):1757–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, et al. 2010. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 59(5):595–604 [DOI] [PubMed] [Google Scholar]
- 98.Sun C-M, Hall JA, Blank RB, Bouladoux N, Oukka M, et al. 2007. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 Treg cells via retinoic acid. J. Exp. Med 204(8):1775–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Edelson BT, KC W, Juang R, Kohyama M, Benoit LA, et al. 2010. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J. Exp. Med 207(4):823–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, et al. 2011. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35(5):780–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, et al. 2009. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med 206(13):3115–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, et al. 2013. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38(5):958–69 [DOI] [PubMed] [Google Scholar]
- 103.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, et al. 2013. IRF4 transcription factor–dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38(5):970–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grainger JR, Askenase MH, Guimont-Desrochers F, da Fonseca DM, Belkaid Y. 2014. Contextual functions of antigen-presenting cells in the gastrointestinal tract. Immunol. Rev 259(1):75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, et al. 2015. Intestinal monocyte-derived macrophages control commensal-specific Th17 responses. Cell Rep. 12(8):1314–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, et al. 2009. Origin of the lamina propria dendritic cell network. Immunity 31(3):513–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, et al. 2013. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38(1):79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ginhoux F, Jung S. 2014. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol 14(6):392–404 [DOI] [PubMed] [Google Scholar]
- 109.Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, et al. 2018. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med 215(6):1507–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Leonardi I, Li X, Semon A, Li D, Doron I, et al. 2018. CX3CR1+ mononuclear phagocytes control immunity to intestinal fungi. Science 359(6372):232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, et al. 2009. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6(5):470–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koscsó B, Kurapati S, Rodrigues RR, Nedjic J, Gowda K, et al. 2020. Gut-resident CX3CR1hi macrophages induce tertiary lymphoid structures and IgA response in situ. Sci. Immunol 5(46):eaax0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Geem D, Medina-Contreras O, McBride M, Newberry RD, Koni PA, Denning TL. 2014. Specific microbiota-induced intestinal Th17 differentiation requires MHC class II but not GALT and mesenteric lymph nodes. J. Immunol 193(1):431–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mayer L 2000. Epithelial cell antigen presentation. Curr. Opin. Gastroenterol 16(6):531–35 [DOI] [PubMed] [Google Scholar]
- 115.Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, et al. 2018. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175(5):1307–20.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hülsdünker J, et al. 2019. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity 51(5):885–98.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bilate AM, London M, Castro TBR, Mesin L, Bortolatto J, et al. 2020. T cell receptor is required for differentiation, but not maintenance, of intestinal CD4+ intraepithelial lymphocytes. Immunity 53(5):1001–14.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. 1995. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol. Immunol 39(8):555–62 [DOI] [PubMed] [Google Scholar]
- 119.Tuganbaev T, Mor U, Bashiardes S, Liwinski T, Nobs SP, et al. 2020. Diet diurnally regulates small intestinal microbiome-epithelial-immune homeostasis and enteritis. Cell 182(6):1441–59.e21 [DOI] [PubMed] [Google Scholar]
- 120.Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, et al. 2015. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria–specific CD4+ T cells. Science 348(6238):1031–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, et al. 2014. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41(2):283–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, et al. 2014. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343(6178):1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, et al. 2018. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554(7691):255–59 [DOI] [PubMed] [Google Scholar]
- 124.Muller PA, Schneeberger M, Matheis F, Wang P, Kerner Z, et al. 2020. Microbiota modulate sympathetic neurons via a gut–brain circuit. Nature 583(7816):441–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yissachar N, Zhou Y, Ung L, Lai NY, Mohan JF, et al. 2017. An intestinal organ culture system uncovers a role for the nervous system in microbe-immune crosstalk. Cell 168(6):1135–48.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yan Y, Ramanan D, Rozenberg M, McGovern K, Rastelli D, et al. 2021. Interleukin-6 produced by enteric neurons regulates the number and phenotype of microbe-responsive regulatory T cells in the gut. Immunity 54(3):499–513.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Teratani T, Mikami Y, Nakamoto N, Suzuki T, Harada Y, et al. 2020. The liver-brain-gut neural arc maintains the Treg cell niche in the gut. Nature 585(7826):591–96 [DOI] [PubMed] [Google Scholar]
- 128.Di Giovangiulio M, Bosmans G, Meroni E, Stakenborg N, Florens M, et al. 2016. Vagotomy affects the development of oral tolerance and increases susceptibility to develop colitis independently of α−7 nicotinic receptor. Mol. Med 22(1):464–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, et al. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504(7480):451–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. 2013. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341(6145):569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, et al. 2013. Commensal microbe–derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–50 [DOI] [PubMed] [Google Scholar]
- 132.Tan J, McKenzie C, Vuillermin PJ, Goverse G, Vinuesa CG, et al. 2016. Dietary fiber and bacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep. 15(12):2809–24 [DOI] [PubMed] [Google Scholar]
- 133.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, et al. 2014. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40(1):128–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wahlström A, Sayin SI, Marschall H-UU, Bäckhed F. 2016. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24(1):41–50 [DOI] [PubMed] [Google Scholar]
- 135.Devlin AS, Fischbach MA. 2015. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol 11(9):685–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paik D, Yao L, Zhang Y, Bae S, D’Agostino GD, et al. 2021. Human gut bacteria produce TH17-modulating bile acid metabolites. bioRxiv 2021.01.08.425913. 10.1101/2021.01.08.425913 [DOI] [Google Scholar]
- 137.Sato Y, Atarashi K, Plichta DR, Arai Y, Sasajima S, et al. 2021. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599:458–64 [DOI] [PubMed] [Google Scholar]
- 138.Li W, Hang S, Fang Y, Bae S, Zhang Y, et al. 2021. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 29(9):1366–77.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Quinn RA, Melnik AV, Vrbanac A, Fu T, Patras KA, et al. 2020. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579:123–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Song X, Sun X, Oh SF, Wu M, Zhang Y, et al. 2020. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577:410–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, et al. 2020. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581:475–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hang S, Paik D, Yao L, Kim E, Jamma T, et al. 2019. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576(7785):143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. 2012. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1821(1):198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cha H-R, Chang S-Y, Chang J-H, Kim J-O, Yang J-Y, et al. 2010. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J. Immunol 184(12):6799–806 [DOI] [PubMed] [Google Scholar]
- 145.Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, et al. 2017. Feedback control of AHR signaling regulates intestinal immunity. Nature 542(7640):242–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iyer SS, Gensollen T, Gandhi A, Oh SF, Neves JF, et al. 2018. Dietary and microbial oxazoles induce intestinal inflammation by modulating aryl hydrocarbon receptor responses. Cell 173(5):1123–34.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Puleston DJ, Baixauli F, Sanin DE, Edwards-Hicks J, Villa M, et al. 2021. Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 184(16):4186–202.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Man K, Kallies A. 2015. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol 15(9):574–84 [DOI] [PubMed] [Google Scholar]
- 149.Almeida L, Lochner M, Berod L, Sparwasser T. 2016. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol 28(5):514–24 [DOI] [PubMed] [Google Scholar]
- 150.MacIver NJ, Michalek RD, Rathmell JC. 2013. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol 31:259–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, et al. 2011. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35(6):871–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, et al. 2002. The CD28 signaling pathway regulates glucose metabolism. Immunity 16(6):769–77 [DOI] [PubMed] [Google Scholar]
- 153.Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, et al. 2013. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153(6):1239–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, et al. 2009. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460(7251):103–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shi LZ, Wang R, Huang G, Vogel P, Neale G, et al. 2011. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med 208(7):1367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, et al. 2011. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 146(5):772–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, et al. 2013. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496(7444):238–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, et al. 2016. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167(2):457–70.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fu G, Guy CS, Chapman NM, Palacios G, Wei J, et al. 2021. Metabolic control of TFH cells and humoral immunity by phosphatidylethanolamine. Nature 595(7869):724–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, et al. 2018. Dietary fiber confers protection against flu by shaping Ly6c− patrolling monocyte hematopoiesis and CD8+ T cell metabolism. Immunity 48(5):992–1005.e8 [DOI] [PubMed] [Google Scholar]
- 161.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, et al. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 13(5):517–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Michaudel C, Sokol H. 2020. The gut microbiota at the service of immunometabolism. Cell Metab. 32(4):514–23 [DOI] [PubMed] [Google Scholar]
- 163.Sun S, Luo L, Liang W, Yin Q, Guo J, et al. 2020. Bifidobacterium alters the gut microbiota and modulates the functional metabolism of T regulatory cells in the context of immune checkpoint blockade. PNAS 117(44):27509–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu L, Hollinshead KER, Hao Y, Au C, Kroehling L, et al. 2020. Niche-selective inhibition of pathogenic Th17 cells by targeting metabolic redundancy. Cell 182(3):641–54.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao Q, Duck LW, Huang F, Alexander KL, Maynard CL, et al. 2021. CD4+ T cell activation and concomitant mTOR metabolic inhibition can ablate microbiota-specific memory cells and prevent colitis. Sci. Immunol 5(54):eabc6373. [DOI] [PubMed] [Google Scholar]
- 166.Li H, Limenitakis JP, Greiff V, Yilmaz B, Schären O, et al. 2020. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature 584(7820):274–78 [DOI] [PubMed] [Google Scholar]
- 167.Pabst O, Slack E. 2020. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol. 13(1):12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, et al. 2015. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17(2):153–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Takeuchi T, Miyauchi E, Kanaya T, Kato T, Nakanishi Y, et al. 2021. Acetate differentially regulates IgA reactivity to commensal bacteria. Nature 595:560–64 [DOI] [PubMed] [Google Scholar]
- 170.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, et al. 2017. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358(6361):eaan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, et al. 2014. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158(5):1000–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, et al. 2014. Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41(1):152–65 [DOI] [PubMed] [Google Scholar]
- 173.Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, et al. 2013. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14(5):571–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Weis AM, Round JL. 2021. Microbiota–antibody interactions that regulate gut homeostasis. Cell Host Microbe 29(3):334–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cahenzli J, Köller Y, Wyss M, Geuking MB, McCoy KD. 2013. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14(5):559–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hong S-W, O E, Lee JY, Lee M, Han D, et al. 2019. Food antigens drive spontaneous IgE elevation in the absence of commensal microbiota. Sci. Adv 5(5):eaaw1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, et al. 2009. Differential roles of interleukin-17A and −17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30(1):108–19 [DOI] [PubMed] [Google Scholar]
- 178.Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, et al. 2019. Effector TH17 cells give rise to long-lived TRM cells that are essential for an immediate response against bacterial infection. Cell 178(5):1176–88.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Harrison OJ, Linehan JL, Shih H-Y, Bouladoux N, Han S-J, et al. 2019. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science 363(6422):eaat6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kim S, Kim H, Yim YS, Ha S, Atarashi K, et al. 2017.Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549(7673):528–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, et al. 2019. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity 51(1):77–89.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]