

Abstract
Background
Radionuclide therapy (RNT) has become a very important treatment modality for cancer nowadays. Comparing with other cancer treatment options, sufficient efficacy could be achieved in RNT with lower toxicity. β− emitters are frequently used in RNT due to the long tissue penetration depth of the β− particles. The dysprosium-166/holmium-166 (166Dy/166Ho) in vivo generator shows great potential for treating large malignancies due to the long half-life time of the mother nuclide 166Dy and the emission of high energy β− from the daughter nuclide 166Ho. However, the internal conversion occurring after β− decay from 166Dy to 166Ho could cause the release of about 72% of 166Ho when 166Dy is bound to conventional chelators. The aim of this study is to develop a nanoparticle based carrier for 166Dy/166Ho in vivo generator such that the loss of the daughter nuclide 166Ho induced by internal conversion is prevented. To achieve this goal, we radiolabelled platinum-gold bimetallic nanoparticles (PtAuNPs) and core–shell structured gold nanoparticles (AuNPs) with 166Dy and studied the retention of both 166Dy and 166Ho under various conditions.
Results
The 166Dy was co-reduced with gold and platinum precursor to form the 166DyAu@AuNPs and 166DyPtAuNPs. The 166Dy radiolabelling efficiency was determined to be 60% and 70% for the two types of nanoparticles respectively. The retention of 166Dy and 166Ho were tested in MiliQ water or 2.5 mM DTPA for a period of 72 h. In both cases, more than 90% of both 166Dy and 166Ho was retained. The results show that the incorporation of 166Dy in AuNPs can prevent the escape of 166Ho released due to internal conversion.
Conclusion
We developed a chelator-free radiolabelling method for 166Dy with good radiolabelling efficiency and very high stability and retention of the daughter nuclide 166Ho. The results from this study indicate that to avoid the loss of the daughter radionuclides by internal conversion, carriers composed of electron-rich materials should be used.
Supplementary Information
The online version contains supplementary material available at 10.1186/s41181-022-00170-3.
Keywords: Radionuclide therapy, Dysprosium-166, Holmium-166, In vivo generator, Internal conversion, Gold nanoparticle
Introduction
Cancer is one of the leading causes of death in the world (Sung et al. 2021; Bray et al. 2018). Nowadays, surgery and external beam radiation therapy (EBRT) are still the most common treatment modalities for localized tumors. In the case of metastases, systemic treatments such as radionuclide therapy (RNT) are preferred. RNT has been proved to be able to significantly prolong the life expectancy of terminal patients without affecting quality of life (Pool et al. 2010; Humm et al. 2015). In RNT, the therapeutic radionuclides are usually linked to chelators conjugated to tumor targeting vectors such as peptides, nucleotides and antibodies. Once distributed to the tumor site, the ionizing radiation emitted by the radionuclides can damage the DNA of the cancer cells and lead to apoptosis (Gudkov et al. 2016; Dash et al. 2013; Sgouros et al. 2020; Tafreshi et al. 2019; Kostelnik and Orvig 2019).
Over the past decades, many radiopharmaceuticals have been developed and some of them have been already applied in the clinic (Suman et al. 2021). Radionuclides that emit β− particles are more commonly applied in the clinic but the interest in α emitters is also growing (Nelson et al. 2021; Tafreshi et al. 2019). Since β− particles have relatively long tissue penetration depth, they are suitable for treating larger metastases (Pouget et al. 2011; Marcu et al., 2018). Moreover, additional benefits can be achieved with β− emitters by the so called “cross-fire” effect, i.e. due to the long range of β− particles, it is not essential to target every single tumor cell to efficiently irradiate the whole tumor (Pouget et al. 2011).
Holmium-166 (166Ho) is a β− emitter that decays to 166Er with a half-life time of 26.8 h and emits β− particles with maximum energy of 1.85 MeV (Fig. 1a). The high energy of the β− particles results in a maximum tissue penetration depth of 8.7 mm which makes 166Ho a promising radionuclide for treating larger malignancies (Klaassen et al. 2019). In addition, 166Ho can also be imaged by single-photon emission computed tomography (SPECT) due to its gamma emission at 80.57 keV (Elschot et al. 2011).166Ho is generally produced by the neutron activation of 165Ho following the (n, γ) reaction. An alternative route for 166Ho production is the 166Dy/166Ho generator (Klaassen et al. 2019). Dysprosium-166 (166Dy) has a half-life time of 81.6 h, decays to 166Ho via β− decay and can be produced by a double neutron capture reaction from 164Dy (Fig. 1b). 166Dy/166Ho can also serve as in vivo generator which is capable of delivering higher radiation dose per administrated activity due to the three times longer half-life time of 166Dy than 166Ho (Poty et al. 2018; Baidoo et al. 2013; Edem et al. 2016). Therefore, better treatment outcome could be expected by using 166Dy/166Ho in vivo generator instead of the direct administration of 166Ho.
Fig. 1.
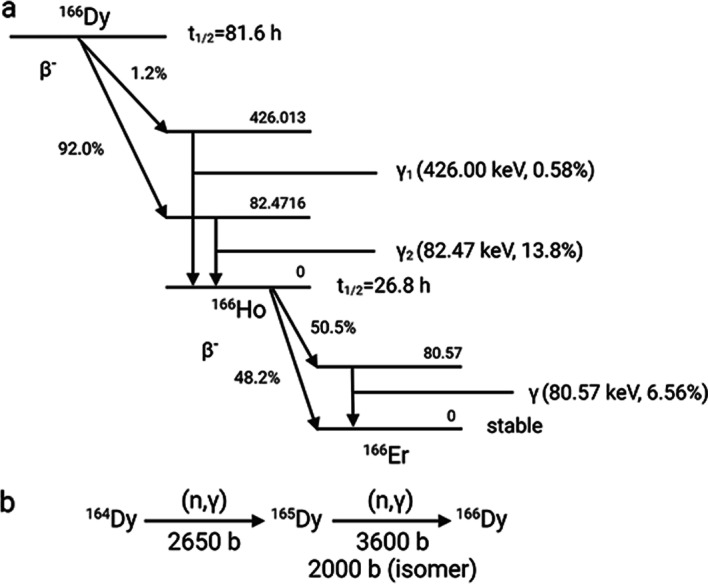
a Decay scheme of 166Dy and 166Ho including the major transitions. b The double-neutron capture nuclear reaction of 164Dy to produce 166Dy and the corresponding cross-sections
However, Zeevaart et al. reported the radiolabelling of 166Dy on dodecane tetraacetic acid (DOTA) and surprisingly found that about 72% of the daughter 166Ho was released from the 166Ho-DOTA complex (Zeevaart et al. 2012). The 166Ho loss was attributed to the de-excitation of 166Ho* via internal conversion instead of γ emission. Internal conversion is a process where the excited daughter nucleus electromagnetically interacts with inner orbital electrons and results in the emission of an inner electron from K shell or L shell along with the creation of electron vacancies. The electrons from the outer shells will be reorganized to fill in the vacancies while emitting auger electrons as well as characteristic X-ray. As the result of the emission of auger electrons, the de-excited 166Ho ions become highly charged and will extract electrons from the surrounding environment (i.e. DOTA). Due to the electron transfer to 166Ho, the DOTA component also becomes positively charged while the 166Ho ion acquires its original oxidation state (+ 3). The repulsion force between the two components having the same charge results in the rupture of the bonds between 166Ho and DOTA. Thus, 166Ho is released as free ion. The theoretical calculation predicts 73.6% 166Ho release which matches well with the published experimental results (Zeevaart et al. 2012). Being an isotope of a lanthanide element, free 166Ho tends to accumulate in liver, kidney, spleen and bone and may cause severe side effect to the patient (Suzuki et al. 1998). Therefore, to implement the 166Dy/166Ho in vivo generator in the clinic, a carrier that can prevent the loss of the internally converted 166Ho has to be developed.
Nowadays, the medical application of different types of nanoparticles has been extensively reported for diagnostics and the treatment of cancer and other diseases (Mitchell et al. 2021; Pelaz et al. 2017; Thomas and Weber 2019; Shi et al. 2017; Wong et al. 2020). Gold nanoparticles (AuNP) have shown great potential as carriers for anti-cancer agents due to their unique properties such as biocompatibility, precisely controlled size and the possibility of easy surface modification (Singh et al. 2018; Boisselier and Astruc 2009). Besides using AuNP as carriers for conventional payloads, multiple reports on the chelator-free labelling of medical radionuclides on AuNP have been published (Ge et al. 2020; Silva et al. 2021). In these studies, radionuclides in the form of metallic ions or halogen ions are either co-reduced into the lattice of AuNP (e.g. 64Cu (Frellsen et al. 2016; Sun et al. 2014; Pretze et al. 2019; Zhao et al. 2014), 111In (Zheng et al. 2021) and 68 Ga (Zheng et al. 2021)) or chemically absorbed on the surface of AuNP (125I, 124I (Lee et al. 2016; Lee et al. 2017) and 211At (Dziawer et al. 2017)). In most cases, the radiolabelling stability and the tumor uptake of the radionuclides appear to be improved after being loaded on AuNPs when compared to the common chelator approaches (Pretze et al. 2019). The improved tumor uptake is likely from the prolonged circulation time of AuNPs comparing with small molecules. However, the toxicity of AuNP itself have to be considered even gold is considered to be biocompatible (Ranjbar Bahadori et al. 2021).
In this study, we developed a chelator-free radiolabelling method to incorporate 166Dy in AuNP. In this radiolabelling method, we co-reduced 166Dy3+ ions with gold and platinum precursors to form either a bimetallic (166DyAuNP) or trimetallic (166DyPtAuNP) nanoparticle. In addition, an extra gold layer was added to the 166DyAuNP to form a core–shell structured 166DyAu@AuNP. We first characterized the physical properties of the DyAu@AuNP and DyPtAuNP with non-radioactive Dy. Then the radiolabelling of 166Dy was performed and the retention of 166Ho on 166DyAu@AuNP and 166DyPtAuNP was evaluated.
Methods and materials
Materials
Gold(III) chloride trihydrate (≥ 99.9%, HAuCl4 · 3H2O), chloroplatinic acid hexahydrate (≥ 37.50% Pt, H2PtCl6 · 6H2O), sodium borohydride (≥ 98.0%, NaBH4), cetyltrimethylammonium bromide (≥ 98%, CTAB), cetyltrimethylammonium chloride solution (25 wt.% in water, CTAC), L-Ascorbic acid (≥ 99%, AA), sodium hydroxide (NaOH) and dysprosium(III) chloride hexahydrate (≥ 99.9%) were purchased from Sigma-Aldrich (Zwijndrecht, the Netherlands). 90% enriched dysprosium-164 oxide powder (164Dy2O3) was obtained from Oak Ridge National Laboratory (sample number 122502, ORNL, Tennessee, USA). Ethylenediaminetetraacetic acid disodium salt dihydrate (Na2EDTA · 2H2O), Diethylenetriamine pentaacetate (DTPA), hydrochloric acid (HCl, 30%, Suprapur®) and nitric acid (HNO3, 69%, Supelco®) was supplied by Merck. All chemicals were used as received without further purification. MiliQ water was obtained from an in-house MiliQ system (Millipore) and used throughout this study.
Production of 166Dy
166Dy was produced by the double neutron capture reaction of 164Dy. 3 mg 90% enriched 164Dy2O3 powder was irradiated in the reactor facilities of the SCK•CEN—BR2 Reactor (Mol, Belgium), the Institute of Energy Security and Environmental Safety Centre for Energy Research (Budapest, Hungary) or the nuclear reactor research facility (HOR, Hoger Onderwijs Reactor) at the Department of Radiation Science and Technology of the Delft University of Technology (Delft, the Netherlands). The obtained 166Dy2O3 powder was dissolved in 5 ml 1 M HCl under mild heating to prepare a stock solution of 166DyCl3. 2.5 ml of the stock solution was transferred to a 20-ml glass vial and the pH of the stock solution was adjusted to ~ 5.5 by adding 2.35 ml of 1 M NaOH solution (checked by pH test paper). The activity of 166Dy and 166Ho in the stock solution was measured on a calibrated well-type HPGe detector (Canberra).
Synthesis of AuNP seed
The synthesis was adapted from a published protocol with some changes (Zheng et al. 2014) and is schematically illustrated in Fig. 2. The AuNP seeds were synthesized by the reduction of HAuCl4 by NaBH4 using CTAB as capping agent. 0.1 ml 25 mM HAuCl4, 4 ml 250 mM CTAB and 5.9 ml MiliQ water was added to a glass vial and mixed for 10 min. 0.6 ml freshly prepared, ice-cold 10 mM NaBH4 solution was added to the mixture dropwise under vigorous stirring. The color of the solution changed from yellow to dark brown rapidly. The obtained AuNP seeds were left undisturbed at 27 °C for 1.5 h before further usage.
Fig. 2.
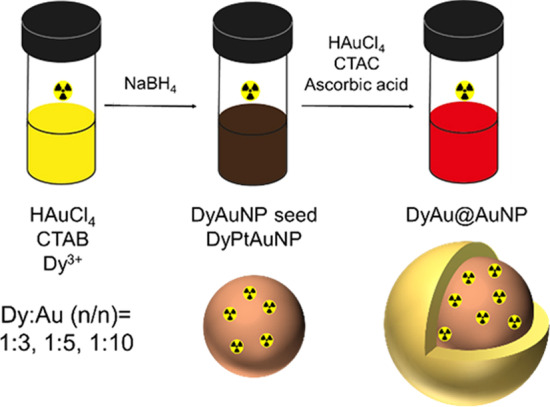
Schematic illustration of the synthesis of 166DyAu@AuNP and 166DyPtAuNP
Growth of AuNP seed to 5 nm AuNP
2 ml 200 mM CTAC, 1.5 ml 100 mM AA and 1 ml AuNP seed dispersion were added to a glass vial and mixed for 5 min at 27 °C. 2 ml 0.5 mM HAuCl4 was then added in one-shot by a pipet. The reaction was continued at 27 °C for another 15 min.
Synthesis of 5 nm non-radioactive DyAu@AuNP
33 μl, 20 μl or 10 μl 25 mM DyCl3 solution (pH 5.5) was mixed with 0.1 ml 25 mM HAuCl4 and 4 ml 250 mM CTAB in a glass vial to achieve the Dy:Au (n:n) feeding ratio of 1:3, 1:5 or 1:10. The total volume was adjusted to 10 ml by MiliQ water. 0.6 ml ice-cold 10 mM NaBH4 solution was then added to the mixture dropwi under vigorous stirring. The growth of the DyAuNP seed to 5 nm core–shell structured DyAu@AuNP was performed in the same way as the growth of AuNP seed to 5 nm AuNP after aging the DyAuNP seed at 27 °C for 1.5 h.
Synthesis of non-radioactive DyPtAuNP
10 μl 25 mM H2PtCl6, 90 μl 25 mM HAuCl4 and 4 ml 250 mM CTAB were mixed in a glass vial. 33 μl or 10 μl 25 mM DyCl3 solution (pH 5.5) was then added to achieve the Dy:(Pt + Au) (n/n) feeding ration of 1:3 or 1:10. MiliQ water was added to adjust the total volume to be 10 ml and stirred for 10 min. 0.6 ml freshly prepared, ice-cold 10 mM NaBH4 solution was added to the mixture dropwise under vigorous stirring. The colour of the solution changed from yellow to dark brown rapidly. The obtained DyPtAuNP was left undisturbed at 27 °C for 1.5 h before purification.
Synthesis of 5 nm 166DyAu@AuNP
88.2 μl stock solution of 166DyCl3 containing approximately 0.134 MBq 166Dy and 0.2 MBq 166Ho was mixed with 0.1 ml 25 mM HAuCl4 and 4 ml 250 mM CTAB in a glass vial. 30.1 μl, 17.1 μl or 7.1 μl of 25 mM DyCl3 solution (pH 5.5) was then added to ensure the Dy:Au (n:n) feeding ratio to be 1:3, 1:5 or 1:10. The synthesis of 166DyAuNP seed and growth to 166DyAu@AuNP was performed in the same way as non-radioactive DyAu@AuNP which was described above.
Synthesis of 166DyPtAuNP
88.2 μl stock solution of 166DyCl3 containing approximately 0.134 MBq 166Dy and 0.2 MBq 166Ho was mixed with 10 μl 25 mM H2PtCl6, 90 μl 25 mM HAuCl4 and 4 ml 250 mM CTAB in a glass vial. 30.1 μl or 7.1 μl of 25 mM DyCl3 solution (pH 5.5) was then added to ensure Dy:Au (n:n) feeding ratio of 1:3 or 1:10. The final volume was then adjusted to 10 ml by MiliQ water and stirred for 10 min. 0.6 ml freshly prepared, ice-cold 10 mM NaBH4 solution was added to the mixture dropwise under vigorous stirring. The obtained 166DyPtAuNP was left undisturbed at 27 °C for 1.5 h before purification.
Characterization of non-radioactive nanoparticles
The morphology and size of the AuNP, DyAu@AuNP and DyAuNP was determined with a JEM-1400 Plus transmission electron microscope (TEM, JEOL) at the acceleration voltage of 120 kV. The UV–vis absorption spectra of AuNPs were measured by a UV–VIS-NIR spectrophotometer (UV-6300PC, VWR). The hydrodynamic radius of the samples was determined by dynamic light scattering (DLS) which consisted of a JDS uniphase 633 nm 35 mW laser source, an ALV sp 125 s/w 93 goniometer, a fibre detector and a Perkin Elmer photo counter. The data was fitted using the CONTIN method and the Stokes–Einstein equation (Eq. 1) was used to determine the hydrodynamic radius of the nanoparticles.
| 1 |
Determination of 166Dy radiolabelling efficiency
100 μl 100 mM EDTA or 100 mM DTPA was added to the 166DyAu@AuNP and 166DyPtAuNP samples and incubated at 27 °C for 30 min to bind with free 166Dy3+ ions. Then the samples were centrifuged (4000 rpm, 10 min) and washed three times using spin filters (MWCO 10 KDa, Amicon). The final volume of the washed samples was adjusted to 4 ml by MiliQ water and stored at 37 °C. The counts of the nanoparticles and filtrates (166Dy-EDTA) of all samples were measured by an automatic gamma counter (Wallac Wizard2 2480, Perkin Elmer) or a low energy Ge-detector (GL2020R, Canberra). 166Dy was measured using its gamma emission at 425.99 keV. The radiolabelling efficiency of 166Dy was calculated by the following formula: Counts(NPs)/[Counts(NPs) + ∑Counts(filtrate)] × 100%.
Determination of 166Ho and 166Dy retention
To assess the stability of 166Ho and 166Dy on nanoparticles, the samples were dispersed in 4 ml MiliQ water or 2.5 mM DTPA (pH 7.5) and incubated at 37 °C for 24, 48 and 72 h. At each time point, the samples were collected and washed by MiliQ water using spin filters under centrifugation (4000 rpm, 10 min). The counts of the nanoparticles and the filtrate was measured to calculate the retention of both 166Ho and 166Dy.
Determination of gold and dysprosium content in 166DyAu@AuNP and 166DyPtAuNP
1 ml of each completely decayed sample was completely destructed in 1 ml aqua regia (HCl/HNO3 = 3:1) and diluted by MiliQ water to a final volume of 10 ml. The concentration of Au and Dy were then measured by ICP-OES (Optima 8000, Perkin Elmer).
Results
Synthesis and characterization of non-radioactive DyAu@AuNP
In this study, we designed a core–shell structured AuNP to function as the carrier for 166Dy /166Ho in vivo generator. The gold precursor was first co-reduced with 166Dy3+ ions to form the 166DyAuNPs. Subsequently, an extra gold shell was grown by reducing gold precursor with ascorbic acid to prevent the possible escape of free 166Ho3+ ions. Besides assisting to retain free 166Ho3+ ions, the growth of an extra gold layer can also improve the colloidal stability of the DyAuNPs (Zheng et al. 2014). It is important that the original physiochemical properties of the AuNPs are not altered upon 166Dy encapsulation. Thus, we first performed a pilot study with non-radioactive DyCl3 to explore the influence of Dy content on the physical properties of the DyAu@AuNPs. The non-radioactive DyCl3 was co-reduced with HAuCl4 by a strong reducing agent NaBH4 to form the bimetallic DyAuNPs. Three samples with different Dy:Au feeding ratios of 1:3, 1:5 and 1:10 were prepared. An extra gold shell was then grown on the seed particles via the reduction of HAuCl4 at lower concentration using ascorbic acid and resulting in the core–shell structured DyAu@AuNPs. The non-incorporated Dy3+ ions were removed by incubating DyAu@AuNPs with EDTA or DTPA, followed by multiple cycles of washing with MiliQ water. Au@AuNP without Dy content was also prepared with the same method and used as the control group.
The size and shape of the DyAu@AuNPs were characterized by transmission electron microscope (TEM). As shown in Fig. 3a–d, DyAu@AuNPs with varying Dy:Au feeding ratios as well as the Au@AuNP all showed a diameter of 4.9 nm. The hydrodynamic radius (RH) of the DyAu@AuNPs and Au@AuNP was measured by dynamic light scattering (DLS). As shown in Fig. 3e, the intensity weighted RH was determined to be within the range of 12 ~ 14 nm for both the DyAu@AuNPs and the Au@AuNP. The hydrodynamic radius of the DyAu@AuNPs was found to be larger than the radius measured by TEM, since DLS measures the hydration layer formed around CTAB/CTAC on the surface of the nanoparticles. Due to the surface plasmon resonance (SPR) effect of AuNPs, the characteristic UV–vis spectrum can be used as an indication of the size of AuNPs (Barbosa et al. 2010). The UV–vis spectrum of the DyAu@AuNPs and Au@AuNP is shown in Fig. 3f. The wavelength of the SPR peak (λSPR) of all samples were detected near 520 nm, indicating that the DyAu@AuNPs all had comparable size to the Au@AuNP. All results of the characterization of the DyAu@AuNPs and Au@AuNP are summarized in Table 1.
Fig. 3.

Characterization of DyAu@AuNPs. a–d Representative TEM image of samples with different Dy:Au feeding ratios: Dy:Au = 1:3 (a), 1:5 (b), 1:10 (c), no Dy addition (d). Scale bar is 20 nm. See supporting information for size distribution histograms (Additional file 1: Fig. S1). e Hydrodynamic radius (RH) of the samples measured by DLS. f) UV–vis spectrum of the DyAu@AuNPs
Table 1.
Summary of the physical properties of DyAu@AuNPs with different Dy:Au feeding ratios
| No Dy | 1:3 | 1:5 | 1:10 | |
|---|---|---|---|---|
| d (nm) | 4.9 ± 0.8 | 4.9 ± 0.6 | 4.9 ± 0.7 | 4.9 ± 0.7 |
| RH (nm) | 12.3 ± 0.3 | 12.9 ± 0.3 | 14.1 ± 0.5 | 11.8 ± 0.3 |
| λSPR (nm) | 518 | 522 | 519 | 520 |
Based on the TEM, UV–vis and DLS measurements, we conclude that incorporating different amounts of Dy into the gold nanoparticle had no influence on the final size and shape of the core–shell structured DyAu@AuNPs.
Synthesis and characterization of non-radioactive DyPtAuNP
To better understand the behavior of 166Ho and 166Dy on nanoparticles and check if internally converted 166Ho can be retained even without the extra gold layer, we attempted to directly use the DyAuNPs as the carrier for 166Dy. However, the DyAuNPs were not stable and aggregated to larger AuNPs within 24 h (Additional file 1: Fig. S3). To improve the colloidal stability of DyAuNPs, we hereby prepared trimetallic DyPtAuNPs by replacing 10% of Au with Pt while the Dy:(Au + Pt) feeding ratios was still set to be 1:3 and 1:10. PtAuNP with no Dy content was also prepared and used as control. The size of the DyPtAuNPs as determined by TEM are shown in Fig. 4a–c. The diameter of the DyPtAuNP with Dy feeding ratio of 1:3 was measured to be 4.4 ± 1.1 nm which is comparable to the PtAuNP (4.0 ± 1.3 nm). However, larger particles (d = 6.6 ± 1.8 nm) were measured for the DyPtAuNP with Dy feeding ratio of 1:10. The UV–vis spectrum of the DyPtAuNPs and PtAuNP is given in Fig. 4d. The absence of SPR peak near 500 nm further confirmed the small size of the nanoparticles (Alric et al. 2013).
Fig. 4.

Characterization of DyPtAuNPs. a–c Representative TEM image of samples with different Dy:Au feeding ratios: Dy:Au = 1:3 (a), 1:10 (b) and no Dy addition (c). Scale bar is 50 nm. See supporting information for size distribution histograms (Additional file 1: Fig. S2). d UV–vis spectrum of the DyPtAuNPs
Radiolabelling of 166Dy on DyAu@AuNP and DyPtAuNP
The radiolabelling of 166Dy was carried out by a similar method as used for the preparation of non-radioactive DyAu@AuNPs and DyPtAuNPs. The Dy source was changed to a mixture of non-radioactive DyCl3 and 166DyCl3 stock solution containing 0.134 MBq 166Dy. Due to the decay of 166Dy, 166Ho was also present in the stock solution of 166DyCl3. Considering the trace amount of 166Ho3+ ions, we expect that this to have negligible influence on the formation of the NPs. Three independent samples of 166DyAu@AuNP and 166DyPtAuNP with different Dy:Au feeding ratios were prepared and washed thoroughly by EDTA/DTPA and MiliQ water to remove all unbounded 166Dy. The 166Dy radiolabelling efficiency was calculated by comparing the counts of nanoparticles and the washing solution at 425.99 keV. The calculated results are shown in Fig. 5. Radiolabelling efficiency of 60% and 70% was achieved for 166DyAu@AuNPs and 166DyPtAuNPs respectively. No significant difference of the radiolabelling efficiency was found among the groups with different Dy:Au feeding ratios.
Fig. 5.
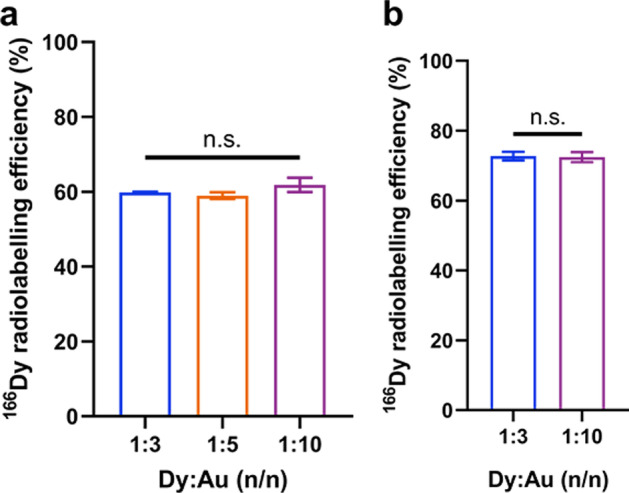
166Dy radiolabelling efficiency of 166DyAu@AuNP (a) and 166DyPtAuNP (b) with different Dy:Au feeding ratios. The error bars represent the standard deviations of three independent experiments ( n.s. indicates non-significant difference, 2way ANOVA test)
Due to the big lattice mismatch (11.9%) and the large difference of reduction potential between Dy (III, − 2.29 V) and Au (III, [AuCl4]−, + 0.93 V), not all initially added Dy was reduced in the AuNP core which resulted in 166Dy radiolabelling efficiency of around 60%. As the same activity of 166DyCl3 was used during the synthesis of 166DyAu@AuNPs and 166DyPtAuNPs, the activity of radiolabelled 166Dy was the same for all samples with different Dy:Au feeding ratios. No improvement of the 166Dy radiolabelling efficiency was achieved by lowering the initial amount of Dy3+.
The completely decayed 166DyAu@AuNP and 166DyPtAuNP samples were also destructed and further analysed by ICP-OES to measure the concentration of Au and Dy. Comparing with the Au concentration of the Au@AuNP and PtAuNP samples, little difference of the Au concentration was found from the 166DyAu@AuNP and 166DyPtAuNP samples (Table S1). Taking ICP-OES measurements together with other characterizations, we further confirmed that the reduction of gold precursor by NaBH4 as well as the formation of nanoparticles was not affected by the addition of Dy3+. The 166Dy radiolabelling efficiency was also calculated using the total concentration of Dy (including both radioactive and non-radioactive Dy) measured by ICP-OES (Additional file 1: Fig. S4). Similar to the results from Ge-detector measurement, the radiolabelling efficiency was not influenced by the Dy:Au feeding ratios. However, we found that the 166Dy radiolabelling efficiency calculated from ICP-OES data was approximately 10% lower than that from the Ge-detector data. Further studies will be carried out to explain this phenomenon.
Retention of 166Ho and 166Dy
In vivo generator of therapeutic radionuclides can generally increase the delivered dose per administrated activity because of the longer half-life time of the mother nuclides (Edem et al. 2016). To make sure the radiation dose is mainly delivered to the tumor while sparing the normal tissues, both the mother and the daughter nuclides should be kept within the carrier. Therefore, we radiolabelled core–shell structured gold nanoparticles, i.e. the 166DyAu@AuNPs with 166Dy. An outer layer of gold was added to prevent the diffusion of free 166Ho if it escapes from the core nanoparticle. On the other hand, nanoparticles without the shell structure, i.e. the 166DyPtAuNPs were also radiolabelled with 166Dy for comparison. 166DyAuNP seeds were not studied because of the low colloidal stability (Additional file 1: Fig. S3).
To measure the retention of the internally converted 166Ho as well as the retention of 166Dy, 166DyAu@AuNPs and 166DyPtAuNPs were incubated in MiliQ water or 2.5 mM DTPA (pH 7.5) at 37 °C for 72 h. Every 24 h, the samples were centrifuged to separate NPs from free 166Dy3+ and 166Ho3+. The counts of the nanoparticles and the washing solution was measured at 65 ~ 90 keV and 340 ~ 460 keV for 166Ho and 166Dy respectively. As the NPs were still capped by CTAB/CTAC, the nanoparticles would form aggregation upon interaction with high concentration salt solution or protein (Zhang and Lin 2014). Thus, the in vitro stability tests were not performed in PBS or serum to avoid the interference of nanoparticle aggregation. As shown in Fig. 6, more than 95% of 166Ho was found to be retained in both 166DyAu@AuNPs and 166DyPtAuNPs for at least 72 h in MiliQ water (Fig. 6a, b). The retention of 166Dy was also found to be more than 95% for both 166DyAu@AuNPs and 166DyPtAuNPs during the 72 h incubation in MiliQ water (Fig. 6c, d). For all the samples challenged by DTPA, about 90% of both 166Ho and 166Dy was still bounded to the nanoparticles even after 72 h incubation (Fig. 7). These results indicate that very high 166Ho and 166Dy retention was achieved independent from the Dy:Au feeding ratio and the extra shell of coating.
Fig. 6.

166Ho and 166Dy retention of 166DyAu@AuNPs (a, c) and 166DyPtAuNPs (b, d) with different Dy:Au feeding ratios in MiliQ water at 37 °C as function of time. The error bars represent the standard deviation of three independent experiments
Fig. 7.

166Ho and 166Dy retention of 166DyAu@AuNPs (a, c) and 166DyPtAuNPs (b, d) with different Dy:Au feeding ratios in 2.5 mM DTPA at 37 °C as function of time. The error bars represent the standard deviation of three independent experiments
Discussion
Surprisingly, the 166DyPtAuNPs were found to be able to retain the same percentage of 166Ho as the 166DyAu@AuNPs. This result suggests that high 166Ho retention could still be achieved even without the addition of an extra gold layer. This finding made us think about a the possible mechanism responsible for the high 166Ho retention on AuNPs. The internal conversion of 166Dy results in highly charged 166Ho ions which tend to seek electrons from the surrounding environment, i.e. the carrier. In the case of 166Dy coupled to a simple chelator composed of low Z elements such as dodecane tetraacetic acid (DOTA), the number of free electrons in the system is low. Therefore, DOTA molecule could be easily altered to be positively charged after the electron migration to the 166Ho ions. Due to the repulsion between the entities having the same charge, i.e. 166Ho3+ and [DOTA]n+, the 166Ho-DOTA complex is ruptured. When a high Z material is used as the carrier for 166Dy, such as AuNP, many more free electrons are available. When the highly positive 166Ho ion extracts electrons from its neighbouring Au atoms, electrons can be quickly redistributed to fill in the new vacancies. The redistribution of electrons might cause a transient change of the surface charge of AuNP, but then electrons from the solvent (i.e. water) will be attracted to the AuNP due to the ultra-high affinity of Au to solvated electrons (Ghandi et al. 2015). Therefore, the colloidal stability of AuNP is preserved while the release of 166Ho is avoided. A similar method was reported to improve the retention of 80Br which was internally converted from 80mBr (49 and 37 keV, α = 1.6 and 300 respectively) by Adamson et al. (Adamson and Grunland 1951; Wexler and Anderson 1960). The authors found that 100% and 86% of 80Br was released from [Co(NH3)5Br]2+ (aq) and solidified [Co(NH3)5Br](NO3)2 (s) while 47% and even 0% of 80Br was released from [PtBr6]2− (aq) and solidified (NH4)2PtBr6 (s). This result supported our hypothesis on the function of AuNP as electron source for the internally converted 166Ho. Besides, the results from these studies also suggest that in our case the reduction of 166Dy3+ into solid state (Dy0) might also contribute to the high retention of 166Ho.
Besides the high retention of the internally converted 166Ho, our radiolabelling method is also simple and quick. The whole procedure can be finished within 8 h without the need of separating 166Dy from 166Ho. The interaction between the β− particle emitted by 166Ho and gold atoms is also favourable for a more efficient dose delivery due to the formation of secondary electrons and free radicals such as · OH radicals (Haume et al. 2016). To make the 166DyPtAuNPs and 166DyAu@AuNPs more applicable for clinical application, the current capping ligand, CTAB/CTAC, has to be exchanged with biocompatible ligands such as PEG. In previous studies it has been shown that small AuNPs not conjugated with targeting agents have tumour uptake around 4–5% ID/g depending on the morphology and surface properties of the nanoparticles (Sun et al. 2014; Zhang et al. 2022). In comparison small molecules such as PSMA can achieve much higher tumour uptake (Banerjee et al. 2014). Therefore, it will be very interesting to determine whether the addition of such targeting moieties will increase tumour accumulation.
Conclusion
In summary, we developed a chelator-free radiolabelling method to obtain a 166Dy/166Ho in vivo generator and prevented the loss of 166Ho that is caused by internal conversion. The explanation for the high 166Ho retention was not experimentally proven but might be related to the high electron density of the gold nanoparticles. To further understand the mechanism of 166Ho retention on gold nanoparticles, the structure of the nanoparticles should be studied by both experiments as well as theoretical simulations. Besides the further research on 166Ho retention mechanism, the capping ligands of the nanoparticles should be replaced to increase the biocompatibility of the nanoparticles and make them suitable for medical applications.
Supplementary Information
Additional file1. Theoretical calculation of 166Ho loss due to internal conversion; Fig. S1. Size distribution histogram of DyAu@AuNPs with different Dy:Au feeding ratios; Fig. S2. Size distribution histogram of DyPtAuNPs with different Dy:Au feeding ratios; Fig. S3. Representative picture of 166DyAuNP (Dy:Au=1:3) after 24 h incubation at 37 °C; Fig. S4. Comparison of 166Dy radiolabelling efficiency calculated from Ge-detector data and ICP-OES data; Table S1. Comparison of Au concentration of Au@AuNP, 166DyAu@AuNP, PtAuNP and 166DyPtAuNP
Acknowledgements
We would like to thank the Institute of Energy Security and Environmental Safety Centre for Energy Research (Budapest, Hungary) for the production of 166Dy. China Scholarship Council is also acknowledged for the financial support of RW.
Abbreviations
- AA
Ascorbic acid
- AuNP
Gold nanoparticle
- CTAB
Cetyltrimethylammonium bromide
- CTAC
Cetyltrimethylammonium chloride
- DLS
Dynamic light scattering
- DOTA
Dodecane tetraacetic acid
- DTPA
Diethylenetriamine pentaacetate
- Dy
Dysprosium
- EBRT
External beam radiation therapy
- EDTA
Ethylenediaminetetraacetic acid
- Ho
Holmium
- RNT
Radionuclide therapy
- SPECT
Single photon emission computed tomography
- SPR
Surface plasmon resonance
- TEM
Transmission electron microscope
- UV–vis
Ultraviolet–visible
Author contributions
AD and HW contributed to the design of the study and oversaw the research project. RW designed and carried out the experiments and analysed the results. BP was involved in the radiochemistry experiments of this work. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
The data associated to this research work are available in this manuscript or in the online supplementary file.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adamson AW, Grunland JM. Retention of Br 80 in comple bromides, following isomeric transition. J Am Chem Soc. 1951;73(11):5508. doi: 10.1021/ja01155a570. [DOI] [Google Scholar]
- Alric C, Miladi I, Kryza D, Taleb J, Lux F, Bazzi R, et al. The biodistribution of gold nanoparticles designed for renal clearance. Nanoscale. 2013;5(13):5930–5939. doi: 10.1039/c3nr00012e. [DOI] [PubMed] [Google Scholar]
- Baidoo KE, Milenic DE, Brechbiel MW. Methodology for labeling proteins and peptides with lead-212 (212Pb) Nucl Med Biol. 2013;40(5):592–599. doi: 10.1016/j.nucmedbio.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee SR, Pullambhatla M, Foss CA, Nimmagadda S, Ferdani R, Anderson CJ, et al. 64Cu-labeled inhibitors of prostate-specific membrane antigen for PET imaging of prostate cancer. J Med Chem. 2014;57(6):2657–2669. doi: 10.1021/jm401921j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S, Agrawal A, Rodríguez-Lorenzo L, Pastoriza-Santos I, Alvarez-Puebla RA, Kornowski A, et al. Tuning size and sensing properties in colloidal gold nanostars. Langmuir. 2010;26(18):14943–14950. doi: 10.1021/la102559e. [DOI] [PubMed] [Google Scholar]
- Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38(6):1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Dash A, Russ Knapp F, Ra Pillai M. Targeted radionuclide therapy-an overview. Curr Radiopharm. 2013;6(3):152–80. doi: 10.2174/18744710113066660023. [DOI] [PubMed] [Google Scholar]
- Dziawer L, Koźmiński P, Mȩczyńska-Wielgosz S, Pruszyński M, Łyczko M, Wąs B, et al. Gold nanoparticle bioconjugates labelled with 211At for targeted alpha therapy. RSC Adv. 2017;7(65):41024–41032. doi: 10.1039/C7RA06376H. [DOI] [Google Scholar]
- Edem P, Fonslet J, Kjaer A, Herth M, Severin G. In vivo radionuclide generators for diagnostics and therapy. Bioinorg Chem Appl. 2016;2016:1–8. doi: 10.1155/2016/6148357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elschot M, Nijsen JFW, Dam AJ, de Jong HWAM. Quantitative evaluation of scintillation camera imaging characteristics of isotopes used in liver radioembolization. PLoS One. 2011;6(11):e26174. doi: 10.1371/journal.pone.0026174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frellsen AF, Hansen AE, Jølck RI, Kempen PJ, Severin GW, Rasmussen PH, et al. Mouse positron emission tomography study of the biodistribution of gold nanoparticles with different surface coatings using embedded Copper-64. ACS Nano. 2016;10(11):9887–9898. doi: 10.1021/acsnano.6b03144. [DOI] [PubMed] [Google Scholar]
- Ge J, Zhang Q, Zeng J, Gu Z, Gao M. Radiolabeling nanomaterials for multimodality imaging: New insights into nuclear medicine and cancer diagnosis. Biomaterials. 2020;228:119553. doi: 10.1016/j.biomaterials.2019.119553. [DOI] [PubMed] [Google Scholar]
- Ghandi K, Findlater AD, Mahimwalla Z, MacNeil CS, Awoonor-Williams E, Zahariev F, et al. Ultra-fast electron capture by electrosterically-stabilized gold nanoparticles. Nanoscale. 2015;7(27):11545–11551. doi: 10.1039/C5NR02291F. [DOI] [PubMed] [Google Scholar]
- Gudkov SV, Shilyagina NY, Vodeneev VA, Zvyagin AV. Targeted radionuclide therapy of human tumors. Int J Mol Sci. 2016 doi: 10.3390/ijms17010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haume K, Rosa S, Grellet S, Śmiałek MA, Butterworth KT, Solov’yov AV, et al. Gold nanoparticles for cancer radiotherapy: a review. Cancer Nanotechnol. 2016;7(1):8. doi: 10.1186/s12645-016-0021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humm JL, Sartor O, Parker C, Bruland OS, Macklis R. Radium-223 in the treatment of osteoblastic metastases: a critical clinical review. Int J Radiat Oncol Biol Phys. 2015;91(5):898–906. doi: 10.1016/j.ijrobp.2014.12.061. [DOI] [PubMed] [Google Scholar]
- Klaassen NJM, Arntz MJ, Gil Arranja A, Roosen J, Nijsen JFW. The various therapeutic applications of the medical isotope holmium-166: a narrative review. EJNMMI Radiopharm Chem. 2019;4(1):1–26. doi: 10.1186/s41181-019-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostelnik TI, Orvig C. Radioactive main group and rare earth metals for imaging and therapy. Chem Rev. 2019;119(2):902–956. doi: 10.1021/acs.chemrev.8b00294. [DOI] [PubMed] [Google Scholar]
- Lee SB, Ahn SB, Lee S-W, Jeong SY, Ghilsuk Y, Ahn B-C, et al. Radionuclide-embedded gold nanoparticles for enhanced dendritic cell-based cancer immunotherapy, sensitive and quantitative tracking of dendritic cells with PET and Cerenkov luminescence. NPG Asia Mater. 2016;8(6):e281–e281. doi: 10.1038/am.2016.80. [DOI] [Google Scholar]
- Lee SB, Lee SW, Jeong SY, Yoon G, Cho SJ, Kim SK, et al. Engineering of radioiodine-labeled gold core-shell nanoparticles as efficient nuclear medicine imaging agents for trafficking of dendritic cells. ACS Appl Mater Interfaces. 2017;9(10):8480–8489. doi: 10.1021/acsami.6b14800. [DOI] [PubMed] [Google Scholar]
- Marcu L, Bezak E, Allen BJ. Global comparison of targeted alpha vs targeted beta therapy for cancer: in vitro, in vivo and clinical trials. Crit Rev Oncol Hematol. 2018;2018(123):7–20. doi: 10.1016/j.critrevonc.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi: 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BJB, Andersson JD, Wuest F. Targeted alpha therapy: progress in radionuclide production, radiochemistry and applications. Pharmaceutics. 2021;13(1):1–28. doi: 10.3390/pharmaceutics13010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz B, Alexiou C, Alvarez-Puebla RA, Alves F, Andrews AM, Ashraf S, et al. Diverse applications of nanomedicine. ACS Nano. 2017;11(3):2313–2381. doi: 10.1021/acsnano.6b06040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool SE, Krenning EP, Koning GA, van Eijck CHJ, Teunissen JJM, Kam B, et al. Preclinical and clinical studies of peptide receptor radionuclide therapy. Semin Nucl Med. 2010;40(3):209–218. doi: 10.1053/j.semnuclmed.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Poty S, Francesconi LC, McDevitt MR, Morris MJ, Lewis JS. Alpha-emitters for radiotherapy: from basic radiochemistry to clinical studies-part 2. J Nucl Med. 2018;59(7):1020–7. doi: 10.2967/jnumed.117.204651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget J-P, Navarro-Teulon I, Bardiès M, Chouin N, Cartron G, Pèlegrin A, et al. Clinical radioimmunotherapy-the role of radiobiology. Nat Rev Clin Oncol. 2011;8(12):720–734. doi: 10.1038/nrclinonc.2011.160. [DOI] [PubMed] [Google Scholar]
- Pretze M, van der Meulen NP, Wängler C, Schibli R, Wängler B. Targeted 64Cu-labeled gold nanoparticles for dual imaging with positron emission tomography and optical imaging. J Label Compd Radiopharm. 2019;62(8):471–482. doi: 10.1002/jlcr.3736. [DOI] [PubMed] [Google Scholar]
- Ranjbar Bahadori S, Mulgaonkar A, Hart R, Wu C-Y, Zhang D, Pillai A, et al. Radiolabeling strategies and pharmacokinetic studies for metal based nanotheranostics. Wires Nanomed Nanobiotechnol. 2021;13(2):e1671. doi: 10.1002/wnan.1671. [DOI] [PubMed] [Google Scholar]
- Sgouros G, Bodei L, McDevitt MR, Nedrow JR. Radiopharmaceutical therapy in cancer: clinical advances and challenges. Nat Rev Drug Discov. 2020;19(9):589–608. doi: 10.1038/s41573-020-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva F, Campello MPC, Paulo A. Radiolabeled gold nanoparticles for imaging and therapy of cancer. Materials (basel) 2021;14(1):1–32. doi: 10.3390/ma14010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Pandit S, Mokkapati VRSS, Garg A, Ravikumar V, Mijakovic I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int J Mol Sci. 2018;19(7):1979. doi: 10.3390/ijms19071979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suman SK, Subramanian S, Mukherjee A. Combination radionuclide therapy: a new paradigm. Nucl Med Biol. 2021;98–99:40–58. doi: 10.1016/j.nucmedbio.2021.05.001. [DOI] [PubMed] [Google Scholar]
- Sun X, Huang X, Yan X, Wang Y, Guo J, Jacobson O, et al. Chelator-free 64Cu-integrated gold nanomaterials for positron emission tomography imaging guided photothermal cancer therapy. ACS Nano. 2014;8(8):8438–8446. doi: 10.1021/nn502950t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Suzuki YS, Momose Y, Higashi N, Shigematsu A, Park K-B, Kim YM, et al. Biodistribution and kinetics of Holmium-166-Chitosan complex (DW-166HC) in rats and mice. J Nucl Med. 1998;39(12):2161–2166. [PubMed] [Google Scholar]
- Tafreshi NK, Doligalski ML, Tichacek CJ, Pandya DN, Budzevich MM, El-Haddad G, et al. Development of targeted alpha particle therapy for solid tumors. Molecules. 2019;24(23):4314. doi: 10.3390/molecules24234314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas OS, Weber W. Overcoming physiological barriers to nanoparticle delivery—are we there yet? Front Bioeng Biotechnol. 2019;7:415. doi: 10.3389/fbioe.2019.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler S, Anderson GR. Dissociation of methyl bromide by nuclear isomeric transition of 4.4-hr Br 80m. J Chem Phys. 1960;33(3):850–63. doi: 10.1063/1.1731274. [DOI] [Google Scholar]
- Wong XY, Sena-Torralba A, Álvarez-Diduk R, Muthoosamy K, Merkoçi A. Nanomaterials for nanotheranostics: tuning their properties according to disease needs. ACS Nano. 2020;14(3):2585–2627. doi: 10.1021/acsnano.9b08133. [DOI] [PubMed] [Google Scholar]
- Zeevaart JR, Szücs Z, Takács S, Jarvis NV, Jansen D. Recoil and conversion electron considerations of the 166Dy/166Ho in vivo generator. Radiochim Acta. 2012;100(2):109–113. doi: 10.1524/ract.2011.1841. [DOI] [Google Scholar]
- Zhang Z, Lin M. Fast loading of PEG–SH on CTAB-protected gold nanorods. RSC Adv. 2014;4(34):17760–17767. doi: 10.1039/c3ra48061e. [DOI] [Google Scholar]
- Zhang X, Detering L, Sultan D, Heo GS, Luehmann H, Taylor S, et al. C-X-C chemokine receptor type 4-targeted imaging in glioblastoma multiforme using 64Cu-radiolabeled ultrasmall gold nanoclusters. ACS Appl Bio Mater. 2022;5(1):235–242. doi: 10.1021/acsabm.1c01056. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sultan D, Detering L, Luehmann H, Liu Y. Facile synthesis, pharmacokinetic and systemic clearance evaluation, and positron emission tomography cancer imaging of 64Cu-Au alloy nanoclusters. Nanoscale. 2014;6(22):13501–13509. doi: 10.1039/C4NR04569F. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Zhong X, Li Z, Xia Y. Successive, seed-mediated growth for the synthesis of single-crystal gold nanospheres with uniform diameters controlled in the range of 5–150 nm. Part Part Syst Charact. 2014;31(2):266–273. doi: 10.1002/ppsc.201300256. [DOI] [Google Scholar]
- Zheng B, Wu Q, Jiang Y, Hou M, Zhang P, Liu M, et al. One-pot synthesis of 68Ga-doped ultrasmall gold nanoclusters for PET/CT imaging of tumors. Mater Sci Eng C. 2021;128:112291. doi: 10.1016/j.msec.2021.112291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file1. Theoretical calculation of 166Ho loss due to internal conversion; Fig. S1. Size distribution histogram of DyAu@AuNPs with different Dy:Au feeding ratios; Fig. S2. Size distribution histogram of DyPtAuNPs with different Dy:Au feeding ratios; Fig. S3. Representative picture of 166DyAuNP (Dy:Au=1:3) after 24 h incubation at 37 °C; Fig. S4. Comparison of 166Dy radiolabelling efficiency calculated from Ge-detector data and ICP-OES data; Table S1. Comparison of Au concentration of Au@AuNP, 166DyAu@AuNP, PtAuNP and 166DyPtAuNP
Data Availability Statement
The data associated to this research work are available in this manuscript or in the online supplementary file.