

Summary
The changes that drive differentiation facilitate the emergence of abnormal cells that need to be removed before they contribute to further development or the germline. Consequently, in mouse in the lead-up to gastrulation, ~35% of embryonic cells are eliminated. This elimination is caused by a hypersensitivity to apoptosis, but how it is regulated is poorly understood. Here we show that upon exit of naïve pluripotency, mouse embryonic stem cells lower their mitochondrial apoptotic threshold and this increases their sensitivity to cell death. We demonstrate that this enhanced apoptotic response is induced by a decrease in mitochondrial fission due to a reduction in the activity of Dynamin-related protein 1 (DRP1). Furthermore, we show that in naive pluripotent cells DRP1 prevents apoptosis by promoting mitophagy. In contrast, during differentiation reduced mitophagy levels facilitates apoptosis. Together, these results indicate that during early mammalian development DRP1 regulation of mitophagy determines the apoptotic response.
Graphical Abstract
eTOC
Pernaute et al show that during the early stages of mouse embryo development there is a decrease in the activity of the mechanisms that repair the mitochondria, which is the powerhouse of the cell. This helps eliminate abnormal cells and prevents them from contributing to the new-born organism.
Introduction
The appropriate regulation of apoptosis during embryonic development is essential for maintaining the right balance between the elimination of suboptimal cells and the availability of sufficient cell numbers to sustain embryo growth (Bowling et al., 2019). The maintenance of this balance is most acutely evident in the lead up to gastrulation, when the mouse epiblast not only undergoes the process of germ layer specification, but also displays a significant expansion in cell numbers, from 150 at E5.5 to 15,000 cells at E7.5 (Snow, 1977). This rapid proliferation of cells is concomitant with the substantial cellular changes that accompany gastrulation, most notably extinction of the pluripotency network and activation of differentiation genes in a lineage-specific manner. Any abnormal or mis-specified cell, which cannot perform these changes appropriately, needs to be removed from the embryo to prevent them from contributing to further development or the germline, which is also specified around E6.5 (Leitch et al., 2013). Hence, a wave of cell death takes place in the mouse embryo at E6.5, most likely reflecting the elimination of these abnormal cells (Manova et al., 1998; Spruce et al., 2010).
One way by which the embryo facilitates the elimination of aberrant cells is through lowering the apoptotic threshold as it proceeds from a pre-implantation to an early post-implantation stage of development. Indeed, whilst low doses of UV irradiation do not induce apoptosis in mouse pre-implantation embryos, these same low doses lead to a strong apoptotic response in the post-implantation epiblast (Heyer et al., 2000). Similarly, although cells with a range of genetic defects, including chromosome fragmentation or chromosome mis-segregation survive pre-implantation development, they are efficiently eliminated by apoptosis during early post-implantation development (Bolton et al., 2016; Brown and Baltimore, 2000; Dobles et al., 2000; Kalitsis et al., 2000). Subsequently, it has been proposed that the post-implantation epiblast is primed for death, and therefore hyper-responsive to apoptotic signals (Heyer et al., 2000; Pernaute et al., 2014).
The mitochondrial apoptotic pathway is tightly regulated by a balance between pro- and anti-apoptotic factors belonging to the BCL-2 family. The binding of the anti-apoptotic proteins (e.g BCL-2, BCL-XL, A1 or MCL-1) to their pro-apoptotic BCL-2 family counterparts (e.g. BIM, BID, PUMA, NOXA or BAD) prevents the induction of apoptosis. When this balance is lost, BIM or BID bind to the apoptotic effector molecules BAX and BAK, which induce mitochondrial outer membrane permeabilization (MOMP) and caspase activation (Tait and Green, 2013). We have previously shown that, in the post-implantation epiblast, this balance is at least in part maintained by microRNAs (miRNAs) of the miR-20, miR-92, and miR-302 families. These miRNAs target Bim (Bcl2l11) and maintain it in a state that is poised for activation (Pernaute et al., 2014). However, it is worth noting that mutation of Bim does not prevent the endogenous wave of cell death occurring during early mouse development, suggesting that other factors must be contributing to making the epiblast hypersensitive to death signals (Pernaute et al., 2014).
The balance between mitochondrial fusion and fission, termed mitochondrial dynamics, has emerged over the last few years as an important regulator of the apoptotic response. Mitochondrial dynamics are facilitated by proteins such as Mitofusin 1 (MFN1) and Mitopfusin 2 (MFN2) and Mitochondrial Dynamin Like GTPase (OPA1), that promote fusion and (Dynamin-related protein 1) DRP1 or Mitochondrial Fission Factor (MFF), that promote fission (Chen et al., 2003; Smirnova et al., 2001; Tilokani et al., 2018). During cell death, remodelling of the cristae network regulated by OPA1 (Frezza et al., 2006) and DRP1 (Estaquier and Arnoult, 2007; Otera et al., 2016) is required for release of cytochrome C and subsequent apoptosis. DRP1-induced mitochondrial fragmentation is also coupled to the later stages of apoptosis (Breckenridge et al., 2003; Frank et al., 2001; Karbowski et al., 2002; Montessuit et al., 2010). Finally, MFN1-induced fusion has been shown to promote a mitochondrial size that is permissive for BAX function (Renault et al., 2015). Therefore, the regulators of mitochondrial dynamics play a variety of different roles in the apoptotic process. In the developing embryo, mitochondrial shape undergoes substantial changes around the time of implantation (Lima et al., 2018). During pre-implantation stages mitochondria are round and have a low cristae density (Stern et al., 1971), but later in development mitochondria elongate and cristae density increases (Lima et al., 2018). These observations raise the possibility that changes in mitochondrial dynamics may play roles in regulating the changes in apoptotic threshold that occur during early development.
Here we have identified that a change in mitochondrial dynamics increases the sensitivity to cell death signals in cells of the post-implantation epiblast. For this, we combined experiments in the embryo with studies using embryonic stem cells (ESCs), which capture the naïve pluripotent state found in the pre-implantation epiblast, and in epiblast stem cells (EpiSCs), which resemble the post-implantation epiblast (Nichols and Smith, 2009). We find that although post-implantation epiblast cells show high mitochondrial apoptotic priming, their increased sensitivity to cell death is not due to differential expression of members of the BCL-2 pro- and anti-apoptotic family. Instead, we observed that during the onset of differentiation decreased mitochondrial fission facilitates apoptosis. We demonstrate that mechanistically, this regulation of apoptosis is mediated by increased DRP1 activity in naïve pluripotent cells, which promotes mitophagy. Accordingly, inhibition of mitophagy in naïve pluripotent cells facilitates the response to cell death stimuli. Together, these results demonstrate a key role for DRP1 in determining the apoptotic response of pluripotent cells.
Results
Pluripotent cells become hypersensitive to cell death upon exit of naïve pluripotency.
We set out firstly to characterise the apoptotic sensitivity of the different pluripotent cell types found during early mouse development. Previous studies have shown that whilst cells from the pre-implantation embryo are relatively resistant to low doses of UV radiation, the early post-implantation cells respond to the same low doses of UV by undergoing apoptosis (Heyer et al., 2000). In line with these findings, we observed no overt apoptosis in E3.5 mouse embryos treated with the DNA damage-inducing drug etoposide for 1.5h, but we detected a strong apoptotic response when the etoposide treatment was applied to E6.5 embryos (Supplementary Figure 1A-B).
We next tested whether the observed differential sensitivity to apoptosis is also apparent in ESCs and EpiSCs, the two in vitro counterparts of the pre- and post-implantation epiblast (Nichols and Smith, 2009). We have previously shown that loss of miRNAs leads to an upregulation of BIM expression and a consequent induction of apoptosis in EpiSCs (Pernaute et al., 2014). To test if miRNAs also regulate apoptosis in ESCs, we induced Dicer deletion in Dicerfx/fx ESCs by tamoxifen administration, as we had previously done for EpiSCs (Pernaute et al., 2014). This led to miRNA depletion (Supplementary Figure 1C) and an up-regulation of BIM expression similar to that found in EpiSCs (Figure 1A). However, in contrast to what was seen in EpiSCs, the increase in BIM expression did not cause apoptosis in ESCs (Figure 1B-C and Supplementary Figure 1D). This suggests that mouse ESCs are more resistant to apoptosis than EpiSCs.
Figure 1. The apoptotic threshold decreases upon pluripotent stem cell differentiation.

A. Increase in BIM expression upon Dicer deletion in EpiSCs (day 6 post-deletion) and ESCs (day 7 post-deletion). Representative western blot and western blot quantification relative to α-TUBULIN (TUB.). B. Change in % cells with mitochondrial outer membrane permeabilization (MOMP) upon Dicer deletion in EpiSCs (day 5 post-deletion) and ESCs (day 6 post-deletion) relative to un-deleted cells as measured by DiOC6 staining. C. Cleaved-CASPASE 3 (c-CASP3) immunostaining in ESCs and EpiSCs upon Dicer deletion at day 5 and 6 respectively. Scale bar =100 μm. D. Change in % Annexin V positive cells after 16h treatment with 1μM sodium arsenate (NaAsO2) relative to untreated cells. E. Cleaved-CASPASE 3 levels in ESCs and EpiSCs upon treatment for 16h with increasing concentrations of thapsigargin (Tg). Representative western blot and graph showing average western blot quantification relative to α-TUBULIN. Average of 3 (A, E) or 4 (B, D) experiments +/− SEM is shown. Students T-Test (A-B and D) or 2-way ANOVA with Šidák correction (E) *p<0.05, **p<0.01, ***p<0.001. ns= not significant. MW= Molecular weight. See also Figure S1.
Importantly, we found that the differential apoptotic response of ESCs and EpiSCs was also evident upon exposing these cells to different sources of stress. Annexin V binding to phosphatidylserine residues that are exposed on the cell surface during apoptosis can be used detect and quantify apoptotic cells. Induction of oxidative stress with 1μM sodium arsenate for 16 hours produced a 3-fold increase in Annexin V positive ESCs, but a 7-fold increase in Annexin V positive EpiSCs (Figure 1D). Similarly, induction of endoplasmic reticulum (ER) stress with increasing doses of thapsigargin for 16 hours led to a small change in the basal levels of cleaved Caspase 3 in ESCs, whereas the same treatment induced a robust apoptotic response in EpiSCs (Figure 1E). Overall, these results indicate that EpiSCs exhibit an increased sensitivity to apoptosis when compared to ESCs, thus recapitulating the features observed in their in vivo counterparts.
The mitochondrial apoptotic pathway is primed for cell death in primed pluripotent stem cells.
We next addressed if the differences in apoptotic response between naïve and primed pluripotent cells were reflected at the level of the mitochondrial apoptotic pathway. This pathway is regulated by the relative expression of pro-apoptotic and anti-apoptotic BCL-2 family members (Tait and Green, 2013). This balance can be artificially changed, for example by inhibiting BCL-2/XL activity with BH3 mimetics such as ABT-737. We observed that 24h treatment of EpiSCs with ABT-737 led to a strong increase in the percentage of cells displaying (1) MOMP (Supplementary Figure 1E), (2) Annexin V positivity (Figure 2A) and (3) cleaved caspase-3 expression (Figure 2B). In contrast, treatment with ABT-737 failed to induce apoptosis in ESCs, even at the highest concentration used (Figure 2A-B and Supplementary Figure 1E). These findings are strengthened by the observation that ABT-737 leads to cytochrome C release from the mitochondria in EpiSCs, but not ESCs (Figure 2B). To test the physiological significance of these observations, we treated pre- and post-implantation mouse embryos with 2μM ABT-737 for 1.5h. Similar to what we observed in vitro, we found that this treatment induced apoptosis in epiblast and the overlaying visceral endoderm cells of E6.5 embryos, but not in inner cell mass cells of E3.5 embryos (Figure 2C and Supplementary Figure 1F).
Figure 2. Enhanced activation of the mitochondrial apoptotic pathway in primed pluripotent cells.

Apoptosis quantified as % Annexin V positive ESCs and EpiSCs treated for 24h with increasing concentrations of ABT-737. Fold change versus DMSO treated cells is shown. B. Cleaved-CASPASE 3 (c-CASP3) levels relative to HEXOKINASE 2 (HK2) in ESCs and EpiSCs treated for 24h with 5μM ABT-737. CYTOCHROME C (CYT-C) levels in the cytosolic and membrane fractions in ESCs and EpiSCs treated with 5μM ABT-737 for 24h relative to ERK1/2 and ATP-b. C. Cleaved-CASPASE 3 immunostaining in E3.5 and E6.5 embryos treated with DMSO (E3.5 n= 3; E6.5 n=5) or 2μM ABT-737 (E3.5 n= 3; E6.5 n=5) for 1.5h. Scale bar E3.5=50 μm; E6.5=100μm. D. Fold change in % Annexin V positive EpiSCs transfected with control siRNA or Bim siRNA and treated with DMSO or 5μM ABT-737 for 24h. E. % membrane depolarization in ESCs and EpiSCs treated with either the BIM (10μM), BID (10μM) or control (10μM) peptides for the indicated amounts of time. Average of 3 (A, E) or 4 (B) experiments +/−SEM (D) or +/−SD (E) is shown. (D) Student T-Test or (A) 2-way ANOVA with Šidák correction *p<0.05, **p<0.01 or ****<0.00001. MW= Molecular weight. See also Figure S1.
Next, we determined whether the enhanced sensitivity of EpiSCs to BCL2/BCLXL inhibition was dependent on BIM. For this we transfected previously tested siRNAs targeting Bim (Pernaute et al., 2014) into EpiSCs and treated them with ABT-737. We observed that this was sufficient to suppress the apoptotic response induced by BCL2/BCL-XL inhibition (Figure 2D), further highlighting the importance of BIM for the apoptotic response of EpiSCs. Given that the differences in apoptotic response between ESCs and EpiSCs appear to be mediated by the mitochondrial apoptotic pathway, we measured the level of mitochondrial apoptotic priming of these cells by analysing the kinetics of mitochondrial membrane depolarization induced by BIM and BID BH3 peptides (Certo et al., 2006). We observed that the depolarization of the mitochondrial membrane was significantly more pronounced in EpiSCs than in ESCs for both these peptides (Figure 2E). Together these results indicate that primed pluripotent cells have a lower apoptotic threshold than naïve cells due to enhanced sensitivity of the mitochondrial pathway.
The relative expression of pro- and anti-apoptotic BCL2 family members does not explain the different apoptotic threshold of ESCs and EpiSCs.
To address if differences in the relative expression of pro- and anti- apoptotic BCL2 family members underpin the different apoptotic response of ESCs and EpiSCs, we compared the expression of these proteins in each cell type. We observed no significant difference in the expression of the pro-apoptotic activator proteins PUMA, BIM, BID or BAD in either whole cell or mitochondrial extracts between these two cell types (Figure 3A). Similarly, we did not observe any difference in the expression of the pro-apoptotic effector proteins BAX or BAK in mitochondrial extracts, although the expression of BAX was lower in whole cell extracts from EpiSCs than in those from ESCs (Figure 3B). In contrast, when anti-apoptotic proteins were analysed, we found little difference in the expression of BCL-XL, MCL1 or A1, but significantly higher expression of BCL-2 in EpiSCs, in both whole cell and mitochondrial extracts (Figure 3C). Whilst a higher expression of anti-apoptotic protein expression may seem counterintuitive given the enhanced sensitivity to death of EpiSCs, this elevated BCL-2 expression is likely part of the adaptation of these cells to their low apoptotic threshold (Korsmeyer, 1992).
Figure 3. Expression levels of BCL-2 apoptotic factors in pluripotent cells.

A. Levels of pro-apoptotic factors in whole cell lysate (WCL) and mitochondrial extracts (Mito) of ESCs an EpiSCs (Epi). Graphs show protein level normalised against α-TUBULIN (TUB.) or β-ACTIN in whole cell lysate and ATP-b in mitochondrial extract. B. Levels of apoptosis effector proteins in whole cell lysate and mitochondrial extract of ESCs an EpiSCs. Graphs show protein level normalised against PCNA for the whole cell lysates and ATP-b for the mitochondrial extracts. C. Levels of anti-apoptotic factors in whole cell lysate and mitochondrial extracts of ESCs and EpiSCs. Graphs show protein level normalised against αTUBULIN or βACTIN in whole cell lysate and ATP-b in mitochondrial extract. Average of 4 independent experiments +/−SEM is shown. Students T-Test *p<0.05, **p<0.01, ***p<0.001. Where no significance is indicated it is because no statistically significant differences were observed between ESCs and EpiSCs. MW= Molecular weight. See also Figure S1.
To determine if the balance of expression of pro- and anti-apoptotic factors changes differently in ESCs and EpiSCs upon induction of apoptosis, we analysed the expression of key BCL-2 family members after treating these cells with 5μM ABT-737 for 24 hours. In these experiments we analysed the expression of the anti-apoptotic BCL-2 family members as well as BIM expression, as BIM is required for the apoptotic response to ABT-737 (Figure 2D). However, we observed no significant change in the levels of expression of BCL-2, BCL-XL, A1, MCL-1 or BIM between ABT-737 treated samples and controls in either whole cell or mitochondrial extracts of ESCs or EpiSCs (Supplementary Figure 1G-H). This suggests that the induction of apoptosis does not significantly shift the balance of pro- and anti-apoptotic BCL-2 family expression. Together, these results indicate that the relative expression of BCL-2 family proteins is not the primary cause of the higher sensitivity to apoptosis of primed pluripotent cells.
High levels of mitochondrial fission are observed in naïve pluripotent cells when compared to primed cells.
Mitochondrial dynamics have been suggested as a mechanism that contributes to the regulation of the mitochondrial apoptotic threshold (Prudent and McBride, 2017). The mitochondria of ESCs and pre-implantation embryos have been shown to be rounded (Stern et al., 1971) and to elongate upon differentiation (Shepard et al., 2000; Zhou et al., 2012). Analysis of mitochondrial morphology in ESCs revealed rounded doughnut shaped mitochondria in agreement with previous studies (Zhou et al., 2012). In contrast, the mitochondria of EpiSCs were more elongated (Figure 4A). Importantly, these differences were also seen in vivo, with E3.5 inner cell mass cells and E4.5 epiblast cells having rounded mitochondria and the mitochondria of E6.5 epiblast cells being elongated and forming networks (Figure 4B-C).
Figure 4. Mitochondria fuse to form complex networks upon differentiation.

A. ATP-b immunostaining showing mitochondrial morphology in ESCs and EpiSCs. Scale bar=10 μm. B. ATP-b immunostaining showing mitochondrial morphology in E3.5, E4.5 and E6.5 mouse embryos. 2x magnification over ICM/Epiblast area. Scale bar=5 μm. C. Quantification of the mitochondria circularity and perimeter of E3.5, E4.5 and E6.5 embryos. D. Basal levels of mitochondrial fusion and fission proteins in ESC and EpiSCs. E. Quantification of (D). Protein levels are normalised against α-TUBULIN (TUB.). The average from 3 independent experiments +/−SEM is shown. 2-way ANOVA with Šidák correction **p<0.01, ***p<0.001 or ****<0.00001. MW= Molecular weight. See also Figure S2.
The differences in mitochondrial shape between naïve and primed pluripotent cells suggest that these cell types have different mitochondrial dynamics. We therefore studied the expression of fusion and fission regulators in ESCs and EpiSCs. While we observed no difference in the expression of the fusion regulators MFN1, MFN2 or OPA1 between these cell types, we noticed that ESCs had significantly higher levels of total DRP1 and p-DRP1 (S616), a phosphorylation form that induces fission (Knott et al., 2008) (Figure 4D-E). Similarly, in E4.5 embryos p-DRP1 was robustly expressed in the epiblast cells, but by E5.5 this expression was restricted to dividing cells (Supplementary Figure 2A), where it is required to segregate the mitochondria during mitosis (Tilokani et al., 2018). This indicates that naïve pluripotent cells have higher fission activity. To determine if the higher p-DRP1 expression is responsible for the rounded mitochondrial shape of ESCs, we knocked-out Drp1 by CRISPR-Cas9. Although Drp1−/− ESCs remained pluripotent, their mitochondria became elongated and hyperfused (Figure 5A-C and Supplementary Figure 3A-C). These results indicate that DRP1-induced mitochondrial fission is required for the fragmented mitochondrial shape observed in ESCs.
Figure 5. Drp1 deletion facilitates early apoptotic events.

A. ATP-b and NANOG immunostaining showing mitochondrial morphology in wild type and Drp1−/− ESCs. Scale bar=5 μm. B. Quantitative RT-PCR showing gene expression levels of naïve and primed pluripotency markers in wild-type and Drp1−/− ESCs. Drp1−/− 1, 2 and 3 represent three independent mutant lines. Gene expression normalised against Gapdh. C. Western blot analysis for OCT4, SOX2 and NANOG expression wild-type and Drp1−/− ESCs in pluripotency culture conditions and after they are induced to differentiate for 3 days. D. % of cells with MOMP detected by TMRM staining in wild-type and Drp1−/− ESCs untreated or treated with 1μM sodium arsenate (NaAsO2) for 16h or E. 1μM thapsigargin (Tg) for 16h. F. % cells with MOMP in ESCs and EpiSCs treated with either BIM (0.5μM), BID (2.5μM) or control (1μM) peptides for the indicated amounts of time. Average of 3 (B, C), 4 (D) or 7 (E) independent experiments +/−SEM (C, D) or +/−SD (E) is shown. 2-way ANOVA with Šidák correction *p<0.05, **p<0.01 or ****<0.00001. Where no significance is indicated it is because no statistically significant difference was observed. MW= Molecular weight. See also Figure S3.
DRP1 plays a key role in determining the apoptotic threshold of naïve and primed pluripotent cells.
To evaluate whether DRP1 levels determine the apoptotic threshold of pluripotent cells we first analysed the effects of loss of Drp1. In the first instance we compared the apoptotic response of wild-type and Drp1−/− ESCs to ER stress or oxidative stress. We observed that a significantly higher proportion of Drp1−/− cells treated with thapsigargin or sodium arsenite show MOMP (Figure 5D-E). We next analysed the kinetics of MOMP in response to BH3 peptides and observed that both BIM and BID BH3 peptides were more efficient at inducing mitochondrial outer membrane depolarization in Drp1−/− ESCs than wild-type cells (Figure 5F). This indicates that loss of Drp1 is sufficient to lower the apoptotic threshold of ESCs. Importantly, this change in sensitivity to cell death induction was unlikely to be a secondary consequence of a disruption of the metabolism of mutant cells, as Drp1−/− ESCs did not show decreased glycolysis or oxidative phosphorylation rates when compared to wild-type cells (Supplementary Figure 3D-G).
DRP1 has been shown to be involved in the remodelling of the mitochondrial cristae and the subsequent release of cytochrome C into the cytoplasm (Estaquier and Arnoult, 2007; Otera et al., 2016), an event downstream of MOMP. We therefore analysed if cytochrome C release is compromised in Drp1−/− ESCs. We observed that Drp1−/− ESCs displayed lower levels of cytoplasmic cytochrome C upon thapsigargin treatment (Supplementary Figure 3H). This suggests that DRP1 is likely to play at least two roles in the apoptotic response in pluripotent cells. First, it reduces the effect of the pro-apoptotic roles of BCL-2 family members and second it promotes cytochrome C release by helping the remodelling of the mitochondrial cristae.
Given the roles of DRP1 in determining the apoptotic response of ESCs, we next wanted to analyse its importance during ESCs differentiation. During differentiation p-DRP1 levels decrease (Figure 4D-E and Supplementary Figure 2) and for this reason we analysed the effects of Drp1 overexpression (Drp1OE) in differentiating ESCs. We have previously shown that culturing ESCs for 3 days in N2B27 leads to a post-implantation epiblast-like state (Sancho et al., 2013). We therefore cultured Drp1OE cells for 3 days in N2B27 and found that this led to sustained DRP1 expression and increase in p-DRP1 levels (Supplementary Figure 4A-D). Importantly, this increase in p-DRP1 did not prevent exit of naïve pluripotency but induced mitochondria to adopt a more rounded morphology (Figure 6A-C and Supplementary Figure 4D-E). We therefore tested the effect on the apoptotic response of a sustained increase in p-DRP1 during the onset of ESC differentiation. We found that after 3 days in differentiation conditions, Drp1OE cells displayed not only lower basal cleaved caspase 3 levels (Figure 6D and Supplementary Figure 4D), but also significantly reduced levels of cleaved Caspase 3 in response to treatment with thapsigargin, sodium arsenate or ABT-737 (Figure 6E-G and Supplementary Figure 4D, F, G). Together with our findings in Drp1−/− ESCs, these results suggest that the decrease in DRP1 and p-DRP1 levels observed during exit of pluripotency play an important role in sensitising cells to apoptosis.
Figure 6. DRP1 over-expression inhibits the apoptotic response during the onset of pluripotent stem cell differentiation.

A. ATP-b immunostaining showing mitochondrial morphology in wild-type and Drp1OE cells during differentiation. Scale bar =10μm. B. Circularity measurement of mitochondrial particles from ATP-b immunostained images of wild-type and Drp1OE cells at day 3 of differentiation. C. Quantitative RT-PCR showing gene expression levels of naïve and primed pluripotency markers in wild-type and Drp1OE ESCs cultured in pluripotency (Plurip.) or differentiation (Diff.) conditions. Gene expression normalised against Gapdh. D. Fold change in basal cleaved-CASPASE 3 levels in wild-type and Drp1OE cells at day 3 of differentiation, quantified from Supplementary Figure 4D, F and G. E. Fold change in cleaved-CASPASE 3 levels in wild-type and Drp1OE cells untreated or treated with 1μM thapsigargin (Tg) for 5h. Quantified from Supplementary Figure 4D. F. Fold change in cleaved-CASPASE 3 levels in wild-type and Drp1OE cells untreated or treated with 1μM sodium arsenate (NaAsO2) for 5h. Quantified from Supplementary Figure 4F. G. Fold change in cleaved-CASPASE 3 levels in wild type and Drp1OE cells untreated or treated with 1μM ABT-737 for 5h at day 3 of differentiation. Quantified from Supplementary Figure 4G. Protein levels in D. E. F. and G. are against α-TUBULIN and graphs show protein expression levels relative to wild-type cells. normalised Average of 3 (C, E, F) or 5 (D) independent experiments +/− SEM is shown. Students T-Test (D) or 2-way ANOVA with Šidák correction (E, F, G) ****p<0.0001. The statistical comparisons are made to untreated cells. Drp1OE 1 and 2 represent two independent DrplOE lines. See also Figure S4.
DRP1 regulation of mitophagy levels determines the apoptotic response of naïve pluripotent stem cells.
Mitochondrial fission is required for efficient mitophagy and inhibition of Drp1 has been shown to impair the execution of mitophagy (Cho et al., 2019; Gomes et al., 2011; Rambold et al., 2011). Given that a slow clearance of defective mitochondria could cause an enhanced apoptotic response, we first analysed if DRP1 regulates mitophagy levels in response to apoptotic stimuli in naïve pluripotent cells. The recruitment of the E3 ubiquitin ligase PARKIN to the mitochondria is a key step in mitophagy (Narendra et al., 2008). We analysed the localization of a GFP-PARKIN fusion protein (Trempe et al., 2013) in wild-type and Drp1−/− ESCs in normal conditions and after treatment with CCCP, that induces mitochondrial membrane depolarization and activates mitophagy, or treatment with thapsigargin. We observed that Drp1 mutant ESCs displayed significantly reduced mitochondrial GFP-PARKIN levels compared to wild-type cells (Supplementary Figure 5). A later step in mitophagy is the co-localization of mitochondria and lysosomes, as this reflects the targeting of mitochondria for degradation. When this co-localization was measured using lysotracker together with TOMM20 immunostaining we observed that there was a significant increase in the localization of the mitochondria to the lysosome after thapsigargin treatment in wild-type ESCs but not in Drp1 mutant cells (Figure 7A-B). Together these results suggest that DRP1 is regulating mitophagy in naïve pluripotent cells.
Figure 7. Mitophagy regulates the apoptotic response of pluripotent stem cells.

A. Immunostaining showing mitochondrial (TOMM20 and ATPb) and lysosomal (LysoTracker) co-localization in wild-type (WT) and Drp1 mutant cells cultured in pluripotency conditions and wild-type cells cultured in differentiation conditions and then left untreated or treated with 1μM thapsigargin for 5h. Scale bar=10 μm. B. Quantification of co-localization area of lysotracker and TOMM20 from whole images taken from (A). C. Immunostaining showing P62 expression in wild-type and Drp1 mutant embryos at E4.5. Epi denotes epiblast localization. Scale bar =10μm. D. TUNEL staining of wild-type and Drp1 mutant embryos at E5.5. Scale bar =10μm. E. Quantification of P62 expression seen in (C) n8 +/+; 8 +/− and 7 −/− embryos . F. Quantification of TUNEL positive foci over the embryonic region area seen in (D) n=4 +/+; 7 +/− and 3 −/− embryos. G. Fold change in basal cleaved-CASPASE 3 levels in wild-type cells cultured in pluripotency or differentiation conditions and then left untreated or treated with 1μM thapsigargin (Tg) +/− 10nM bafilomycin (Baf.) for 5h. This is a quantification of Supplementary Figure 7C. Protein levels are normalised against αTUBULIN and the graph shows protein expression levels relative to wild-type cells. (A-C and G). Average of 3 independent experiments +/− SEM is shown. 2-way ANOVA with Šidák correction. For (E) and (F) a Tuckey’s correction was used. *p<0.05, **p<0.01, ***p<0.001 or ****p<0.0001. The statistical comparisons are made to untreated cells. See also Figure S5, Figure S6 and Figure S7.
We next analysed if the lower levels of mitophagy observed in Drp1−/− ESCs and in wild-type ESCs induced to differentiate led to an accumulation of damaged mitochondria. It has been previously shown that in the absence of DRP1 mitophagy is decreased leading to an accumulation of damaged and ubiquitinated proteins as well as to an accumulation of P62 (Kageyama et al., 2014; Kageyama et al., 2012; Yamada et al., 2016; Yamada et al., 2018a; Yamada et al., 2018b). In agreement with these observations we observed that Drp1 mutant ESCs showed accumulation of P62 at the mitochondria (Supplementary Figure 6A-B). We next analysed how this affected mitochondrial health by studying mitochondrial membrane potential with TMRM. Again, we found that Drp1−/− ESCs had lower mitochondrial membrane potential than control cells (Supplementary Figure 6C-D). These results suggest that the lower mitophagy of Drp1 mutant cells leads to an increase in damaged mitochondria.
Given that DRP1 levels decrease during the onset of differentiation, we also analysed if mitophagy levels also change in the transition from naïve to primed pluripotency. We first treated ESCs and cells cultured for 3 days in N2B27 with CCCP, and measured the levels of the mitochondrial DNA encoded protein mt-CO1 to establish the rate of mitochondrial clearance. We found that while both low and high doses of CCCP reduced mt-CO1 levels in ESCs, only the highest concentration had this effect in cells cultured in N2B27 (Supplementary Figure 6E-F). To further test if mitophagy levels are different between naïve and primed pluripotent cells we analysed GFP-PARKIN localization in ESCs and in cells cultured in N2B27 for 3 days. We observed that both in the basal condition as well as when cells were treated with CCCP or thapsigargin, the mitochondrial localization of GFP-PARKIN was higher in cells cultured in ESC conditions than in cells induced to differentiate in N2B27 (Supplementary Figure 5). Similarly, we also observed that thapsigargin treatment led to higher levels of mitochondria localization to the lysosomes in naïve pluripotency culture conditions than during differentiation (Figure 7A-B and Supplementary Figure 6G). This indicates that as occurs with DRP1 activity, mitophagy also decreases upon differentiation. To analyse the consequences of this lower mitophagy we studied the mitochondrial localization of P62 and the mitochondrial membrane potential of cells cultured in pluripotency conditions compared with those induced to differentiate. We found that differentiating cells showed lower mitochondrial membrane potential (Supplementary Figure 6C-D), lower mitophagy levels (Supplementary Figure 6E-F) and an accumulation of P62 in the mitochondria (Supplementary Figure 6A-B). These observations support that upon exit of naïve pluripotency there is a less efficient clear-up of defective mitochondria.
We have shown that compared to Drp1−/− ESCs or differentiated cells, wild-type ESCs show lower levels of mitochondrial P62 and there is a lower proportion of cells with reduced mitochondrial membrane potential. In addition, higher mitochondria/lysosome and GFP-PARKIN/mitochondria co-localization is seen in wild-type ESCs after thapsigargin treatment, when compared to Drp1 mutant cells or differentiated cells. Together, these results suggest that high expression of DRP1 in ESCs allows for a more efficient clearing of dysfunctional mitochondria in these cells.
To further explore the role of DRP1 in regulating mitophagy and the cell death response of pluripotent cells we analysed Drp1 mutant embryos (Wakabayashi et al., 2009). We found that at E4.5 Drp1−/− embryos exhibited an accumulation in P62 protein levels (Figure 7C and E), pointing to impaired clearing of damaged proteins in the absence of DRP1. Furthermore, when we examined cell death we found that by E5.5 Drp1−/− embryos displayed significantly higher apoptosis levels (Figure 7D and F). These results suggest that DRP1 is required to clear damage mitochondrial proteins in naïve pluripotent cells and that the failure to do so sensitizes embryonic cells to cell death.
Finally, to study the relationship between mitophagy and the apoptotic response, we analysed how inhibition of mitophagy affects the apoptotic response of pluripotent cells. First ESCs cultured in conditions that promote naive pluripotency or cultured for 3 days in N2B27 were left untreated or treated with thapsigargin. Bafilomycin was used to block the autophagic clearance of mitochondria in these cells. We observed that whilst bafilomycin increased the apoptotic response in naive pluripotency conditions, it had no effect on those cells cultured in N2B27 (Figure 7G and Supplementary Figure 7A). We also tested the effect of mutating Atg5, a critical component of the autophagy pathway (Qu et al., 2007). For this we compared the apoptotic response of Atg5−/− ESCs cultured in naïve pluripotency conditions to their response when cultured for 3 days in N2B27. We found that whilst Atg5−/− cells displayed an enhanced apoptotic response in ESC media, they showed lower apoptotic levels than control cells in N2B27 (Supplementary Figure 7B-C). Together these results indicate that the autophagic clearance of defective mitochondria is promoting cell survival in naïve pluripotent cells. In contrast to this, the observation that inhibiting autophagy in cells induced to differentiate has little effect on their apoptotic response, suggests that the low mitophagy levels found in primed pluripotent cells likely accounts for the enhanced response to cell death stimuli of these cells.
Discussion
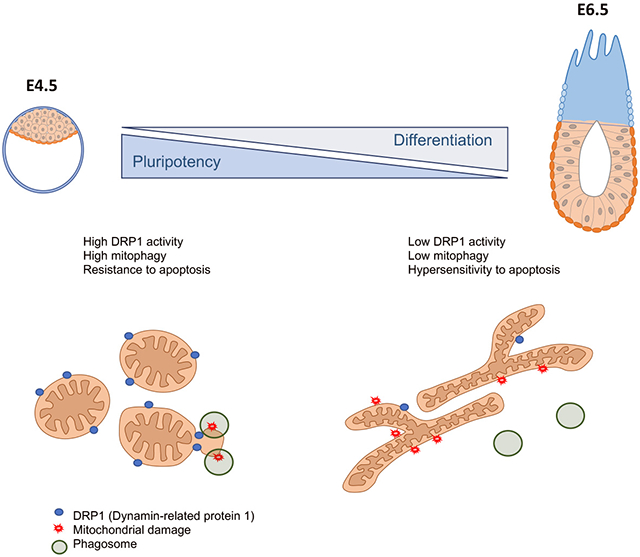
The removal of aberrant cells during development is important to prevent them from contributing to further development and the germline. To facilitate this elimination cells become hypersensitive to apoptosis during the onset of differentiation (Heyer et al., 2000; Laurent and Blasi, 2015; Liu et al., 2013; Pernaute et al., 2014). Here we show that the regulator of mitochondrial fission DRP1 regulates apoptotic priming in these cells via influencing mitochondrial dynamics. We show that upon exit of naïve pluripotency mitochondria form complex networks due to a decrease in mitochondrial fission induced by a loss of DRP1 phosphorylation. We also demonstrate that this loss of DRP1 activity changes the apoptotic threshold, as deletion of Drp1 facilitates the early stages of apoptosis and its over-expression protects against apoptosis. Together, these findings highlight the pivotal role that DRP1 plays in modulating the response to cell death during differentiation.
Our work has also uncovered one mechanism by which DRP1 affects the apoptotic response of pluripotent cells. We find not only that DRP1 is required for mitophagy in these cells, but also that concomitant with the decrease in DRP1 activity that occurs during the onset differentiation, there is a paralleled decrease in mitophagy levels. This conclusion is supported by a number of lines of evidence. For example, we observe low levels of PARKIN recruitment to the mitochondria and low levels of mitochondria/lysosome co-localization in cells cultured in N2B27 and in Drp1 mutant cells treated with thapsigargin compared to cells cultured in ESC media. We also find that during differentiation or after Drp1 mutation there is an increase in the proportion of cells showing P62 accumulation in the mitochondria and low mitochondrial membrane potential. This indicates that upon exit of naïve pluripotency there is a less efficient clearance of defective mitochondria due to the decrease in DRP1 activity. Our finding that DRP1 is promoting mitophagy in pluripotent cells is consistent with the observations made in several other tissues (Gomes et al., 2011; Kageyama et al., 2014; Rambold et al., 2011; Tanaka et al., 2010; Twig et al., 2008). In contrast to this, other reports find that Drp1 is either not required for mitophagy (Mendl et al., 2011; Song et al., 2015; Yamashita et al., 2016) or that Drp1 mutant cells display constitutive recruitment of PARKIN to the mitochondria and increased mitophagy (Burman et al., 2017). Thus, it is likely that the role of DRP1 in mitophagy is cell type/context dependant and that additional factors determine if it promotes or inhibits mitophagy.
Here we provide evidence for a role of DRP1 in promoting mitophagy that fits with the notion that DRP1-dependent fission serves as a mitochondrial quality surveillance system by providing selective pressure for eliminating “bad sectors” of the mitochondrial network (Cho et al., 2019; Twig et al., 2008). Furthermore, our data indicates that the levels of mitophagy that are set by DRP1 activity determine the apoptotic response of pluripotent cells, therefore providing a physiological consequence for this surveillance system. We show that upon exit from naïve pluripotency the decrease in mitophagy levels caused by the lowering of DRP1 activity contribute to sensitising the cells to apoptosis, allowing the elimination of cells that may have accumulated defects during the onset of differentiation.
Regarding the mechanism by which DRP1 could promote PARKIN recruitment to the mitochondria, we speculate that DRP1 may regulates mitophagy at multiple points. As well as by ensuring the fission of damaged parts of the mitochondria, it is possible that DRP1 also has roles upstream of this step. DRP1 has been shown to interact with ZIP1, a Zn2+ transporter, and this interaction has been demonstrated to be important for PARKIN recruitment to the mitochondria (Cho et al., 2019). As increased mitochondrial Zn2+ has also been shown to promote PINK1/PARKIN mitophagy (Bian et al., 2018), we propose that the interaction of DRP1 and ZIP1 promotes the influx of Zn2+ into the mitochondria and this in turn promotes PINK1/PARKIN recruitment (Cho et al., 2019).
Our observations do not preclude other roles for mitochondrial fission in setting the apoptotic threshold. For example fission would promote a rounded mitochondrial shape, that has been found to be refractory to BAX permeabilization of the outer mitochondrial membrane (Renault et al., 2015). It could also determine the degree of contact between the mitochondria and endoplasmic reticulum (Csordas et al., 2018; Prudent and McBride, 2017) and in this affect cell death by modulating the entry of calcium into the mitochondria.
Our results contrast with the roles that others have identified for mitochondrial dynamics in pluripotency. For example over-expression of Mff, a fission regulator, inhibits the expression of neural markers during pluripotent stem cell differentiation (Zhong et al., 2019), suggesting that inhibiting mitochondrial elongation disrupts differentiation. Similarly, mutation of Mtch2, a potential fusion regulator, delays exit from naïve pluripotency (Bahat et al., 2018), leading to the suggestion that mitochondrial fusion promotes differentiation. In contrast to this we find that Drp1 null ESCs, which have hyper-fused mitochondria, show normal pluripotency gene expression and over-expression of Drp1 does not affect exit from naïve pluripotency. What these results suggest, is that mitochondrial elongation per se is not linked to exit of pluripotency, but rather it is likely that other mitochondrial processes regulated by Mff and Mtch2 have an impact on the onset of differentiation.
Finally, our work has implications that transcend early mammalian development as DRP1 has been shown to play roles in tumour progression. For example, in glioblastoma DRP1 activation is correlated with poor prognosis. Mechanistically this is explained as brain tumour initiating cells have fragmented mitochondria and require high p-DRP1 levels for their survival (Xie et al., 2015). Similarly, in pancreatic ductal adenocarcinomas, oncogenic Ras mutations induces mitochondrial fragmentation. Reversion of this phenotype by knock-down of Drp1 inhibits tumour growth (Kashatus et al., 2015; Nagdas et al., 2019), further highlighting the potential importance of DRP1 for tumour progression. Our findings that in embryonic stem cells DRP1 promotes cell survival, raises the possibility that part of this oncogenic role is through the regulation of the apoptotic response. Understanding the players downstream of DRP1 will therefore likely open new avenues for our understanding of transformation.
Limitations of the Study
In the present study, mitophagy was analysed by studying GFP-PARKIN localization to the mitochondria or colocalization of mitochondrial markers (TOMM20 or ATPb) with the lysosomal marker lysotracker. A more comprehensive analysis of mitophagy could involve using the mtKeima mitophagy reporter and inhibiting the activity of PARKIN by Crispr/Cas9 or siRNA approaches.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and reagents’ requests should be addressed to the lead contact, Tristan Rodriguez (tristan.rodriguez@imperial.ac.uk).
Materials availability
All unique materials generated in this paper are available upon request to the lead contact without restriction.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
There is no original code reported in this paper.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Stem Cell culture
All cells were cultured at 37°C in an atmosphere with 5% CO2. Reagents used for tissue culture were obtained from Invitrogen unless otherwise stated. Mouse embryonic stem cells (ESCs) were cultured on 0.1% gelatin-coated flasks (Nunc, Thermo Fisher) in GMEM containing with 10% (v/v) foetal calf serum (FCS; Seralab), 1X non-essential amino acids, 2 mM L-glutamine, 0.1 mM β-mercaptoethanol and supplemented with homemade leukaemia inhibitory factor (1:500, LIF). ESCs were routinely dissociated with trypsin and cryopreserved in 10%DMSO in FCS. Atg5−/− ESCs (Qu et al., 2007), Dicerfx/fx ESCs and EpiSCs (Nesterova et al., 2008; Pernaute et al., 2014), E14 ESCs (a kind gift from Prof. Austin Smith, Univ. Exeter) and Drp1−/− ESCs (this study) were used.
Epiblast Stem Cells (EpiSCs) were cultured on FCS coated dishes in N2B27 medium (100mL DMEM F12, 100mL Neurobasal, 1mL N2, 2mL B27 without retinoic acid, 2mM L-Glutamine, 50mM -mercaptoethanol) containing 20μg/ml Activin A (R&D Systems) and 12ng/ml bFGF (R&D Systems). Cells were passaged by mechanical disruption as previously described (Brons et al., 2007). E14 EpiSC were derived from E14 mESCs as previously described (Guo et al., 2009). All experiments were performed using cells in passage between 20 and 30.
Mice husbandry and embryo collection
Mice were maintained and treated in accordance with the Home Office’s Animals (Scientific Procedures) Act 1986 and covered by the Home Office project license PBBEBDCDA. All mice were housed on a 10 hr-14 hr light-dark cycle with access to water and food ad libitum. Mattings were generally set up in the afternoon. Noon of the day of finding a vaginal plug was designated embryonic day 0.5 (E0.5). Embryo dissection was performed at appropriate timepoints in M2 media (Sigma), using Dumont No.5 forceps (11251-10, FST). No distinction was made between male and female embryos during the analysis. Drp1 mutant embryos were generated by crossing Drp1Fx/Fx conditional animals (Wakabayashi et al., 2009) to Sox2-Cre mice (Hayashi et al., 2002).
METHOD DETAILS
Stem cell differentiation and treatments
To induce ESCs differentiation cells were seeded onto plates coated with fibronectin (Merk) and cultured in N2B27 media (Neurobasal media; DMEM F12 media, 0.5 x B27 supplement; 0.5 x N2 supplement; 0.1mM 2-mercaptoetanol; 2mM glutamine; all Thermo Fisher Scientific) during 3 days to allow differentiation.
To induce Dicer deletion Dicerfx/fx ESCs and EpiSCs (Nesterova et al., 2008; Pernaute et al., 2014) were cultured in the presence of 0.3 mM 4-OH-Tamoxifen for three days and left untreated from the third day onwards as previously described (Pernaute et al., 2014). Atg5−/− ESCs have been previously described (Qu et al., 2007).
For BCL2/BCL-XL inhibition ABT-737 (Selleck Chemicals) was added to the media for 24 h at the stated concentration. Oxidative stress and ER stress were induced by adding sodium arsenite (Sigma Aldrich) or Thapsigargin (Sigma Aldrich) for 16 h at the stated concentrations. Bafilomycin (Sigma) was used at a 10nM concentration. To induce Bim knockdown, a previously tested Bim siRNA (Mm_Bcl2l11_2 FlexiTube siRNA, Qiagen) (Pernaute et al., 2014) was transfected into EpiSCs at a final concentration of 75nM using HiPerFect transfection reagent (Qiagen) according to manufacturers’ instructions. Transfection of Flexi-Tube Negative Control siRNA (Qiagen) at a final concentration of 75nM was used as negative control.
Flow cytometry analysis: Annexin V staining and MMP measurement
For apoptosis detection by flow cytometry, Annexin V-APC (Thermo Fisher Scientific) was used in combination with Propidium Iodide (Sigma) according to manufacturer’s instructions. Briefly, approximately 2x105 cells were stained in 100μl of Annexin V Binding Buffer (0.1% BSA in 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH7.4) containing APC conjugated Annexin V for 15 minutes in the dark, after which 0.1 mg/ml propidium iodide was added and the samples immediately analysed by flow cytometry. Loss of mitochondrial membrane potential was measured using the fluorescent dyes DiOC6 or TMRM. Briefly, 2x105 cells were re-suspended in PBS containing 40 nM DiOC6 (Sigma) or 100nM TMRM (T668, ThermoFisher Scientific), incubated for 15 min at 37°C and analyzed by flow cytometry. Data was acquired with a BD LSRII cytometer and analyzed with the FlowJo software (BD).
Stem cells immunofluorescence
For immunostaining ESCs and EpiSCs were fixed for 10min in 4%PFA at room temperature, permeabilised in 0,4% Triton-X100/PBS for 5 minutes at room temperature, blocked in 10%BSA/ 0,1% Triton X-100/PBS and incubated overnight at 4°C in primary antibody diluted in 1%BSA/0,1%Triton X-100 (anti-Cleaved Caspase 3 Asp175, Cell Signalling, 1:100; anti-NANOG (eBioscience, 1:100), anti ATP-b (Abcam, 1:200), anti-P62 (BD, 1:100). Alexa-Fluor conjugated secondary antibodies (Thermo Fisher Scientific, 1:600) were used at 1/500 dilution in 1%BSA/0,1%Triton X-100. Cells were mounted for visualization in Vectashield with DAPI (Vector Laboratories). Images were acquired with a Zeiss confocal microscope and analysed with the Fiji software (Schindelin et al., 2012).
Mitochondrial Staining
Cells were washed with PBS and fixed with 3.7% formaldehyde diluted in serum free media for 15 min at 37°C, 5% CO2. Cells were washed two times with PBS and incubated with pre-cooled acetone for 5 min at −20°C. Cells were washed two times with PBS and incubated with blocking/permeabilization (5% BSA, 0.4% Triton-X in PBS) solution for 30 min at RT before incubating with the primary antibody O/N at 4°C. Excess antibody was removed and cells washed three times in PBS, then incubated with the secondary antibody for 45 min at RT. Before mounting with Vectashield with DAPI, secondary antibody was removed and cell washed again three times in PBS.
Images were acquired in a LSM Z800 Confocal microscope and processed with Fiji. Images for deconvolution were acquired with the same microscope and further processed with the software Huygens. Deconvolution analysis was performed with the support and advice from Mr. Stephen Rothery from the Facility for Imaging by Light Microscopy (FILM) at Imperial College London.
Mitochondria circularity measurements were done with a plugin from Fiji that calculates object circularity according to the formula circularity=4π(area/perimeter2). A circularity value of 1.0 indicates a perfect circle. As the value approaches 0.0, it indicates an increasingly elongated polygon. The calculations were done on ATP-b immunostained images.
To generate cells transiently expressing GFP-PARKIN, ESCs were transfected with 2ug of a pEGFP-Parkin plasmid (pEGFP-PARKIN WT was a gift from Edward Fon -Addgene plasmid # 45875) using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. GFP-PARKIN was detected by direct GFP fluororescence. For lysosome staining cells were treated for 4h with 60nM LysoTracker Red DND-99 (Thermo Fisher Scientific). Co-localization of mitochondria and lysosomes was measured using Fiji as the area shared by lysotracker and TOMM20 in square pixels (px^2) divided by the total area occupied by both proteins. P62 mitochondrial accumulation was measured using Fiji as the integrated density of P62 staining where it co-localizes with TOMM20 staining. For both types of quantifications several images were quantified in 3 independent experiments.
Western blot analysis
Western blot was performed according to established protocols described elsewhere (Pernaute et al., 2014). Briefly, protein samples were collected in Laemmli buffer and denatured for 10 minutes at 95°C. All samples were run in polyacrylamide gels and transferred to nitrocellulose membranes. Blocking was performed in 5% milk in TBST buffer and primary antibody incubation was done overnight at 4°C in TBST containing 5% BSA. The following antibodies were used at the stated concentration: anti-BIM/BOD (Enzo, 1:1000), anti-PUMA (Abcam, 1:1000), anti- BID (Cell Signaling, 1:1000), anti- BAD (Santa Cruz Biotechnology, 1:500), anti-BAX N20 (Santa Cruz Biotechnology, 1:1000), anti-BCL2 (BioLegend, 1:500), anti-BCL-XL (Santa Cruz Biotechnology, 1:1000), anti- MCL1 (Rockland, 1:10000), anti-Bcl2A1 (R&D Systems, 1:500), anti-HEXOKINASE II (Cell Signaling, 1:1000), anti-ATP-b (Abcam, 1:1000), anti-CYTOCHROME C (BD Pharmigen, 1:1000), anti-ERK1/2 (Sigma, 1:20000), anti-Cleaved CASPASE 3 Asp175 (Cell Signaling, 1:000), anti-alpha TUBULIN (Cell Signaling, 1:2000), anti- beta ACTIN (Santa Cruz Biotechnology, 1:1000), anti-BAK (Millipore, 1:1000) anti-OCT3/4 (Santa Cruz Biotechnology, 1:1000), anti-NANOG (eBiosciences, 1:1000), anti-TOM20 (Santa Cruz Biotechnologies, 1:1000), anti-DRP11 (Cell Signalling, 1:1000) anti- pDRP1 (S616) (Cell Signaling 1:1000), anti-MFN1 (Abcam, 1:1000), anti-MFN2 (Abcam, 1:1000), anti-OPA1 (BD, 1:1000), anti-mt-CO1 (Abcam, 1:1000), anti-VINCULIN (Sigma, 1:100), anti-PCNA (Abcam, 1:5000), anti-SOX2 (Abcam, 1:1000). Western blot quantification was performed using the Fiji software (Schindelin et al., 2012).
Mitochondria purification and cytochrome C release assay
For mitochondria purification cells were trypsinized and washed twice in 10 packed cell volumes of 1mM TrisHCl pH 7.4, 0.13M NaCl, 5mM KCl, 7.5 mM MgCl2 followed by centrifugation at 370g for 10 minutes. After the second wash pelleted cells were re-suspended in 6 packed cell volumes of homogenization buffer (10mM TrisHCl pH6.7, 10mM KCl, 0.15 mM MgCl2, 1mM PMSF, 1mM DTT) and incubated for 10 minutes on ice. Cells were homogenized in a glass homogenizer until achieving approximately 60% cell breakage. Homogenate was poured into a tube containing 1 packed cell volume of 2M sucrose solution, mixed and centrifuged at 1200g for 5 minutes to pellet unbroken cells, nuclei and debris. This treatment was repeated twice discarding the pellet, followed by 10 minutes centrifugation at 7000g in order to pellet the mitochondria. Mitochondrial pellet was re-suspended in 3 packed cell volumes of mitochondrial suspension buffer (10mM TrisHCl pH6.7, 0.15mM MgCl2, 0.25% sucrose, 1mM PMSF, 1mM DTT) and centrifuged for 5 minutes at 9500g. Final mitochondrial pellet was re-suspended in 1x Laemmli Buffer and boiled at 95°C for 10 minutes for western blot analysis.
For the separation of cytosolic and membrane fractions in order to investigate cytochrome C release, cells were washed twice with ice cold PBS and incubated for 10 minutes rocking on ice in Digitonin Buffer (20mM HEPES/KOH pH7.5, 100mM sucrose, 2.5mM MgCl2, 100mM KCL, 1mM DTT, 0.0025% digitonin) supplemented with Complete protease inhibitors (Roche). The supernatant was collected as cytosolic fraction and Triton X-100 Extraction Buffer (Ramsby and Makowski, 2011) was added to the plates followed by 30 minute incubation rocking on ice. The resulting supernatant was taken as membrane fraction.
Embryo culture, treatment and immunofluorescence
Wild-type E3.5 embryos were obtained by flushing of the uterus at 3.5 days post coitum and cultured in M16 media containing DMSO, 1μM Etoposide (Sigma) or 2μM ABT-737 (Selleckchem). E6.5 embryos were dissected from pregnant females at 6.5 days post coitum and cultured in N2B27 media (see Epiblast Stem Cell culture conditions) containing DMSO, 1μM Etoposide or 2μM ABT-737.
For immunostaining, embryos were fixed in 4% PFA/0.1%Tween/0.01% Triton X-100/PBS (10 min for E3.5 and E4.5, 15min for E5.5 and 20min for E6.5) at room temperature, permeabilized in 0.5% Triton X-100/PBS for 15min (E3.5 and E4.5) or 20min (E5.5 and E6.5), washed in 0.1% Triton X-100/PBS and blocked in 2% Horse serum in 0.1% Triton X-100/PBS for 45 minutes at room temperature. Primary antibodies: anti-Cleaved Caspase 3 (Asp175) Cell Signaling 1/100, anti ATP-b, (Ab14730) Abcam 1:200, anti-pDrp1 (S616) Cell Signalling 1/2000, anti-P62 BD 1:200, were incubated overnight at 4°C in 0.2% Horse serum in 0.01% Triton X-100/PBS. Embryos were incubated with Alexa-Fluor conjugated secondary antibodies (Thermo Fisher Scientific) for 1h at 4°C and counterstained with 6ug/ml Hoechst for nuclear visualization. Images from the p-Drp1 immunostaining in embryos were acquired with a Leica SP8 inverted confocal microscope. All other images were acquired with a Zeiss confocal microscope. All images were analysed with the Fiji software (Schindelin et al., 2012).
Whole-mount TUNEL staining was carried out using the ApopTag® Plus Peroxidase In Situ Apoptosis Kit (Sigma Aldrich).
Embryo image quantification
Corrected Total Cellular Fluorescence (CTCF) was measured using (Fiji, Image J) as previously described (reference below). An outline was drawn around each embryo. Area, mean fluorescence and integrated density were measured. In addition, adjacent areas were also selected an measured as background readings. Corrected total cellular fluorescence was calculated using the formula: CTCF = integrated density − (area of selected cell × mean fluorescence of background readings). Cleaved Caspase 3 signal was normalised to DAPI (McCloy et al., 2014). The percentage of P62 positive cells at E4.5 was calculated by counting the number of P62 positive epiblast cells and dividing it by the total epiblast number. Apoptotic levels at E5.5 was calculated by counting the number of TUNEL positive foci and dividing it by the embryonic region area.
BH3 profiling
The assay was performed following the protocol described by Anthony Letai’s laboratory (Certo et al., 2006). Briefly, 15μL of 2x peptides diluted in DTEB buffer (135mM threalose, 10mM HEPES KOH pH 7.5, 50mM KCl, 20μM EGTA, 20μM EDTA, 0.10% BSA, 5mM succinic acid) were added to each well of a dark 384 well plate (Nunc). Cells were dissociated with trypsin, counted and resuspended at 2.67x106 cells/mL. Equal volume of cells and dying mix (4x digitonin (0.01%), 4x JC-1 (4mM) and Oligomycin (40μg/mL)) were incubated for 10 minutes at room temperature protected from light. 15μL of this mix was added per well of the 384 well plate. DMSO and CCCP (Sigma) were used as no-depolarization and full depolarization controls. BID, BIM, BMF and a peptide control were tested in this profile. Each sample was loaded by triplicate and at least three biological replicates were analysed. Fluorescence was measured every 5 minutes for a period of 70 minutes, with a 544/590 filter in a FluoStar Omega plate reader (BMG Omega). Percentage of depolarization was calculated by normalizing the data to the membrane potential of cells that have not been exposed to peptides but have been treated with DMSO (vehicle) or CCCP, a protonophore that causes complete mitochondrial depolarization (Certo et al., 2006).
Peptide Sequences: BIM Ac-MRPEIWIAQELRRIGDEFNA-NH2; BID Ac-EDIIRNIARHLAQVGDSMDRY-NH2; Control Ac-EQWAREIGAQARRMAADLNA-NH2.
Generation of Drp1 Knock-Out and Drp1 Overexpressing cells
Drp1 knockout ESCs were generated by CRISPR-Cas9 mediated deletion of Drp1 exon 2. sgRNA guides flanking Drp1 exon2 were cloned into the PX459 vector (Addgene) (Ran et al., 2013) : Drp1 exon 2 upstream sgRNA: 5’ TGGAACGGTCACAGCTGCAC 3’; Drp1 exon 2 downstream sgRNA: 5’ TGGTCGCTGAGTTTGAGGCC 3’. E14 ESCs were co-transfected with 1ug of each sgRNA expression using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. As control E14 ESCs were transfected in parallel with equal amount of empty PX459 plasmid. Following 6 days of Puromycin selection, single colonies were picked from both Drp1 sgRNA and empty vector transfected ESCs and screened for mutations. Drp1 exon 2 deletion was confirmed by PCR genotyping using the following primers: Drp1_genot F: 5’ GGATACCCCAAGATTTCTGGA 3’; Drp1_genot R: 5’ AGTCAGGTAATCGGGAGGAAA 3’, followed by Sanger Sequencing.
Drp1 overexpressing cells were generated by transfecting ESCs with a pCAG-Drp1 plasmid. To generate this plasmid the Drp1 cDNA (Addgene plasmid 45160) was cloned into a pyCAGIP mammalian expression plasmid (Chambers et al., 2003). E14 ESCs were transfected with 2 ug of the pCAG-Drp1 plasmid using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. Following 6 days of Puromycin selection, single colonies were picked and analysed for DRP1 expression by western blot.
RNA Extraction and Quantitative RT-PCR
RNA was extracted with the RNeasy mini kit (Qiagen) and Superscript III reverse transcriptase (Thermo Fisher Scientific) was used for cDNA synthesis according to manufacturer’s instructions. Quantitative RT-PCR was performed by amplification with Lightcycler 480 SYBR Green Master (Roche). The primers used are listed in the key resources table. RNA samples from wild type and mutant clones were collected from 3 independent experiments.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat#9661; RRID: AB_2341188 |
| Nanog Monoclonal Antibody | eBioscience | Cat#14-5761; RRID: AB_763613 |
| anti ATP-b | Abcam | Cat# Ab14730 RRID:AB_301438 |
| anti-SQSTM1/p62 | BD | Cat#610833 RRID:AB_398152 |
| Bim/BOD polyclonal antibody | Enzo | Cat#ADI-AAP-330; RRID: AB_10616974 |
| Anti-PUMA antibody | Abcam | Cat#ab9643; RRID:AB_296537 |
| BID Antibody (Mouse Specific) | Cell Signaling Technology | Cat#2003; RRID:AB_10694562 |
| Bad Antibody (C-20) | Santa Cruz Biotechnology | Cat#sc-943; RRID:AB_630896 |
| Bax (N-20) | Santa Cruz Biotechnology | Cat#sc-493; RRID:AB_2227995 |
| Purified anti-Bcl-2 Antibody | BioLegend | Cat#633501; RRID:AB_961017 |
| Bcl-xS/L Antibody (S-18) | Santa Cruz Biotechnology | Cat#sc-634; RRID:AB_630917 |
| anti- MCL1 | Santa Cruz | Cat#sc-819 RRID:AB_2144105 |
| Mouse Bcl-2 related protein A1 Antibody | R&D Systems | Cat#MAB851; RRID:AB_2064577 |
| Hexokinase II (C64G5) Rabbit mAb | Cell Signaling Technology | Cat#2867; RRID:AB_2232946 |
| α-Tubulin Antibody | Cell Signaling Technology | Cat#2144; RRID:AB_2210548 |
| Actin Antibody (C-2) | Santa Cruz Biotechnology | Cat#sc-47778; RRID:AB_2714189 |
| Purified Mouse Anti-Cytochrome C | BC Pharmigen | Cat#556433; RRID:AB_396417 |
| Anti-MAP Kinase (ERK-1, ERK-2) antibody produced in rabbit | Sigma Aldrich | Cat#M5670; RRID:AB_477216 |
| anti-BAK | Merck Millipore | Cat#ABC12 RRID: AB_10563091 |
| Anti-Oct-3/4 Antibody (C-10) | Santa Cruz Biotechnology | Cat#sc-5279; RRID:AB_628051 |
| TOM20 Antibody (FL-145) | Santa Cruz Biotechnology | Cat#sc-11415; RRID:AB_2207533 |
| anti-DRP1 | Cell Signaling Technology | Cat#8570S RRID: AB_10950498 |
| Phospho-DRP1 (Ser616) (D9A1) Rabbit mAb | Cell Signaling Technology | Cat#4494; RRID:AB_11178659 |
| anti-OPA1 | BD | Cat#612606 RRID: AB_399888 |
| anti-MFN1 | Abcam | Cat#Ab57602 RRID: AB_2142624 |
| anti-MFN2 | Abcam | Cat#Ab56889 RRID: AB_2142629 |
| anti-mt-CO1 | Abcam | Cat#Ab14705 RRID: AB_2084810 |
| anti-VINCULIN | Sigma Aldrich | Cat#V9131 RRID: AB_477629 |
| Anti-PCNA antibody | Abcam | Cat#ab18197; RRID:AB_444313 |
| anti-SOX2 | Abcam | Cat#ab79351 RRID:AB_10710406 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987 |
| Chemicals, peptides, and recombinant proteins | ||
| LIF | Home made | N/A |
| Gelatine Solution | Sigma-Aldrich | Cat# G1393 |
| G-MEM BK-21 (1x) | Gibco | Cat# 21710025 |
| Embryonic Stem Cell-tested Foetal Calf Serum | Seralab | Cat# EU-000-H |
| Neurobasal medium (1x) | Gibco | Cat# 21103049 |
| DMEM/F12 (1:1) 1x | Gibco | Cat# 21331020 |
| MEM NEAA (100x) | Gibco | Cat# 11140050 |
| B27 Supplement minus Vitamin A | Gibco | Cat# 12587010 |
| M16 Medium | Sigma Aldrich | Cat# M7292 |
| N2 Supplement | Gibco | Cat# 17502048 |
| Sodium Pyruvate | Gibco | Cat# 11360070 |
| 2-Mercaptoethanol | Gibco | Cat# 31350010 |
| L-Glutamine 200mM (100x) | Gibco | Cat# 25030024 |
| Basic Fgf | R&D Systems | Cat# 3139-FB-025 |
| Activin A | R&D Systems | Cat# 338-AC-050 |
| Puromicin | Sigma Aldrich | Cat# P8833 |
| HiPerFect Transfection Reagent | Qiagen | Cat# 301705 |
| Lipofectamine 2000 Trasfection Reagent | Thermo Fisher Sceintific | Cat# 11668027 |
| 4-OH Tamoxifen | Sigma | Cat# H7904 |
| ABT-737 | Selleck Chemicals | Cat# S1002 |
| Sodium Arsenite | Sigma Aldrich | 1062771000 |
| Thapsigargin | Sigma Aldrich | T9033-1MG |
| Etoposide | Sigma Aldrich | Cat# E1383 |
| Bafilimocin | Sigma Aldrich | C2759-100MG |
| DMEM, no glucose, no glutamine, no pyruvate,no carbonate, no phenol red | Sigma Aldrich | D5030-10X1L |
| CCCP | Sigma Aldrich | C2759-100MG |
| Rotetone | Sigma Aldrich | R8875-1G |
| Antimycin | Sigma Aldrich | A8674-25MG |
| FCCP | Sigma Aldrich | C2920-10MG |
| Oligomycin | Sigma Aldrich | 75351-5MG |
| 2-DG | Sigma Aldrich | D3179-250MG |
| Glucose | Sigma Aldrich | G8644 |
| Propidium Iodide | Sigma Aldrich | P4864-10ML |
| Annexin V-APC | Thermo Fisher Scientific | Cat# R37176 |
| DiOC6 | Thermo Fisher Scientific | Cat# D273 |
| TMRM | Sigma Aldrich | Cat# T5428-25MG |
| Vectashield with Dapi | Vector Laboratories | Cat# H-1200 |
| Paraformaldehyde | Sigma Aldrich | Cat# 158127 |
| Formaldehyde | Sigma Aldrich | Cat# F8775 |
| Triton X-100 | Sigma Aldrich | Cat# T8787 |
| Bovine Serum Albumin (lyophilized powder, ≥96%) | Sigma Aldrich | Cat# A2153 |
| Horse serum | Sigma Aldrich | Cat# H1270 |
| BIM peptide: Ac-MRPEIWIAQELRRIGDEFNA-NH2 | Peptide Protein Research Ltd | Newly synthesized |
| BID peptide: Ac-EDIIRNIARHLAQVGDSMDRY-NH2 | Peptide Protein Research Ltd | Newly synthesized |
| BMF peptide: Ac-HQAEVQIARKLQLIADQFHRY-NH2 | Peptide Protein Research Ltd | Newly synthesized |
| Control peptide: Ac-EQWAREIGAQARRMAADLNA-NH2 | Peptide Protein Research Ltd | Newly synthesized |
| Critical commercial assays | ||
| LysoTracker Red DND-99 | Thermo Fisher Scientific | Cat#L7528 |
| ApopTag® Plus Peroxidase In Situ Apoptosis Kit | Sigma Aldrich | Cat#S7100 |
| RNeasy mini kit | Qiagen | Cat#74106 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080044 |
| Lightcycler 480 SYBR green Master | Roche | Cat#04707516001 |
| mirVana miRNA isolation kit | Ambion | Cat#AM1560 |
| TaqMan miRNA reverse transcription kit | Applied Biosystems | Cat#4366596 |
| TaqMan™ Universal PCR Master Mix | Applied Biosystems | Cat#4326708 |
| Experimental models: Cell lines | ||
| Dicer Floxed mouse ESC | (Nesterova et al., 2008) | N/A |
| Dicer Floxed mouse EpiSCs | (Pernaute et al., 2014) | N/A |
| E14 mouse ESCs | Gift from Prof A. Smith (Exeter Univ.). | RRID: CVCL_C320 |
| E14 mouse EpiSCs | This study | N/A |
| Atg5−/− mouse ESCs | (Qu et al., 2007) | N/A |
| Drp1−/− mouse ESCs | This study | N/A |
| Experimental models: Organisms/strains | ||
| Drp1fx/fx mice | (Wakabayashi et al., 2009) | N/A |
| Sox2Cre mice | (Hayashi et al., 2002) | N/A |
| Oligonucleotides | ||
| Mm_Bcl2l11_2 FlexiTube siRNA | Qiagen | Cat#1027417 |
| Flexi-Tube Negative Control siRNA | Qiagen | Cat#1022076 |
| Drp1 genotyping primer F: 5’ GGATACCCCAAGATTTCTGGA 3’ | This study | N/A |
| Drp1_genotyping primer R: 5’ AGTCAGGTAATCGGGAGGAAA 3’ | This study | N/A |
| qPCR primer Gapdh F: 5’CATGGCCTTCCGTGTTCCTA 3’ | This study | N/A |
| qPCR primer Gapdh R: 5’ GCGGCACGTCAGATCCA 3’ | This study | N/A |
| qPCR primer Fgf5 F: 5’ AAAGTCAATGGCTCCCACGAA 3’ | This study | N/A |
| qPCR primer Fgf5 R: 5’ CTTCAGTCTGTACTTCACTGG 3’ | This study | N/A |
| qPCR primer Esrrb F: 5’ GGACACACTGCTTTGAAGCA 3’ | This study | N/A |
| qPCR primer Esrrb R: 5’ ACAGATGTCTCTCATCTGGC 3’ | This study | N/A |
| qPCR primer Nanog F: 5’CTTACAAGGGTCTGCTACTGAGATGC 3’ | This study | N/A |
| pPCR primer Nanog R: 5’ TGCTTCCTGGCAAGGACCTT 3’ | This study | N/A |
| qPCR primer Rex1 F: 5’ CGAGTGGCAGTTTCTTCTTGG 3’ | This study | N/A |
| qPCR primer Rex1 R: 5’ GACTCACTTCCAGGGGGCAC 3’ | This study | N/A |
| TaqMan™ MicroRNA Assay: mmu-miR-291-3p | Applied Biosystems | Cat# 001135 |
| TaqMan™ MicroRNA Assay: mmu-miR-292-3p | Applied Biosystems | Cat# 001054 |
| TaqMan™ MicroRNA Assay: mmu-miR-294 | Applied Biosystems | Cat# 001056 |
| TaqMan™ MicroRNA Assay: snoRNA135 | Applied Biosystems | Cat# 001230 |
| Recombinant DNA | ||
| pEGFP-parkin WT | (Trempe et al., 2013) | Addgene Cat# 45875; RRID:Addgene_458 75 |
| pSpCas9(BB)-2A-Puro (PX459) | (Ran et al., 2013) | Addgene Cat# 48139; RRID:Addgene_481 39 |
| PX459-Drp1_exon2_upstream_sgRNA | This study | N/A |
| PX459-Drp1_exon2_downstream_sgRNA | This study | N/A |
| pEYFP-C1-Drp1 | PMID: 11703942 | Cat# 45160; RRID:Addgene_451 60 |
| pCAG-Drp1 plasmid | This study | N/A |
| pyCAGIP plasmid | (Chambers et al., 2003) | N/A |
| Software and algorithms | ||
| FlowJo™ Software | BD Life Sciences | https://www.flowjo.com/solutions/flowjo; RRID:SCR_008520 |
| Fiji Software | (Schindelin et al., 2012) | https://fiji.sc; RRID:SCR_002285 |
| Huygens Software | Scientific Volume Imaging, Hilversum, Netherlands | https://svi.nl/HuygensSoftware; RRID:SCR_014237 |
| Seahorse assay report generator | Agilent Technologies, UK | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-xf-cell-mito-stress-test-report-generators-740899 https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-xf-glycolytic-rate-assay-report-generators-740900 |
| GraphPad Prism Software | GraphPad Software, Inc. | http://www.graphpad.com/; RRID:SCR_002798 |
For miRNA q-PCR total RNA was extracted using the mirVana miRNA isolation kit (Ambion) and cDNA was synthesized with the TaqMan miRNA reverse transcription kit (Applied Biosystems). qPCR for individual miRNAs was performed using TaqMan probes and TaqMan universal PCR master mix (Applied Biosystems). miRNA expression was normalised against sno135.
Seahorse Analysis
The metabolic function of cells was assessed by extracellular flux analysis using Seahorse XF24 (Agilent Technologies, UK). On the day prior to the assay, 5x104 cells per well were seeded on 0.1% gelatin-coated (Sigma, UK) 24-well plates and grown in 300 μL of pluripotency maintenance conditions. Cells were washed, just before the assay, with the assay media and left with a final volume of the 600 μL per well. The plate was then equilibrated on a non-CO2 incubator at 37°C for 30 min. The assay media consisted in unbuffered DMEM (D5030 – Sigma, UK), reconstituted with 1.83 g.L−1 of NaCl in dH2O, that was supplemented on the day of the assay according to the test performed. For the OCR measurements the assay media was supplemented with 0.5 g.L−1 of glucose (Sigma, UK) and 2 mM of L-glutamine (Life Technologies, UK), while for the ECAR measurements the media was supplemented with 1 mM of Sodium Pyruvate and 2 mM of L- glutamine (both from Life Technologies, UK), pH 7.4 at 37°C.
Assays were performed with 5 biological replicates of each cell type and 4 wells were left without cells for background correction measurements. Both ECAR and OCR measurements were performed on the same plate. The assay parameters for both tests were calculated following the Seahorse assay report generator (Agilent Technologies, UK).
The protocol for the assay consisted of 4 baseline measurements and 3 measurements after each compound addition. Compounds (all from Sigma, UK) used in OCR and ECAR tests were prepared in the supplemented assay media. For the OCR, test the following compounds were added: 2.5 μM oligomycin (OM), 300 nM Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) and a mixture of rotenone and antimycin A at 6 μM each (R&A). For the ECAR test, the following compounds were added: 2.5 mM and 10 mM of glucose, 2.5 μM of oligomycin (OM), and a 50 mM of 2-deoxyglucose (2-DG).
At the end of the assay, cells were fixed and stained with Hoechst. Both OCR and ECAR were normalised to cell number, determined by manual cell counts using Fiji software. The normalisation of the data was processed on Wave Desktop software (Agilent Technologies, UK) and data exported to Prism 8 (GraphPad) for statistical analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification was performed using Microsoft excel and GraphPad Prism software. Statistical analysis was performed using GraphPad Prism software. Statistical methods are indicated in the relevant figure legends, including details of the statistical tests used and sample size (n). No randomization or blinding was used in the experiments. Sample size (n) represents independent replicate experiments and is described in each figure legend. Quantifications are shown as the average +/− SEM, where the average is measured as the sum of quantified values divided by the sample size (n); standard deviation (SD) accounts for the dispersion of data points relative to the average and is measured as the square root of the variance; standard error of the mean (SEM) is calculated as the SD divided by the square root of n.
Supplementary Material
Research Highlights.
Upon differentiation embryonic stem cells become hypersensitive to apoptosis.
DRP1 promotes mitophagy in pluripotent stem cells.
During the onset of differentiation DRP1 levels and mitophagy activity decreases.
Decreased mitophagy during the onset of differentiation primes cells for apoptosis.
Acknowledgements
We would like to thank Massimo Signore and Juan Pedro Martinez Barbera for critical discussion. We thank Stephen Rothery for guidance and advice with confocal microscopy. Gratitude also goes to James Elliot for performing cell sorting. Research in the Tristan Rodriguez lab was supported by an MRC project grant (MR/N009371/1 and MR/T028637/1) and by the BBSRC project grant (BB/S008284/1). Barbara Pernaute was supported by an EMBO Long Term Fellowship (1340-2010) and a Marie Curie Intra-European Fellowship (FP7-PEOPLE-2010-IEF n° 273884). Salvador Perez Montero has been a recipient of an EMBO long-term Fellowship (846-2015) and a Marie Curie Intra-European Fellowship (H2020-MSCA-IF-2015 n° 709010). Ana Lima was supported by a BHF centre for research excellence PhD studentship. Katerina Lawlor was supported by an NHLI PhD studentship. Sarah Bowling was supported by an MRC-DTP PhD studentship. Work in the Meier lab is funded by Breast Cancer Now (CTR-QR14-007). Guy A. Rutter was supported by Wellcome Trust Senior Investigator (WT098424AIA ) and Investigator (212625/Z/18/Z) Awards, MRC Programme grants (MR/R022259/1, MR/J0003042/1, MR/L020149/1) and Experimental Challenge Grant (DIVA, MR/L02036X/1), MRC (MR/N00275X/1), and Diabetes UK (BDA/11/0004210, BDA/15/0005275, BDA 16/0005485) grants. This project has received funding from the European Union via the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115881 (RHAPSODY) to Guy A. Rutter. We acknowledge NHS funding to the NIHR Biomedical Research Centre. Alejandra Tomás is funded by the MRC project grant MR/R010676/1. Hiromi Sesaki is funded by the NIH grant number (GM144103).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Bahat A, Goldman A, Zaltsman Y, Khan DH, Halperin C, Amzallag E, Krupalnik V, Mullokandov M, Silberman A, Erez A, et al. (2018). MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in embryonic stem cells. Nat Commun 9, 5132. 10.1038/s41467-018-07519-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X, Teng T, Zhao H, Qin J, Qiao Z, Sun Y, Liun Z, and Xu Z (2018). Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic Res 52, 80–91. 10.1080/10715762.2017.1414949. [DOI] [PubMed] [Google Scholar]
- Bolton H, Graham SJ, Van der Aa N, Kumar P, Theunis K, Fernandez Gallardo E, Voet T, and Zernicka-Goetz M (2016). Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploid cells and normal developmental potential. Nat Commun 7, 11165. ncomms11165 [pii] 10.1038/ncomms11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling S, Lawlor K, and Rodriguez TA (2019). Cell competition: the winners and losers of fitness selection. Development 146. 10.1242/dev.167486. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, and Shore GC (2003). Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160, 1115–1127. 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, and Vallier L (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. nature05950 [pii] 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Brown EJ, and Baltimore D (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, and Youle RJ (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 216, 3231–3247. 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, and Letai A (2006). Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. S1535-6108(06)00113-9 [pii] 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, and Smith A (2003). Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113, 643–655. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, and Chan DC (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160, 189–200. 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HM, Ryu JR, Jo Y, Seo TW, Choi YN, Kim JH, Chung JM, Cho B, Kang HC, Yu SW, et al. (2019). Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol Cell 73, 364–376 e368. 10.1016/j.molcel.2018.11.009. [DOI] [PubMed] [Google Scholar]
- Csordas G, Weaver D, and Hajnoczky G (2018). Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol 28, 523–540. 10.1016/j.tcb.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, and Sorger PK (2000). Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 101, 635–645. S0092-8674(00)80875-2 [pii]. [DOI] [PubMed] [Google Scholar]
- Estaquier J, and Arnoult D (2007). Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14, 1086–1094. 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, and Youle RJ (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1, 515–525. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, and Scorrano L (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, and Scorrano L (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13, 589–598. 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Yang J, Nichols J, Hall JS, Eyres I, Mansfield W, and Smith A (2009). Klf4 reverts developmentally programmed restriction of ground state pluripotency.Development 136, 1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, and McMahon AP (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev 119 Suppl 1, S97–S101. 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Heyer BS, MacAuley A, Behrendtsen O, and Werb Z (2000). Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev 14, 2072–2084. [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, et al. (2014). Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33, 2798–2813. 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, and Sesaki H (2012). Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197, 535–551. 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalitsis P, Earle E, Fowler KJ, and Choo KH (2000). Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev 14, 2277–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, and Youle RJ (2002). Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159, 931–938. 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, and Kashatus DF (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57, 537–551. S1097-2765(15)00003-9 [pii] 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, and Bossy-Wetzel E (2008). Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9, 505–518. 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ (1992). Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood 80, 879–886. [PubMed] [Google Scholar]
- Laurent A, and Blasi F (2015). Differential DNA damage signalling and apoptotic threshold correlate with mouse epiblast-specific hypersensitivity to radiation. Development 142, 3675–3685. dev.125708 [pii] 10.1242/dev.125708. [DOI] [PubMed] [Google Scholar]
- Leitch HG, Tang WW, and Surani MA (2013). Primordial germ-cell development and epigenetic reprogramming in mammals. Curr Top Dev Biol 104, 149–187. B978-0-12-416027-9.00005-X [pii] 10.1016/B978-0-12-416027-9.00005-X. [DOI] [PubMed] [Google Scholar]
- Lima A, Burgstaller J, Sanchez-Nieto JM, and Rodriguez TA (2018). The Mitochondria and the Regulation of Cell Fitness During Early Mammalian Development. Curr Top Dev Biol 128, 339–363. 10.1016/bs.ctdb.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Liu JC, Guan X, Ryan JA, Rivera AG, Mock C, Agarwal V, Letai A, Lerou PH, and Lahav G (2013). High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13, 483–491. S1934-5909(13)00363-9 [pii] 10.1016/j.stem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manova K, Tomihara-Newberger C, Wang S, Godelman A, Kalantry S, Witty-Blease K, De Leon V, Chen WS, Lacy E, and Bachvarova RF (1998). Apoptosis in mouse embryos: elevated levels in pregastrulae and in the distal anterior region of gastrulae of normal and mutant mice. Dev Dyn 213, 293–308. [pii]. [DOI] [PubMed] [Google Scholar]
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, and Burgess A (2014). Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400–1412. 10.4161/cc.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendl N, Occhipinti A, Muller M, Wild P, Dikic I, and Reichert AS (2011). Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 124, 1339–1350. 10.1242/jcs.076406. [DOI] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, and Martinou JC (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagdas S, Kashatus JA, Nascimento A, Hussain SS, Trainor RE, Pollock SR, Adair SJ, Michaels AD, Sesaki H, Stelow EB, et al. (2019). Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep 28, 1845–1859 e1845. 10.1016/j.celrep.2019.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, and Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803. 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesterova TB, Popova BC, Cobb BS, Norton S, Senner CE, Tang YA, Spruce T, Rodriguez TA, Sado T, Merkenschlager M, and Brockdorff N (2008). Dicer regulates Xist promoter methylation in ES cells indirectly through transcriptional control of Dnmt3a. Epigenetics Chromatin 1, 2. 10.1186/1756-8935-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, and Smith A (2009). naive and primed pluripotent states. Cell Stem Cell 4, 487–492. S1934-5909(09)00224-0 [pii] 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Otera H, Miyata N, Kuge O, and Mihara K (2016). Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212, 531–544. 10.1083/jcb.201508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernaute B, Spruce T, Smith KM, Sanchez-Nieto JM, Manzanares M, Cobb B, and Rodriguez TA (2014). MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes Dev 28, 1873–1878. 28/17/1873 [pii] 10.1101/gad.245621.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, and McBride HM (2017). The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr Opin Cell Biol 47, 52–63. 10.1016/j.ceb.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, and Levine B (2007). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946. S0092-8674(07)00178-X [pii] 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, and Lippincott-Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 108, 10190–10195. 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsby M, and Makowski G (2011). Differential detergent fractionation of eukaryotic cells. Cold Spring Harb Protoc 2011, prot5592. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault TT, Floros KV, Elkholi R, Corrigan KA, Kushnareva Y, Wieder SY, Lindtner C, Serasinghe MN, Asciolla JJ, Buettner C, et al. (2015). Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Molecular cell 57, 69–82. S1097-2765(14)00863-6 [pii] 10.1016/j.molcel.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M, Di-Gregorio A, George N, Pozzi S, Sanchez JM, Pernaute B, and Rodriguez TA (2013). Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell 26, 19–30. S1534-5807(13)00351-1 [pii] 10.1016/j.devcel.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard TH, Muffley LA, and Smith LT (2000). Mitochondrial ultrastructure in embryos after implantation. Hum Reprod 15 Suppl 2, 218–228. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, and van der Bliek AM (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12, 2245–2256. 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow MHL (1977). Gastrulation in the mouse: growth and regionalization of the epiblast. J. Embryol. exp. Morph 42, 293–303. [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, and Dorn GW 2nd (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21, 273–285. S1550-4131(14)00562-2 [pii] 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce T, Pernaute B, Di-Gregorio A, Cobb BS, Merkenschlager M, Manzanares M, and Rodriguez TA (2010). An early developmental role for miRNAs in the maintenance of extraembryonic stem cells in the mouse embryo. Dev Cell 19, 207–219. S1534-5807(10)00342-4 [pii] 10.1016/j.devcel.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Stern S, Biggers JD, and Anderson E (1971). Mitochondria and early development of the mouse. J Exp Zool 176, 179–191. 10.1002/jez.1401760206. [DOI] [PubMed] [Google Scholar]
- Tait SW, and Green DR (2013). Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 5. 5/9/a008706 [pii] 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, and Youle RJ (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191, 1367–1380. 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilokani L, Nagashima S, Paupe V, and Prudent J (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62, 341–360. 10.1042/EBC20170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446. 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, and Sesaki H (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186, 805–816. 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, et al. (2015). Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18, 501–510. nn.3960 [pii] 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Adachi Y, Fukaya M, Iijima M, and Sesaki H (2016). Dynamin-Related Protein 1 Deficiency Leads to Receptor-Interacting Protein Kinase 3-Mediated Necroptotic Neurodegeneration. Am J Pathol 186, 2798–2802. 10.1016/j.ajpath.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Adachi Y, Yanagawa T, Iijima M, and Sesaki H (2018a). p62/sequestosome-1 knockout delays neurodegeneration induced by Drp1 loss. Neurochem Int 117, 77–81. 10.1016/j.neuint.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM, et al. (2018b). Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab 28, 588–604 e585. 10.1016/j.cmet.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T,Yoshimori T, Sakai Y, Mihara K, and Kanki T (2016). Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215, 649–665. 10.1083/jcb.201605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Cui P.M.-m.m.f.d.e.f.n.p.i.e.s.c., Cai Y, Wang L, He X, Long P, Lu K, Yan R, Zhang Y, Pan X, et al. (2019). Mitochondrial Dynamics Is Critical for the Full Pluripotency and Embryonic Developmental Potential of Pluripotent Stem Cells. Cell Metab 29, 979–992 e974. 10.1016/j.cmet.2018.11.007. [DOI] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, and Ruohola-Baker H (2012). HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31, 2103–2116. emboj201271 [pii] 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
There is no original code reported in this paper.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.