

Abstract
Biliary tract cancers (BTCs) have poor prognosis and limited therapeutic options. The impact of O 6‐methylguanine‐DNA methyltransferase (MGMT) inactivation in advanced BTC patients is not established. We investigated the prevalence, prognostic, and predictive impact of MGMT inactivation in two multicenter cohorts. MGMT inactivation was assessed through PCR and immunohistochemistry (IHC) in an Italian cohort; the results were then externally validated using RNA sequencing (RNA‐seq) data from the BTC subcohort of the Molecularly Aided Stratification for Tumor Eradication Research (MASTER) precision oncology program of the National Center for Tumor Diseases Heidelberg and the German Cancer Consortium. Among 164 Italian cases, 18% presented MGMT promoter hypermethylation (> 14%) and 73% had negative MGMT protein expression. Both were associated with worse overall survival (OS; HR 2.31; P < 0.001 and HR 1.99, P = 0.012, respectively). In the MASTER cohort, patients with lower MGMT mRNA expression showed significantly poorer OS (median OS [mOS] 20.4 vs 31.7 months, unadjusted HR 1.89; P = 0.043). Our results suggest that MGMT inactivation is a frequent epigenetic alteration in BTC, with a significant prognostic impact, and provide the rationale to explore DNA‐damaging agents in MGMT‐inactivated BTCs.
Keywords: biliary tract cancer, biomarker, cholangiocarcinoma, MGMT, molecular profiling, temozolomide
The impact of MGMT inactivation in patients with advanced biliary tract cancer (BTC) is not established. We investigated its prevalence, prognostic, and predictive impact, assessed through methylation‐specific PCR and IHC, in a large Italian cohort. Then we evaluated MGMT inactivation through RNA‐seq and DNA methylation analysis in an independent German cohort. Overall, MGMT inactivation or reduced expression consistently resulted in poorer survival.
Abbreviations
- 5‐FU
5‐fluorouracil
- 1L
first‐line
- BTCs
biliary tract cancers
- CI
confidence interval
- dCCA
distal cholangiocarcinoma
- DKTK
German Cancer Consortium
- ECOG PS
Eastern Cooperative Oncology Group performance status
- GBC
Gall bladder cancer
- HR
hazard ratio
- iCCA
intrahepatic cholangiocarcinoma
- IHC
immunohistochemistry
- INT
Fondazione IRCCS Istituto Nazionale Tumori Of Milan
- IQR
interquartile range
- MASTER
Molecularly Aided Stratification For Tumor Eradication Research
- MGMT
O6‐methylguanine DNA methyltransferase
- NA
not available
- NCT
Nationale Centrum Für Tumorerkrankungen
- OS
overall survival
- pCCA
perihilar cholangiocarcinoma
- PD
progressive disease
- PFS
progression‐free survival
- RNA‐seq
RNA sequencing
- TMZ
temozolomide
- TPM
transcripts per kilobase million
- VIMP
variable importance
1. Introduction
Biliary Tract Cancer (BTC) is a rare disease with overall poor prognosis and limited therapeutic options [1, 2]. Emerging evidence reveals that BTC is heterogeneous from a pathological and molecular perspective, with significant differences between intrahepatic cholangioarcinoma (iCCA), extrahepatic cholangiocarcinoma (eCCA), and gallbladder cancer (GBC) [3, 4, 5, 6, 7]. There is a growing evidence in favor of biomarker‐directed treatments in BTC, such as pemigatinib and infigratinib for CCA with FGFR2 fusions [8, 9], ivosidenib for IDH1 mutated CCAs [10], and BRAF plus MEK inhibitors in BRAF V600E mutated or HER2 inhibitors in ERRB2 amplified/mutated BTCs [11, 12, 13]. Finally, there is an increasing amount of data regarding the identification of DNA damage repair aberrations and distinct DNA hypermethylation patterns in these cancers [3, 4, 14].
O6‐methylguanine‐DNA methyltransferase (MGMT) encodes for a key DNA repair enzyme, responsible for the elimination of alkyl groups from the O6‐position of guanine. MGMT promoter methylation, leading to reduction of MGMT expression, ultimately results in diminished DNA‐repair of O6‐alkylguanine adducts and enhanced sensitivity to alkylating agents, such as temozolomide (TMZ) [15]. MGMT promoter hypermethylation is a validated biomarker for the efficacy of TMZ in glioblastoma [15]. Similarly, both MGMT promoter hypermethylation and reduced/absent MGMT expression are described in a variety of gastrointestinal malignancies, including colorectal cancers [16, 17]. In patients with metastatic colorectal cancer, several trials showed that MGMT silencing is a potential biomarker to select patients for TMZ‐based treatment [18, 19, 20].
In the present study, we investigate the prevalence, as well as the prognostic and predictive impact of MGMT inactivation in two independent series of advanced BTC cases. Moreover, we report the first evidence on the activity of TMZ in patients with BTC.
2. Materials and methods
2.1. Patient population and study objectives
We first conducted a multicenter observational study at Fondazione IRCCS Istituto Nazionale Tumori of Milan (INT) and Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola. From October 2017 to November 2020, we included all patients fulfilling the following eligibility criteria: (a) histologically/cytologically confirmed diagnosis of BTC; (b) unresectable primary tumor and/or evidence of metastases; and (c) available archival tumor tissue for molecular profiling.
Baseline demographic, clinical, and biological data were collected through an electronic database. All patients were followed up until death, loss to follow up or data cut‐off date (December 10, 2020).
The primary aim was to investigate the frequency and percentage of MGMT promoter hypermethylation in advanced BTC patients. Secondary aims were: (a) to investigate the prognostic impact of MGMT promoter hypermethylation in terms of overall survival (OS); (b) to investigate the predictive role of MGMT promoter hypermethylation in first‐line (1L) of standard chemotherapy in terms of 1L‐progression‐free survival (PFS); (c) to explore the potential effect on 1L‐PFS of the interaction between MGMT promoter hypermethylation and use of platinum‐based chemotherapy; (d) to evaluate the prognostic and predictive role of MGMT expression as evaluated by immunohistochemistry (IHC), given the growing evidence of the role of IHC as complementary assessment tool of MGMT status [18, 19, 21]; (e) to explore the association between MGMT promoter hypermethylation and the tumor molecular profile; and (f) to report safety and efficacy data of a case series of patients treated with TMZ‐related regimens.
The study was approved by the Institutional Review Boards of the two institutions and was conducted in accordance with the Declaration of Helsinki; patients provided written informed consent.
2.2. External validation cohort
To externally validate the proof‐of‐concept results of the Italian cohorts with different omics layers, data from the BTC subcohort of the Molecularly Aided Stratification for Tumor Eradication Research (MASTER) study conducted by NCT Heidelberg and the German Cancer Consortium (DKTK) were interrogated [22]. NCT/DKTK MASTER is a registry trial and analytical platform for prospective, multi‐omics‐guided stratification of patients with advanced cancers diagnosed at a young age (< 51 years) or with rare cancers, including BTC, and comprises broad molecular profiling including RNA sequencing (RNA‐seq) and DNA methylation analysis. For the analyses presented here, all patients enrolled in MASTER from March 2012 to February 2021 with histologically/cytologically confirmed diagnosis of BTC and available clinical and RNA‐seq data were considered. The study was approved by the Ethics Committee of the Medical Faculty of Heidelberg University and was conducted in accordance with the Declaration of Helsinki; patients provided written informed consent.
2.3. MGMT status assessment
For samples from the two Italian centers, hematoxylin–eosin slides were reviewed by an expert pathologist to select an area comprising at least 50% tumor cells. DNA was extracted as previously described [20]. MGMT promoter methylation was assessed by MGMT plus® Diatech Pharmacogenetics (Jesi, Italy), which analyzes 10 CpG islands spanning the promoter region (chr10:131, 256, 507–131, 256 556). Briefly, after bisulfite conversion of the extracted DNA (range: 200–500 ng) and its amplification by using primers specific for methylated and unmethylated DNA sequences, a pyrosequencing of the obtained templates was performed. The final result provided the number of methylated CpG islands present in the promoter region of the MGMT gene, expressed as a percentage. When sufficient residual tumor tissue was available, MGMT expression was assessed by immunohistochemistry (IHC), as previously described [19]. Additional tumor molecular characterization was non‐uniformly performed through the Ion Torrent Personal Genome platform (50 genes ‘Hotspot Cancer Panel, Ion Torrent®’; Life Technologies®, Waltham, MA, USA), as in [23], or the FoundationOne®CDx panel [24].
In the Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers (MASTER BTC) cohort, processing of tumor specimens and technical details of RNA‐seq analyses are described in the study done by Horak et al. [22]. MGMT mRNA expression was measured as transcripts per kilobase million (TPM). The methylation status of the MGMT promoter derived from available Illumina Infinium EPIC array data (n = 50) using the MGMT‐STP27 prediction model [25] as implemented in the mgmtstp27 r package (https://github.com/badozor/mgmtstp27).
2.4. Statistical methods
MGMT promoter methylation was first analyzed as a continuous variable (i.e. as number of methylated CpGs present in the promoter region of the MGMT gene, expressed as a percentage), with non‐linear effects assessed by means of restricted cubic splines. Subsequently, the optimal cutoff for OS prediction was calculated using the maximally selected rank statistic, as described by Hothorn and Lausen [26]. The cutoff was then adopted also in 1L‐PFS analyses for consistency. Cases from the MASTER BTC validation cohort were grouped according to MGMT mRNA expression (TPM value), using the median value as cutoff. The Fisher's exact test, Chi‐squared test, and Wilcoxon–Mann–Whitney test were used to study the distribution of categorical and continuous variables, respectively, according to dichotomized MGMT status, as appropriate. Cohen's kappa was used to measure the agreement of PCR and IHC assays assessing MGMT status.
Median follow‐up was quantified with the reverse Kaplan–Meier estimator [27]. Survival analysis methods were used to analyze OS and 1L‐PFS. OS was calculated from the date of advanced disease diagnosis to death or last follow‐up, while 1L‐PFS was calculated from the date of first‐line treatment start to the first event [i.e. progressive disease (PD, as defined as according to RECIST v1.1) or death]. Patients who had not undergone PD or death at the time of data cut‐off were censored at their last disease evaluation. Survival curves and related descriptive statistics were obtained with the Kaplan–Meier method and comparisons between curves were performed with the logrank test. Multivariate analyses in the Italian cohorts were performed with a two‐step strategy: at first, covariates were modeled with a random forest method [28] according to the following endpoints: (a) imputation of missing data at random, using adaptive tree imputation; (b) selection of relevant covariates, by taking the top‐ranked variables by matching the variable importance (VIMP) and Minimal Depth statistics. Then, multivariable Cox proportional models were designed using the selected variables and imputed missing data, and results were summarized using hazard ratios (HRs), together with the corresponding 95% confidence intervals (CI). Interaction terms were used to investigate the interplay between MGMT methylation status and use of platinum‐based first‐line chemotherapy in terms of PFS.
In the MASTER BTC cohort, OS was calculated from the date of first disease diagnosis, and available data on patients' age, gender, and primary tumor location, prior tumor resection and use of adjuvant therapy were used as covariates in multivariate Cox proportional models.
A threshold of significance of 0.05 was set for all statistical evaluations. Statistical analyses were performed in the r (Version 4.0.3) and rstudio (Version 1.3.1073) software [R Foundation for Statistical Computing, Vienna, Austria].
3. Results
3.1. Italian cohort
3.1.1. Patient population
Of a total number of 230 patients, 164 cases were successfully profiled for MGMT promoter methylation status (patients' flow depicted in Fig. S1). Baseline characteristics are displayed in Table 1.
Table 1.
Patients' characteristics in the Italian study cohort. 5‐FU, 5‐fluorouracil; dCCA, distal cholangiocarcinoma; ECOG PS, Eastern Cooperative Oncology Group Performance Status; iCCA, intrahepatic cholangiocarcinoma; IHC, immunohistochemistry; IQR, interquartile range; MGMT, O6‐methylguanine DNA methyltransferase; NA, not available; pCCA, perihilar cholangiocarcinoma.
| Characteristic | N (%) |
|---|---|
| Total number of patients | 164 (100.0) |
| Age in years [median (IQR)] | 66 (57–72) |
| Gender | |
| Male | 71 (43.3) |
| Female | 93 (56.7) |
| ECOG PS | |
| 0–1 | 128 (85.3) |
| ≥ 2 | 22 (14.7) |
| NA | 14 |
| Primary tumor location | |
| iCCA | 100 (61.0) |
| pCCA | 7 (4.3) |
| dCCA | 23 (14.0) |
| Gall bladder | 34 (20.7) |
| Primary tumor resected | |
| Yes | 107 (65.2) |
| No | 57 (34.8) |
| Adjuvant treatment | |
| Yes | 46 (28.0) |
| No | 61 (37.2) |
| Not applicable | 57 (34.8) |
| Diagnosis of advanced disease a | |
| Synchronous | 85 (51.8) |
| Metachronous | 79 (48.2) |
| Liver‐limited disease | 48 (29.3) |
| Sites of metastatic disease | |
| Lymph nodes | 78 (47.6) |
| Bones | 11 (6.7) |
| Liver | 90 (54.9) |
| Lungs | 34 (20.7) |
| Peritoneum | 31 (18.9) |
| Total lines of treatment for unresectable/metastatic disease | |
| Best supportive care | 4 (2.4) |
| 1 | 43 (26.2) |
| 2 | 40 (24.4) |
| > 2 | 42 (25.6) |
| NA | 35 |
| First line treatment regimen | |
| Best Supportive Care | 4 (2.4) |
| Capecitabine/5‐FU | 12 (7.3) |
| Gemcitabine | 12 (7.3) |
| Capecitabine/5‐FU + Oxaliplatin | 15 (9.1) |
| Gemcitabine + Oxaliplatin | 11 (6.7) |
| Gemcitabine + Cisplatin | 88 (53.7) |
| Other | 18 (11.0) |
| NA | 4 |
| Platinum‐based first line regimen b | |
| Yes | 115 (73.7) |
| No | 41 (26.3) |
| NA | 8 |
| MGMT promoter methylation [median (IQR)] | 5 (3–10) |
| MGMT expression by IHC | |
| Positive | 27 (27.0) |
| Weakly positive | 32 (32.9) |
| Negative | 41 (41.0) |
| NA | 64 |
Diagnosis of advanced disease was considered synchronous if occurring < 6 months from primary tumor detection, metachronous if ≥ 6 months.
One patient was treated with Carboplatin‐based chemotherapy.
At data cut‐off date, 138 patients had experienced disease progression on first‐line treatment, and 132 patients had died. The median follow‐up was 58.0 months [interquartile range (IQR): 34.9–79.9], with a median 1L‐PFS of 4.9 months (IQR: 2.7–9.1) and a median OS of 17.3 months (IQR: 8.0–36.1). First‐line‐PFS was longer in patients treated with 1‐line platinum‐based chemotherapy (5.4 vs 3.5 months, P = 0.05), while mOS did not differ according to use of platinum (19.8 vs 10.7, P = 0.2).
3.1.2. Prognostic and predictive impact of MGMT promoter hypermethylation
We first investigated the impact of MGMT promoter methylation, as a continuous variable, on OS. In a univariate Cox regression model, higher MGMT promoter methylation values were associated with an increased risk of death (unadjusted HR 1.02 per 1% increase in methylation value, 95% CI 1.01–1.04, P = 0.009, Fig. S2).
Then, we identified 14% as the best percentage cutoff of MGMT promoter methylation for the prediction of OS. Overall, 135 (82%) and 29 (18%) patients were thus aggregated in the low (≤ 14%) and high (> 14%) MGMT promoter methylation groups, respectively. According to each tumor entity, high MGMT hypermethylation was observed in 10 (10%) iCCA, 6 (26%) dCCA, and 13 (38%) of GBC patients.
Patients and disease characteristics according to MGMT promoter methylation are summarized in Table S1. Of note, highly methylated cases more frequently had worse baseline Eastern Cooperative Oncology Group Performance Status [ECOG PS ≥ 2 in 11/29 (39%) vs 11/135 (9%), respectively] and did not receive first‐line platinum‐based chemotherapy [16/29 (57%) vs 25/135 (19%)].
Patients with MGMT hypermethylation experienced worse median OS (mOS 8.0 vs 18.4 months, unadjusted HR 2.16; 95% CI 1.41–3.29; P < 0.001, Fig. 1A).
Fig. 1.
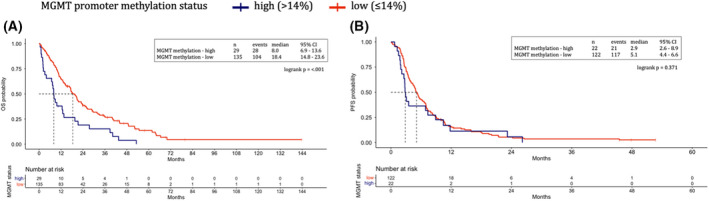
MGMT promoter methylation status. Overall survival (A) and first‐line progression‐free survival (B) represented through Kaplan–Meier curves according to MGMT promoter methylation status in the Italian cohort. Patients with MGMT hypermethylation (blue) experienced worse median overall survival as compared with patients with low MGMT methylation (red) (A). MGMT promoter methylation values did not impact the progression‐free survival of patients treated with first‐line systemic treatment for advanced disease (B). Dotted lines indicate the median survival time. MGMT, O 6‐methylguanine‐DNA methyltransferase. [Colour figure can be viewed at wileyonlinelibrary.com]
Then, we investigated the impact of MGMT promoter methylation on patient OS, as evaluated as a dichotomous variable with the 14% cutoff, in a multivariable model. Via a Random Survival Forest approach, the following covariates were selected as the most relevant, together with MGMT status (i.e. variables in the lower left quadrant of Fig. S3): ECOG PS, patient age, previous adjuvant treatment, presence of bone and lung metastases, and number of treatment lines for advanced disease. In a multivariable Cox model including these variables, MGMT status was confirmed as an independent negative prognostic factor for OS (adjusted HR 2.31; 95% CI 1.44–3.71; P < 0.001, Table 2).
Table 2.
Multivariable cox proportional hazards model for overall survival. The HR for continuous variables is expressed as the HR variation per unit increase of the variable value (i.e. per 1 year increase). CI, confidence interval; ECOG PS, Eastern Cooperative Oncology Group Performance Status; HR, Hazard Ratio; MGMT, O6‐methylguanine DNA methyltransferase.
| Variables | HR | 95% CI | P | |
|---|---|---|---|---|
| MGMT promoter methylation | High (> 14%) vs low (≤ 14%) | 2.31 | 1.44–3.71 | < 0.001 |
| ECOG PS | ≥ 2 vs 0–1 | 1.68 | 1.01–2.79 | 0.044 |
| Age | Continuous | 1.03 | 1.01–1.05 | < 0.001 |
| Adjuvant treatment | Not applicable (no tumor resection) vs no | 1.51 | 0.99–2.30 | 0.054 |
| Yes vs no | 1.51 | 0.96–2.40 | 0.071 | |
| Lines of treatment for unresectable/metastatic disease | 1 vs best supportive care | 1.12 | 0.32–3.88 | 0.860 |
| 2 vs best supportive care | 0.82 | 0.24–2.79 | 0.755 | |
| > 2 vs best supportive care | 0.70 | 0.20–2.40 | 0.570 | |
| Presence of lung metastases | Yes vs No | 1.43 | 0.93–2.19 | 0.101 |
| Presence of bone metastases | Yes vs No | 2.00 | 1.01–3.95 | 0.045 |
Concerning 1L‐PFS, outcome data were available for 144 (88%) patients. MGMT promoter methylation values were not significantly associated with 1L‐PFS, neither as a continuous variable (unadjusted HR 1.01 per 1% increase in methylation value, 95% CI 0.99–1.03, P = 0.219, Fig. S4), nor as dichotomized variable (mPFS 2.9 vs 5.1 months, HR 1.24, 95% CI 0.78–1.98, P = 0.363, Fig. 1B).
Next, the potential interaction between MGMT promoter hypermethylation and use of platinum‐based chemotherapy as first‐line regimen was assessed. In a multivariate Cox model including the most relevant covariates affecting PFS (i.e. the use of platinum‐based chemotherapy, patient age, previous adjuvant treatment, and presence of lung metastases; Fig. S5), a significant interaction between these two factors was observed (P for interaction = 0.038, Table S2).
First‐line‐PFS Kaplan–Meier curves of patients with high (> 14%) and low (≤ 14%) MGMT promoter methylation, stratified according to the use of platinum‐based regimens as first‐line chemotherapy, are shown in Fig. S6a,b: patients in the high MGMT methylation subgroup treated with first‐line platinum‐based chemotherapy showed longer 1L‐PFS compared to those treated with platinum‐free regimens (mPFS 8.1 vs 2.8 months, unadjusted HR 0.28; 95% CI 0.09–0.85; P = 0.018), while no difference was highlighted in the low MGMT methylation subgroup (mPFS 5.4 vs 4.5 months, unadjusted HR 0.86; 95% CI 0.55–1.36; P = 0.530). As an alternative representation of the interaction, the impact of MGMT promoter methylation on 1L‐PFS was assessed according to the chemotherapy regimen: in patients not treated with platinum salts, patients with high MGMT promoter methylation had significantly poorer 1L‐PFS (mPFS 2.8 vs 4.5 months, unadjusted HR 3.95; 95% CI 1.69–9.25; P < 0.001), while this effect was neutralized in patients treated with platinum‐based chemotherapy (mPFS 8.1 vs 4.5 months, unadjusted HR 0.83; 95% CI 0.44–1.55; P = 0.550).
3.1.3. Exploratory analysis of MGMT expression as a biomarker
Within the study cohort, 100 (61%) patients had evaluable tumor tissue for MGMT expression by IHC. In detail, MGMT expression was positive in 27 (27%), weakly positive in 32 (32%), and negative in 41 (41%) patients. The impact of MGMT expression on patient OS and PFS was first explored in the three‐level definition, showing that weakly positive and negative cases had similar outcomes (see Fig. S7a,b). For further analyses, these two groups were thus merged and considered as negative.
MGMT methylation and IHC expression had poor agreement (Cohen's κ −0.003, 95%CI ‐0.068–0.061). Patient and disease characteristics according to MGMT expression by IHC are summarized in Table S3.
Patients with negative MGMT expression showed significantly worse OS (mOS 17.3 vs 31.6 months, unadjusted HR 1.99; 95% CI 1.17–3.41; P = 0.012) and 1L‐PFS (mPFS 4.7 vs 9.1 months, unadjusted HR 2.24; 95% CI 1.36–3.68; P = 0.001; Fig. 2A,B). No significant interaction with use of platinum‐based chemotherapy regimens was observed (P for interaction = 0.898).
Fig. 2.
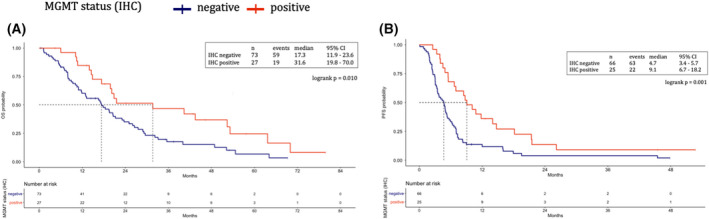
MGMT expression assessed by immunohistochemistry. Overall survival (A) and first‐line progression‐free survival (B) represented through Kaplan–Meier curves according to MGMT status assessed by IHC in the Italian cohort. Patients with negative MGMT expression (blue) showed significantly worse overall survival (A) and progression‐free survival with first‐line systemic treatment (B), as compared with patients with positive MGMT expression (red). Dotted lines indicate the median survival time. MGMT, O 6‐methylguanine‐DNA methyltransferase. [Colour figure can be viewed at wileyonlinelibrary.com]
3.1.4. Molecular profiling
Among the study cohort, 130 (79%) cases treated at INT underwent additional tumor molecular profiling, as depicted in Fig. S8. The most frequent alterations identified were TP53 mutations (29%), KRAS mutations (18%), followed by FGFR2 alterations (11% and 4% had FGFR2 fusions and mutations, respectively), IDH1 mutations (10%), and PIK3CA mutations (9%). Among cases in the MGMT promoter hypermethylation subgroup, only 9/29 (31%) were profiled. In this group, no cases harbored IDH1/2 mutations, while 3/9 (33%) were found as FGFR2 rearranged.
3.2. MASTER BTC cohort
3.2.1. External validation of the prognostic role of MGMT
We analyzed 76 BTC cases enrolled in the NCT/DKTK MASTER trial with available RNA‐seq data. Patients' characteristics are reported in Table 3.
Table 3.
Patient characteristics of the MASTER BTC cohort according to MGMT expression (TPM value). The P value of the χ2 test, Fisher's exact test (for categorical variables), or Mann–Whitney test (for continuous variables) assessing the association between each characteristic and MGMT status is indicated in the right column of the table. MASTER: Molecularly Aided Stratification for Tumor Eradication Research; eCCA: extrahepatic cholangiocarcinoma; iCCA: intrahepatic cholangiocarcinoma; IQR: interquartile range; MGMT: O6‐methylguanine DNA methyltransferase; NA: not available; NOS: not otherwise specified.
| Characteristic | Total, N (%) | MGMT high, N (%) | MGMT low, N (%) | P value |
|---|---|---|---|---|
| Total number of patients | N = 76 | N = 38 | N = 38 | |
| Age in years [median (IQR)] | 47 (38–50) | 45 (35–49) | 48 (44–52) | 0.079 |
| Gender | ||||
| Female | 31 (40.8) | 17 (44.7) | 14 (36.8) | 0.641 |
| Male | 45 (59.2) | 21 (55.3) | 24 (63.2) | |
| Primary tumor location | ||||
| CCA NOS | 7 (9.2) | 4 (10.5) | 3 (7.9) | 0.937 |
| iCCA | 44 (57.9) | 22 (57.9) | 22 (57.9) | |
| eCCA | 16 (21.1) | 7 (18.4) | 9 (23.7) | |
| Gallbladder | 9 (11.8) | 5 (13.2) | 4 (10.5) | |
| Primary tumor resected | ||||
| No | 40 (52.6) | 18 (47.4) | 22 (57.9) | 0.491 |
| Yes | 36 (47.4) | 20 (52.6) | 16 (42.1) | |
| Adjuvant treatment | ||||
| Yes | 9 (11.8) | 3 (7.9) | 6 (15.8) | 0.230 |
| No | 27 (35.5) | 17 (44.7) | 10 (26.3) | |
| Not applicable | 40 (52.6) | 18 (47.4) | 22 (57.9) | |
| Total lines of treatment for unresectable/metastatic disease | ||||
| 1 | 19 (26.4) | 11 (30.6) | 8 (22.2) | 0.687 |
| 2 | 19 (26.4) | 8 (22.2) | 11 (30.6) | |
| > 2 | 34 (47.2) | 17 (47.2) | 17 (47.2) | |
| NA | 4 | 2 | 2 | |
| First line treatment regimen | ||||
| Capecitabine/5‐FU | 1 (1.9) | 1 (3.8) | / | 0.554 |
| Capecitabine/5‐FU + Oxaliplatin | 4 (7.5) | 2 (7.7) | 2 (7.4) | |
| Gemcitabine | 2 (3.8) | 1 (3.8) | 1 (3.7) | |
| Gemcitabine + Cisplatin | 35 (66.0) | 19 (73.1) | 16 (59.3) | |
| Gemcitabine + Oxaliplatin | 2 (3.8) | 1 (3.8) | 1 (3.7) | |
| Other | 9 (17.0) | 2 (7.7) | 7 (25.9) | |
| NA | 23 | 12 | 11 | |
| Platinum‐based first line regimen | ||||
| Yes | 49 (92.4) | 24 (92.3) | 25 (92.6) | 1.000 |
| No | 4 (7.5) | 2 (7.7) | 2 (7.4) | |
| NA | 23 | 12 | 11 | |
| MGMT promoter methylation status | ||||
| Yes | 5 (10.6) | 3 (12.5) | 2 (8.7) | 1.000 |
| No | 42 (89.4) | 21 (87.5) | 21 (91.3) | |
| NA | 29 | 14 | 15 | |
Cases were grouped according to the median MGMT mRNA expression value (27.9). No significant differences were observed between patients with high and low MGMT expression in terms of baseline characteristics. Methylation data were available for 47 (62%) cases, of which 5 (11%) had MGMT promoter hypermethylation. No association between MGMT expression and promoter methylation was observed (Wilcoxon P = 0.44, Fig. S9).
At data cut off, the median follow‐up was 39.4 months (18.0–66.5) and the median OS was 25.4 months (IQR 16.4–60.7); 53 (70%) patients received first‐line chemotherapy, with a median 1L‐PFS of 5.0 months (IQR 3.0–9.0); median 1L‐PFS and mOS were not affected by use of platinum in 1‐line chemotherapy (mPFS 5.0 vs 5.7, P = 0.8; mOS 20.9 vs 71.4, P = 0.1).
Patients with lower MGMT mRNA expression showed significantly poorer OS (mOS 20.4 vs 31.7 months, unadjusted HR 1.89; 95% CI 1.01–3.56; P = 0.043), which was confirmed in a multivariable model accounting for patients' age, gender, and primary tumor location, prior tumor resection and use of adjuvant therapy (Fig. 3A and Table S4). For 1L‐PFS, MGMT mRNA expression showed no significant impact (mPFS 5.0 vs 5.2 months, unadjusted HR 1.27; 95% CI 0.72–2.22; P = 0.405), and a multivariate model showed only a trend for interaction (P = 0.115) between MGMT expression and use of platinum‐based chemotherapy (Fig. 3B and Table S5). Concerning DNA methylation data, no reliable association with survival outcomes could be observed, both in terms of OS and PFS, as this analysis was limited by the low number of cases and events (n = 2) in the subgroup of patients with MGMT promoter hypermethylation.
Fig. 3.
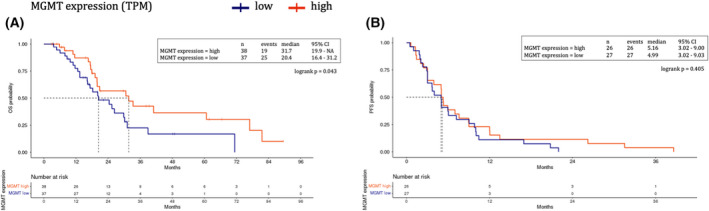
MGMT expression assessed by RNAseq (TPM values). Overall survival (OS) (A) and first‐line progression‐free survival (B) represented through Kaplan–Meier curves according to MGMT expression in the MASTER BTC cohort. Patients with lower MGMT mRNA expression (blue) showed significantly poorer OS as compared with patients with high MGMT expression (red) (A). MGMT mRNA expression had no significant impact on progression‐free survival of of patients treated with first‐line systemic treatment (B). Dotted lines indicate the median survival time. MGMT, O 6‐methylguanine‐DNA methyltransferase; TPM, transcripts per kilobase million; RNAseq, RNA sequence; MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers. [Colour figure can be viewed at wileyonlinelibrary.com]
3.3. Case series of temozolomide‐treated BTC patients
Based on the published clinical activity of TMZ in MGMT methylated colorectal cancers [18, 20], four patients with no other therapeutic options were treated at INT with TMZ‐based therapies from December 2018 until September 2019. Among these, two patients were treated with TMZ plus irinotecan (TEMIRI regimen) [18], while two patients received single‐agent TMZ. The clinical characteristics of these patients are detailed in Table S6. Of note, all but one cases were affected by iCCA, and all were previously treated with at least one platinum‐based chemotherapy. Median MGMT promoter methylation value was 12 (range 9–49), and three patients were evaluated for MGMT expression by IHC, all resulting negative. Figure S10 shows the swimmer plot of PFS during TMZ‐based treatment: three patients had stable disease as best response, with a PFS ranging from 2.0 to 6.1 months overall. No new safety signals were observed.
4. Discussion
Here we showed that both a high MGMT promoter hypermethylation and low/absent MGMT expression are associated with worse OS in patients with advanced BTC. Our evidence was consistently validated in two independent cohorts of patients and by investigating different assays including methylation‐specific (MSP) PCR, IHC, and RNA‐seq. Previous studies investigated the frequency of MGMT promoter hypermethylation in BTC [29, 30] and its role in tumor progression [31, 32] with inconclusive results, mostly due to the small sample size, the lack of clinical information, and the variability of assays used for testing this biomarker. Whether MGMT inactivation may be associated with a more aggressive behavior and which are the mechanisms causing this is not yet fully elucidated. However, MGMT loss is linked with an increased susceptibility to acquiring other mutations in both oncogenes and tumor suppressor genes, thus potentially stimulating tumor progression and poorer prognosis [17].
The prevalence of MGMT promoter hypermethylation in the Italian cohort was 18%, which is clinically relevant in these rare cancers. Furthermore, despite the over‐representation of iCCAs in the Italian cohort, we found a greater proportion of MGMT hyper‐methylated samples in eCCA and GBC. Given the heterogeneity of the disease, there is an unmet need for new therapeutic targets, especially for patients lacking targeted options such as those with eCCA and GBC, since most of the known actionable molecular alterations are usually found in iCCA. Therefore, our finding is potentially important from a clinical point of view, given the increasing body of evidence on efficacy for TMZ‐based regimens in MGMT‐methylated gastrointestinal tumors and especially colorectal cancer [19, 20, 33, 34]. TMZ may indeed be considered as a ‘targeted chemotherapy’ and even an agnostic investigational option in cancers with MGMT inactivation.
MGMT hypermethylation did not show an impact in terms of 1L‐PFS, probably due to the heterogeneity of treatments administered in this retrospective series. However, we found a significant interaction between the use of platinum‐based chemotherapy and MGMT promoter hypermethylation, despite these data should be interpreted with caution, given the non‐randomized nature of our treatment groups. From a biological perspective, our data are consistent with the growing evidence suggesting that MGMT may be involved in platinum‐induced DNA damage response (DDR) by playing a role in the homologous recombination signaling in cancer cells [35]. The latter consideration is particularly interesting given the amount of new evidence regarding possible combinations of TMZ with DDR inhibitors such as Poly‐ADP‐ribose polymerase (PARP) [36, 37] and ataxia telangiectasia mutated and Rad3‐related (ATR) inhibitors [38]. Furthermore, various preclinical and clinical observations have linked acquired resistance to TMZ to the emergence of alterations in the mismatch repair system and increased tumor mutational burden, that could be exploited in combinations of TMZ and immune checkpoint inhibitors [21, 39]. Here, we reported four cases that were treated with TMZ‐based regimens, with three patients experiencing stable disease as best response. Though we cannot derive conclusions from such a small number of patients, these patients were heavily pretreated and not strictly selected according to a MGMT methylation cutoff, thus highlighting the opportunity to better investigate the potential activity of TMZ in molecularly hyper‐selected subgroups and as part of combination regimens with potentially synergic drugs [18, 40]. Interestingly, the patient achieving the longest PFS had a lower methylation value compared to others; in this regard, the results may be explained by the fact that tumor MGMT expression was negative, other than that the patient was less pretreated than the others and received a combination regimen (TEMIRI).
In the Italian cohort, we explored whether any molecular alteration was enriched in cases with MGMT inactivation, but no clear pattern was observed. The lack of significant association with IDH1/2 mutations in the high MGMT methylation group is somewhat unexpected, given that these mutations ultimately lead to the formation of oncometabolite 2‐hydroxyglutarate (2HG), which has been associated with hypermethylator phenotype [CpG island methylator phenotype (CIMP)] and MGMT silencing [41, 42, 43, 44, 45]. However, given the overall rarity of individual molecular alterations and the non‐uniformity of the molecular characterization performed in this cohort, further studies on larger datasets are required.
Our analysis has limitations, mainly originating in the retrospective nature of the investigation in both cohorts, which included heterogeneous series of patients in terms of primary tumor origin, molecular profiling, and adopted treatments. Concerning the Italian cohort, limited evidence could be derived from IHC analysis, which was performed only on ~ 60% of cases. As for the MASTER BTC cohort, OS was available from the first tumor diagnosis instead of diagnosis of advanced disease, data regarding the classification of different subtypes of eCCA (pCCA and dCCA) were not available and only a small number of cases were profiled with methylation analysis, thus limiting the evidence derived from this data layer.
Both cohorts included only patients with advanced disease, either at initial diagnosis or at metachronous relapse after surgery. However, while the Italian cohort also included patients with rapidly progressing disease and treated with best supportive care, as per clinical practice, all patients in the MASTER BTC cohort were pretreated with standard chemotherapy options and had optimal ECOG PS at enrolment. Of note, highly methylated cases in the Italian cohort more frequently had worse baseline ECOG PS and did not receive first‐line platinum‐based chemotherapy. This does not seem to influence the results of our study, since MGMT status was confirmed as an independent negative prognostic factor for OS in the multivariable models that considered other prognostic variables in both cohorts. Overall, despite these differences, the two cohorts are well representative of this rare tumor entity: median 1L‐PFS was in line with literature and real‐life data, while the OS is coherent with the availability of new treatment options in later lines of treatment and the patients' selection that inevitably occurs in high‐volume cancer centers. While detailed data about later lines of treatment are not available, 26% and 47% of Italian and German patients were treated with more than two lines, respectively, and this likely had an impact on the OS.
Finally, in both cohorts, MGMT promoter methylation and expression did not show an optimal concordance, as previously reported in other works [17]. There is no unique explanation to the discordance between methylation and protein expression results and the regulation of MGMT inactivation is far from being fully clarified. Methylation is probably not the only mechanisms behind MGMT protein silencing, as several transcription factors, such as secreted protein 1 (SP1), CCAAT‐enhancer‐binding proteins (CEBP), activator protein 1 (AP1), hypoxia inducible factor‐1α (HIF‐1α), and the p65 (RELA) subunit of the nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) [46], bind to the MGMT promoter to induce or suppress MGMT expression [47]. Furthermore, MGMT promoter CpG islands may present a differential pattern of methylation along the region, with some CpGs being more important in terms of gene transcription and correlation with protein expression. Finally, methylation data in both cohorts may be influenced by contamination of the tumor sample from surrounding normal tissue, and spatial and temporal tumoral heterogeneity may influence the results of IHC.
Given these considerations, our findings are in line with previous evidence, suggesting that these methodologies should be used as complementary and not interchangeably in clinical practice [17] and that IHC could be regarded as a method for refining the discriminative ability of MGMT assessment in terms of survival. This approach has shown promising results in metastatatic colorectal cancer, as proved by the recently published MAYA trial [21].
Overall, despite these limitations, in these large series of BTCs cases treated at three European comprehensive cancer centers and tested with three different methodologies, our results are a convincing proof of concept of the negative prognostic impact of MGMT inactivation in BTCs.
5. Conclusions
We provide the first clear evidence that MGMT inactivation is a frequent epigenetic alteration in BTC patients, with a relevant prognostic impact. Based on our data, we believe that MGMT is indeed a new piece of the molecular puzzle of BTCs and could serve to prospectively explore the efficacy of alkylating agents in MGMT‐silenced BTCs in future trials, possibly in combinations with DNA damaging agents or novel DDR inhibitors.
Conflict of interest
MN: Travel expenses from Celgene, speaker honorarium from Accademia della Medicina; honoraria from Sandoz, Medpoint SRL for editorial collaboration. Consultant honoraria from EMD Serono, Basilea Pharmaceutica, Incyte and MSD Italia. FM: honoraria from SERVIER, research grant from Incyte. SP: Consultant/Advisory role: Ipsen, Novartis, Pfizer, Advanced accelerator Application AAA, Merk. GP: honoraria from Foundation Medicine. FP: honoraria from Amgen, Roche, Sanofi, Bayer, Servier, Merck‐Serono, Lilly, MSD, Astrazeneca; advisory role with Amgen, Bayer, Servier, Merk‐Serono, MSD and research grants from Bristol‐Myers Squibb, AstraZeneca and Incyte. AIRC under IG 2019 – ID. 23 624 project – P.I. Pietrantonio Filippo. MDB: honoraria from Lilly, MSD Oncology, Servier, consulting/advisory role with Lilly, MSD oncology, research grant from Lilly, travel/accommodation expenses from Roche and Sanofi. SF: Consulting or advisory board membership: Bayer, Illumina, Roche; honoraria: Amgen, Eli Lilly, PharmaMar, Roche; research funding: AstraZeneca, Pfizer, PharmaMar, Roche; travel or accommodation expenses: Amgen, Eli Lilly, Illumina, PharmaMar, Roche. FdB: Consultant Advisory Board: Roche, EMD Serono, NMS Nerviano Medical Science, Sanofi, MSD, Novartis, Incyte, BMS, Menarini. Speaker: BMS, Healthcare Research & Pharmacoepidemiology, Merck Group, ACCMED, Nadirex, MSD, Pfizer, Servier, Sanofi, Roche, AMGEN, Incyte, Dephaforum. Principal Investigator for Novartis, F.Hoffmann‐LaRoche Ltd, BMS, Ignyta Operating INC, Merck Sharp & Dohme Spa, Kymab, Pfizer, Tesaro, MSD, MedImmune LCC, Exelixis Inc., LOXO Oncology Incorporated, DAICHI SANKIO Dev. Limited, Basilea Pharmaceutica International AG, Janssen‐Cilag International NV, Merck KGAA. All other authors declare no conflict of interest.
Author contributions
MN, FdB, MDB, FN, FP: Study concept and design; All authors: Acquisition of data; MN, FN, JH, AD, DBL, FdB, FP: Analysis and interpretation of data; MN, FN, DBL, FP, FM: Drafting of the manuscript; All authors: Manuscript revision and input; FN, LA, AV: Statistical analysis; MN, BCK, DBL, DH, SF, FdB: Study supervision.
Supporting information
Fig. S1. Study flowchart for the Italian cohort.
Fig. S2. Graphical representation of Cox regression model evaluating MGMT promoter methylation impact on patients' OS, with non‐linear effects handled by restricted cubic splines (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase; OS, overall survival.
Fig. S3. Graphical comparison of Minimal Depth and VIMP rankings for OS prediction. Covariates ranking in the lower left quadrant were included in multivariable Cox Proportional Hazard Model (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase; VIMP, variable importance; OS, overall survival.
Fig. S4. Graphical representation of Cox regression model evaluating MGMT promoter methylation on impact on patients' 1L‐PFS, with non‐linear effects handled by restricted cubic splines (Italian cohort). 1L‐PFS; first‐line progression‐free survival; MGMT, O 6‐methylguanine‐DNA methyltransferase.
Fig. S5. Graphical comparison of Minimal Depth and VIMP rankings for PFS prediction. Covariates ranking in the lower left quadrant were included in multivariable Cox Proportional Hazard Model (Italian cohort). VIMP, variable importance; PFS, progression‐free survival.
Fig. S6. (a,b). Progression‐free survival represented through Kaplan–Meier curves according to use of platinum‐based chemotherapy (CT) in patients with high (>14%, 6a) and low (≤14%, 6b) MGMT promoter methylation status (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase.
Fig. S7. (a,b). Overall survival and progression‐free survival represented through Kaplan–Meier curves according to MGMT status assessed by IHC reported on three levels. MGMT, O 6‐methylguanine‐DNA methyltransferase; IHC, immunohistochemistry.
Fig. S8. Oncoplot of molecular alterations in patients profiled with the IonTorrent® or FoundationOne®CDx panel in the INT cohort.
Fig. S9. MGMT mRNA expression (TPM values) according to MGMT promoter methylation in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers; MGMT, O 6‐methylguanine‐DNA methyltransferase; TPM, transcripts per kilobase million.
Fig. S10. Swimmer plot of patients treated with temozolomide‐based regimens. MGMT promoter methylation values are reported right to each patients' bar. MGMT, O 6‐methylguanine‐DNA methyltransferase.
Table S1. Patients' characteristics according to MGMT promoter methylation (with the 14% cutoff) in the Italian cohort. MGMT, O 6‐methylguanine‐DNA methyltransferase.
Table S2. Multivariable Cox proportional hazards model for progression‐free survival in the Italian cohort. Missing data of covariates included in the model were imputed.
Table S3. Patients' characteristics according to MGMT expression by IHC in the Italian cohort. MGMT, O 6‐methylguanine‐DNA methyltransferase; IHC, immunohistochemistry.
Table S4. Multivariable Cox proportional hazards model for overall survival in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers.
Table S5. Multivariable Cox proportional hazards model for progression‐free survival in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers.
Table S6. Clinical characteristics of patients treated with temozolomide.
Acknowledgements
The authors would like to thank the patients participating in the study and their families. This research was partially funded by the Italian Ministry of Health “Ricerca Corrente” Funds. LA was supported by the Accelerator Award #29374 through CRUK‐AIRC partnership. The MASTER program was supported by the NCT Molecular Precision Oncology, grant H021 from the DKFZ‐Heidelberg Center for Personalized Oncology, and the DKTK Joint Funding Program.
Data availability statement
Clinical data of the Italian and German cohorts were collected through an electronic database and are available upon reasonable request to the corresponding author. Sequencing data of the MASTER program have been deposited in the European Genome‐phenome Archive (https://www.ebi.ac.uk/ega/datasets) under accession EGAS00001004813.
References
- 1. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second‐line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol. 2014;25:2328–38. 10.1093/annonc/mdu162 [DOI] [PubMed] [Google Scholar]
- 2. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–81. 10.1056/NEJMoa0908721 [DOI] [PubMed] [Google Scholar]
- 3. Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH‐mutant molecular profiles. Cell Rep. 2017;19:2878–80. 10.1016/j.celrep.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole‐Genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017;7:1116–35. 10.1158/2159-8290.CD-17-0368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic Cholangiocarcinomas: potential targets for intervention. Clin Cancer Res. 2018;24:4154–61. 10.1158/1078-0432.CCR-18-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montal R, Sia D, Montironi C, Leow WQ, Esteban‐Fabro R, Pinyol R, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol. 2020;73:315–27. 10.1016/j.jhep.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakamura H. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003–10. 10.1038/ng.3375 [DOI] [PubMed] [Google Scholar]
- 8. Abou‐Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al‐Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open‐label, phase 2 study. Lancet Oncol. 2020;21:671–84. 10.1016/S1470-2045(20)30109-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open‐label, single‐arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6:803–15. 10.1016/S2468-1253(21)00196-5 [DOI] [PubMed] [Google Scholar]
- 10. Abou‐Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1‐mutant, chemotherapy‐refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol. 2020;21:796–807. 10.1016/S1470-2045(20)30157-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hainsworth JD, Meric‐Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open‐label, phase IIa multiple basket study. J Clin Oncol. 2018;36:536–42. 10.1200/JCO.2017.75.3780 [DOI] [PubMed] [Google Scholar]
- 12. Meric‐Bernstam F, Hanna DL, El‐Khoueiry AB, Kang Y‐K, Oh D‐Y, Chaves JM, et al. Zanidatamab (ZW25) in HER2‐positive biliary tract cancers (BTCs): results from a phase I study. J Clin Oncol. 2021;39:299–9. 10.1200/JCO.2021.39.3_suppl.299 [DOI] [Google Scholar]
- 13. Subbiah V, Lassen U, Elez E, Italiano A, Curigliano G, Javle M, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)‐mutated biliary tract cancer (ROAR): a phase 2, open‐label, single‐arm, multicentre basket trial. Lancet Oncol. 2020;21:1234–43. 10.1016/S1470-2045(20)30321-1 [DOI] [PubMed] [Google Scholar]
- 14. Lamarca A, Barriuso J, McNamara MG, Valle JW. Biliary tract Cancer: state of the art and potential role of DNA damage repair. Cancer Treat Rev. 2018;70:168–77. 10.1016/j.ctrv.2018.09.002 [DOI] [PubMed] [Google Scholar]
- 15. Hegi ME. Gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 16. Bae SI. Inactivation of O6‐methylguanine‐DNA methyltransferase by promoter CpG Island hypermethylation in gastric cancers. Br J Cancer. 2002;86:1888–92. 10.1038/sj.bjc.6600372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pietrantonio F, Randon G, Romagnoli D, Di Donato S, Benelli M, de Braud F. Biomarker‐guided implementation of the old drug temozolomide as a novel treatment option for patients with metastatic colorectal cancer. Cancer Treat Rev. 2020;82:101935. 10.1016/j.ctrv.2019.101935 [DOI] [PubMed] [Google Scholar]
- 18. Morano F, Corallo S, Niger M, Barault L, Milione M, Berenato R, et al. Temozolomide and irinotecan (TEMIRI regimen) as salvage treatment of irinotecan‐sensitive advanced colorectal cancer patients bearing MGMT methylation. Ann Oncol. 2018;29:1800–6. 10.1093/annonc/mdy197 [DOI] [PubMed] [Google Scholar]
- 19. Pietrantonio F, de Braud F, Milione M, Maggi C, Iacovelli R, Dotti KF, et al. Dose‐dense temozolomide in patients with MGMT‐silenced Chemorefractory colorectal Cancer. Target Oncol. 2016;11:337–43. 10.1007/s11523-015-0397-2 [DOI] [PubMed] [Google Scholar]
- 20. Pietrantonio F, Perrone F, de Braud F, Castano A, Maggi C, Bossi I, et al. Activity of temozolomide in patients with advanced chemorefractory colorectal cancer and MGMT promoter methylation. Ann Oncol. 2014;25:404–8. 10.1093/annonc/mdt547 [DOI] [PubMed] [Google Scholar]
- 21. Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, et al. Temozolomide followed by combination with low‐dose ipilimumab and nivolumab in patients with microsatellite‐stable, O(6)‐methylguanine‐DNA methyltransferase‐silenced metastatic colorectal Cancer: the MAYA trial. J Clin Oncol. 2022;40:1562–73. 10.1200/jco.21.02583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hullein J, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov. 2021;11:2780–95. 10.1158/2159-8290.CD-21-0126 [DOI] [PubMed] [Google Scholar]
- 23. Cremolini C, Morano F, Moretto R, Berenato R, Tamborini E, Perrone F, et al. Negative hyper‐selection of metastatic colorectal cancer patients for anti‐EGFR monoclonal antibodies: the PRESSING case‐control study. Ann Oncol. 2017;28:3009–14. 10.1093/annonc/mdx546 [DOI] [PubMed] [Google Scholar]
- 24. FoundationOne CDX . https://www.foundation‐medicine.com/genomic‐testing/foundation‐one‐cdx (2018).
- 25. Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP‐status. Acta Neuropathol. 2012;124:547–60. 10.1007/s00401-012-1016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hothorn T, Lausen B. On the exact distribution of maximally selected rank statistics. Comput Stat Data Anal. 2003;43:121–37. 10.1016/S0167-9473(02)00225-6 [DOI] [Google Scholar]
- 27. Schemper M, Smith TL. A note on quantifying follow‐up in studies of failure time. Control Clin Trials. 1996;17:343–6. 10.1016/0197-2456(96)00075-x [DOI] [PubMed] [Google Scholar]
- 28. Ishwaran H, Kogalur UB. Consistency of random survival forests. Stat Probab Lett. 2010;80:1056–64. 10.1016/j.spl.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S, Kim WH, Jung HY, Yang MH, Kang GH. Aberrant CpG Island methylation of multiple genes in intrahepatic cholangiocarcinoma. Am J Pathol. 2002;161:1015–22. 10.1016/S0002-9440(10)64262-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang B, House MG, Guo M, Herman JG, Clark DP. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod Pathol. 2005;18:412–20. 10.1038/modpathol.3800287 [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Li Z, Chen J, Du Y, Song W, Xuan Z, et al. Downregulation of MGMT promotes proliferation of intrahepatic cholangiocarcinoma by regulating p21. Clin Transl Oncol. 2020;22:392–400. 10.1007/s12094-019-02140-9 [DOI] [PubMed] [Google Scholar]
- 32. Koga Y, Kitajima Y, Miyoshi A, Sato K, Kitahara K, Soejima H, et al. Tumor progression through epigenetic gene silencing of O(6)‐methylguanine‐DNA methyltransferase in human biliary tract cancers. Ann Surg Oncol. 2005;12:354–63. 10.1245/ASO.2005.07.020 [DOI] [PubMed] [Google Scholar]
- 33. Amatu A, Sartore‐Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, et al. Promoter CpG Island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res. 2013;19:2265–72. 10.1158/1078-0432.CCR-12-3518 [DOI] [PubMed] [Google Scholar]
- 34. Calegari MA, Inno A, Monterisi S, Orlandi A, Santini D, Basso M, et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br J Cancer. 2017;116:1279–86. 10.1038/bjc.2017.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen SH, Huang WT, Kao WC, Hsiao SY, Pan HY, Fang CW, et al. O6‐methylguanine‐DNA methyltransferase modulates cisplatin‐induced DNA double‐strand breaks by targeting the homologous recombination pathway in nasopharyngeal carcinoma. J Biomed Sci. 2021;28:2. 10.1186/s12929-020-00699-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farago AF, Yeap BY, Stanzione M, Hung YP, Heist RS, Marcoux JP, et al. Combination Olaparib and temozolomide in relapsed small‐cell lung Cancer. Cancer Discov. 2019;9:1372–87. 10.1158/2159-8290.Cd-19-0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanna C, Kurian KM, Williams K, Watts C, Jackson A, Carruthers R, et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro Oncol. 2020;22:1840–50. 10.1093/neuonc/noaa104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jackson CB, Noorbakhsh SI, Sundaram RK, Kalathil AN, Ganesa S, Jia L, et al. Temozolomide sensitizes MGMT‐deficient tumor cells to ATR inhibitors. Cancer Res. 2019;79:4331–8. 10.1158/0008-5472.Can-18-3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–20. 10.1038/nature24673 [DOI] [PubMed] [Google Scholar]
- 40. Pietrantonio F, Lobefaro R, Antista M, Lonardi S, Raimondi A, Morano F, et al. Capecitabine and temozolomide versus FOLFIRI in RAS‐mutated, MGMT‐methylated metastatic colorectal Cancer. Clin Cancer Res. 2020;26:1017–24. 10.1158/1078-0432.Ccr-19-3024 [DOI] [PubMed] [Google Scholar]
- 41. Figueroa ME. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hughes LAE, Melotte V, de Schrijver J, de Maat M, Smit VTHBM, Bovée JVMG, et al. The CpG Island methylator phenotype: what's in a name? Cancer Res. 2013;73:5858–68. 10.1158/0008-5472.CAN-12-4306 [DOI] [PubMed] [Google Scholar]
- 43. Noushmehr H. Identification of a CpG Island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. 10.1016/j.ccr.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Turcan S. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (London). 2012;483:479–83. 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van den Bent MJ. MGMT‐STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19:5513–22. 10.1158/1078-0432.CCR-13-1157 [DOI] [PubMed] [Google Scholar]
- 46. Rahman MA, Gras Navarro A, Brekke J, Engelsen A, Bindesbøll C, Sarowar S, et al. Bortezomib administered prior to temozolomide depletes MGMT, chemosensitizes glioblastoma with unmethylated MGMT promoter and prolongs animal survival. Br J Cancer. 2019;121:545–55. 10.1038/s41416-019-0551-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lv W, Li Q, Jia B, He Y, Ru Y, Guo Q, et al. Differentiated embryonic chondrocyte‐expressed gene 1 promotes temozolomide resistance by modulating the SP1‐MGMT axis in glioblastoma. Am J Transl Res. 2021;13:2331–49. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Study flowchart for the Italian cohort.
Fig. S2. Graphical representation of Cox regression model evaluating MGMT promoter methylation impact on patients' OS, with non‐linear effects handled by restricted cubic splines (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase; OS, overall survival.
Fig. S3. Graphical comparison of Minimal Depth and VIMP rankings for OS prediction. Covariates ranking in the lower left quadrant were included in multivariable Cox Proportional Hazard Model (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase; VIMP, variable importance; OS, overall survival.
Fig. S4. Graphical representation of Cox regression model evaluating MGMT promoter methylation on impact on patients' 1L‐PFS, with non‐linear effects handled by restricted cubic splines (Italian cohort). 1L‐PFS; first‐line progression‐free survival; MGMT, O 6‐methylguanine‐DNA methyltransferase.
Fig. S5. Graphical comparison of Minimal Depth and VIMP rankings for PFS prediction. Covariates ranking in the lower left quadrant were included in multivariable Cox Proportional Hazard Model (Italian cohort). VIMP, variable importance; PFS, progression‐free survival.
Fig. S6. (a,b). Progression‐free survival represented through Kaplan–Meier curves according to use of platinum‐based chemotherapy (CT) in patients with high (>14%, 6a) and low (≤14%, 6b) MGMT promoter methylation status (Italian cohort). MGMT, O 6‐methylguanine‐DNA methyltransferase.
Fig. S7. (a,b). Overall survival and progression‐free survival represented through Kaplan–Meier curves according to MGMT status assessed by IHC reported on three levels. MGMT, O 6‐methylguanine‐DNA methyltransferase; IHC, immunohistochemistry.
Fig. S8. Oncoplot of molecular alterations in patients profiled with the IonTorrent® or FoundationOne®CDx panel in the INT cohort.
Fig. S9. MGMT mRNA expression (TPM values) according to MGMT promoter methylation in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers; MGMT, O 6‐methylguanine‐DNA methyltransferase; TPM, transcripts per kilobase million.
Fig. S10. Swimmer plot of patients treated with temozolomide‐based regimens. MGMT promoter methylation values are reported right to each patients' bar. MGMT, O 6‐methylguanine‐DNA methyltransferase.
Table S1. Patients' characteristics according to MGMT promoter methylation (with the 14% cutoff) in the Italian cohort. MGMT, O 6‐methylguanine‐DNA methyltransferase.
Table S2. Multivariable Cox proportional hazards model for progression‐free survival in the Italian cohort. Missing data of covariates included in the model were imputed.
Table S3. Patients' characteristics according to MGMT expression by IHC in the Italian cohort. MGMT, O 6‐methylguanine‐DNA methyltransferase; IHC, immunohistochemistry.
Table S4. Multivariable Cox proportional hazards model for overall survival in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers.
Table S5. Multivariable Cox proportional hazards model for progression‐free survival in the MASTER BTC cohort. MASTER BTC Molecularly Aided Stratification For Tumor Eradication Research on Biliary Tract Cancers.
Table S6. Clinical characteristics of patients treated with temozolomide.
Data Availability Statement
Clinical data of the Italian and German cohorts were collected through an electronic database and are available upon reasonable request to the corresponding author. Sequencing data of the MASTER program have been deposited in the European Genome‐phenome Archive (https://www.ebi.ac.uk/ega/datasets) under accession EGAS00001004813.