

Abstract
The increasing availability of short‐read whole genome sequencing (WGS) provides unprecedented opportunities to study ecological and evolutionary processes. Although loci of interest can be extracted from WGS data and combined with target sequence data, this requires suitable bioinformatic workflows. Here, we test different assembly and locus extraction strategies and implement them into secapr, a pipeline that processes short‐read data into multilocus alignments for phylogenetics and molecular ecology analyses. We integrate the processing of data from low‐coverage WGS (<30×) and target sequence capture into a flexible framework, while optimizing de novo contig assembly and loci extraction. Specifically, we test different assembly strategies by contrasting their ability to recover loci from targeted butterfly protein‐coding genes, using four data sets: a WGS data set across different average coverages (10×, 5× and 2×) and a data set for which these loci were enriched prior to sequencing via target sequence capture. Using the resulting de novo contigs, we account for potential errors within contigs and infer phylogenetic trees to evaluate the ability of each assembly strategy to recover species relationships. We demonstrate that choosing multiple sizes of kmer simultaneously for assembly results in the highest yield of extracted loci from de novo assembled contigs, while data sets derived from sequencing read depths as low as 5× recovers the expected species relationships in phylogenetic trees. By making the tested assembly approaches available in the secapr pipeline, we hope to inspire future studies to incorporate complementary data and make an informed choice on the optimal assembly strategy.
Keywords: de novo assembly, loci extraction, low‐coverage whole genome sequencing, secapr, target sequence capture
1. INTRODUCTION
Until recently, the most cost‐efficient approaches to obtain genome‐wide data for phylogenomic and molecular ecology studies relied on genomic subsampling using size selection or enrichment prior to the sequencing process (genomic partitioning and reduced‐representation sequencing; Lemmon & Lemmon, 2013); such approaches include restriction site‐associated DNA sequencing (RADseq) and target sequence capture of conserved genetic regions, such as exons or ultraconserved elements (UCEs; e.g., Andermann et al., 2020; Burbano et al., 2010; Davey & Blaxter, 2010; Faircloth et al., 2012; Lemmon & Lemmon, 2013). The continuing decrease of sequencing costs (currently, one megabase [Mbp] of raw DNA sequences costs ~0.01 US$; https://www.genome.gov/sequencingcostsdata) have made low‐coverage whole‐genome sequencing (WGS) more economically feasible, and WGS data are also becoming widely applied in ecological and evolutionary studies (e.g., Li et al., 2019; Olofsson et al., 2019). Although the implementation of flexible and user‐friendly post‐sequencing bioinformatic pipelines has flourished within the past 5 years, there is still a gap when it comes to integrating data coming from different sequencing approaches (e.g., WGS and reduced‐representation sequencing).
Loci of interest are often enriched in the laboratory prior to sequencing (in vitro enrichment; Albert et al., 2007; Gnirke et al., 2009) to achieve a sufficiently high read coverage for its processing into multilocus data sets. Moreover, the increasing volume of publicly available low read coverage WGS data provides a source for the bioinformatic (in silico) harvesting of these loci of interest. However, a guideline is still missing on how to assemble short reads most efficiently into contigs de novo (i.e., which parameters to use for contig assembly) when simultaneously working with data derived from low and high read genomic coverages of loci of interest, especially in cases in which the genome size of an organism is unknown or reference genomes are not available.
Some bioinformatic pipelines have been developed for the processing of unassembled WGS data into multiple sequence alignments. For example, the Phylogenomics from Low‐coverage Whole‐genome Sequencing pipeline (PLWS; Zhang, Ding, et al., 2019) runs iterative de novo contig assemblies using the minia3 assembler (Chikhi & Rizk, 2013). Although the PLWS pipeline is computationally efficient (e.g., 21 hexapod genomes spanning from 0.1 to 2 gigabases [Gbp] were assembled in a period of 2 to 24 h on 16 [GB] or 32 [GB] of RAM PCs), it is unclear whether other contig assemblers using multi‐kmer strategies (see Box 1) can recover more complete multilocus alignments. Alternatively, aTRAM (Allen et al., 2017) uses iterative blast searches (Altschul et al., 1990) to find matching reads within a library of loci of interest (references) and subsequently assemble them with different contig assemblers relying on single‐kmer strategies (Box 1). Other pipelines can extract and assemble repetitive and high‐copy number genomic regions such as mitochondrial loci and rDNA repeat regions. For instance, GRAbB (Brankovics et al., 2016) uses computationally efficient assemblers such as edena (Hernandez et al., 2008, 2014), whereas mitofinder (Allio et al., 2020) maximizes the use of UCE data by retrieving, assembling, and annotating nonenriched mitochondrial loci using multi‐kmer assemblers such as metaspades (Nurk et al., 2017). There are other software that extract loci of interest from metagenomes (anvi'o; Eren et al., 2021; also see: https://merenlab.org/2019/10/17/export‐locus), from assembled genomes (Costa et al., 2016; Jarvis et al., 2014), or even from DNA data archived in VCF files (seqtailor; Zhang, Boisson, et al., 2019). To our knowledge, however, no studies have yet attempted to integrate WGS data of various underlying read coverages into multilocus data sets that have comparable quality to those resulting from in vitro target capture data, while comparing assemblers and assembly strategies in the same pipeline.
BOX 1. Brief description of the assembly process.
Most assembler programs carry out three essential steps in order to assemble short reads into longer contigs: (1) Decomposing read sequences into kmers to improve efficiency of contig assembly by eliminating redundant short read overlaps; (2) Build a de Bruijn graph from the kmer overlapping information, which facilitates the connection of short reads; and (3) de Bruijn graph simplification. Decomposing read sequences into kmers requires the user to specify a kmer‐size lower than the short‐read length. A read is decomposed into n kmers by extracting the substring of kmer size length at each nucleotide position of the read. Then, all possible kmer are evaluated and two kmers connected if both overlap in kmer length size—1. Connections between kmers create a graph (de Bruijn graph) where nodes represent kmer sequences and edges represent connections. In the last step, the graph is simplified using information extracted from the reads and kmers themselves: coverage, distances, and pairing of reads. More details on the challenges of contig assembly and strategies to tackle those challenges are described in Sohn & Nahm (2018) and Liao et al. (2019).
abyss (Figure 2, left): Starting from paired‐end reads (PE reads), abyss version 2 (Jackman et al., 2017) implements two modes to extract the kmers using a single kmer‐size provided by the user. The first mode, implemented in abyss version 1 (Simpson et al., 2009), which is the one used in this study, builds a hash table from kmers across all reads. A hash table is a data structure that efficiently stores large amounts of information. Unlike spades, abyss distributes sections of the hash table using a Message‐Passing Interface (mpi) to independent cluster nodes in order to parallelize the process of building the de Bruijn graph. The graph is extended by evaluating the overlap between all kmers and later simplified by using read pairing information. The second mode (abyss version 2, Jackman et al., 2017) stores all possible kmers of length = kmer‐size and their relative position using a bloom filter (Bloom, 1970) with one or more hashing functions used for indexing the information. Then, abyss applies a user‐provided threshold to flag infrequent kmers as an attempt to remove potential sequencing errors. Reads that do not contain flagged kmers are then used to build and extend the de Bruijn graph, which is simplified using the paired‐end information to trim off branches (a series of kmers connected to the graph only at one end) and bubbles (alternative paths on the graph joint at both ends that arise from single nucleotide polymorphisms or sequencing errors).
spades (Figure 2, right): Contrary to abyss, spades simultaneously uses all possible kmers extracted from a range of kmer sizes (the default values are 21, 33, 55, 77, 99) to build the initial de Bruijn graph. This multi‐kmer approach capitalizes on the advantages of building a graph from short and long kmer‐sizes. Smaller kmer‐sizes minimize contig fragmentation by increasing the probability of finding overlapping kmers. However, small kmer‐sizes might face difficulties in resolving repetitive regions, and longer kmer‐sizes further improve the graph (Bankevich et al., 2012). spades runs a read correction step before assembling the first de Bruijn. The initial graph architecture is extracted, and a series of graph operations take place on it, leveraging information about kmer coverage, kmer‐to‐kmer distances, and paired information to simplify the graph and remove branches and bubbles. Once the graph is simplified, the reads retained are mapped back onto the graph in order to extract the extended contigs.
To improve best‐practices in de novo contig assembly from low‐coverage WGS reads and to demonstrate its integration with other types of reduced‐representation data, we expand the sequence capture processor pipeline (secapr – Andermann et al., 2018) to include the assembler spades (Bankevich et al., 2012). We also implement a new iterative assembly approach with the software abyss (Jackman et al., 2017; Simpson et al., 2009), in which contigs assembled with different sizes of read substrings—kmers—are combined and their orthology with reference sequences assessed to obtain new sets of contigs (which here we call a multi‐kmer approach; Box 1; Figure 2). secapr is a Python pipeline, available as a conda package for Linux, Windows, and MacOS, that automatically installs and executes software dependencies to obtain multilocus alignments from raw short sequencing reads. secapr was originally designed to process target sequence capture data of multisample data sets (see Figure 1 for pipeline workflow), inspired by the Phyluce pipeline workflow for UCEs (Faircloth, 2016).
FIGURE 2.
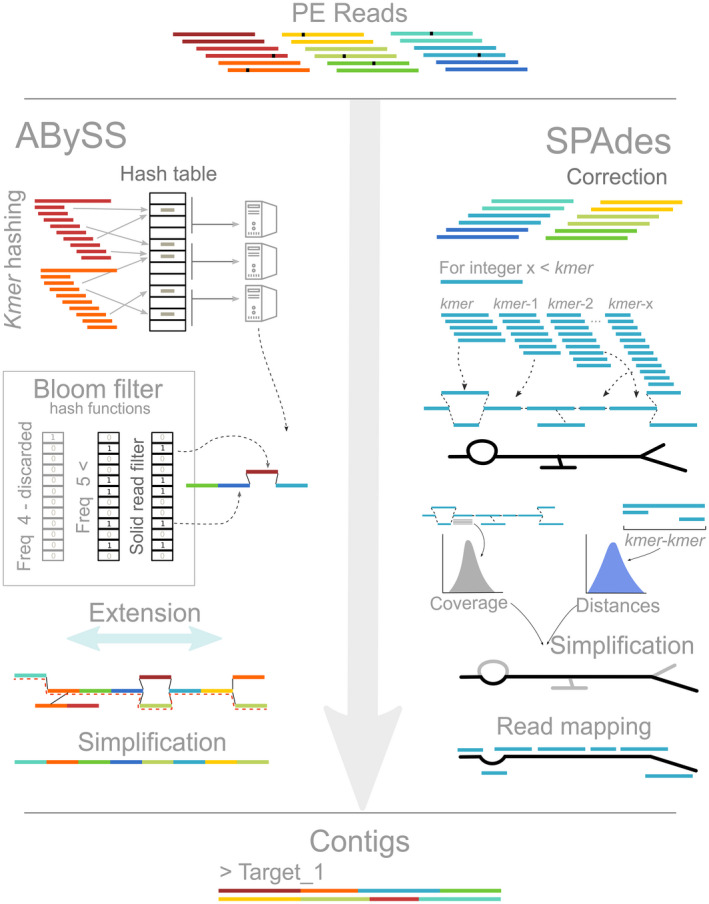
Flowchart summarizing the process of contig assemblage implemented in abyss (left, Jackman et al., 2017; Simpson et al., 2009) and spades (right, Bankevich et al., 2012). The chart briefly describes the de Bruijn graph construction and the differences between both assemblers. See Box 1 for more details
FIGURE 1.
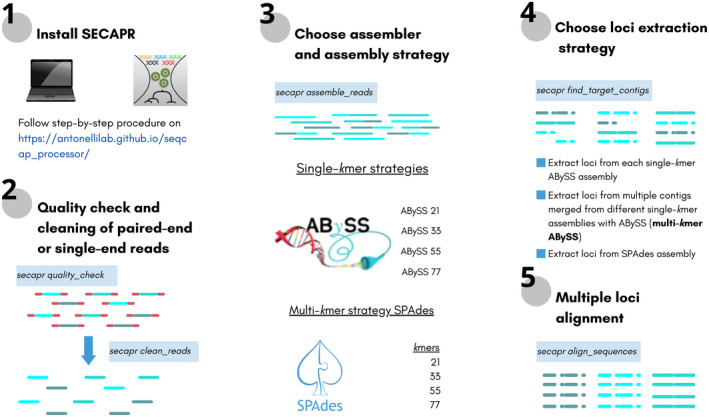
Schematic representation of the workflow implemented in this study using the secapr version 2.2.3 pipeline. The bash commands used in each step are shown inside coloured (blue) boxes
Starting from unassembled low coverage WGS data, the bioinformatic steps for their processing are: (1) sequence quality filtering and cleaning, (2) de novo contig assembly (when no reference genome is available for read mapping), (3) identification and extraction of loci of interest from assembled contigs and (4) alignment of multiple sequences. In the new version of secapr, we enhance its efficiency for processing WGS data by modifying steps (2)—assembly using abyss and now, spades—and (3) ‐ identification (orthology assessment) and extraction of loci of interest using blastz (Schwartz et al., 2003). We allow for parallelization of multiple jobs in these steps and concomitant processing of short sequencing reads derived from different types of library preparations (target sequence capture and WGS). We tested the updated secapr pipeline (now called secapr version 2.2.3) on target sequence capture and WGS data and assessed the efficiency of using both single‐kmer and multi‐kmer de novo contig assembly (see Box 1 for detailed information on genome assembly using spades and abyss) to recover more, and more complete, multilocus alignments. Our study proposes a way forward to process data from different sequencing approaches in a single bioinformatic pipeline, assessing the performance of different de novo contig assemblers and strategies to enhance the extraction of loci of interest from WGS data.
2. MATERIALS AND METHODS
2.1. Data
We used published low‐coverage WGS data (Li et al., 2019) and newly generated target sequence capture data from skipper butterflies (Lepidoptera: Hesperiidae: Eudaminae). The study group was representative of commonly studied taxa in molecular ecology, given the lack of annotated reference genomes and the need to integrate closely related (species within genera) and highly divergent (lineages within a subfamily) samples. We downloaded low‐coverage whole‐genome sequences of 10 butterfly species representing three Eudaminae tribes (Table S1; Li et al., 2019; Bioproject PRJNA464409). The WGS data were generated via 150 bp paired‐end Illumina sequencing (HiSeq X Ten platform) at an average of ~10× genome coverage, which is about 5 Gbp sequencing data per sample considering a representative genome size of Eudaminae ~500–600 Mbp (Shen et al., 2017).
To demonstrate how to integrate data from different sequencing approaches in a single bioinformatic pipeline, we generated target sequence capture data for different individuals representing the same 10 species in the low‐coverage WGS data (Table S1; BioProject PRJNA681152). We targeted 406 exons from protein‐coding genes using the butterfly 1.0 probe kit, which consists of 56,470 baits of 120 bp size each (Espeland et al., 2018). DNA was isolated from two to three butterfly legs using Qiagen DNA extraction kits following the manufacturer's instructions. RapidGenomics (FL, USA) prepared target enrichment libraries and conducted high‐throughput sequencing using paired‐end 150 bp on an Illumina NovaSeq platform.
Raw reads from the publicly available WGS and the newly generated target sequence capture data were jointly processed using secapr version 2.2.3. We used a Linux centos version 7.9 system on a dedicated cluster provided by the Czech National Computing Infrastructure Metacentrum. secapr version 2.2.3 can be freely downloaded and installed following the detailed documentation at https://github.com/AntonelliLab/seqcap_processor (Open Research section at the end).
2.2. Sequencing quality check and cleaning
The quality of raw Illumina reads from both WGS, and target sequence capture was checked using fastqc version 0.11.9 (Andrews, 2010; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) via the secapr version 2.2.3 pipeline using the command secapr quality_check. Target sequence capture data were filtered by removing low‐quality reads and trimming Illumina adapters using Trimmomatic (Bolger et al., 2014) via the secapr version 2.2.3 command secapr clean_reads. The average quality of the filtered, adapter‐free sequence capture data was assessed again using the command secapr quality_check.
2.3. De novo contig assembly
We evaluated the performance of abyss version 1.3.7 (Jackman et al., 2017; Simpson et al., 2009) and spades version 3.14.1 (Bankevich et al., 2012) with WGS and target sequence capture data. By default, spades uses six kmer values concurrently (21, 33, 55, 77, 99, and 127) and abyss uses a single‐kmer in each run with values of kmer up to 97. To make comparable evaluations of both assemblers, we ran abyss four times, each with a single‐kmer value of 21, 33, 55 or 77 and ran spades for the same kmer values concurrently with the command secapr assemble_reads.
2.4. Extraction of contigs containing loci of interest
We created 406 reference sequences representing each of the target sequence capture loci. The references are the consensus sequences of targeted exons from 129 unpublished Eudaminae samples to enhance matching with the assembled contigs.
We extracted contigs of interest from the WGS and target sequence capture assemblies using the alignment algorithm blastz (Schwartz et al., 2003) via secapr version 2.2.3 using secapr find_target_contigs. For secapr version 2.2.3, we developed a new approach to automatically extract loci of interest from multiple abyss runs, here termed the multi‐kmer abyss approach. For this, we combined contigs from all individual kmer assemblies with abyss (using kmers = 21, 33, 55, and 77) and used the command secapr find_target_contigs to identify matched contigs against the reference sequences. The approach consists of removing redundant contig matches by selecting the longest contig among multiple single‐kmer runs in abyss, and for each targeted locus using the argument ‐‐keep_paralogs. This approach is different from the automated de novo contig assembly implemented within spades. spades outputs the assembled contigs resulting from a de Bruijn graph incorporating all different kmers across all kmer sizes at once. For abyss multi‐kmer, the selection of targeted contigs from multiple assemblies under different kmer values is done after the assembly run, thus, specifically targeting the step (3) in the bioinformatic pipeline.
2.5. DNA alignments and performance assessment
We generated six multilocus data sets, each from a different assembly and locus extraction strategy (spades, four single‐kmer abyss—21, 33, 55, and 77—and multi‐kmer abyss) and then we aligned each locus within the data sets using mafft version 7.130 (Katoh & Standley, 2013) in secapr version 2.2.3 with the command secapr align_sequences. We produced final alignment data sets of the same length as the reference sequences by trimming the exon boundaries using the ‐‐addfull option in mafft.
To assess the performance of both assemblers and of their specific assembly strategies, we counted the number of recovered loci per sample across the six multilocus data sets. In addition, we evaluated the completeness of final alignments generated by each assembly strategy by comparing the alignments before and after excluding samples with more than 50% missing data (N bases) from the extracted loci alignments using Sequence_Cleaner (here named processed alignments) (https://github.com/metageni/Sequence‐Cleaner). Alignments for which we did not remove these sequences are named unprocessed alignments.
2.6. Producing data sets of different genome coverage
To evaluate the performance of assemblers and assembly strategies under different coverage depths, we randomly subsampled the original average 10× WGS coverage using BBTools/BBMap (Bushnell, 2021). We used the reformat.sh script to subsample 50% and 20% of the original WGS data set to generate new data sets with average genome coverages of 5× and 2×, respectively. These data sets were used in subsequent analyses to estimate the limitations of contig assembly and the variability in recovering loci of interest under lower genome sequencing coverages. We used reduced coverages in which differences in assembly performance may be more evident.
2.7. Statistical analysis
To statistically assess significant differences in the recovered number of loci amongst assembly approaches, we used linear mixed models using the r (R Core Team, 2020) package lme4 (Bates et al., 2015). We performed model selection using the corrected Akaike information criterion (AICc) to assess the best‐fit model that explains the estimated marginal means of recovered loci.
Our analyses involved two approaches. First, we compared the 10× coverage WGS data set and the target sequence capture data set to test if, overall, there are statistically significant differences between estimated means of recovered loci. We considered the 10 Eudaminae samples as random variables and assembly strategies (single‐kmer abyss—21, 33, 55, and 77, multi‐kmer abyss and spades), sequencing strategy (WGS or target sequence capture), and processing of final alignments (whether sequences with more than 50% missing data are excluded or not) as fixed variables. The number of recovered loci per sample was used as the effect variable. Second, we compared the performance of the assembly strategies under different average coverages of WGS data. For this, we followed the same protocol described above and considered the subsampled coverages (10×, 5× or 2×) as fixed variables. For both approaches, we made a full model considering the whole set of fixed variables, models with every possible combination of fixed variables and a null model with only our random variable. After checking that the residuals fulfilled the assumptions of linear models, we also calculated a full model considering the interactions between the fixed variables.
2.8. Phylogenetic inference
We inferred species trees for each sequencing data set and assembly strategy to assess if they produced phylogenetically informative alignments that were congruent with the expected phylogenetic relationships among species. First, we inferred gene trees from the alignment of each targeted locus using iq‐tree version 2.0.7 (Minh et al., 2020). We used ModelFinder (Kalyaanamoorthy et al., 2017) as implemented in iq‐tree to estimate the best substitution models, and we performed 1000 ultrafast bootstrap replicates (Hoang et al., 2017). Second, we used the sets of maximum‐likelihood gene trees to infer coalescent species trees in ASTRAL III (Zhang et al., 2018). Support was calculated as local posterior probabilities from quartet frequencies (Sayyari & Mirarab, 2016). Species trees were also inferred using each of the data sets with different sequencing coverages (WGS under 10×, 5×, and 2×) to evaluate the informativeness of multilocus alignments under decreasing amount of raw WGS data. We considered a species tree as well‐resolved if it successfully recovered the expected species relationships (Li et al., 2019). We also considered branches as well‐supported when they presented a local posterior probability support higher than 0.95. To assess discordance among species trees, we calculated symmetric distances (Robinson & Foulds, 1981) using the r package phangorn (Schliep, 2011). Since the symmetric distances only consider tree topology, we used a reference species tree generated with our WGS data set and under 5× depth of coverage, assembled with the multi‐kmer abyss approach, which retrieved the expected tree topology (as in Li et al., 2019). This reference was then compared against each of the species’ trees obtained with ASTRAL III.
2.9. Assessment of potential errors in de novo contig assemblies
To evaluate the accuracy of the de novo contig assembly, we aligned queries of the assembled target loci sequences for each species of our study, against subject reference sequences of the same species using blast (Altschul et al., 1990). Both queries and subject references were generated by our own alignments so we could assess the performance of our own implemented assembly strategies. We then calculated the percentage of errors as the number of nucleotide mismatches, multiplied by 100, and divided by the total length of the alignment for each locus. We used the number of mismatches as reported by blast because ambiguous nucleotides and Ns are not considered errors, and the alignment length only counts the positions in which the queries and the subject sequences match (but it includes potential gaps). This procedure allowed us to interpret any observed differences (mismatches) between alignments as potential assembly errors.
Our target sequence capture data set comes from different individuals of the same species present in the WGS data set. Therefore, differences from comparing WGS versus target sequence capture may as well result from within‐species polymorphisms and not necessarily due to assembly errors. We therefore carried out the analyses for the WGS and target sequence capture data sets separately. For the WGS data set, first we created the blast database of subject sequences for each species, each database including all the exons derived from the WGS 10× data set and assembled with either our multi‐kmer abyss approach or spades. We only used the 10× average coverage data as subjects since this data set generated the more complete alignments in terms of total recovered loci and number of present species within alignments, and because they produced well‐resolved species trees (see Section 3.3). Second, we performed per‐species blast alignments between the query sequences and the corresponding species’ subject sequences. The query sequences are those generated by multi‐kmer abyss and spades, and under different depths of coverage (10×, 5×, and 2×). Third, we calculated the percentage of error for each blast alignment (mismatches relative to matches), grouped the results by locus, averaged the percentage of error across species, and plotted the kernel density for every alignment. For the target sequence capture data set, we chose only the sequences assembled with spades to create subject sequences, since only these sequences generated well‐resolved species trees (see Section 3). Query sequences were generated for the multi‐kmer abyss approach and aligned against the subject. We also calculated the percentage of error for the blast alignment and plotted the averaged percentages. For all alignments, we used blast’s default values for blastn searches. Our factorial pairwise comparisons allowed us to quantify any differences in sequences coming from the same individual resulting from differences in assembly strategy (same data set, different assembly approach) and depths of coverage in the case of the WGS data set (same assembly approach, different coverages).
3. RESULTS
3.1. Sequence quality and computing performance
fastqc reports showed that per base sequence quality and per sequence quality score for clean and trimmed reads for both the WGS and target sequence capture data sets were above 28 Phred score. WGS data available from NCBI already contained adapter‐free Illumina reads.
In general, de novo contig assemblies for the target sequence capture data were faster in terms of CPU time and wall time in comparison to the WGS data set even at 2× average read depth coverage, and they also required less memory usage. Also, single‐kmer abyss runs used less computational resources than spades for CPU time, wall time and memory usage regardless of the type of data. A comprehensive table with running times and memory usage for assemblies can be found in Table S2.
3.2. Recovery of loci of interest and statistical analyses
First, we determined which of the six assembly strategies (four single‐kmer runs with abyss—kmers 21, 33, 55, 77, our novel multi‐kmer approach with abyss, and spades) maximized the recovery of loci of interest from low‐coverage WGS (average 10× read depth) and target sequence capture data. Our multi‐kmer abyss approach significantly recovered more loci of interest per sample in both data sets compared to all other strategies, including spades (Table 1). Nevertheless, spades significantly recovered more loci than all other single‐kmer strategies (Table 1). A summary of estimated marginal means for every assembly strategy and data set can be found in Table S3. AICc‐based model selection indicated that the best fit model considered the interactions among all our fixed variables (Tables S4 and S5).
TABLE 1.
Pairwise contrasts of the marginal estimated means of recovered loci per sample between multi‐kmer and single‐kmer strategies and between both multi‐kmer strategies
| Pairwise comparisons among strategies | Completeness of alignments | WGS 10× read depth | Target sequence capture | ||
|---|---|---|---|---|---|
| Estimate | p‐value | Estimate | p‐value | ||
| abyss Multikmer ‐ abyss 21 | Unprocessed | 136 | <.0001 | 126 | <.0001 |
| abyss Multikmer ‐ abyss 33 | 99 | <.0001 | 120 | <.0001 | |
| abyss Multikmer ‐ abyss 55 | 143 | <.0001 | 115 | <.0001 | |
| abyss Multikmer ‐ abyss 77 | 191 | <.0001 | 104 | <.0001 | |
| abyss Multikmer ‐ spades | 68 | <.0001 | 85 | <.0001 | |
| spades ‐ abyss 21 | 68 | <.0001 | 41 | .012 | |
| spades ‐ abyss 33 | 31 | .1311 | 35 | .0535 | |
| spades ‐ abyss 55 | 75 | <.0001 | 30 | .1386 | |
| spades ‐ abyss 77 | 123 | <.0001 | 19 | .6389 | |
| abyss Multikmer ‐ abyss 21 | Processed | 117 | <.0001 | 107 | <.0001 |
| abyss Multikmer ‐ abyss 33 | 81 | <.0001 | 102 | <.0001 | |
| abyss Multikmer ‐ abyss 55 | 129 | <.0001 | 100 | <.0001 | |
| abyss Multikmer ‐ abyss 77 | 180 | <.0001 | 92 | <.0001 | |
| abyss Multikmer ‐ spades | 27 | .2523 | 44 | .006 | |
| spades ‐ abyss 21 | 90 | <.0001 | 63 | <.0001 | |
| spades ‐ abyss 33 | 54 | .0002 | 58 | <.0001 | |
| spades ‐ abyss 55 | 102 | <.0001 | 56 | .0001 | |
| spades ‐ abyss 77 | 153 | <.0001 | 48 | .0015 | |
Contrasts are made considering an average of 10× coverage for the low‐coverage WGS and the target sequence capture data sets. Standard error = 12.4 for all pairwise comparisons; degrees of freedom = 477 for all pairwise comparisons
Second, we analysed the performance of our different assembly strategies using low‐coverage WGS data under varying degrees of read depth. Our multi‐kmer abyss strategy significantly recovered more loci of interest per sample than any other strategy for the 10× and the 5× coverage data sets, in both unprocessed and processed final alignments. spades, on the other hand, significantly recovered more loci of interest than any other strategy for the 2× coverage data set, and more than the single‐kmer strategies across all the data sets of varying average coverages (Table 2). Overall, the multi‐kmer strategies (multi‐kmer approach with abyss and spades) were significantly better than single‐kmer strategies in recovering more loci of interest (Tables 1 and 2; Figures 3 and 4).
TABLE 2.
Pairwise contrasts of the marginal estimated means of recovered loci per sample between multi‐kmer and single‐kmer strategies and between both multi‐kmer strategies for each of the subsets of depths of coverage of low‐coverage WGS data set
| Pairwise comparisons among strategies | Completeness of alignments | WGS 10× read depth | WGS 5× read depth | WGS 2× read depth | |||
|---|---|---|---|---|---|---|---|
| Estimate | p‐value | Estimate | p‐value | Estimate | p‐value | ||
| abyss Multikmer ‐ abyss 21 | Unprocessed | 149.1 | <.0001 | 133.55 | <.0001 | 42.55 | .0005 |
| abyss Multikmer ‐ abyss 33 | 116.02 | <.0001 | 126.17 | <.0001 | 61.62 | <.0001 | |
| abyss Multikmer ‐ abyss 55 | 166.22 | <.0001 | 172.62 | <.0001 | 95.97 | <.0001 | |
| abyss Multikmer ‐ abyss 77 | 217.75 | <.0001 | 229.5 | <.0001 | 109.85 | <.0001 | |
| abyss Multikmer ‐ spades | 70.38 | <.0001 | 69.28 | <.0001 | −33.97 | .0118 | |
| spades ‐ abyss 21 | 78.72 | <.0001 | 64.27 | <.0001 | 76.52 | <.0001 | |
| spades ‐ abyss 33 | 45.63 | .0001 | 56.88 | <.0001 | 95.58 | <.0001 | |
| spades ‐ abyss 55 | 95.83 | <.0001 | 103.33 | <.0001 | 129.93 | <.0001 | |
| spades ‐ abyss 77 | 147.37 | <.0001 | 160.22 | <.0001 | 143.82 | <.0001 | |
| abyss Multikmer ‐ abyss 21 | Processed | 117.8 | <.0001 | 102.25 | <.0001 | 11.25 | .8786 |
| abyss Multikmer ‐ abyss 33 | 82.78 | <.0001 | 92.93 | <.0001 | 28.38 | .061 | |
| abyss Multikmer ‐ abyss 55 | 131.48 | <.0001 | 137.88 | <.0001 | 61.23 | <.0001 | |
| abyss Multikmer ‐ abyss 77 | 182.45 | <.0001 | 194.2 | <.0001 | 74.55 | <.0001 | |
| abyss Multikmer ‐ spades | 33.12 | .0155 | 32.02 | <.0001 | −71.23 | <.0001 | |
| spades ‐ abyss 21 | 84.68 | <.0001 | 70.23 | <.0001 | 82.48 | <.0001 | |
| spades ‐ abyss 33 | 49.67 | <.0001 | 60.92 | <.0001 | 99.62 | <.0001 | |
| spades ‐ abyss 55 | 98.37 | <.0001 | 105.87 | <.0001 | 132.47 | <.0001 | |
| spades ‐ abyss 77 | 149.33 | <.0001 | 162.18 | .022 | 145.78 | <.0001 | |
Standard error for all pairwise contrasts = 10.2, degrees of freedom for all pairwise contrasts = 375.
FIGURE 3.
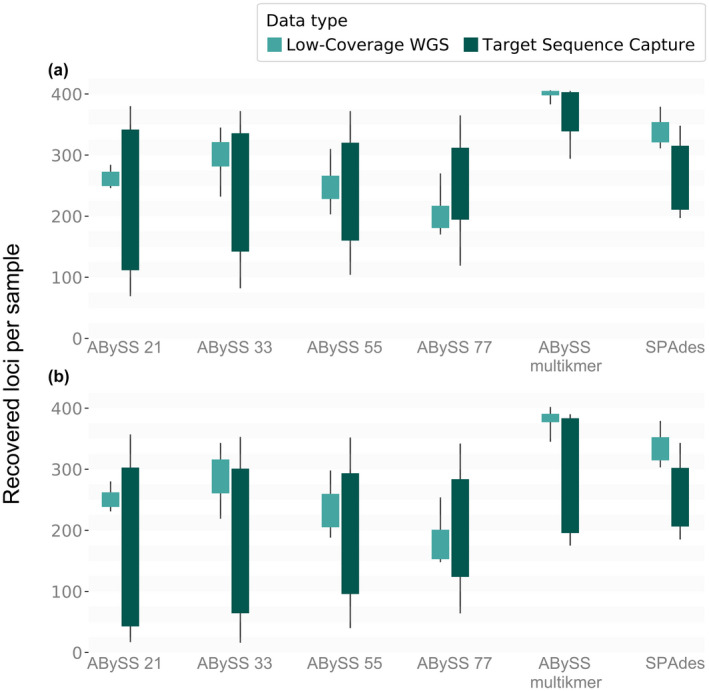
Boxplot of the median recovered loci per sample for each assembly strategy for the 10× average coverage WGS and target sequence capture data sets. (a) Unprocessed data, without the exclusion of sequences with more than 50% missing information – Ns. (b) Processed data (when sequences with more than 50% missing information ‐ Ns ‐ are excluded). Different colours represent the two different types of sequencing approaches that our data are derived from, target sequence capture and low‐coverage WGS at 10× coverage
FIGURE 4.
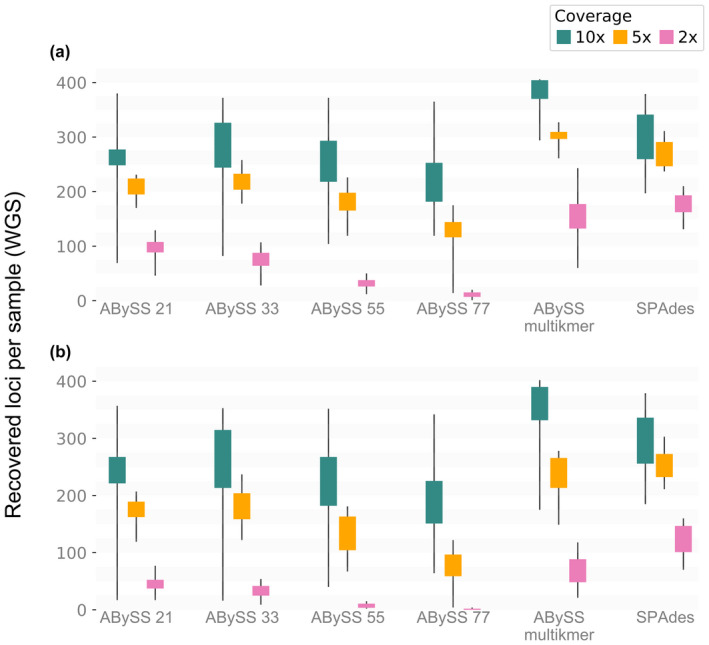
Boxplot of the median recovered loci per sample for each assembly strategy for the three subsets of different average coverages (10×, 5× and 2×). (a) Unprocessed data, without the exclusion of sequences with more than 50% missing information – Ns. (b) Processed data (when sequences with more than 50% missing information ‐ Ns ‐ are excluded). Different colours represent the different average depths of coverage for the WGS data sets
3.3. Phylogenetic inference
For the target sequence capture data set, only spades generated alignments that led to well‐resolved species trees with well‐supported branches, that were consistent with the expected phylogenetic hypothesis (Li et al., 2019; Figure 5). For the low‐coverage WGS data sets at 10× and 5× coverage, both multi‐kmer abyss and spades resulted in alignments that recovered species trees with the expected tree topology. Single‐kmer abyss 21, 33, and 55 resulted in well‐resolved trees for the 10× coverage subset, although the single‐kmer abyss 55 strategy did not recover a well‐resolved species tree for the processed alignments (with sequences with more than 50% missing data removed). Only abyss 33 and 55 recovered well‐resolved species trees for the 5× coverage subset. Single‐kmer abyss 77 did not produce useful alignments for any of the data sets and did not result in well‐resolved species trees. None of our implemented assemblers and strategies using the 2× WGS coverage subset resulted in well‐supported branches in species trees nor recovered the expected tree topology in terms of species relationships. Finally, Robinson‐Fould distances showed that the trees with most incongruences were obtained from the 2× coverage subset. Single‐kmer abyss 77 also presented high levels of incongruences in comparison with the expected tree topology (Table S6). All species trees inferred in this study can be found at the Zenodo repository (https://doi.org/10.5281/zenodo.5515798).
FIGURE 5.
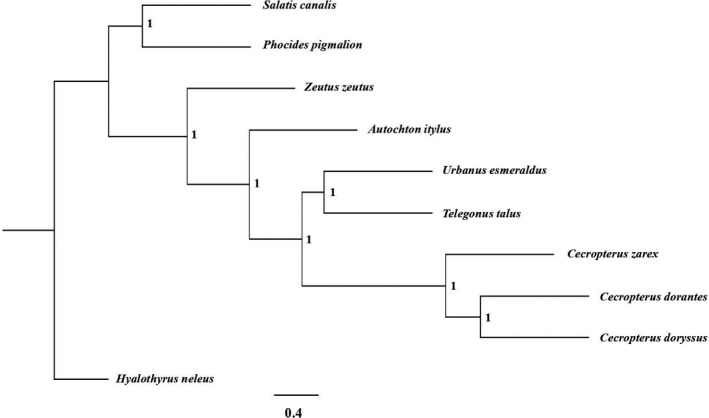
Species tree obtained with ASTRAL III (Zhang et al., 2018) by using gene trees estimated with iq‐tree version 2.0.7 (Minh et al., 2020). Tips represent each of the studied species and numbers represent local posterior probabilities inferred by ASTRAL III for the specific node. This species tree was obtained using the alignment derived from our abyss multi‐kmer strategy for the WGS data set with an average of 10× read depth coverage. Only this tree is shown since topology is the same for other well‐resolved species tree
3.4. Assessment of potential errors in de novo contig assemblies
When estimating the errors introduced by each assembly strategy using multi‐kmer abyss 10× as a query and spades 10× as the subject reference, most alignments resulted in a low percentage of errors (<1%; Figure 6). The same was observed when multi‐kmer abyss was used as query and spades as subject reference but considering the target sequence capture data set. This suggests that both assembly strategies produced very similar sequences for both data sets. When using the WGS 10× average coverage as subject reference (multi‐kmer or spades), and reduced depth coverage sequences as queries, more alignments resulted in a higher percentage of errors (~10%), both for 5× and 2× coverage and using either assembly strategy (Figure 6). This indicates that errors are more likely to appear as coverage decreases, regardless of the assembly strategy (Figure 6). Averaged results for the performed blast alignments can be found in Table S8.
FIGURE 6.
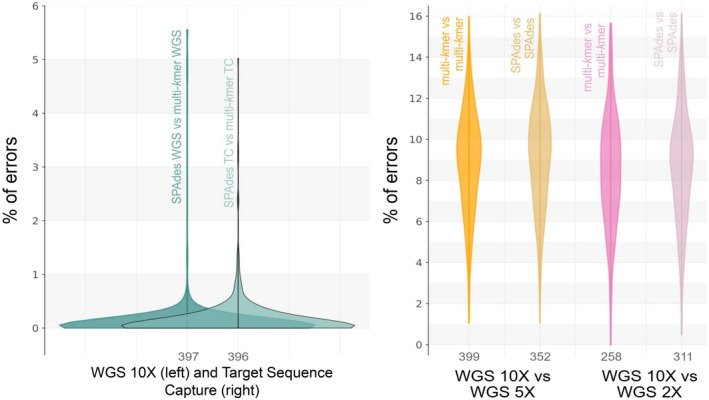
Kernel density of the percentage of error per locus, averaged across species. Percentage of error was calculated from the mismatches and alignment lengths in the blast results. (a) Comparisons between spades and our multi‐kmer abyss approach, showing the density of the average percentage of error in assembly strategy (same data set, different assembly approach); WGS indicates our whole genome sequence data set with 10× average depth of coverage and TC indicates the target sequence capture data set. (b) Comparisons showing the density of the average percentage of error from comparing the same assembly approach but different coverages. Multi‐kmer indicates the multi‐kmer abyss approach. In each density graph, the term before “vs” indicates the subject and the term after “vs” indicates the query. Numbers under each density graph indicate the average number of loci aligned per species in the blast alignments. Graphs show that the percentage of errors tend to increase when using reduced read coverage, regardless of the used assembly strategy
4. DISCUSSION
Our study exemplifies how short reads from WGS can be efficiently processed and integrated in multiple sequence alignments. By doing this within a single pipeline, we provide a way forward for the integration of sequencing strategies and the use of low‐coverage genomic data in phylogenomics and molecular ecology.
We compared the performance of two de novo contig assembly methods and assembly strategies (single‐kmer vs. multi‐kmer) to recover 406 loci that represent exons of protein‐coding genes from low‐coverage WGS and target sequence capture data. We also developed a new approach to extract loci of interest from multiple single‐kmer contig assemblies and compared its performance with other assembly strategies including single and multi‐kmer de novo contig assembly. We implemented this approach in an expanded secapr pipeline, which was originally designed to process target sequence capture data, but it is now able to jointly process different types of sequencing approaches including low‐coverage WGS.
4.1. Multi‐kmer approaches most efficiently recover loci of interest
We showed that multi‐kmer approaches significantly recovered more loci of interest from our set of targeted loci than single‐kmer approaches from both low‐coverage WGS (average depth of coverages ~10×, 5×, and 2×) and target sequence capture data. More loci, however, does not always translate into better phylogenetic inferences. For example, for the target sequence capture data, the multi‐kmer approach with abyss recovered more loci than spades (Table 1), but only the alignments from spades assemblies recovered well‐resolved trees with the expected phylogenetic relationships among species (Li et al., 2019). Since the percentage of errors within target sequence capture alignments was similar to the percentage of errors for the 10× WGS data set (Figure 6), the well‐resolved trees only for spades may be the result of spades assemblies generating less N bases within sequences in the target sequence capture data (Table S7). Alternatively, our target sequence capture may have recovered a smaller number of alignments compared to the WGS at 10× average coverage, due to the unspecific nature of probe sequences used for in vitro capture. The butterfly probe kit aims to target protein‐coding genes of all major butterfly lineages (families); thus, probe sequences were not designed specifically to target our study organisms, species within the butterfly family Hesperiidae. This scenario is widespread in target sequence capture studies where the design of sequence probes tends to be more universal to increase the cost‐efficiency of designing baits for library preparation.
spades performs better than multi‐kmer abyss with decreasing amounts of genomic coverage, since this assembler statistically outperformed all the other strategies for the 2× coverage subset in recovering more loci of interest per sample. Nevertheless, all the alignments for the 2× average coverage data set contained a large proportion of missing data in terms of nucleotide calls (more N bases in the alignments), and a more pronounced reduction in the mean of recovered loci per sample after processing of the data (exclusion of sequences with more than 50% N), especially for the abyss strategies (Table S7). In fact, it has been shown that abyss consistently generates more incomplete sequences with reduced genomic coverage (Allen et al., 2017). Since spades simultaneously uses different kmer sizes during the building of the de Bruijn graph, performing multiple graph reduction and correction steps, the final contig assemblies are expected to be more complete compared to single‐kmer approaches (Bankevich et al., 2012).
We advocate the use of multi‐kmer approaches, which retrieve significantly more loci of interest from assembled contigs, resulting in more complete alignments from both target sequence capture and low‐coverage WGS data. We highlight that our new implementation of the multi‐kmer abyss strategy was the most efficient approach for WGS with averages of 10× and 5× read depth for both extraction of loci and phylogenetic inference. A similar multi‐kmer approach (post contig assembly) was described by Zhang, Ding, et al. (2019) using the minia3 assembler. However, it is unclear how the performance of this approach compares to multi‐kmer approaches when all kmers are processed during the contig assembly step as in spades. The new expansion of secapr version 2.2.3 including spades (Bankevich et al., 2012) and our newly developed multi‐kmer abyss strategy, therefore, represent a significant way forward for molecular ecology and phylogenomics.
4.2. Sequencing approaches and their impact on phylogenetic trees
Average coverages of ~10× from small to medium sized genomes (<1 [Gbp]) have been shown to be optimal for extracting single‐copy orthologs amenable to phylogenomics (Allen et al., 2017), while a coverage of 5× is sufficient for retrieving UCEs (Zhang, Ding, et al., 2019). We showed that multi‐kmer approaches significantly recover more target loci from both 10× and 5× genomes, and that those loci were useful for the recovery of expected and well‐supported phylogenetic relationships compared to single‐kmer assembly approaches.
On average, sequencing a 1 [Gbp] genome at 5× is ~65% the cost of sequencing the same genome at 10× read depth and is similar to target sequence capture of hundreds of loci for phylogenomics. However, library preparations for WGS are more straightforward (Allen et al., 2017; Lemmon & Lemmon, 2013) and require less initial DNA material compared to target sequence capture (Zhang, Ding, et al., 2019). Nevertheless, in cases where the expected genome size is large (e.g., 2, 3 [Gbp]), target sequence capture might still be the most cost‐efficient approach to obtain phylogenomic markers. Taken together, this shows that low‐coverage WGS aiming for at least 5× read depths and for genomes as large as 1 [Gbp] is currently the most cost‐efficient approach for phylogenomics.
4.3. Potential errors within de novo contig assemblies
We found that with decreasing depths of coverage in WGS data, the percentage of errors generated during de novo contig assembly increases, which might ultimately generate biases in branch length and divergence time estimations (Andermann et al., 2019; Simion et al., 2020). At lower coverages, individual reads containing sequencing errors and those resulting from contamination have a higher impact on the assembled contigs, leading to the observed increase of assembly errors in low coverage samples. We advise caution when using 5× or lower coverages for analyses that require the estimation of within‐species polymorphisms, variant calling, and population genetic studies (Lou et al., 2021; Menelaou & Marchini, 2013), unless a reference genome is provided (e.g., Bizon et al., 2014; Rustagi et al., 2017). However, we demonstrate that 5× is sufficient for phylogenomic inference in terms of retrieving accurate species relationships. Finally, our results add further evidence showing that a minimum of 10× average coverage is suitable for obtaining high numbers of single‐copy target loci shared between samples of varying evolutionary distances (Zhang, Ding, et al., 2019) and to enable accurate phylogenetic inference (Allen et al., 2017; Li et al., 2019).
Low‐coverage WGS data might bias the estimation of population‐level parameters, genotyping and phasing of alleles in which cases the use of genomic imputation to estimate missing genotypes more confidently is needed (Lou et al., 2021). Although it is possible to phase alleles with secapr version 2.2.3, the implementation of new models and software dealing with genotyping and phasing of alleles at low WGS coverage (e.g., Lou et al., 2021; Menelaou & Marchini, 2013; Rubinacci et al., 2021; Zan et al., 2019) would represent an important advance to fully integrate different short‐read library preparations and research scopes (e.g., phylogenomics and population genomics using the same WGS data in a single bioinformatic pipeline).
5. CONCLUSION
Our assessment of assemblers, assembly strategies and WGS sequencing depth of coverage provides a guide for improving the extraction of more loci of interest from WGS and target sequence capture data. With further increases in the cost‐benefit of low‐coverage WGS sequencing, researchers are now able to address questions in molecular ecology and evolutionary biology using more taxa, even in the absence of reference genomes. By using multi‐kmer approaches, either spades or by merging the assembled contigs of interest from different single‐kmer abyss assemblies (multi‐kmer abyss), we were able to generate alignments with more samples and of better quality to infer robust species trees. Also, other available types of data, such as RADseq and UCEs, can be tested within our pipeline so users are able to recycle these data by extracting other specific regions of genomes from them.
For years to come, both types of sequencing techniques will likely remain at the center of a myriad of questions in evolutionary biology and molecular ecology. Our freely available bioinformatic platform and guidelines allow researchers to make informed choices on the generations of contigs, be them from low‐coverage WGS data, specifically enriched target sequence capture data, or a combination of both.
AUTHOR CONTRIBUTIONS
Pedro G. Ribeiro, María Fernanda Torres Jiménez, Tobias Andermann, Christine D. Bacon, and Pável Matos‐Maraví conceived/designed the study, Alexandre Antonelli and Pável Matos‐Maraví secured funding, Tobias Andermann created the bioinformatic pipeline, Pedro G. Ribeiro, María Fernanda Torres Jiménez and Pável Matos‐Maraví conducted analyses, Pedro G. Ribeiro and María Fernanda Torres Jiménez interpreted the results, Pedro G. Ribeiro and María Fernanda Torres Jiménez created figures, Pedro G. Ribeiro wrote the first draft, and all authors contributed to the final version of the article.
Supporting information
Table S1‐S7
Table S8
ACKNOWLEDGEMENTS
We would like to thank editors Bridget O’Boyle, Benjamin Sibbet and Sangeet Lamichhaney, and the three anonymous reviewers, for their constructive suggestions that helped improve this manuscript. We are grateful to Rayner Núñez (Instituto de Ecología y Sistemática, Cuba), Yves Basset (Smithsonian Tropical Research Institute, STRI, Panama) and André Freitas (UNICAMP, Brazil) for providing samples that were used for target enrichment sequencing. We thank Leonardo Ré Jorge for thorough help with statistical analyses. We thank the Servicio Nacional Forestal y de Fauna Silvestre (SERFOR), Peru, for assistance in obtaining research and export permits (Permit no. 223‐2017‐SERFOR/DGGSPFFS). We acknowledge the computational resources for this study provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, under the programme “Projects of Large Research, Development, and Innovations Infrastructures”. The Swedish Research Council (2017‐04980) provided funding to M.F.T.J and C.D.B. T.A. received funding from the Swedish Research Council (2019‐04739) and funding to A.A. was provided by the Swedish Research Council, the Swedish Foundation for Strategic Research, and the Royal Botanic Gardens, Kew. The Grant Agency of the Czech Republic (GAČR grant: GJ20‐18566Y) and the Marie Skłodowska‐Curie Fellowship of the European Commission (MARIPOSAS‐704035) provided funding to P.M.M.
Ribeiro, P. G. , Torres Jiménez, M. F. , Andermann, T. , Antonelli, A. , Bacon, C. D. , & Matos‐Maraví, P. (2021). A bioinformatic platform to integrate target capture and whole genome sequences of various read depths for phylogenomics. Molecular Ecology, 30, 6021–6035. 10.1111/mec.16240
DATA AVAILABILITY STATEMENT
Low‐coverage WGS data is accessible on the SRA Archive at NCBI (https://www.ncbi.nlm.nih.gov/sra) under BioProject PRJNA464409 and SRA accession number SRP147939. Target sequence capture data is assigned to BioProject PRJNA681152 and SRA accession number SRP342699. All specific accession numbers for samples used in this study are supplied in Table S1. All the phylogenetic species trees generated with ASTRAL III are available at DOI 10.5281/zenodo.5515798 as .tre files (https://doi.org/10.5281/zenodo.5515798). SECAPR v2.2.3 can be freely downloaded and installed following the detailed documentation at https://github.com/AntonelliLab/seqcap_processor. We also used the Sequence Cleaner program available at https://github.com/metageni/Sequence‐Cleaner, as well as reformat.sh script of the BBTools/BBmaps set of programs and scripts, which are available at https://sourceforge.net/projects/bbmap.
REFERENCES
- Albert, T. J. , Molla, M. N. , Muzny, D. M. , Nazareth, L. , Wheeler, D. , Song, X. , Richmond, T. A. , Middle, C. M. , Rodesch, M. J. , Packard, C. J. , Weinstock, G. M. , & Gibbs, R. A. (2007). Direct selection of human genomic loci by microarray hybridization. Nature Methods, 4(11), 903–905. 10.1038/nmeth1111 [DOI] [PubMed] [Google Scholar]
- Allen, J. M. , Boyd, B. , Nguyen, N.‐P. , Vachaspati, P. , Warnow, T. , Huang, D. I. , Grady, P. G. S. , Bell, K. C. , Cronk, Q. C. B. , Mugisha, L. , Pittendrigh, B. R. , Soledad Leonardi, M. , Reed, D. L. , & Johnson, K. P. (2017). Phylogenomics from whole genome sequences using aTRAM. Systematic Biology, 66(5), 786–798. 10.1093/sysbio/syw105 [DOI] [PubMed] [Google Scholar]
- Allio, R. , Schomaker‐Bastos, A. , Romiguier, J. , Prosdocimi, F. , Nabholz, B. , & Delsuc, F. (2020). MitoFinder: Efficient automated large‐scale extraction of mitogenomic data in target enrichment phylogenomics. Molecular Ecology Resources, 20(4), 892–905. 10.1111/1755-0998.13160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Andermann, T. , Cano, Á. , Zizka, A. , Bacon, C. , & Antonelli, A. (2018). SECAPR—A bioinformatics pipeline for the rapid and user‐friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ, 2018(7), 1–15. 10.7717/peerj.5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann, T. , Fernandes, A. M. , Olsson, U. , Töpel, M. , Pfeil, B. , Oxelman, B. , Aleixo, A. , Faircloth, B. C. , & Antonelli, A. (2019). Allele phasing greatly improves the phylogenetic utility of ultraconserved elements. Systematic Biology, 68(1), 32–46. 10.1093/sysbio/syy039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann, T. , Torres Jiménez, M. F. , Matos‐Maraví, P. , Batista, R. , Blanco‐Pastor, J. L. , Gustafsson, A. L. S. , Kistler, L. , Liberal, I. M. , Oxelman, B. , Bacon, C. D. , & Antonelli, A. (2020). A guide to carrying out a phylogenomic target sequence capture project. Frontiers in Genetics, 10(1407), 1–20. 10.3389/fgene.2019.01407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC. Retrieved from https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, D. , Mächler, M. , Bolker, B. M. , & Walker, S. C. (2015). Fitting linear mixed‐effects models using lme4. Journal of Statistical Software, 67(1), 1–48. 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- Bizon, C. , Spiegel, M. , Chasse, S. A. , Gizer, I. R. , Li, Y. , Malc, E. P. , Mieczkowski, P. A. , Sailsbery, J. K. , Wang, X. , Ehlers, C. L. , & Wilhelmsen, K. C. (2014). Variant calling in low‐coverage whole genome sequencing of a Native American population sample. BMC Genomics, 15(85), 1–10. 10.1186/1471-2164-15-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, B.H. (1970). Space/time trade‐offs in hash coding with allowable errors. Communications of the ACM, 13(7), 422–426. 10.1145/362686.362692 [DOI] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankovics, B. , Zhang, H. , van Diepeningen, A. D. , van der Lee, T. A. J. , Waalwijk, C. , & de Hoog, G. S. (2016). GRAbB: Selective assembly of genomic regions, a new niche for genomic research. PLoS Computational Biology, 12(6), 1–9. 10.1371/journal.pcbi.1004753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbano, H. A. , Hodges, E. , Green, R. E. , Briggs, A. W. , Krause, J. , Meyer, M. , Good, J. M. , Maricic, T. , Johnson, P. L. F. , Xuan, Z. , Rooks, M. , Bhattacharjee, A. , Brizuela, L. , Albert, F. W. , de la Rasilla, M. , Fortea, J. , Rosas, A. , Lachmann, M. , Hannon, G. J. , & Paabo, S. (2010). Targeted investigation of the neandertal genome by array‐based sequence capture. Science, 723, 723–726. 10.1126/science.1188046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell, B. (2021). BBTools/BBMap. [Google Scholar]
- Chikhi, R. , & Rizk, G. (2013). Space‐efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms for Molecular Biology, 8(1), 1–9. 10.1186/1748-7188-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, I. R. , Prosdocimi, F. , & Jennings, W. B. (2016). In silico phylogenomics using complete genomes: A case study on the evolution of hominoids. Genome Research, 26(9), 1257–1267. 10.1101/gr.203950.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, J. L. , & Blaxter, M. W. (2010). RADseq: Next‐generation population genetics. Briefings in Functional Genomics, 9(5–6), 416–423. 10.1093/bfgp/elq031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren, A. M. , Kiefl, E. , Shaiber, A. , Veseli, I. , Miller, S. E. , Schechter, M. S. , Fink, I. , Pan, J. N. , Yousef, M. , Fogarty, E. C. , Trigodet, F. , Watson, A. R. , Esen, Ö. C. , Moore, R. M. , Clayssen, Q. , Lee, M. D. , Kivenson, V. , Graham, E. D. , Merrill, B. D. , … Willis, A. D. (2021). Community‐led, integrated, reproducible multi‐omics with anvi’o. Nature Microbiology, 6(1), 3–6. 10.1038/s41564-020-00834-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeland, M. , Breinholt, J. , Willmott, K. R. , Warren, A. D. , Vila, R. , Toussaint, E. F. A. , Maunsell, S. C. , Aduse‐Poku, K. , Talavera, G. , Eastwood, R. , Jarzyna, M. A. , Guralnick, R. , Lohman, D. J. , Pierce, N. E. , & Kawahara, A. Y. (2018). A comprehensive and dated phylogenomic analysis of butterflies. Current Biology, 28(5), 770–778.e5. 10.1016/j.cub.2018.01.061 [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. (2016). PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics, 32(5), 786–788. 10.1093/bioinformatics/btv646 [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. , McCormack, J. E. , Crawford, N. G. , Harvey, M. G. , Brumfield, R. T. , & Glenn, T. C. (2012). Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Systematic Biology, 61(5), 717–726. 10.1093/sysbio/sys004 [DOI] [PubMed] [Google Scholar]
- Gnirke, A. , Melnikov, A. , Maguire, J. , Rogov, P. , LeProust, E. M. , Brockman, W. , Fennell, T. , Giannoukos, G. , Fisher, S. , Russ, C. , Gabriel, S. , Jaffe, D. B. , Lander, E. S. , & Nusbaum, C. (2009). Solution hybrid selection with ultra‐long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology, 27(2), 182–189. 10.1038/nbt.1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, D. , François, P. , Farinelli, L. , Østerås, M. , & Schrenzel, J. (2008). De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Research, 18(5), 802–809. 10.1101/gr.072033.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, D. , Tewhey, R. , Veyrieras, J. B. , Farinelli, L. , Østerås, M. , François, P. , & Schrenzel, J. (2014). De novo finished 2.8 Mbp Staphylococcus aureus genome assembly from 100 bp short and long range paired‐end reads. Bioinformatics, 30(1), 40–49. 10.1093/bioinformatics/btt590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D. T. , Chernomor, O. , von Haeseler, A. , Minh, B. Q. , & Vinh, L. S. (2017). UFBoot2: Improving the ultrafast bootstrap approximation. BioRxiv, 35(2), 518–522. 10.1101/153916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman, S. D. , Vandervalk, B. P. , Mohamadi, H. , Chu, J. , Yeo, S. , Hammond, S. A. , & Birol, I. (2017). ABySS 2.0: Resource‐efficient assembly of large genomes using a bloom filter effect of bloom filter false positive rate. Genome Research, 27, 768–777. 10.1101/gr.214346.116.Freely [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, E. D. , Mirarab, S. , Aberer, A. J. , Li, B. O. , Houde, P. , Li, C. , Ho, S. Y. W. , Faircloth, B. C. , Nabholz, B. , Howard, J. T. , Suh, A. , Weber, C. C. , da Fonseca, R. R. , Li, J. , Zhang, F. , Li, H. , Zhou, L. , Narula, N. , Liu, L. , … Zhang, G. (2014). Whole‐genome analyses resolve early branches in the tree of life of modern birds. Science, 346(6215), 1320–1331. 10.1126/science.1253451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B. Q. , Wong, T. K. F. , Von Haeseler, A. , & Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods, 14(6), 587–589. 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, E. M. , & Lemmon, A. R. (2013). High‐throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics, 44, 99–121. 10.1146/annurev-ecolsys-110512-135822 [DOI] [Google Scholar]
- Li, W. , Cong, Q. , Shen, J. , Zhang, J. , Hallwachs, W. , Janzen, D. H. , & Grishin, N. V. (2019). Genomes of skipper butterflies reveal extensive convergence of wing patterns. Proceedings of the National Academy of Sciences of the United States of America, 116(13), 6232–6237. 10.1073/pnas.1821304116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, X. , Li, M. , Zou, Y. , Wu, F‐X. , Yi, P. , & Wang, J. (2019). Current challenges and solutions of de novo assembly. Quantitative Biology, 7(2), 90–109. 10.1007/s40484-019-0166-9 [DOI] [Google Scholar]
- Lou, R. N. , Jacobs, A. , Wilder, A. P. , & Therkildsen, N. O. (2021). A beginner’s guide to low‐coverage whole genome sequencing for population genomics. Molecular Ecology, 1–68. 10.1111/mec.16077 [DOI] [PubMed] [Google Scholar]
- Menelaou, A. , & Marchini, J. (2013). Genotype calling and phasing using next‐generation sequencing reads and a haplotype scaffold. Bioinformatics, 29(1), 84–91. 10.1093/bioinformatics/bts632 [DOI] [PubMed] [Google Scholar]
- Minh, B. Q. , Schmidt, H. A. , Chernomor, O. , Schrempf, D. , Woodhams, M. D. , Von Haeseler, A. , & Teeling, E. (2020). IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530–1534. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Meleshko, D. , Korobeynikov, A. , & Pevzner, P. A. (2017). MetaSPAdes: A new versatile metagenomic assembler. Genome Research, 27(5), 824–834. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson, J. K. , Cantera, I. , Van de Paer, C. , Hong‐Wa, C. , Zedane, L. , Dunning, L. T. , Alberti, A. , Christin, P.‐A. , & Besnard, G. (2019). Phylogenomics using low‐depth whole genome sequencing: A case study with the olive tribe. Molecular Ecology Resources, 19(4), 877–892. 10.1111/1755-0998.13016 [DOI] [PubMed] [Google Scholar]
- R Core Team . (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing. Retrieved from https://www.r‐project.org/ [Google Scholar]
- Robinson, D. F. , & Foulds, L. R. (1981). Comparison of phylogenetic trees. Mathematical Biosciences, 53(1–2), 131–147. 10.1016/0025-5564(81)90043-2 [DOI] [Google Scholar]
- Rubinacci, S. , Ribeiro, D. M. , Hofmeister, R. J. , & Delaneau, O. (2021). Efficient phasing and imputation of low‐coverage sequencing data using large reference panels. Nature Genetics, 53(1), 120–126. 10.1038/s41588-020-00756-0 [DOI] [PubMed] [Google Scholar]
- Rustagi, N. , Zhou, A. , Watkins, W. S. , Gedvilaite, E. , Wang, S. , Ramesh, N. , Muzny, D. , Gibbs, R. A. , Jorde, L. B. , Yu, F. , & Xing, J. (2017). Extremely low‐coverage whole genome sequencing in South Asians captures population genomics information. BMC Genomics, 18(1), 1–12. 10.1186/s12864-017-3767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari, E. , & Mirarab, S. (2016). Fast coalescent‐based computation of local branch support from quartet frequencies. Molecular Biology and Evolution, 33(7), 1654–1668. 10.1093/molbev/msw079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep, K. P. (2011). phangorn: Phylogenetic analysis in R. Bioinformatics, 27(4), 592–593. 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S. , Kent, W. J. , Smit, A. , Zhang, Z. , Baertsch, R. , Hardison, R. C. , & Miller, W. (2003). Human‐mouse alignments with BLASTZ. Genome Research, 13(1), 103–107. 10.1101/gr.809403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Cong, Q. , Borek, D. , Otwinowski, Z. , & Grishin, N. V. (2017). Complete genome of achalarus lyciades, the first representative of the eudaminae subfamily of skippers. Current Genomics, 18(4), 366–374. 10.2174/1389202918666170426113315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simion, P. , Delsuc, F. , Philippe, H. , Simion, P. , Delsuc, F. , Philippe, H. , Philippe, H. (2020). To what extent current limits of phylogenomics can be overcome? No Commercial Publisher | Authors Open Access Book. Retrieved from https://hal.archives‐ouvertes.fr/hal‐02535366/document [Google Scholar]
- Simpson, J. T. , Wong, K. , Jackman, S. D. , Schein, J. E. , Jones, S. J. M. , & Birol, I. (2009). ABySS: A parallel assembler for short read sequence data. Genome Research, 19(6), 1117–1123. 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, J‐I. , & Nam, J.W. (2016). The present and future of de novo whole‐genome assembly. Briefings in Bioinformatics, 19(1), 23–40. 10.1093/bib/bbw096 [DOI] [PubMed] [Google Scholar]
- Zan, Y. , Payen, T. , Lillie, M. , Honaker, C. F. , Siegel, P. B. , & Carlborg, Ö. (2019). Genotyping by low‐coverage whole‐genome sequencing in intercross pedigrees from outbred founders: A cost‐efficient approach. Genetics Selection Evolution, 51(1), 1–11. 10.1186/s12711-019-0487-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Rabiee, M. , Sayyari, E. , & Mirarab, S. (2018). ASTRAL‐III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19(Suppl. 6), 15–30. 10.1186/s12859-018-2129-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Ding, Y. , Zhu, C. D. , Zhou, X. , Orr, M. C. , Scheu, S. , & Luan, Y. X. (2019). Phylogenomics from low‐coverage whole‐genome sequencing. Methods in Ecology and Evolution, 10(4), 507–517. 10.1111/2041-210X.13145 [DOI] [Google Scholar]
- Zhang, P. , Boisson, B. , Stenson, P. D. , Cooper, D. N. , Casanova, J. L. , Abel, L. , & Itan, Y. (2019). SeqTailor: A user‐friendly webserver for the extraction of DNA or protein sequences from next‐generation sequencing data. Nucleic Acids Research, 47(W1), W623–W631. 10.1093/nar/gkz326 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S7
Table S8
Data Availability Statement
Low‐coverage WGS data is accessible on the SRA Archive at NCBI (https://www.ncbi.nlm.nih.gov/sra) under BioProject PRJNA464409 and SRA accession number SRP147939. Target sequence capture data is assigned to BioProject PRJNA681152 and SRA accession number SRP342699. All specific accession numbers for samples used in this study are supplied in Table S1. All the phylogenetic species trees generated with ASTRAL III are available at DOI 10.5281/zenodo.5515798 as .tre files (https://doi.org/10.5281/zenodo.5515798). SECAPR v2.2.3 can be freely downloaded and installed following the detailed documentation at https://github.com/AntonelliLab/seqcap_processor. We also used the Sequence Cleaner program available at https://github.com/metageni/Sequence‐Cleaner, as well as reformat.sh script of the BBTools/BBmaps set of programs and scripts, which are available at https://sourceforge.net/projects/bbmap.