

Abstract
Aims
Examine relationships between the systemic exposure of acalabrutinib, a highly selective, next‐generation Bruton tyrosine kinase inhibitor, and its active metabolite (ACP‐5862) vs. efficacy and safety responses in patients with B‐cell malignancies who received acalabrutinib as monotherapy or in combination with obinutuzumab.
Methods
For exposure–efficacy analyses, patients with untreated chronic lymphocytic leukaemia were assessed for best overall response, progression‐free survival and tumour regression. For exposure–safety analyses, incidences of grade ≥2 adverse events (AEs), grade ≥3 AEs and grade ≥2 events of clinical interest were assessed in patients with B‐cell malignancies. Acalabrutinib and ACP‐5862 pharmacokinetic (PK) parameter estimates were obtained from population PK modelling. Exposure calculations were based on study dosing regimens. Total active moieties were calculated to account for contributions of ACP‐5862 to overall efficacy/safety.
Results
A total of 573 patients were included (exposure–efficacy analyses, n = 274; exposure–safety analyses, n = 573). Most patients (93%) received acalabrutinib 100 mg twice daily. Median total active area under the concentration–time curve (AUC24h,ss) and total active maximal concentration at steady‐state (Cmax,ss) were similar for patients who received acalabrutinib as monotherapy or in combination with obinutuzumab, and for responders and nonresponders. No relationship was observed between AUC24h,ss/Cmax,ss and progression‐free survival or tumour regression. Acalabrutinib AUC24h,ss and Cmax,ss were generally comparable across groups regardless of AE incidence.
Conclusion
No clinically meaningful correlations between acalabrutinib PK exposure and efficacy and safety outcomes were observed. These data support the fixed acalabrutinib dose of 100 mg twice daily in the treatment of patients with B‐cell malignancies.
Keywords: acalabrutinib, ACP‐5862, B‐cell, BTK inhibitor, Calquence, chronic, exposure–efficacy, exposure–response, exposure–safety, leukaemia, lymphocytic, obinutuzumab, pharmacokinetics
1. What is already known about this subject
Acalabrutinib is a highly selective, potent, next‐generation, covalent Bruton tyrosine kinase (BTK) inhibitor approved for the treatment of previously treated mantle cell lymphoma as well as previously untreated and relapsed/refractory chronic lymphocytic leukaemia/small lymphocytic lymphoma.
ACP‐5862, the major pharmacologically active metabolite of acalabrutinib, has exposures approximately 2‐fold that of acalabrutinib, is approximately half as potent in terms of BTK inhibition, and has an analogous kinase selectivity profile.
What this study adds
No clinically meaningful relationships were observed between acalabrutinib/ACP‐5862 exposures and efficacy and safety outcomes following treatment with acalabrutinib monotherapy or in combination with obinutuzumab in patients with B‐cell malignancies.
The lack of relationship between exposure and efficacy and safety endpoints indicates that the acalabrutinib 100‐mg twice‐daily regimen produced robust and consistent therapeutic effects across the exposure range observed.
See Short Communication article here .
1. INTRODUCTION
Bruton tyrosine kinase (BTK) signalling is essential for B‐cell differentiation, proliferation and survival, and critical for the survival of leukaemic cells. 1 Therefore, inhibition of BTK is an established therapeutic intervention for the treatment of B‐cell malignancies. Acalabrutinib (Calquence, AstraZeneca, Gaithersburg, MD, USA) is a highly selective, potent, next‐generation BTK inhibitor approved for the treatment of previously treated mantle cell lymphoma (MCL) as well as previously untreated and relapsed/refractory chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma. 2 , 3 , 4 , 5 , 6 , 7 , 8 In nonclinical and clinical studies, acalabrutinib inhibited BTK‐mediated activation of downstream signalling proteins and inhibited malignant B‐cell proliferation and survival. 4 , 9 Acalabrutinib displays minimal off‐target inhibition of kinases such as epidermal growth factor receptor and interleukin 2–inducible T cell kinase (ITK) relative to other BTK inhibitors. 2
After oral administration, acalabrutinib is rapidly absorbed and has a short pharmacokinetic (PK) half‐life; median time to peak plasma concentrations (Tmax) was 0.9 hours (range, 0.5–1.9 h). 3 Following a single oral dose (100 mg), the terminal elimination half‐life of acalabrutinib was 1–2 hours. 10 ACP‐5862, the major pharmacologically active metabolite from acalabrutinib, is approximately half as potent as acalabrutinib in terms of BTK inhibition, has a similar kinase selectivity profile and has a terminal elimination half‐life of approximately 7 hours. 10 ACP‐5862 formation is also characterized by a Tmax of approximately 1 hour and a mean exposure twice that of acalabrutinib. 10 Both acalabrutinib and ACP‐5862 are irreversible, covalent, kinase inhibitors and the inhibition of signal transduction is maintained until functional levels of the target kinase are restored. 11 Median steady‐state BTK occupancy (≥95%) in peripheral blood mononuclear cells was maintained over 12 hours with 100 mg twice‐daily (BID) acalabrutinib administration in patients with B‐cell malignancies. 3 Acalabrutinib is metabolized to ACP‐5862, primarily by CYP3A‐mediated oxidation of its pyrrolidine ring. In a human [14C] absorption, distribution, metabolism and excretion study, 95.7% of total radioactivity was recovered (12% in urine, 83.5% in faeces). In human faeces, parent acalabrutinib accounted for 1.2% of the excreted dose. The results from these analyses indicate that metabolic clearance is a major route of drug elimination of acalabrutinib in humans. 12
Because acalabrutinib and ACP‐5862 inhibit BTK and contribute to efficacy and safety in the clinical setting, we sought to further assess the relationship between plasma exposure to acalabrutinib and ACP‐5862 and clinical efficacy and safety via exposure–response analyses in patients with B‐cell malignancies. We evaluated the relationship between acalabrutinib and ACP‐5862 PK exposure and clinical efficacy in patients with untreated CLL treated with acalabrutinib alone or in combination with obinutuzumab (ACE‐CL‐007). We also evaluated the relationship between acalabrutinib and ACP‐5862 PK exposure and selected safety measures in patients with B‐cell malignancies from 8 clinical studies.
2. MATERIALS AND METHODS
2.1. Compliance with ethics guidelines
All analyses were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. The study protocols were approved by all institutional review boards and informed consent was obtained from all patients.
2.2. Study populations
Data were pooled from 8 phase 1–3 trials in adult patients with B‐cell malignancies (Table 1). All patients who received ≥1 dose of acalabrutinib and had estimated individual acalabrutinib or ACP‐5862 exposures were included.
TABLE 1.
Summary of studies included in the analysis
| Study | Study description | PK sample collection a | Acalabrutinib n with PK data (all doses/100 mg BID b ) | ACP‐5862 n with PK data a (all doses/100 mg BID b ) |
|---|---|---|---|---|
| ACE‐CL‐001 4 , 26 (NCT02029443) | Phase 1/2 study in patients with R/R or previously untreated CLL, Richter's transformation, or PLL | Cycle 1: d 1, 8, 15, 22 and 28 | 161/130 | 18/18 |
| ACE‐CL‐003 27 (NCT02296918) | Phase 1b study in patients with R/R CLL | Cycle 1: d 1 | 8/8 | 0/0 |
| ACE‐CL‐007 8 (NCT02475681) | Phase 3 study in patients with previously untreated CLL | Cycles 1 and 2: d 1 | 273/263 | 274/264 |
| ACE‐LY‐002 28 (NCT02112526) | Phase 1b study in patients with R/R de novo activated B‐cell subtype of DLBCL |
Cycle 1: d 1, 8, 15 and 22 Cycle 2: d 1 |
15/15 | 0/0 |
| ACE‐LY‐003 29 (NCT02180711) | Phase 1b study of acalabrutinib alone or in combination with rituximab in patients with FL | Cycle 1: d 1, 8, 15, 22 and 28 | 7/7 | 0/0 |
| ACE‐LY‐004 6 (NCT02213926) | Phase 2 study in patients with MCL | Cycle 1: d 1, 8, 15, 22 and 28 | 45/45 | 0/0 |
| ACE‐MY‐001 (NCT02211014) | Phase 1b study of acalabrutinib alone or in combination with dexamethasone in patients with MM | Cycle 1: d 1, 8, 15, 22 and 28 | 13/13 | 0/0 |
| ACE‐WM‐001 30 (NCT02180724) | Phase 1/2 in patients with WM | Cycle 1: d 1, 8, 15, 22 and 28 | 50/49 | 0/0 |
| Total | 572/530 | 292/282 |
BID, twice daily; CLL, chronic lymphocytic leukaemia; DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma; PK, pharmacokinetic; PLL, prolymphocytic leukaemia; R/R, relapsed/refractory; WM, Waldenström macroglobulinemia.
For patients without observed acalabrutinib or ACP‐5862 concentrations, exposures were predicted based on the typical population PK parameter values.
Dose is based on the most prevalent dose/dosing regimen administered to individuals for the duration of the clinical study.
For the exposure–efficacy analysis, data from the pivotal phase 3 study in previously untreated patients with CLL (ACE‐CL‐007) were evaluated. 8 The 2 acalabrutinib‐treatment arms (acalabrutinib monotherapy and acalabrutinib+obinutuzumab) were analysed separately as well as pooled.
For the exposure–safety analyses, 6 subpopulations were considered, including: (i) a TN CLL population composed of previously untreated CLL patients who received acalabrutinib either as monotherapy or in combination with obinutuzumab; (ii) a total CLL population composed of all CLL patients, including both previously untreated and relapsed/refractory CLL patients, who received acalabrutinib either as monotherapy or in combination with obinutuzumab; (iii) a CLL mono population composed of CLL patients who were treated with acalabrutinib monotherapy; (iv) a CLL combo population composed of CLL patients who were treated with acalabrutinib in combination with obinutuzumab; (v) a Haem mono population composed of patients with haematological malignancies treated with acalabrutinib monotherapy; and (vi) an overall population composed of all evaluable patients (Table S1). These safety sub‐populations were created to focus on the CLL population, while also adding a broader haematological (i.e., non‐CLL) safety data set and enabling the identification of less‐common adverse events (AEs). All patients with available (observed) exposures and corresponding safety/efficacy data were included in the analysis; no patients were removed due to toxicities.
2.3. Acalabrutinib and ACP‐5862 exposure assessments
Individual estimates of the acalabrutinib and ACP‐5862 concentration–time profiles at steady‐state were obtained from the population PK model, using the individual empirical Bayes estimates and each patient's dosing regimen. 13 For patients without observed concentrations of either acalabrutinib or ACP‐5862 (but not both), the nonobserved profiles were predicted based on the mean parameter values in the population. Exposure calculations were based on the most prevalent dosing regimen administered to the individuals for the duration of the respective clinical study.
To account for contribution of the major active metabolite ACP‐5862 to overall efficacy or safety, acalabrutinib and ACP‐5862 molar concentrations were adjusted with respective BTK potency and protein binding [Equation 1] and used to estimate the total active moiety.
| (1) |
Cparent and Cmetabolite are molar concentrations of acalabrutinib and ACP‐5862, respectively. The free fractions for acalabrutinib (fuparent) and for ACP‐5862 (fumetabolite) are 0.025 and 0.013, respectively (data on file). Compared with acalabrutinib, ACP‐5862 has an approximately 0.5‐fold potency for BTK inhibition. The sum of the potency‐adjusted molar concentrations were then converted to ng/mL scale using the molecular weight of acalabrutinib (MWacalabrutinib = 465.5). Further, the correlation between acalabrutinib and ACP‐5862 exposure metrics were assessed to determine what metric(s) to use in the exposure–response analyses. In the case of a very strong correlation (ρ ≥ 0.9) between parent and metabolite exposures, the analysis was to be conducted using parent exposure metrics. If the correlation was strong (≥0.7 but <0.9), analyses were to be conducted separately for parent and metabolite exposure metric and both results were to be presented. In the case of a weaker correlation (<0.7), the parent and metabolite were to be included simultaneously in the analyses (total active). Interpretation of the correlation coefficient, as applied in this analysis, is discussed by Mukaka. 14
Some studies contain more data for the parent drug than the metabolite, namely ACE‐CL‐001 and studies for non‐CLL indications. For that reason, the safety analyses performed for the pooled haematological malignancies population, and potentially the pooled CLL population, were performed using parent exposure only, irrespective of the correlation coefficient.
Systemic exposures in the form of daily area under the concentration–time curve (AUC24h,ss) and maximal concentration (Cmax,ss) at steady‐state were considered in this analysis and their correlation was assessed using Spearman's correlation.
2.4. Efficacy and safety assessments
Efficacy variables evaluated in the analysis included: (i) best overall response (BOR) assessed by an independent review committee, categorized as complete response, partial response, stable disease, progressive disease and unknown (any patient missing response information); (ii) progression‐free survival (PFS) assessed by independent review committee; and (iii) tumour regression assessed by change from baseline in the sum of the products of the greatest diameters (SPD) of index lesions.
Safety variables evaluated in the analysis included: (i) incidence of any grade ≥2 AEs; (ii) incidence of any grade ≥3 AEs; and (iii) incidence of grade ≥2 AEs of clinical interest experienced by ≥5% of the overall population. The 5% incidence threshold and assessment of AEs of grades 2 and higher ensured inclusion of a sufficient number of events in the exposure–safety analysis. AEs of clinical interest were identified based on nonclinical findings, emerging data from clinical studies relating to acalabrutinib and pharmacological effects of approved BTK inhibitors, following Medical Dictionary for Regulatory Activities (MedDRA) v21.1 terminology and included anaemia, cardiac events, diarrhoea, headache, haemorrhage, hepatic events, hypertension, infections, neutropenia and thrombocytopenia.
2.5. Exposure–response analyses
Data were first evaluated through summary statistics and graphical assessments. Exploratory assessments included comparison of acalabrutinib and ACP‐5862 exposures in relevant outcome groups including treatment groups (efficacy analyses) and histology/treatment regimen subpopulations (safety analyses). For continuous outcome variables, the AUC24h,ss and Cmax,ss values were categorized by binning into quartiles. Further, Kaplan–Meier representations, stratified by exposure quartiles, were generated for the time‐to‐event outcomes.
Model‐based statistical analyses were performed if a potential relationship between exposure and response outcome was suggested by the graphical analysis; logistic regression was used to investigate potential relationships between exposure and the probability of outcomes/events [Equation 2].
| (2) |
pi is the probability of the considered event for patient i, α is an intercept parameter and β0 is a slope parameter for the effect of exposure. Ci is the exposure metric for patient i.
If there was no evidence of an exposure dependency of the considered events based on the logistic regression modelling, the relationship was not further investigated. However, if a significant exposure–response relationship was identified, other candidate predictors were investigated in addition to exposure.
Assessment of model adequacy and decisions about increasing model complexity were guided by goodness‐of‐fit criteria, including: (i) predicted probabilities of the events compared with the number of events observed for each quartile of the exposure metric; (ii) plausibility of parameter estimates; (iii) precision of parameter estimates; and (iv) likelihood ratio test.
2.6. Analytical method
Acalabrutinib and ACP‐5862 samples were assayed across studies using validated, sensitive and specific liquid chromatography–tandem mass spectrometry methods. The lower limit of quantification (linear range) was 1 (1–1000) ng/mL and 5 (5–5000) ng/mL. Intra‐assay and interassay variability (% coefficient of variance) were ≤8.2 and ≤8.8%, respectively, across both analytes.
2.7. Software
Preparation of the datasets was conducted using SAS (v. 9.4, SAS Institute, TS1M3). R‐3.5.1 (R Core Team [2018]. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/) statistical data analysis software was used for data wrangling and all exposure–response analyses.
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 15
3. RESULTS
Distributions of acalabrutinib, ACP‐5862 and total active exposure metrics were similar in the overall efficacy and safety populations by quartile (Tables S2 and S3) and across subpopulations (i.e., across the 2 acalabrutinib‐containing treatment arms of ACE‐CL‐007 and the 6 safety populations; Figures S1 and S2), as were the distributions of baseline patient demographics (Tables S4–S7). Further, the exposure–response results were consistent for all subpopulations. Therefore, only results based on pooled ACE‐CL‐007 for the efficacy–response analysis and the overall population for the safety–response analysis are described in the sections below.
3.1. Patients
A total of 573 patients were included in the exposure–response analyses (Table 1). Patients from study ACE‐CL‐007 (n = 274) who received acalabrutinib as monotherapy (n = 140) or in combination with obinutuzumab (n = 134) were evaluated in the exposure–efficacy analyses. All 573 patients were included in the overall safety population evaluated in the exposure–safety analyses (subpopulations: TN CLL, n = 328; total CLL, n = 443; CLL mono, n = 301; CLL combo, n = 142; and Haem mono, n = 431; Table S1). Demographic characteristics are presented in Table 2. Distributions of baseline patient demographics were similar across the 2 acalabrutinib‐containing arms of ACE‐CL‐007 (Tables S4 and S5) and the 6 safety subpopulations (Tables S6 and S7).
TABLE 2.
Patient demographics and baseline characteristics
| Characteristic | Efficacy population (n = 274) | Safety population (n = 573) |
|---|---|---|
| Categorical variables a | ||
| Sex | ||
| Female | 104 (38.0) | 197 (34.4) |
| Male | 170 (62.0) | 376 (65.6) |
| Race | ||
| White | 254 (95.5) | 511 (90.9) |
| Black/African American | 9 (3.4) | 24 (4.3) |
| Asian | 3 (1.1) | 6 (1.1) |
| Other | 0 (0) | 21 (3.7) |
| Ethnicity | ||
| Hispanic/Latino | 11 (4.0) | 19 (3.3) |
| Not Hispanic/Latino | 248 (90.5) | 520 (90.8) |
| Not reported | 15 (5.5) | 19 (3.3) |
| Unknown | 0 (0) | 15 (2.5) |
| ECOG performance status | ||
| 0 | 141 (51.5) | 253 (44.2) |
| 1 | 117 (42.7) | 293 (51.1) |
| 2 | 16 (5.8) | 25 (4.4) |
| 3 | 0 (0) | 2 (0.3) |
| Line of therapy | ||
| Treatment naïve | 274 (100) | 364 (63.5) |
| Relapsed/refractory | 0 (0) | 209 (36.5) |
| Chromosomal abnormality: 17p deletion | ||
| No | 250 (91.2) | 361 (86.8) |
| Yes | 24 (8.8) | 55 (13.2) |
| Chromosomal abnormality: 11q deletion | ||
| No | 220 (80.3) | 324 (77.7) |
| Yes | 54 (19.7) | 93 (22.3) |
| Continuous variables b | ||
| Age, y | 69.5 (7.8) | 66.9 (9.5) |
| Body weight, kg | 80.4 (18.4) | 81.1 (17.7) |
| SPD at baseline | 38.7 (50.9) | NA c |
| CD19+ cells, count | 3985 (15 152) | 18 160 (45 420) |
| Absolute neutrophil count, 109/L | 11.3 (15.4) | 7.6 (11.4) |
| Haemoglobin, g/L | 117 (20.0) | 116.0 (20.9) |
| Thrombocyte count, 109/L | 147 (67.0) | 150 (81.1) |
CD19+, cluster of differentiation 19 expressing B‐cells; ECOG, Eastern Cooperative Oncology Group; SPD, sum of the products of the greatest diameters.
Values are n (%).
Values are mean (standard deviation).
Not relevant for the safety population.
For the majority of patients (n = 531, 92.7%), the most prevalent dose/dosing regimen received over the study duration was acalabrutinib 100 mg BID. Other regimens included 100 mg daily (QD; n = 16, 2.8%), 175 mg QD (n = 3, 0.5%), 200 mg QD (n = 4, 0.7%), 250 mg QD (n = 1, 0.2%) and 200 mg BID (n = 18, 3.1%). In ACE‐CL‐007, 264 (96.4%) patients were treated with acalabrutinib 100 mg BID and 10 (3.6%) with 100 mg QD. The average daily dose across the efficacy/safety populations was approximately 200 mg/d; the regimen in the majority of subjects was 100 mg BID (Tables S2 and S3). Minimal dose reductions (~3%; overall median time to first dose reduction >260 days), dose interruptions (~15%; overall median time to first dose reduction >253 days) and discontinuations (~9%; overall median time to first dose reduction >26 mo) due to AEs were observed. The majority of safety events of clinical interest occurred in the first 6 months of treatment, with minimal dose modifications due to AEs reported during that period (≤23.2% of subjects across studies), over a total follow‐up period of up to 48 months.
3.2. Acalabrutinib and ACP‐5862 exposure
Acalabrutinib and ACP‐5862 AUC24h,ss values demonstrated weak to moderate correlation; Spearman's correlation coefficients (ρ) were 0.474 for the efficacy population (Figure S3) and 0.633 for the overall population (Figure S4). Similar results were noted for Cmax,ss values (data not shown). Based on the lack of collinearity, total active exposure would be the preferable exposure metric. For the exposure–efficacy analysis, total active exposure was used because ACP‐5862 observations were available in all ACE‐CL‐007 patients. All efficacy analyses were also conducted for acalabrutinib and ACP‐5862 separately. However, ACP‐5862 exposure measurements were only collected in a subset of the safety population (n = 292, 51%) and therefore were estimated based on the typical population PK parameters for approximately half of the population. Acalabrutinib exposure measurements were collected in all but 1 patient (n = 572); therefore, acalabrutinib exposure was used as the primary exposure metric for the exposure–safety analysis to avoid bias.
3.3. Effect of exposure on efficacy and safety outcomes
3.3.1. Exposure–efficacy analysis
The evaluation of BOR as a function of AUC24h,ss quartiles indicated overlapping total active exposures and similar median values across all response categories (Figure 1A). Similar results were observed when BOR was evaluated as a function of total active Cmax,ss (Figure 1B). When Kaplan–Meier estimates of PFS were stratified by AUC24h,ss quartiles, the curves for all 4 AUC24h,ss quartiles overlapped, with a p‐value across quartiles of 0.4 (Figure 2A). Similarly, when PFS was evaluated by total active Cmax,ss quartiles, substantial overlap in PFS curves was observed, with a P‐value across quartiles of 0.71 (Figure 2B). Additionally, when the relationship between change from baseline lesion size and total active AUC24h,ss quartiles was evaluated, the change from baseline in SPD was comparable across quartiles (Figure 3A). The change from baseline SPD as a function of total active Cmax,ss was also comparable across quartiles (Figure 3B). Similar results were demonstrated across all efficacy parameters when evaluated as a function of acalabrutinib and ACP‐5862 AUC24h,ss and Cmax,ss (Figure S5). Cmin,ss is very strongly correlated to AUC and will thus generate the same result (Figure S6).
FIGURE 1.
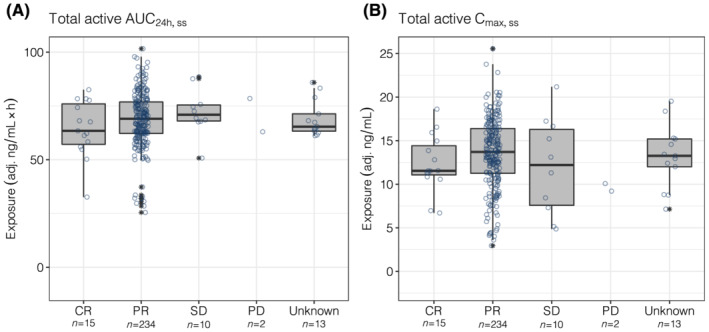
Total active AUC24h,ss (A) and Cmax,ss (B) by best overall response. Information about response was missing for 13 patients (categorized as unknown). The numbers below the categories on the x‐axis indicate the number of patients in each category. Open circles show the individual data. The ends of the box are the lower and upper quartiles, the middle line shows the median. The whiskers indicate 1.5 × interquartile range. Data above/below the whiskers are shown as asterisks. AUC24h,ss, area under the plasma concentration–time curve from time 0 to 24 hours (2 dosing intervals) at steady‐state; Cmax,ss, maximum concentration at steady‐state; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease; total active, acalabrutinib + ACP‐5862 exposure adjusted for molecular weight, potency and protein binding
FIGURE 2.
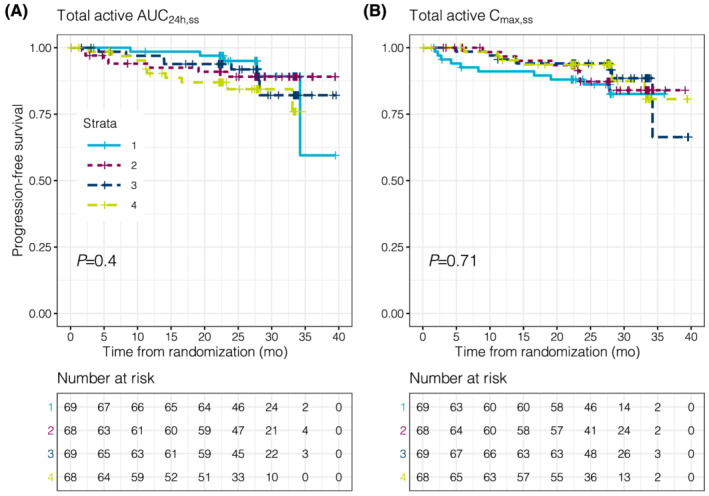
Kaplan–Meier estimates of PFS stratified by quartiles of total active AUC24h,ss (A) and Cmax,ss (B). The P‐value represents the results of Cox‐regression analyses comparing the probabilities across quartiles. Within the strata, 1 signifies the lowest quartile of total active exposure and 4 signifies the highest quartile of total active exposure. The table below the Kaplan–Meier curves represents the number of patients available for the analyses (i.e., not censored or discontinued). AUC24h,ss, area under the plasma concentration–time curve from time 0 to 24 hours (2 dosing intervals) at steady‐state; Cmax,ss, maximum concentration at steady‐state; PFS, progression‐free survival; total active, acalabrutinib + ACP‐5862 exposure adjusted for molecular weight, potency and protein binding
FIGURE 3.
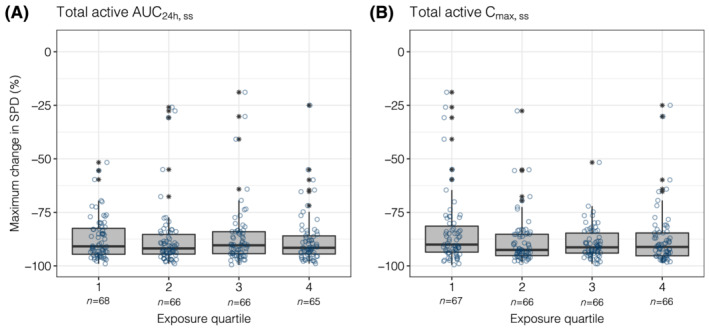
Maximum change from baseline in SPD stratified by quartiles of total active AUC24h,ss (A) or Cmax,ss (B). The numbers below the categories on the x‐axis indicate the number of patients in each category. Open circles show the individual data. The ends of the box are the lower and upper quartiles, the middle line shows the median. The whiskers indicate 1.5 × interquartile range. Data above/below the whiskers are shown as asterisks. AUC24h,ss, area under the plasma concentration–time curve from time 0 to 24 hours (2 dosing intervals) at steady‐state; Cmax,ss, maximum concentration at steady‐state; SPD, sum of the products of the greatest diameters; total active, acalabrutinib + ACP‐5862 exposure adjusted for molecular weight, potency and protein binding
3.3.2. Exposure–safety analysis
Overall, 518 patients (90.4%) experienced a grade ≥2 AE, and 350 patients (67.6%) experienced a grade ≥3 AE. The incidences of selected grade ≥2 AEs of clinical interest were: neutropenia, n = 133 (23.2%); infections, n = 101 (17.6%); anaemia, n = 57 (9.9%); thrombocytopenia, n = 41 (7.2%); cardiac events, n = 32 (5.6%); hypertension, n = 31 (5.4%); hepatic events, n = 16 (2.8%); haemorrhage, n = 12 (2.1%); diarrhoea, n = 11 (1.9%); and headache, n = 4 (0.7%). Based on the prespecified incidence threshold for inclusion of ≥5%, grade ≥2 AEs included in the exposure–response analyses were anaemia, cardiac events, hypertension, infection, neutropenia and thrombocytopenia.
Acalabrutinib AUC24h,ss was generally comparable across patient populations with and without grade ≥2 AEs, grade ≥3 AEs and selected grade ≥2 AEs of clinical interest (Figure 4). A slightly higher AUC24h,ss was noted in the populations of patients with grade ≥2 anaemia, infection and thrombocytopenia. However, the overlaps in the exposures were large between patients with and without these AEs. No such trends were noted for acalabrutinib Cmax,ss (Figure S7).
FIGURE 4.
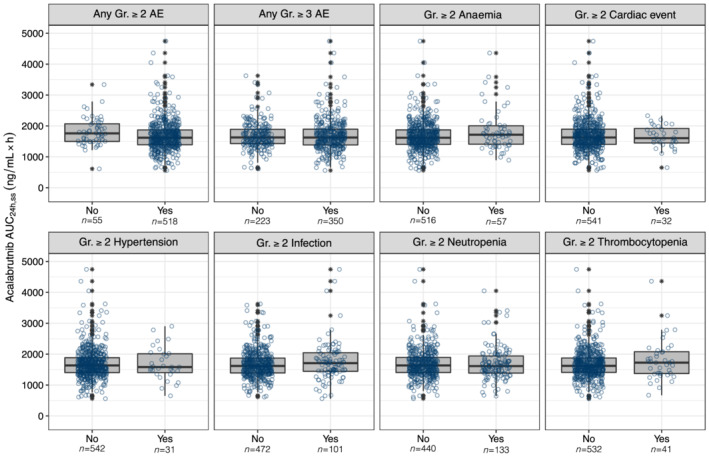
Acalabrutinib AUC24h,ss stratified by adverse event. The numbers below the categories on the x‐axis indicate the number of patients in each category. Open circles show the individual data. The ends of the box are the lower and upper quartiles, the middle line shows the median. The whiskers indicate 1.5 × interquartile range. Data above/below the whiskers are shown as asterisks. AE, adverse event; AUC24h,ss, area under the concentration–time curve at steady‐state conditions for a 24‐hour dosing interval; Gr, grade
When the incidences of grade ≥2 AEs, grade ≥3 AEs and selected grade ≥2 AEs of clinical interest were stratified by quartiles of acalabrutinib AUC24h,ss, no trends were noted except for a potential relationship between grade ≥2 infection and acalabrutinib AUC24h,ss (Figure 5). The incidence of infection was higher in the fourth AUC24h,ss quartile relative to other quartiles; however, no such trends were noted in Cmax,ss (Figure S8).
FIGURE 5.
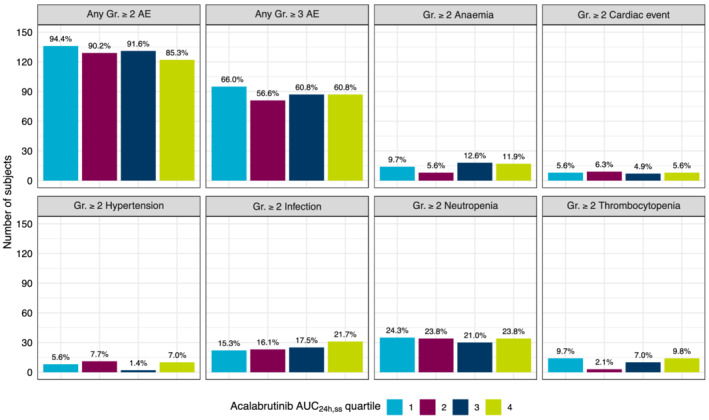
Adverse events of clinical interest by acalabrutinib AUC24h,ss quartiles. Bars represent number of patients, and the percentage of patients in each group is calculated from the total number of patients within the quartile and noted above the bars. AE, adverse event; AUC24h,ss, area under the concentration–time curve at steady‐state conditions for a 24‐hour dosing interval; Gr, grade
Because the graphical analysis suggested a potential relationship between AUC24h,ss and grade ≥2 infection, a logistic regression analysis was performed. The model predictions did not identify a statistically significant relationship between the probability of infection and acalabrutinib AUC24h,ss (P=0.123; Figure S9) and no further investigation was performed.
4. DISCUSSION
This comprehensive efficacy and safety exposure–response analysis investigated the relationship between PK exposure and selected efficacy and safety outcomes in patients with B‐cell malignancies following treatment with acalabrutinib administered as monotherapy or in combination with obinutuzumab. For the exposure–efficacy analysis, no clinically meaningful relationships were observed between total active moiety exposures and efficacy outcomes including BOR, PFS and change from baseline SPD following treatment with acalabrutinib monotherapy or in combination with obinutuzumab in patients with previously untreated CLL. Similarly, no relationships were observed between acalabrutinib exposure and incidences of grade ≥2 AEs, grade ≥3 AEs or select grade ≥2 AEs of clinical interest following treatment with acalabrutinib monotherapy or in combination with obinutuzumab in patients with B‐cell malignancies. The lack of relationship between exposure and efficacy and safety endpoints indicates that the acalabrutinib 100‐mg BID dosing regimen elicits potent and consistent therapeutic effects across the exposure range observed.
Evaluation of the absorption, distribution, metabolism and excretion properties of acalabrutinib demonstrated metabolic clearance to the major, pharmacologically active, circulating metabolite ACP‐5862, which was observed at higher plasma exposure levels than parent drug 12 ; ACP‐5862 has a longer half‐life than acalabrutinib, leading to approximately 2‐fold higher mean AUC24h levels. In addition, based on the results of biochemical kinase assays and kinome‐wide screens, acalabrutinib and ACP‐5862 are highly selective, covalent inhibitors of BTK with very similar activity profiles. 16 Therefore, systemic exposures of both acalabrutinib and ACP‐5862 are expected to contribute towards the overall activity of acalabrutinib, adjusted for the difference in potency (between the parent and metabolite) towards BTK inhibition. The potency (kinact/KI) of ACP‐5862 for BTK inhibition was estimated to be ~0.5‐fold lower than that of acalabrutinib 3 ; kinact/KI is a rate constant that describes the efficiency of covalent bond formation resulting from the potency (KI) of the first reversible binding event and the maximum potential rate (kinact) of inactivation, which is employed as a critical parameter to identify covalent inhibitors, interpret structure–activity relationships, translate activity from biochemical assays to the cell and more accurately define selectivity. In the current exposure–response analyses, acalabrutinib and ACP‐5862 molar exposures were adjusted with respective BTK potencies (and protein binding) to estimate a composite metric (total active moiety exposure), which was used for the analysis. The simulated steady‐state plasma concentration–time profile for the total active moiety in comparison with acalabrutinib and ACP‐5862 is shown in Figure 6. In general, the total active moiety PK profile is expected to provide prolonged target coverage relative to acalabrutinib alone and appears to be comparable to the PK profiles reported for other BTK inhibitors that are either approved or under investigation for the treatment of B‐cell malignancies, such as ibrutinib and zanubrutinib (Figure 6). 13 , 17 , 18
FIGURE 6.
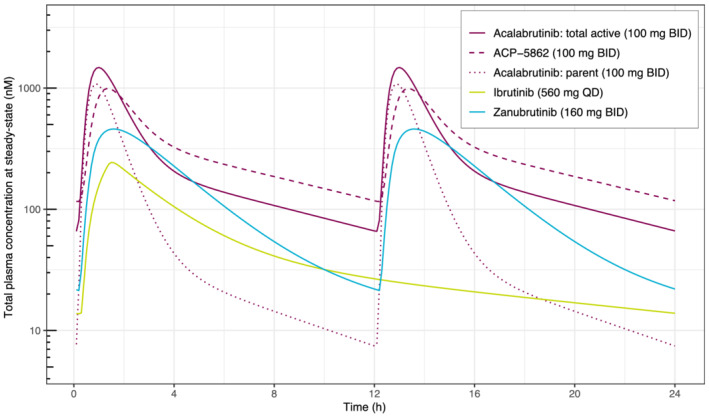
Plasma concentration–time profiles for BTK inhibitors. PK profiles were simulated using reported population PK models for acalabrutinib, ACP‐5862 and the total active moiety, 13 ibrutinib 17 and zanubrutinib. 18 Total simulated drug concentrations are shown, not adjusting for differences in protein binding or BTK potency between substrates. BID, twice daily; BTK, Bruton tyrosine kinase; PK, pharmacokinetic; QD, once daily
The exposure–efficacy relationship was evaluated in a patient population from 1 phase 3 trial (n = 274) with 1 disease histology (previously untreated CLL) while the exposure–safety analysis was performed in a pooled patient population across several clinical trials involving various B‐cell malignancies (n = 573). In the exposure–efficacy analysis in patients with previously untreated CLL, no clinically meaningful relationships were observed between total active exposures and selected efficacy outcomes (including BOR, PFS and change from baseline in SPD) following treatment with acalabrutinib as monotherapy or in combination with obinutuzumab. The exposure–efficacy analyses represented a wide range of acalabrutinib/total active exposures, with an ~3–4‐fold difference between the 2.5th percentile of the first quartile (Q1) and 97.5th percentile of the fourth quartile (Q4) in AUC24h,ss. Similarly, no relationships were observed between acalabrutinib exposure and incidences of grade ≥2 AEs, grade ≥3 AEs, or select grade ≥2 AEs of clinical interest following treatment with acalabrutinib as monotherapy or in combination with obinutuzumab. The overall safety population represented a wide range of acalabrutinib exposures with an ~5‐fold range between the 2.5th percentile for Q1 and 97.5th percentile of Q4 in AUC24h,ss. Overall, the lack of relationship between exposure and efficacy and safety outcomes indicates that the acalabrutinib 100‐mg BID regimen elicited strong and consistent therapeutic effects across the wide range of PK exposures.
These results are consistent with those reported for ibrutinib and zanubrutinib, 19 and indicate a comparable or more favourable risk–benefit profile for acalabrutinib. For ibrutinib, while no exposure‐driven relationships were identified for individual toxicities (grade ≥3 neutropenia or grade ≥3 infections and infestations), a slight increase in the proportion of patients with any grade ≥3 AEs was noted with increasing ibrutinib mean steady‐state trough concentrations in patients with MCL or CLL in 2 pivotal trials. 20 Additionally, a population PK model of ibrutinib and its metabolite in lymphoid malignancies demonstrated significantly higher ibrutinib exposure, as measured by AUC, among patients who discontinued treatment due to toxicity. 21 For zanubrutinib, results from an exposure–response analysis in patients with MCL, CLL and Waldenström macroglobulinemia suggested no relationships between zanubrutinib exposure and the probability of experiencing AEs of interest including neutropenia, thrombocytopenia, anaemia, infections, second primary malignancies, atrial fibrillation/flutter and bleeding events. 22 In general, fewer toxicities related to off‐target kinase inhibition are expected because of the increased kinase selectivity of acalabrutinib relative to ibrutinib or zanubrutinib. 19 However, given the overlapping toxicities observed across BTK inhibitors, some of these events could be related, at least in part, to the inhibition of BTK rather than off‐target kinases.
Because both acalabrutinib and ACP‐5862 are irreversible, covalent inhibitors, the inhibition of B‐cell receptor signal transduction is maintained until functional levels of BTK are restored, following BTK re‐synthesis. 11 Administration of 100 mg acalabrutinib BID has been shown to maintain a near complete and continuous BTK inhibition in CLL. 4 Median BTK occupancy of ≥95% was maintained over 12 hours in patients with B‐cell malignancies following acalabrutinib 100 mg BID. 3 These results, based on measurements conducted in PBMCs, are consistent across tumour‐protective reservoirs such as bone marrow and lymph nodes. 23 Similar results for BTK occupancy have been reported for ibrutinib (>95% BTK occupancy in PBMCs following QD regimens) and zanubrutinib (>95% BTK occupancy in PBMCs and lymph nodes in the majority of patients following 160‐mg BID regimen). 24 , 25
The current study has a few potential limitations. One limitation of the analysis is the lack of data assessing the relationship between accumulated exposure (e.g., accumulated AUC) and safety; however, it would be unlikely to result in a conclusion different from that in the current analysis, which assesses the relationship between steady‐state exposure and safety. Cumulative exposure is not as feasible when pooling data with different durations of treatment; because the data would need to be normalized to days on treatment or similar, the results would probably be similar to the steady‐state metrics currently used. The average daily dose across the efficacy/safety populations was ~200 mg/d and is consistent with the minimal dose reductions, dose interruptions and discontinuations that were observed due to AEs. Overall, the observed dose modifications are not expected to alter the conclusions of the current analysis, and the current strategy of using exposures predicted at the most prevalent dosing regimen is considered appropriate. In addition, a multivariate analysis was not used; however, several efficacy and safety variables were included in the overall assessments of responses to acalabrutinib exposure. Additionally, continuous variables were converted into categorical variables (i.e., cytopenias were assessed by AE grade rather than cell counts) and patients were classified by the highest observed grade of laboratory abnormality for the purposes of this study. This conversion made the results of the assessments more clinically interpretable. The study was limited to patients who were enrolled in clinical trials; in real‐world practice, the presence of baseline characteristics and/or concomitant medications that would have excluded a patient from clinical trial participation could potentially influence the occurrence of the safety events of interest in the current study. However, as the study evaluated a large cohort of patients and included multiple B‐cell histologies for the safety analyses, these results can still be considered applicable to a more general population of patients. Although ACP‐5862 PK data were only available in a subset of patients, the sample size was considered adequate for characterizing the exposure–response profile. Finally, this study did not account for the timing of the events or the possibility that an observed event occurrence could have resolved with continued treatment. However, these potential limitations do not impact the overall conclusions of these analyses.
5. CONCLUSION
No exposure–response relationships were apparent between PK exposures and efficacy outcomes in patients with untreated CLL or safety outcomes in patients with B‐cell malignancies following treatment with acalabrutinib. Across various phase 2 and 3 studies, the 100‐mg BID acalabrutinib regimen has demonstrated an acceptable safety profile with robust efficacy in patients with CLL. 5 , 7 , 8 Taken together, these data support the favourable risk–benefit profile of acalabrutinib 100 mg BID in the treatment of subjects with B‐cell malignancies, including CLL.
TWITTER STATEMENT
In this analysis, acalabrutinib 100 mg twice daily produced robust and consistent therapeutic effects across the exposure range.
COMPETING INTERESTS
Helena Edlund, Karthick Vishwanathan, Miné de Kock, Zhongqing He, Huan Liu and Helen Tomkinson are employees of AstraZeneca. Priti Patel and Shringi Sharma are employees and stockholders of AstraZeneca. Helen Wei, Marshall Baek and Núria Buil‐Bruna are former employees of AstraZeneca. Rakesh Raman and Joseph Ware are former employees of AstraZeneca and retain AstraZeneca stock. The study was funded by Acerta Pharma, South San Francisco, CA, a member of the AstraZeneca Group.
CONTRIBUTORS
H.E. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
N.B.B. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
K.V. contributed to the interpretation of the data, and to writing, reviewing and approving the manuscript.
H.W. contributed to the interpretation of the data, and to reviewing and approving the manuscript.
R.R. contributed to the interpretation of the data, and to reviewing and approving the manuscript.
M.d.K. contributed to the analysis of the data, and to reviewing and approving the manuscript.
Z.H. contributed to the analysis, and to reviewing and approving the manuscript.
H.L. contributed to the analysis, and to reviewing and approving the manuscript.
M.B. contributed to the analysis, and to reviewing and approving the manuscript.
J.A.W. contributed to the study, analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
P.P. contributed to the interpretation of the data, and to reviewing and approving the manuscript.
H.T. contributed to the interpretation of the data, and to writing, reviewing and approving the manuscript.
S.S. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
.
Supporting information
Table S1. Details of studies included in the pooled safety subpopulations
Table S2. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure by quartiles (efficacy population)
Table S3. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure by quartiles (safety population)
Table S4. Patient demographics and baseline characteristics (continuous variables, efficacy population by treatment arms)
Table S5. Patient demographics and baseline characteristics (categorical variables, efficacy population by treatment arms)
Table S6. Patient demographics and baseline characteristics (continuous variables, safety subpopulations)
Table S7. Patient demographics and baseline characteristics (categorical variables, safety subpopulations)
Figure S1. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure in the efficacy population across treatment arms
Figure S2. Distribution of acalabrutinib, ACP‐5862 and total active exposure metrics by safety subpopulations
Figure S3. Distribution and pairwise correlation of acalabrutinib AUC24h,ss and ACP‐5862 AUC24h,ss in the efficacy population (n = 274)
Figure S4. Distribution and pairwise correlation of acalabrutinib AUC24h,ss and ACP‐5862 AUC24h,ss in the overall safety population (n = 573)
Figure S5. AUC24h,ss (A, C) and Cmax,ss (B, D) by best overall response for acalabrutinib (A, B) and ACP‐5862 (C, D)
Figure S6. Correlation between Ctrough (Cmin) and AUC
Figure S7. Acalabrutinib Cmax,ss stratified by adverse event
Figure S8. Relevant adverse events by acalabrutinib Cmax,ss quartiles
Figure S9. Probability of grade ≥2 infection by acalabrutinib AUC24h,ss quartiles
ACKNOWLEDGEMENTS
The authors acknowledge the Acerta Pharma study team. They also thank the patients who participated in these studies as well as their friends and family who supported them. The study was funded by Acerta Pharma, South San Francisco, CA, a member of the AstraZeneca Group. Medical writing assistance, funded by Acerta Pharma, was provided by Tracy Diaz, PhD, and Cindy Gobbel, PhD, of Peloton Advantage, LLC, an OPEN Health company.
Finally, the authors wish to dedicate this manuscript to the memory of Dr Nataliya Chernyukhin, former Head of Medical Safety Science at Acerta Pharma. Nataliya's mentorship, wisdom and passion for patient‐focused drug development will be missed but not forgotten.
Edlund H, Buil‐Bruna N, Vishwanathan K, et al. Exposure–response analysis of acalabrutinib and its active metabolite, ACP‐5862, in patients with B‐cell malignancies. Br J Clin Pharmacol. 2022;88(5):2284-2296. doi: 10.1111/bcp.15087
Helena Edlund and Núria Buil‐Bruna: equal contribution.
A principal investigator was not included in the author byline because the reported data are from a pooled exposure–response analysis of data from 8 different studies. The principal investigators of the original studies were not involved in performing these analyses or interpreting the data; therefore, they did not qualify for authorship according to ICMJE criteria.
DATA AVAILABILITY STATEMENT
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
REFERENCES
- 1. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton's tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP‐196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240‐252. [DOI] [PubMed] [Google Scholar]
- 3. Calquence [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2019. [Google Scholar]
- 4. Byrd JC, Harrington B, O'Brien S, et al. Acalabrutinib (ACP‐196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Byrd JC, Wierda WG, Schuh A, et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: updated phase 2 results. Blood. 2020;135(15):1204‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang M, Rule S, Zinzani PL, et al. Durable response with single‐agent acalabrutinib in patients with relapsed or refractory mantle cell lymphoma. Leukemia. 2019;33(11):2762‐2766. [DOI] [PubMed] [Google Scholar]
- 7. Ghia P, Pluta A, Wach M, et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2849‐2861. [DOI] [PubMed] [Google Scholar]
- 8. Sharman JPE, Egyed M, Jurczak W, Skarbnik A, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment‐naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herman SEM, Montraveta A, Niemann CU, et al. The Bruton tyrosine kinase (BTK) inhibitor acalabrutinib demonstrates potent on‐target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. 2017;23(11):2831‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calquence [summary of product characteristics]. Bedfordshire, United Kingdom: AstraZeneca UK Limited; 2020. [Google Scholar]
- 11. Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55(14):6243‐6262. [DOI] [PubMed] [Google Scholar]
- 12. Podoll T, Pearson PG, Evarts J, et al. Bioavailability, biotransformation, and excretion of the covalent Bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab Dispos. 2019;47(2):145‐154. [DOI] [PubMed] [Google Scholar]
- 13. Edlund H, Bellanti F, Liu H, et al. Improved characterization of the pharmacokinetics of acalabrutinib and its pharmacologically active metabolite, ACP‐5862, in patients with B‐cell malignancies and in healthy subjects using a population pharmacokinetic approach. Br J Clin Pharmacol. 2021. Online ahead of print. 10.1111/bcp.14988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukaka MM. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med J. 2012;24(3):69‐71. [PMC free article] [PubMed] [Google Scholar]
- 15. Alexander SPH, Kelly E, Mathie A, et al. The concise guide to PHARMACOLOGY 2019/20: introduction and other protein targets. Br J Pharmacol. 2019;176(Suppl 1):S1‐s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaptein A, Podoll T, de Bruin G, et al. Preclinical pharmacological profiling of ACP‐5862, the major metabolite of the covalent BTK inhibitor acalabrutinib, displays intrinsic BTK inhibitory activity [poster]. Presented at: Annual Meeting of the American Association for Cancer Research; March 29‐April 3, 2019; Atlanta, GA
- 17. Marostica E, Sukbuntherng J, Loury D, et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol. 2015;75(1):111‐121. [DOI] [PubMed] [Google Scholar]
- 18. NDA/BLA Multi‐disciplinary review and evaluation NDA 213217 Brukinsa (zanubrutinib). Center for Drug Evaluation and Research. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213217Orig1s000MultidisciplineR.pdf. Accessed August 23, 2021.
- 19. Kaptein A, de Bruin G, Emmelot‐van Hoek M, van de Kar B, de Jong A, Gulrajani M, Demont D, Covey T, Mittag D, Barf T Potency and selectivity of BTK inhibitors in clinical development for B‐cell malignancies [poster]. Presented at: Annual Meeting of the American Society of Hematology; December 1‐4, 2018; San Diego, CA.
- 20. Clinical Pharmacology and Biopharmaceutics Review(s): Imbruvica™ (ibrutinib). Center for Drug Evaluation and Research. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205552Orig1s000ClinPharmR.pdf. Accessed August 23, 2021.
- 21. Gallais F, Ysebaert L, Despas F, et al. Population pharmacokinetics of ibrutinib and its dihydrodiol metabolite in patients with lymphoid malignancies. Clin Pharmacokinet. 2020;59(9):1171‐1183. [DOI] [PubMed] [Google Scholar]
- 22. Ou Y, Wang K, Liu L, Jindal A, Gao Y, Sahasranaman S. Exposure‐response relationship of the bruton tyrosine kinase inhibitor, zanubrutinib (BGB‐3111) in patients with hematologic malignancies [abstract]. Blood. 2019;134(Suppl 1):5063. [Google Scholar]
- 23. Sun CCL, Nierman PK, Kendall EK, et al. Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood. 2020;136(1):93‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Byrd JC, Woyach JA, Furman RR, et al. Acalabrutinib in treatment‐naïve chronic lymphocytic leukemia: mature results from phase II study demonstrating durable remissions and long‐term tolerability [abstract]. Presented at: Annual Meeting of the American Society of Clinical Oncology; May 29‐31, 2020; Chicago, Il
- 27. Woyach JA, Blachly JS, Rogers KA, et al. Acalabrutinib plus obinutuzumab in treatment‐naive and relapsed/refractory chronic lymphocytic leukemia. Cancer Discov. 2020;10(3):394‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dyer MJS, De Vos S, Ruan J, et al. Acalabrutinib monotherapy in patients (pts) with relapsed/refractory (R/R) diffuse large B‐cell lymphoma (DLBCL) [abstract]. J Clin Oncol. 2018;36(suppl 15):7547. [Google Scholar]
- 29. Fowler NH, Coleman M, Stevens DA, et al. Acalabrutinib alone or in combination with rituximab (R) in follicular lymphoma (FL) [abstract]. J Clin Oncol. 2018;36(suppl 15):7549. [Google Scholar]
- 30. Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenstrom macroglobulinemia: a single‐arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112‐e121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Details of studies included in the pooled safety subpopulations
Table S2. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure by quartiles (efficacy population)
Table S3. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure by quartiles (safety population)
Table S4. Patient demographics and baseline characteristics (continuous variables, efficacy population by treatment arms)
Table S5. Patient demographics and baseline characteristics (categorical variables, efficacy population by treatment arms)
Table S6. Patient demographics and baseline characteristics (continuous variables, safety subpopulations)
Table S7. Patient demographics and baseline characteristics (categorical variables, safety subpopulations)
Figure S1. Summary of the distribution of acalabrutinib, ACP‐5862 and total active exposure in the efficacy population across treatment arms
Figure S2. Distribution of acalabrutinib, ACP‐5862 and total active exposure metrics by safety subpopulations
Figure S3. Distribution and pairwise correlation of acalabrutinib AUC24h,ss and ACP‐5862 AUC24h,ss in the efficacy population (n = 274)
Figure S4. Distribution and pairwise correlation of acalabrutinib AUC24h,ss and ACP‐5862 AUC24h,ss in the overall safety population (n = 573)
Figure S5. AUC24h,ss (A, C) and Cmax,ss (B, D) by best overall response for acalabrutinib (A, B) and ACP‐5862 (C, D)
Figure S6. Correlation between Ctrough (Cmin) and AUC
Figure S7. Acalabrutinib Cmax,ss stratified by adverse event
Figure S8. Relevant adverse events by acalabrutinib Cmax,ss quartiles
Figure S9. Probability of grade ≥2 infection by acalabrutinib AUC24h,ss quartiles
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.