

Abstract
Background and Purpose
There is a scarcity of information regarding the role of prothrombin kringle‐2 (pKr‐2), which can be generated by active thrombin, in hippocampal neurodegeneration and Alzheimer's disease (AD).
Experimental Approach
To assess the role of pKr‐2 in association with the neurotoxic symptoms of AD, we determined pKr‐2 protein levels in post‐mortem hippocampal tissues of patients with AD and the hippocampi of five familial AD (5XFAD) mice compared with those of age‐matched controls and wild‐type (WT) mice, respectively. In addition, we investigated whether the hippocampal neurodegeneration and object memory impairments shown in 5XFAD mice were mediated by changes to pKr‐2 up‐regulation.
Key Results
Our results demonstrated that pKr‐2 was up‐regulated in the hippocampi of patients with AD and 5XFAD mice, but was not associated with amyloid‐β aggregation in 5XFAD mice. The up‐regulation of pKr‐2 expression was inhibited by preservation of the blood–brain barrier (BBB) via addition of caffeine to their water supply or by treatment with rivaroxaban, an inhibitor of factor Xa that is associated with thrombin production. Moreover, the prevention of up‐regulation of pKr‐2 expression reduced neurotoxic symptoms, such as hippocampal neurodegeneration and object recognition decline due to neurotoxic inflammatory responses in 5XFAD mice.
Conclusion and Implications
We identified a novel pathological mechanism of AD mediated by abnormal accumulation of pKr‐2, which functions as an important pathogenic factor in the adult brain via blood brain barrier (BBB) breakdown. Thus, pKr‐2 represents a novel target for AD therapeutic strategies and those for related conditions.
Keywords: Alzheimer's disease, blood–brain barrier, hippocampus, microglia, prothrombin kringle‐2
Abbreviations
- 5XFAD
five familial Alzheimer's disease
- AD
Alzheimer's disease
- AF
Alexa Fluor
- BBB
blood–brain barrier
- CA1
cornu ammonis 1
- CD31
cluster of differentiation 31
- DAB
3,3′‐diaminobenzidine
- Iba1
ionized calcium‐binding adapter molecule 1
- pKr‐2
prothrombin kringle‐2
- TLR4
toll‐like receptor 4
- WT
wild‐type
- ZO‐1
zonula occludens‐1
What is already known
pKr‐2 up‐regulation can induce microglia‐mediated neurotoxic inflammation in the adult murine brain.
What does this study add
We identified the major source and cause for pKr‐2 up‐regulation in the adult brain.
We identified a novel pathological mechanism in AD mediated by hippocampal up‐regulation of pKr‐2.
What is the clinical significance
pKr‐2 represents a novel target for AD therapeutic strategies and those for related conditions.
1. INTRODUCTION
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease causing dementia characterized by cognitive impairment and memory loss associated with hippocampal neurodegeneration (Cummings, 2004; Kawas, 2003; Querfurth & LaFerla, 2010). Although the aetiology of AD is not yet defined and therefore remains unclear, an accumulation of evidence suggests that neuroinflammatory processes, particularly those mediated by activated microglia, are important in the initiation and progression of the disease (Choi et al., 2005; Merlini et al., 2019; Morales et al., 2014). There were reports that the hippocampal up‐regulation of blood proteins, such as thrombin and fibrinogen, could contribute to microglial activation, which resulted in neurodegeneration in the hippocampus in adult mice (Choi et al., 2005; Merlini et al., 2019). In addition, cerebrovascular alteration, which is a pathological hallmark in the hippocampi of patients with early AD (Giannoni et al., 2016; Sweeney et al., 2018; van de Haar et al., 2016), leads to blood–brain barrier (BBB) disruption resulting in the infiltration of circulating substances into the brain (Merlini et al., 2019; Sweeney et al., 2018; van de Haar et al., 2016). Hence, the investigation of endogenous molecules in the adult brain, up‐regulated by BBB disruption, which can cause neurotoxic inflammation is essential for the developement useful treatment strategies for AD.
Prothrombin kringle‐2 (pKr‐2) is a domain of prothrombin generated by active thrombin, which itself originates by cleavage of prothrombin by factor Xa, the active form of the coagulation factor X (Leem et al., 2016; Mann, 1976; Shin et al., 2015; Taneda et al., 1994). We previously reported that pKr‐2 administration could activate microglia, resulting in neuronal death in the substantia nigra in vivo, and the induction of toll‐like receptor 4 (TLR4) contributed to the pKr‐2‐induced microglial activation (Leem et al., 2016; Kim et al., 2010; Shin et al., 2015). In addition, the increased levels of prothrombin and thrombin have been reported in the post‐mortem brains of patients with neurodegenerative diseases, including AD and Parkinson's disease (PD) (Arai et al., 2006; Ishida et al., 2006; Leem et al., 2016; Sokolova & Reiser, 2008). These previous results suggest that pKr‐2 levels may also be up‐regulated in the brains of patients with neurodegenerative diseases. Further, pKr‐2‐induced microglial activation and neuroinflammatory responses may be closely associated with the diverse pathogeneses of neurodegenerative diseases, including AD. However, to our knowledge the role of pKr‐2 in association with hippocampal neurodegeneration and its mechanisms in AD, including possible pKr‐2 up‐regulation and its cause in the adult brain, has yet to be reported.
In the present study, we determined the pKr‐2 protein levels in post‐mortem hippocampal tissues from patients with AD and compared them with levels from age‐matched controls. We also investigated whether pKr‐2 up‐regulation is derived from disruption of the BBB in five familial AD (5XFAD) mice (Eimer & Vassar, 2013; Giannoni et al., 2016; Oakley et al., 2006) and whether inhibition of this up‐regulation diminishes pathogenic processes, including hippocampal neurodegeneration by microglial activation and object cognitive impairments, in 5XFAD mice.
2. METHODS
2.1. Animals
Male C57BL/6 mice (aged 8 weeks) for experiments related to amyloid‐β aggregation, oligomerization and object cognitive impairments following pKr‐2 administration in wild‐type (WT) mice (Figures 6c and S8) were obtained from Daehan Biolink (Eumseong, RRID:IMSR_JAX:000664, Korea). Littermate WT mice and 5XFAD mice (both male and female) with a C57BL/6 background as an AD model, provided by the Korea Brain Research Institute, were used for other experiments except Figure 6c. Experiments using a sample size of n = 5 included male and female mice at a ratio of 3:2, and experiments using a sample size of n = 6 had a male:female ratio of 3:3.
FIGURE 6.

Effect of prothrombin kringle‐2 (pKr‐2) administration on amyloid‐β (Aβ) expression, neuronal nuclei (NeuN) expression and neuroinflammatory molecules in the hippocampi of five familial Alzheimer's disease (5XFAD) mice. (a) Thioflavin S (protein aggregation marker) and Aβ immunostaining in the hippocampi of WT and 5XFAD mice aged 6 months. Scale bar, 200 μm. (b) Western blotting for the levels of Aβ expression in the hippocampus of WT and 5XFAD mice. (c) Western blotting for the protein levels of Aβ in the hippocampi of WT mice at 1, 7, 14 and 28 days after pKr‐2 injection. CON indicates the hippocampi of intact mice. pKr‐2 administration had no change in Aβ expression in WT mice. (d) Experimental schematic for pKr‐2 treatment in the hippocampi of 5XFAD mice. (e) Western blotting for the protein levels of Aβ in the hippocampi at 7 days and 3 months after pKr‐2 administration. *P < 0.05 versus age‐matched WT controls; NS, no significance (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (f) Immunohistochemistry for analysis of Aβ (dark brown dots) in the hippocampi at 7 days (upper panels) and 3 months (lower panels) after pKr‐2 injection. Scale bar, 500 μm. (g) Western blotting for the protein levels of NeuN and Iba1 in the hippocampi of mice at 7 days and 3 months after pKr‐2 administration. *P < 0.05 versus nontreated WT mice; # P < 0.05 and # P < 0.05 versus nontreated 5XFAD mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (h) Western blotting for the protein levels of TLR4, TNF‐α, IL‐1β and iNOS in the hippocampi of mice at 7 days and 3 months after pKr‐2 administration. *P < 0.05 versus nontreated WT mice; # P < 0.05 versus nontreated 5XFAD mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). Quantitative values are expressed as the mean ± SEM
2.2. Human brain tissues
Frozen brain tissues were obtained from the Victorian Brain Bank Network (VBBN), supported by The Florey Institute of Neuroscience and Mental Health and The Alfred and the Victorian Forensic Institute of Medicine and funded by Australia's National Health and Medical Research Council. Details of the human post‐mortem hippocampal tissues are shown in Figure 1a.
FIGURE 1.
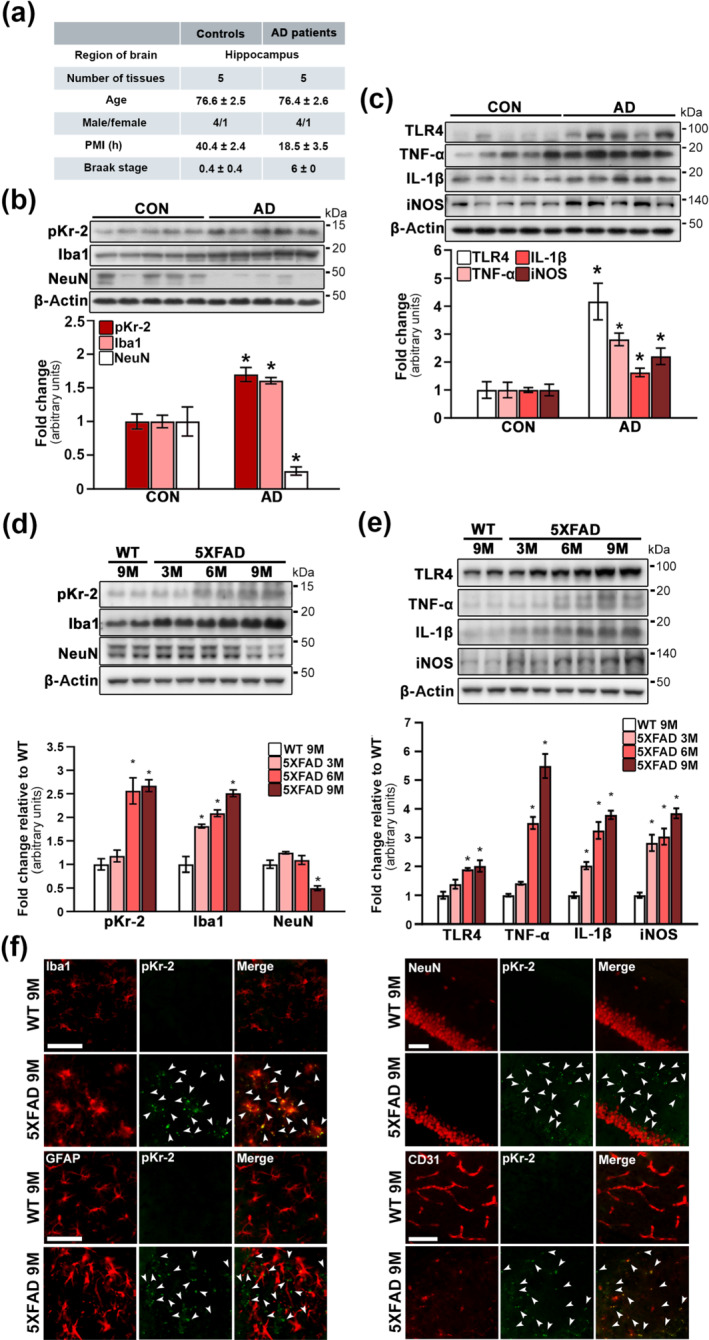
Up‐regulation of prothrombin kringle‐2 (pKr‐2) and neuroinflammatory molecules in the hippocampi of patients with Alzheimer's disease (AD) and five familial Alzheimer's disease (5XFAD) mice. (a) Details of human post‐mortem hippocampal tissues presented from Victorian Brain Bank Network. PMI, post‐mortem interval. (b) Western blotting for the expression levels of pKr‐2, Iba1 and neuronal nuclei (NeuN) in the human hippocampi of age‐matched controls (CON) and patients with AD. *P < 0.05 vs. CON (Unpaired Student's t‐test; n = 5 for each group). (c) Western blotting for the protein levels of TLR4, TNF‐α, IL‐1β and iNOS in the human hippocampi of age‐matched controls (CON) and patients with AD. *P < 0.05, versus CON (Unpaired Student's t‐test; n = 5 for each group). (d) Western blotting for the protein levels of pKr‐2, Iba1 and NeuN in the hippocampal tissues obtained from 3‐, 6‐ and 9‐month‐old 5XFAD and 9‐month‐old WT mice. *P < 0.05 versus 9‐month‐old WT mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (e) Western blotting for the protein levels of TLR4, TNF‐α, IL‐1β and iNOS in the hippocampal tissues obtained from 5XFAD and WT mice. *P < 0.05 and versus 9‐month‐old WT mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). All quantitative values are presented as the mean ± SEM. (f) Location of pKr‐2 expression in the hippocampi of 5XFAD mice. The red fluorescent signals represent Iba1, GFAP, NeuN and CD31, and the green fluorescent signal represents pKr‐2, respectively, in the respective hippocampal section of 9‐month‐old WT and 5XFAD mice. The white arrowheads indicate pKr‐2 expression. Scale bar, 50 μm
2.3. Ethics approval
All animal experimental procedures were conducted according to the Guidelines for Animal Care and Use of Kyungpook National University approved by the Animal Care and Use Committee of Kyungpook National University (Nos. KNU 2016‐0042 and 2019‐0002). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). The animals used in the present study were acquired and cared for according to the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animals were group housed with an inverse 12‐h light–dark cycle in a temperature‐controlled room and were provided with free access to food and water. Three to four mice were kept in a individually ventilated cage containing dried wood chips as bedding. The animals were randomly divided into treatment groups. Human tissue experiments were approved by the Bioethics Committee, Institutional Review Board, Kyungpook National University Industry Foundation (IRB Nos. 2016‐0011 and 2018‐0207‐1).
2.4. Intrahippocampal injection of pKr‐2
The animals were anaesthetized with intraperitoneal injections of ketamine (115 mg·kg−1; Yuhan, Seoul, Korea) and xylazine (23 mg·kg−1; Bayer Korea Ltd., Seoul, Korea) and then placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). During anesthesia, the mice were spontaneously breathing and the body temperature of mice was maintained close to 37°C throughout the procedure by using a heating pad. Using a 10‐μl Hamilton syringe (30 S needle), WT and 5XFAD mice were administered with a unilateral injection of pKr‐2 (24 μg in 2 μl of PBS) (Kim et al., 2010; Leem et al., 2016; Shin et al., 2015) or PBS (as a vehicle control) into the right hippocampal cornu ammonis 1 (CA1) region (anterior–posterior [AP]: −2.0 mm; medial–lateral [ML]: −1.2 mm; and dorsal–ventral [DV]: −1.5 mm; all relative to the bregma) according to the mouse brain atlas (Paxinos & Franklin, 2004). To investigate whether pKr‐2 injection induces object cognitive impairment, WT mice were administered with bilateral injections of pKr‐2 or PBS into the hippocampal CA1 region (AP: −2.0 mm, ML: ±1.2 mm and DV: −1.5 mm, relative to the bregma). Using an automated syringe pump, 2.0 μl of pKr‐2 or PBS injection was infused at a rate of 0.5 μl·min−1 over 4 min, and the injection needle was left in place for an additional 5 min before being slowly retracted. The skull and skin were wetted with PBS and the incision was sutured with a medical surgical suture needle. During recovery, the mice were placed on the heated surface and monitored by investigator until mobility regained.
2.5. Caffeine supply
Caffeine was supplied as previously described by Chen et al. (2008, 2010) but with some modifications. Briefly, caffeine was dissolved in their drinking water (0.06 or 0.6 mg·ml−1). Given that mice drink, on average, 2.5 ml·day−1, we administered a daily dose of 0.15‐ or 1.5‐mg caffeine to each mouse. The caffeinated water was changed every 2 days to ensure that caffeine remained completely dissolved. We designed two experimental conditions. In the first, we aimed to collect data on the anti‐neurodegeneration and anti‐inflammatory effects of inhibiting pKr‐2 up‐regulation in the hippocampi of 5XFAD mice. WT and 5XFAD mice were provided with caffeinated water daily from the age of 2 months for a period of 7 months and data were analysed from mice aged 9 months. In the second, we aimed to investigate preservation of BBB integrity in 5XFAD mice so we provided them caffeinated water daily from the age of 2 months for a period of 4 months and for control this was also carried out in WT mice.
2.6. Blood‐brain barrier (BBB) permeability analysis
For BBB assessment, 5XFAD mice were given caffeine dissolved in their drinking water (0.06 or 0.6 mg·ml−1) from the age of 2 months for a period of 4 months. To measure the permeability of the BBB, these mice were treated with 100 μg of 10‐kDa fluorescein isothiocyanate (FITC)‐dextran (Thermo Fisher Scientific, Cat#: D1820) via intravenous injection into the tail vein and then killed 10 min after the injection. They were then perfused with ice‐cold PBS and further perfused with 1% paraformaldehyde (PFA)/PBS. The brain from each mouse was collected and further fixed with MeOH or 4% PFA/PBS overnight at 4°C. The fixed brains were sectioned to a thickness of 50 μm using a vibratome. Each section was blocked with 10% goat serum diluted in PBS. For the MeOH‐fixed tissue, the sections were incubated with rat anti‐laminin (1:500) and AF488‐conjugated rabbit anti‐claudin‐5 (1:500, Thermo Fisher Scientific, Cat#: 352588, RRID:AB_2532189) antibodies overnight at room temperature. For the PFA‐fixed tissue, the sections were incubated with anti‐CD31 (1:500) and AF568‐conjugated anti‐mouse‐IgG (1:500, Cat#: A‐21235, RRID:AB_2535804) antibodies. These antibodies were diluted in 1% BSA/0.2% Triton X/PBS. The sections were then washed with 0.2% Triton X/PBS three times (each for 30 min), stained with AF647‐conjugated anti‐rat, anti‐rabbit or anti‐goat secondary antibody (Thermo Fisher Scientific, Cat#: A‐21447, RRID:AB_2535864) diluted in 1% BSA/0.2% Triton X/PBS at 4°C overnight and washed again with 0.2% Triton X/PBS three times (each for 30 min). Finally, the sections were coverslipped with a mounting solution (Fluormount G; Electron Microscopy Science, Hatfield, PA) and visualized under a LECIA SP 8.0 confocal microscope (Leica Microsystems, Wetzlar, Germany).
To analyse IgG penetration into the brain, mouse brains were first homogenized and centrifuged at 14,000× g and 4°C for 15 min. The resulting supernatant was transferred to a fresh tube, and the protein concentration was determined using a Bicinchoninic Acid Assay Kit (Bio‐Rad Laboratories, Cat#: 5000116). In total, 50 μg of protein was electrophoresed on a sodium dodecyl sulfate/polyacrylamide gel (Bio‐Rad Laboratories) and transferred to PVDF membranes (Millipore) using an electrophoretic transfer system (Bio‐Rad Laboratories). The membranes were incubated with HRP‐conjugated mouse secondary antibody (1:2000) at room temperature for 1 h. They were then washed, after which they were developed using enhanced chemiluminescence western blot detection reagents (GE Healthcare Life Sciences, Little Chalfont, UK). The resultant signal was analysed using a LAS‐500 image analyzer (GE Healthcare Life Sciences). The density of bands was measured using Multi Gauge Version 3.0 (Fujifilm, RRID:SCR_014299, Tokyo, Japan), and the density of target proteins was normalized to the β‐actin band for each sample.
2.7. Immunostaining procedures
Brain tissues were prepared for immunostaining analysis using previously described methods (Jeon et al., 2020; Shin et al., 2015). As above, mice were initially transcardially perfused and fixed with 4% PFA in PBS. Fixed brains were frozen before being cut into 30‐μm coronal sections using a cryostat microtome (Thermo Fisher Scientific). Immunostaining was performed in a free‐floating manner in 12‐well plates. Brain sections were washed with cold PBS and blocked using a blocking solution (0.5% BSA in PBS). They were then incubated at 4°C for 48 h with the primary antibodies rabbit anti‐amyloid‐β (1:500) and rabbit anti‐CD31 (1:500). After this incubation, the sections were incubated with biotinylated secondary antibodies, including anti‐rabbit IgG (1:400), for 1 h at room temperature. Subsequently, they were processed using an avidin–biotin reagent (Vectastain ABC Kit) for 1 h at room temperature. The signal was then detected by incubating the sections in a peroxidase substrate solution (0.5 mg·ml−1 of 3,3′‐diaminobenzidine [DAB; Sigma, Cat#: D5637] in 0.1‐M PB containing 0.015% H2O2). The stained sections were mounted on positively charged slides (Paul Marienfeld, Baden‐Württemberg, Germany), covered using a cover slip (Paul Marienfeld) and then analysed under a bright‐field microscope (Axio Imager, Carl Zeiss, Jena, Germany). The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
For immunofluorescence staining, sections were first incubated at 4°C for 48 h with the following primary antibodies: rabbit anti‐neuronal nuclei (1:500), rabbit anti‐Iba1 (1:2000), rabbit anti‐GFAP (1:2000), rabbit anti‐CD31 (1:500) and mouse anti‐prothrombin (1:500). The sections were then rinsed and incubated with fluorescence‐conjugated secondary antibodies, which included Texas Red‐conjugated anti‐rabbit IgG (1:400) and fluorescein isothiocyanate‐conjugated anti‐mouse IgG (1:400) antibodies, for 1 h at room temperature, after which the sections were washed and mounted with VECTASHIELD mounting medium (Vector Laboratories). The stained sections were imaged using a fluorescence microscope (Carl Zeiss).
For Nissl staining, the sections were mounted on positively charged slides (Paul Marienfeld), stained with 0.5% cresyl violet (Sigma, Cat#: 10510‐54‐0) and then analysed under a bright‐field microscope (Carl Zeiss).
2.8. Thioflavin S staining
Brain sections were mounted on positively charged slides (Paul Marienfeld) and dried before staining. The sections were washed with 80% ethanol, incubated first with thioflavin S solution (1% in 80% ethanol) for 15 min and then with 70% ethanol for 5 min, and finally washed with distilled water. The stained sections were coverslipped (Paul Marienfeld) and imaged using a fluorescence microscope (Carl Zeiss).
2.9. Counting of hippocampal CA1 neurons
The number of hippocampal CA1 neurons was quantified as described previously, with some modifications (Jeon et al., 2015, 2020; Kim et al., 2017). Alternate sections were prepared from the coronal brain slices of each animal at 1.7, 1.8, 2.0 and 2.1 mm posterior to the bregma. To ensure consistency in tissue sampling, a rectangular box (1.5 × 0.5 mm) was centred over the CA1 cell layer, beginning at 0.5 mm contralateral and ipsilateral to the midline. Neurons with visible nuclei in the CA1 cell layer were counted exclusively under a light microscope (Carl Zeiss) at a magnification of 200×.
2.10. Western blot analysis
Western blotting was performed using previously described methods (Jeon et al., 2020; Shin et al., 2015). Briefly, mouse or human hippocampal tissues were homogenized and centrifuged at 14,000× g and 4°C for 15 min. The supernatant was transferred to a fresh tube, and the protein concentration was determined using a Bicinchoninic Acid Assay Kit (Bio‐Rad Laboratories). Protein (50 μg) was then electrophoresed on a sodium dodecyl sulfate/polyacrylamide gel (Bio‐Rad Laboratories) and transferred to PVDF membranes (Millipore) using an electrophoretic transfer system (Bio‐Rad Laboratories). The membranes were incubated at 4°C for 48 h with the following primary antibodies: sheep anti‐prothrombin fragment 2 (1:500), rabbit anti‐prothrombin (1:1000), rabbit anti‐thrombin (1:1000), mouse anti‐neuronal nuclei (1:1000), rabbit anti‐Iba1 (1:1000), rabbit anti‐TLR4 (1:1000), rabbit anti‐IL‐1β (1:500), mouse anti‐TNF‐α (1:500), rabbit anti‐iNOS (1:1000), rabbit anti‐ZO‐1 (1:1000), rabbit anti‐CD31 (1:1000), rabbit anti‐amyloid‐β (1:1000) and mouse anti‐β‐actin (1:1000). The membranes were subsequently washed and incubated with HRP‐conjugated secondary antibodies, including anti‐mouse IgG (1:4000), anti‐rabbit IgG (1:4000) and anti‐sheep IgG (1:2000), at room temperature for 1 h, after which they were developed using enhanced chemiluminescence western blot detection reagents (GE Healthcare Life Sciences). The resultant signal was analysed using a LAS‐500 image analyser (GE Healthcare Life Sciences), and band density was measured using Multi Gauge Version 3.0 (Fujifilm) with the target protein density normalized to the β‐actin band for each sample. For the analysis of human protein expression levels, western blotting was performed once using all human control and AD lysate samples (n = 5 per group) on a single gel. For analysis of protein expression changes among mouse experimental groups, western blotting was usually conducted once on each of five samples (from five animals) per group (n = 5). In some experiments, however, western blotting was performed using a single sample per experimental group or two different samples per experimental group. Thus, the reported results include at least one expression measurement for all samples and all experimental groups.
2.11. Novel objects and object location recognition tests
Novel object recognition and object location recognition tests were conducted using previously described methods (Bevins & Besheer, 2006; Chao et al., 2016) but with some modifications. The experiments were performed according to the schematic presented in Figure 3a,b. Briefly, mice were preadapted to the open‐field testing arena (40 × 40 × 40 cm: a white, opaque, acrylic open field) for 10 min·day−1 for 3 days before the behavioural tests began. To minimize stress levels, the tests were performed under a low‐illumination light. The arena was wiped clean between trials with 70% ethyl alcohol. In the object recognition training, the mice were allowed to explore two identical objects for 10 min, after which they were returned to their home cages for 1 or 24 h depending on their group. In the object recognition test, the mice were exposed for 5 min to one familiar object and one novel object (a different shape and colour), or one familiar object and one novel placed object (moved from the original location). Object exploration time was recorded using a video camera beginning when the mouse directly or indirectly touched the object with its nose, mouth or forepaws. The mouse was evaluated as ‘interacting’ with the object when its nose was in contact with the object or directed at the object within a minimally defined distance (≤2 cm being most commonly used). In addition, to measure whether there is a difference of the total exploration time between WT and 5XFAD mice, we compared the total time to recognize two objects in the training stage for 10 min and the test stage for 5 min obtained from each mouse. Results of the object recognition tests were analysed using SMART 3.0 video tracking software (Panlab, Barcelona, Spain). To avoid observer biases, the behavioural experiments were scored with the help of a blinded experimenter, who was completely unaware of the identity or the treatment group of their subjects while conducting the research.
FIGURE 3.
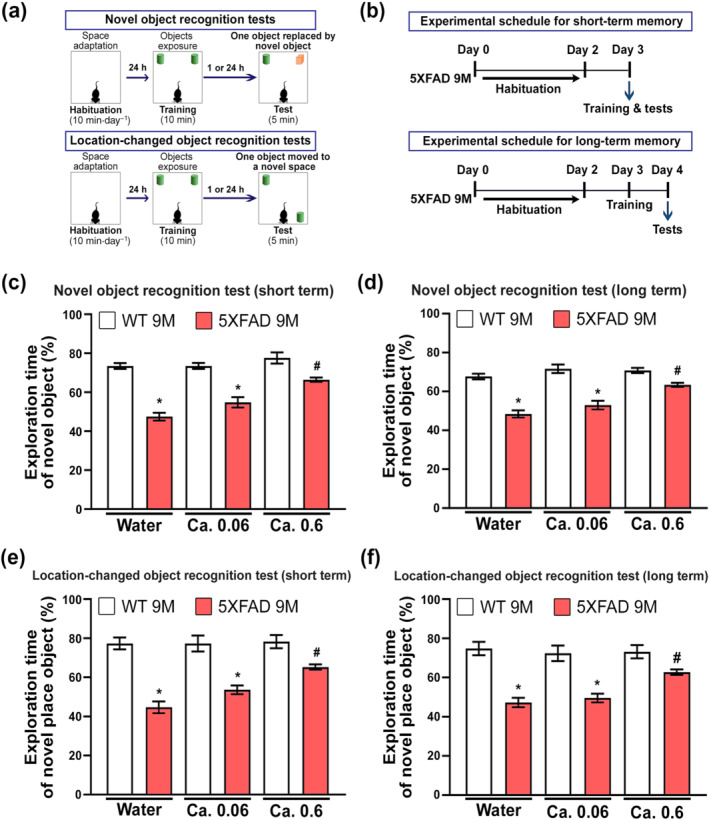
Enhancement of the blood‐brain barrier (BBB) by caffeine supply attenuates cognitive impairments in five familial Alzheimer's disease (5XFAD) mice. (a) Schematic of the experimental design for each object recognition test. (b) Schematic of the experimental design for short‐term and long‐term object recognition tests. (c–f) Results of novel object and location‐changed object recognition tests. The results of the novel object and location‐changed object recognition tests are represented as the time ratios associated with exploring the novel object (= novel object exploration time∕total object exploration time) and the location‐changed object (= location‐changed object exploration time∕total object exploration time), respectively, for short‐term (c, e) and long‐term (d, f) recognition memory. *P < 0.05 versus WT mice supplied with noncaffeinated water; # P < 0.05 versus 5XFAD mice supplied with noncaffeinated water (two‐way ANOVA with Tukey's post hoc analysis; n = 6 for each group). Quantitative values are presented as the mean ± SEM
2.12. Rivaroxaban treatment
Treatment with rivaroxaban, an inhibitor of factor Xa (Perzborn et al., 2010; Verma & Brighton, 2009), was conducted in vivo using previously described methods (Ghorpade et al., 2018; Ichikawa et al., 2019) with some modifications. WT and 5XFAD mice received daily oral administration of rivaroxaban (2 mg·kg−1) or 1% ethanol (vehicle control) for 3 months starting at 6 months of age. Mice were then examined on the object recognition test and killed for western blotting experiments.
2.13. Primary microglial cell cultures and quantitative RT‐PCR
Primary microglial cell cultures were prepared from 1‐ to 2‐day‐old postnatal mice from a C57BL/6 strain according to the method described by Saura et al. (2003). As above, Mice were were anaesthetized with CO2 and killed by dislocation of the neck. Whole brains were homogenized in a 70‐μm strainer. Cells were then seeded into 100‐mm culture dishes and cultured at 37°C in a humidified atmosphere with 5% CO2. The culture media were changed initially after 5 days and then every 2 days, and cells were used after being cultured for 14–21 days. Secondary pure microglial cell cultures were incubated in a prewarmed mild‐trypsinization solution (0.2‐mM EDTA, 0.5‐mM CaCl2 and 0.05% trypsin–EDTA) for 30–60 min at 37°C, after which the astrocyte layer was discarded. Attached microglial cells were dissociated using trypsin–EDTA (Life Technologies, Carlsbad, CA) and then centrifuged at 783 x g for 30 min. The obtained microglial cells were seeded onto plates containing DMEM (Life Technologies) supplemented with 10% heat‐inactivated FBS (Thermo Fisher Scientific) and penicillin–streptomycin (Thermo Fisher Scientific).
For quantitative RT‐PCR, primary microglial cells (50 × 104 cells ml−1) were grown in six‐well plates and treated for 30 min with 1‐μM caffeine, which is higher than the expected concentration in the brain based on the pharmacokinetic properties of the titre supplied by caffeinated water in vivo (Madeira et al., 2017; McLellan et al., 2016; White et al., 2016). Subsequently, the cells were treated with 50‐μg·ml−1 pKr‐2 for 8 h, which dose neurotoxicity was not observed (Kim et al., 2010). RNA was then extracted from the primary microglial cells using TRIzol reagent (Life Technologies) before it was purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Using 100 ng of RNA, cDNA was synthesized at 37°C for 120 min with a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster, CA). Finally, quantitative RT‐PCR was performed using the one‐step SYBR® PrimeScript™ RT‐PCR Kit (Perfect Real Time; Takara Bio Inc., Kusatsu, Japan) according to the manufacturer's instructions and an Applied Biosystems 7500 Real‐Time PCR system for detection. RT‐PCR was performed using the following primers (5′ to 3′): IL‐1β (F): TTG ACG GAC CCC AAA AGA TG, (R): TGG ACA GCC CAG GTC AAA G; TNF‐α (F): TCC AGG CGG TGC CTA TGT, (R): GCC CCT GCC ACA AGC A; iNOS (F): GGA TCT TCC CAG GCA ACC A, (R): TCC ACA ACT CGC TCC AAG ATT; CXCL10 (F): GAT GAC GGG CCA GTG AGA A, (R): GCT CGC AGG GAT GAT TTC AA; LCN2 (F): TGA TCC CTG CCC CAT CTC T, (R): AAC TGA TCG CTC CGG AAG TCT; IL‐23 (F): CCT TCT CCG TTC CAA GAT CCT, (R): GGG CAG CTA TGG CCA AAA; IL‐12 (F): ACG CAG CAC TTC AGA ATC ACA, (R): CAC CAG CAT GCC CTT GTC TA; and IL‐6 (F): CCA CGG CCT TCC CTA CTT C, (R): TTG GGA GTG GTA TCC TCT GTG A. 18S rRNA and β‐actin served as the internal controls. The 2−ΔΔCt method was used to calculate relative changes in gene expression, as determined by real‐time PCR experiments (Livak & Schmittgen, 2001).
2.14. In vitro cytotoxicity assay
Primary microglial cells (10 × 104 cells ml−1) were cultured in 96‐well plates and treated with pKr‐2 protein for 24 h. Cells cultured in an equal volume of PBS were used as controls. To measure cytotoxicity, a Cell Counting Kit‐8 (CCK‐8; Enzo Life Sciences, Cat#: ALX‐850‐039‐0100) was used according to the manufacturer's instructions. Briefly, 10 μl of the CCK‐8 reagent was added to each well, and the plate was incubated at 37°C for 2 h. Subsequently, the absorbance at 450 nm was measured using a microplate reader (Tecan, Männedorf, Switzerland). Cell viability was expressed as a percentage of control (PBS‐treated) cell viability. All experiments were performed in triplicate.
2.15. Statistical analysis
All values are expressed as either the mean ± SD or mean ± SEM. The experimental results were evaluated a priori using the Shapiro–Wilk normality test for Gaussian distribution. Differences between groups were assessed using unpaired Student's t‐test, one‐way ANOVA or two‐way ANOVA followed by Tukey's post hoc analysis. The Kruskal–Wallis and Friedman tests were used for non‐parametric analyses. Statistical values for all figures are summarized in Table S1. The level of statistical significance chosen throughout was P < 0.05. All statistical analyses were conducted using the IBM SPSS software (Version 27.0.1.0; International Business Machines Corporation, RRID:SCR_019096, Armonk, NY) and GraphPad Prism (Version 8.30; GraphPad Software, RRID:SCR_002798, San Diego, CA). The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.16. Materials
The following materials were used in this study (suppliers are shown in parentheses): pKr‐2 (Haematologic Technologies, Inc., Cat#: HCP2‐0010, Essex Junction, VT), rivaroxaban (Selleckchem, Cat#: 366789‐02‐8, Houston, TX), caffeine (Sigma, Cat#: 58‐08‐2, St. Louis, MO), sheep anti‐prothrombin fragment 2 (Fitzgerald, Cat#: 70R‐10587, RRID:AB_1120040, Acton, MA), mouse anti‐prothrombin (which recognizes an epitope in the pKr‐2 domain of prothrombin; Haematologic Technologies, Inc., Cat#: AMP‐9013), rabbit anti‐prothrombin (Thermo Fisher Scientific, Cat#: PA5‐77976, RRID:AB_2736466, Rockford, IL), rabbit anti‐thrombin (Abcam, Cat#: ab92621, RRID:AB_10711179, Cambridge, UK), mouse anti‐neuronal nuclei (NeuN; Millipore, Cat#: MAB377, RRID:AB_2298772, Billerica, MA), rabbit anti‐neuronal nuclei (Cell Signaling, Cat#: 24307, RRID:AB_2651140, Beverly, MA), rabbit anti‐ionized calcium‐binding adapter molecule 1 (Iba1; Wako Pure Chemical Industries, Cat#: 019‐19741, RRID:AB_839504, Osaka, Japan), rabbit anti‐glial fibrillary acidic protein (GFAP; Millipore, Cat#: AB5804, RRID:AB_2109645), rabbit anti‐TLR4 (Santa Cruz Biotechnology, Cat#: sc‐10741, RRID:AB_2240715, Santa Cruz, CA), rabbit anti‐IL‐1β (Santa Cruz Biotechnology, Cat#: sc‐7884, RRID:AB_2124476), mouse anti‐TNF‐α (Santa Cruz Biotechnology, Cat#: sc‐52746, RRID:AB_630341), rabbit anti‐inducible NOS (iNOS; Abcam, Cat#: ab3523, RRID:AB_303872), rabbit anti‐zonula occludens‐1 (ZO‐1; Thermo Fisher Scientific, Cat#: 61‐7300, RRID:AB_2533938), rabbit anti‐cluster of differentiation 31 (CD31; Thermo Fisher Scientific, Cat#: PA5‐16301, RRID:AB_10981955), rat anti‐laminin (Abcam, Cat#: ab2466, RRID:AB_303085), rabbit anti‐claudin‐5 (Thermo Fisher Scientific, Cat#: 352588, RRID:AB_2532189), goat Alexa Fluor (AF) 568‐conjugated anti‐mouse IgG (Thermo Fisher Scientific, Cat#: A‐21235, RRID:AB_2535804), rabbit anti‐β‐amyloid (Aβ; Cell Signaling, Cat#: 2454, RRID:AB_2056585, Beverly, MA), mouse anti‐β‐actin (Santa Cruz Biotechnology, Cat#: sc‐47778, RRID:AB_626632), thioflavin S (Sigma, Cat#: 1326‐12‐1), biotinylated anti‐rabbit IgG (Vector Laboratories, Cat#: BA‐1000, RRID:AB_2313606, Burlingame, CA), AF647‐conjugated anti‐rat IgG (Thermo Fisher Scientific, Cat#: A‐21247, RRID:AB_141778), AF647‐conjugated anti‐rabbit IgG (Thermo Fisher Scientific, Cat#: ab150075, RRID:AB_2752244), HRP‐conjugated anti‐rabbit IgG (Enzo Life Sciences, Cat#: ADI‐SAB‐300, RRID:AB_10998727, Farmingdale, NY), HRP‐conjugated anti‐mouse IgG (Thermo Fisher Scientific, Cat#: 61‐6520, RRID:AB_2533933) and HRP‐conjugated anti‐sheep IgG (GenWay Biotech Inc., Cat#: GWB‐3E18C4, RRID:AB_10517147, San Diego, CA). Sterile PBS was used as a vehicle to dissolve pKr‐2
2.17. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/2022 (Alexander et al., 2021).
3. RESULTS
3.1. Up‐regulation of pKr‐2 and neuroinflammatory molecules in the hippocampi of patients with AD and 5XFAD mice
To investigate the pKr‐2 protein levels and neurotoxic inflammatory molecules in the hippocampi of patients with AD compared with those in the hippocampi of age‐matched controls. Western blotting was performed using the post‐mortem hippocampal tissues presented from the Victorian Brain Bank Network (Figure 1a). Similar to that observed in the substantia nigra of patients with Parkinson's disease (Leem et al., 2016; Shin et al., 2015), the pKr‐2 protein levels were significantly increased in the hippocampi of patients with AD compared with those in the hippocampi of age‐matched controls (Figure 1b). The results presented in Figure 1b revealed an increase in the levels of Iba1 as evidence of microglial activation. Furthermore, the results presented in Figure 1b also indicated a decrease in the level of neuronal nuclei, which is a neuronal marker, in the case of patients with AD compared with their age‐matched controls.
TLR4, which recognizes various ligands such as LPS, envelope proteins, heat shock proteins, fibrinogen and hyaluronan, is a key receptor for activation of immune cells, resulting in the increases in inflammatory cytokines (Leem et al., 2016; Shin et al., 2015; Walter et al., 2007). In the brain, TLR4 is also well known as an important receptor for microglia‐mediated neuroinflammation (Leem et al., 2016; Shin et al., 2015; Walter et al., 2007). In the hippocampi of patients with AD, TLR4 expression levels were significantly increased compared with those in the hippocampi of age‐matched controls (Figure 1c). In addition, increased levels of iNOS, IL‐1β and TNF‐α associated with neuroinflammation were observed in the hippocampi of patients with AD (Figure 1c), indicating microglial activation in the AD brain.
To measure pKr‐2 protein levels in the hippocampi of 5XFAD and WT mice, hippocampal tissues were obtained from 5XFAD mice aged 3, 6 and 9 months and from WT mice aged 9 months. Western blot analysis showed that pKr‐2 protein levels were significantly increased in the hippocampi of 5XFAD mice aged 6 and 9 months compared with those in the hippocampi of WT mice (Figure 1d). Increased levels of Iba1, indicating microglia, were also observed in the hippocampi of 5XFAD mice, and these were increased in a time‐dependent manner with age (Figure 1d). In contrast, the expression levels of neuronal nuclei was significantly decreased in the hippocampi of 5XFAD mice aged 9 months compared with the hippocampi of WT mice (Figure 1d). Moreover, TLR4 protein levels and neuroinflammatory molecules, such as TNF‐α, IL‐1β and iNOS, increased in the hippocampi of 5XFAD from the age of 3 months in a time‐dependent manner (Figure 1e). However, a non‐significant difference of the levels of pKr‐2, Iba1, neuronal nuclei and inflammatory molecules was observed in the WT mice during aging (Figure S1). These results suggest that pKr‐2 up‐regulation and microglial activation occur earlier than the significant loss of neuronal nuclei in the hippocampi of 5XFAD mice. Next, we performed co‐immunofluorescence staining using anti‐pKr‐2 together with antibodies for Iba1 (marker or microglial activation), GFAP (an astrocyte marker; Jeon et al., 2020), neuronal nuclei and CD31 (an endothelial cell marker for BBB; Giannoni et al., 2016; Song et al., 2017), to assess the localization of pKr‐2 in the hippocampi of WT and 5XFAD mice (Figure 1f). Our observations using 9 months aged mice showed that the location of pKr‐2 expression was clustered to Iba1 and CD31 expression, but not localized with GFAP and neuronal nuclei expression (Figure 1f).
3.2. BBB preservation by caffeine reduces pKr‐2 up‐regulation in the hippocampi of 5XFAD mice
We next analysed the degree of BBB disruption in the hippocampi of 5XFAD by the measurement of the expression levels of zonula occludens‐1 (tight junction protein‐1, ZO‐1) as a tight‐junction protein marker (Kook et al., 2012; Song et al., 2017) and CD31 in the hippocampi of 5XFAD mice (Eimer & Vassar, 2013; Giannoni et al., 2016; Oakley et al., 2006) aged 3, 6 and 9 months (Figure 2a). At 3 months, when there was no change in pKr‐2 expression (Figure 1d), there was no significant difference in the expression of ZO‐1 and CD31 in the hippocampi of 5XFAD mice and the hippocampi of WT mice (Figures 2a and S2). However, ZO‐1 and CD31 protein levels were both decreased in the hippocampi of 5XFAD mice aged 6 and 9 months relative to WT mice (Figures 2a and S2). Furthermore, BBB disruption in the hippocampi of 5XFAD mice aged 6 months was confirmed by treatment with fluorescein isothiocyanate‐dextran, a BBB permeability marker, and immunostaining to assess for IgG penetration (Michalak et al., 2012), laminin (a vascular matrix protein) (Russo et al., 2018) and claudin‐5 (a tight‐junction protein) (Kook et al., 2012; Song et al., 2017) (Figure 2b,c). Moreover, the changes in BBB disruption markers in the hippocampi of 5XFAD mice with non‐caffeinated water were significantly inhibited by a caffeine (0.6 mg·ml−1), which can block BBB disruption in animal models of neurodegenerative disease (Chen et al., 2008; Chen et al., 2010), in their drinking water for 4 months (Figure 2b–e). The pKr‐2 up‐regulation and BBB disruption in the hippocampi of 5XFAD mice suggested the possibility that hippocampal up‐regulation of pKr‐2 was induced by blood leakage following BBB disruption. We examined this possibility by supplying 2‐month‐old WT and 5XFAD mice with non‐caffeinated or caffeinated drinking water (0.06 and 0.6 mg·ml−1) for 7 months (Figure 2f), subjecting them to behavioural tests for object recognition when aged 9 months and collecting brain samples to measure the protein levels of prothrombin, thrombin, pKr‐2, Iba1, neuronal nuclei and the neuroinflammatory molecules, ZO‐1 and CD31. WT mice treated with caffeinated drinking water for 7 months showed no significant change in the levels of these proteins when compared with level of these protein in WT mice supplied with non‐caffeinated drinking water (Figure 2g–i). However, in the hippocampi of 9‐month‐old 5XFAD mice, which exhibited neuronal loss as demonstrated by western blotting for neuronal nuclei (Figures 1d and 2g) and counting of hippocampal CA1 neurons (Figure S3), prothrombin protein levels were significantly increased, which in turn resulted in an increase in thrombin and pKr‐2 expression (Figure 2g). However, caffeine supply inhibited these increased levels of prothrombin, resulting in the control of hippocampal expression of thrombin and pKr‐2. In addition, increased levels of Iba1 (Figure 2g) and neuroinflammatory molecules (Figure 2h) in the hippocampi of 5XFAD mice were significantly attenuated by caffeine supply. Moreover, the decrease in neuronal nuclei levels observed in the hippocampi of 5XFAD mice aged 9 months was significantly inhibited by caffeine (Figure 2g), indicating hippocampal neuroprotection in 5XFAD mice. The preservation of BBB integrity by caffeine observed in the hippocampi of 5XFAD mice aged 6 months (Figure 2b–e) was confirmed by western blotting for ZO‐1 and CD31 protein levels in 5XFAD mice aged 9 months (Figure 2i).
FIGURE 2.
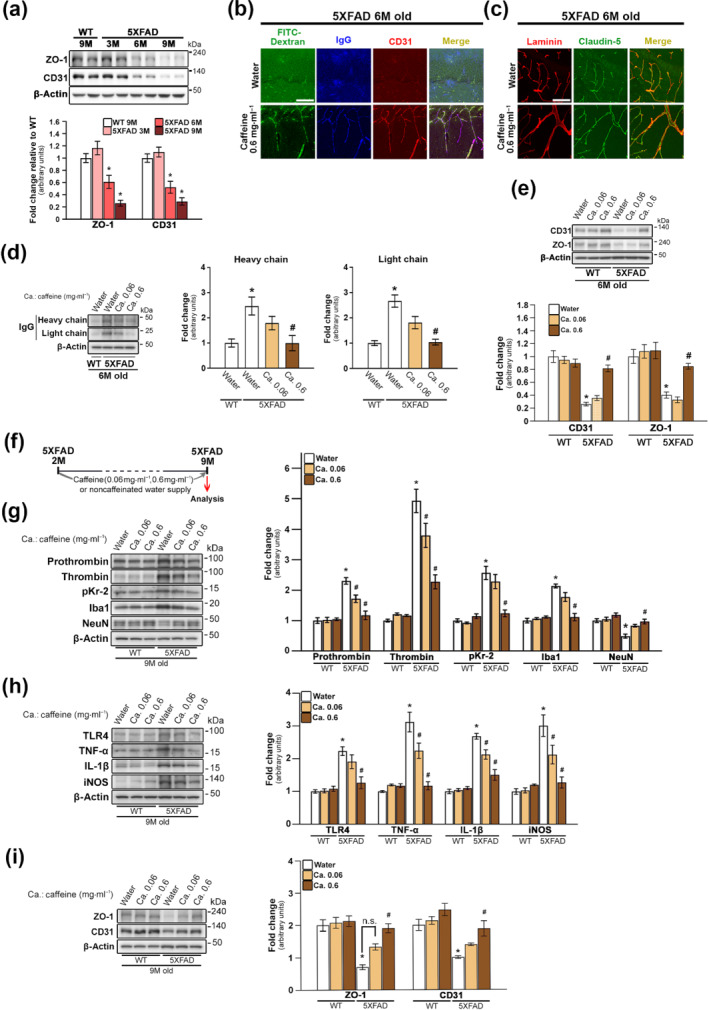
Inhibition of hippocampal pKr‐2 up‐regulation, neuroinflammation and neurodegeneration in five familial Alzheimer's disease (5XFAD) mice by preservation of blood‐brain barrier (BBB) integrity using caffeine supply. (a) Western blotting for the protein levels of ZO‐1 and CD31, representing markers of tight‐junction and endothelial cells, respectively. *P < 0.05 versus 9‐month‐old WT mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (b–e) Comparison of BBB integrity in 5XFAD mice by supply with noncaffeinated and caffeinated water (0.06 and 0.6 mg·ml−1) for 4 months from the age of 2 months. (b) BBB leakage shown as fluorescein isothiocyanate‐dextran permeability (green; BBB permeability marker) and IgG staining (blue) was observed in the hippocampal parenchyma of 5XFAD mice; this leakage was apparently inhibited by a 0.6‐mg·ml−1 caffeine supply. Immunostaining for CD31 (red) was used as a marker for blood vessels in the hippocampus of 5XFAD mice. Scale bar, 50 μm. (c) Immunostaining with anti‐laminin (red; vascular matrix protein) and anti‐claudin‐5 (green; tight‐junction protein). Scale bar, 50 μm. (d) Western blotting for IgG expression in the hippocampal parenchyma of WT and 5XFAD mice. Statistical analysis of western blotting results for IgG. *P < 0.05 versus WT mice supplied with noncaffeinated water; # P < 0.05 versus 5XFAD mice supplied with noncaffeinated water (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (e) Western blotting for the protein levels of CD31 and ZO‐1. *P < 0.05, respectively, versus WT mice supplied with noncaffeinated water; # P < 0.05, respectively, versus 5XFAD mice supplied with noncaffeinated water (two‐way ANOVA with Tukey's post hoc analysis; Friedman test for CD31 analysis; n = 5 for each group). (f) Experimental schematic demonstrating caffeine supply. (g) Western blotting for the protein levels of prothrombin, thrombin, pKr‐2, Iba1 and neuronal nuclei (NeuN) in the hippocampi of WT and 5XFAD mice aged 7 months after caffeine supply. *P < 0.05 versus WT mice supplied with noncaffeinated water; # P < 0.05 versus 5XFAD mice supplied with noncaffeinated water (two‐way ANOVA with Tukey's post hoc analysis; Friedman test for NeuN analysis; n = 5 for each group). (h) Western blotting for the protein levels of TLR4, TNF‐α, IL‐1β and iNOS. *P < 0.05 versus WT mice supplied with noncaffeinated water; # P < 0.05, ## P < 0.01 and # P < 0.05 versus 5XFAD mice supplied with noncaffeinated water (two‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (i) Western blotting for the protein levels of ZO‐1 and CD31. *P < 0.05 versus WT mice supplied with noncaffeinated water;, # P < 0.05 versus 5XFAD mice WT mice supplied with noncaffeinated water (two‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). All quantitative values are presented as the mean ± SEM
3.3. Effects of caffeine in the drinking water on the decline of object recognition memory in 5XFAD mice
To investigate whether the control of pKr‐2 up‐regulation leads to preserve object cognitive impairments in 5XFAD mice, we conducted two recognition behavioural tests, novel object recognition and object location recognition (Figure 3a), and measured two types of memory, short‐term and long‐term, depending on the delay time after habituation (Figure 3b). In addition, to compare whether there is a difference of the total exploration time between WT and 5XFAD mice, we measured the time to explore the objects spent by each group (Figure S4). Our results showed that there was no significant difference of the total exploration time between WT and 5XFAD mice (Figure S4). In the novel object recognition tests of short‐term and long‐term memory, the results revealed that a 0.06‐mg·ml−1 caffeine supply provided for 7 months had no significant effect on the decline of recognition memory in 9‐month‐old 5XFAD mice (Figures 3c,d and S5). However, 0.6‐mg·ml−1 caffeine supply significantly preserved the recognition memory of these 5XFAD mice when their performance was compared with that of 5XFAD mice supplied noncaffeinated water (Figures 3c,d and S5). Similar to the results of the novel object recognition tests, the test for location‐changed object recognition demonstrated that the 0.6‐mg·ml−1 caffeine supply inhibited the decline of object recognition memory in 9‐month‐old 5XFAD mice (Figures 3e,f and S5).
3.4. Reduction of hippocampal pKr‐2 attenuates neurotoxic symptoms in 5XFAD mice
To determine whether inhibition of pKr‐2 generation reduces the neurotoxic symptoms in 5XFAD mice, rivaroxaban (dissolved in 1% ethanol), an inhibitor of factor Xa (Perzborn et al., 2010; Verma & Brighton, 2009), was orally administered (2 mg·kg−1·day−1) to WT and 5XFAD mice for 3 months from the age of 6 months (Figure 4a). We observed that rivaroxaban prevented the decline in object recognition memory observed in 5XFAD mice (Figures 4b–e and S6). Consistent with these results of object recognition tests, western blotting results revealed that the increased levels of thrombin, pKr‐2 and pro‐inflammatory microglial activation markers in the hippocampi of 5XFAD mice were significantly attenuated by rivaroxaban treatment (Figure 4f–i). However, the increased levels of hippocampal prothrombin shown in 5XFAD mice were not affected by rivaroxaban treatment. The results indicate that oral gavage of rivaroxaban may inhibit the cleavage of thrombin from prothrombin, consequently resulting in the inhibition of pKr‐2 production.
FIGURE 4.
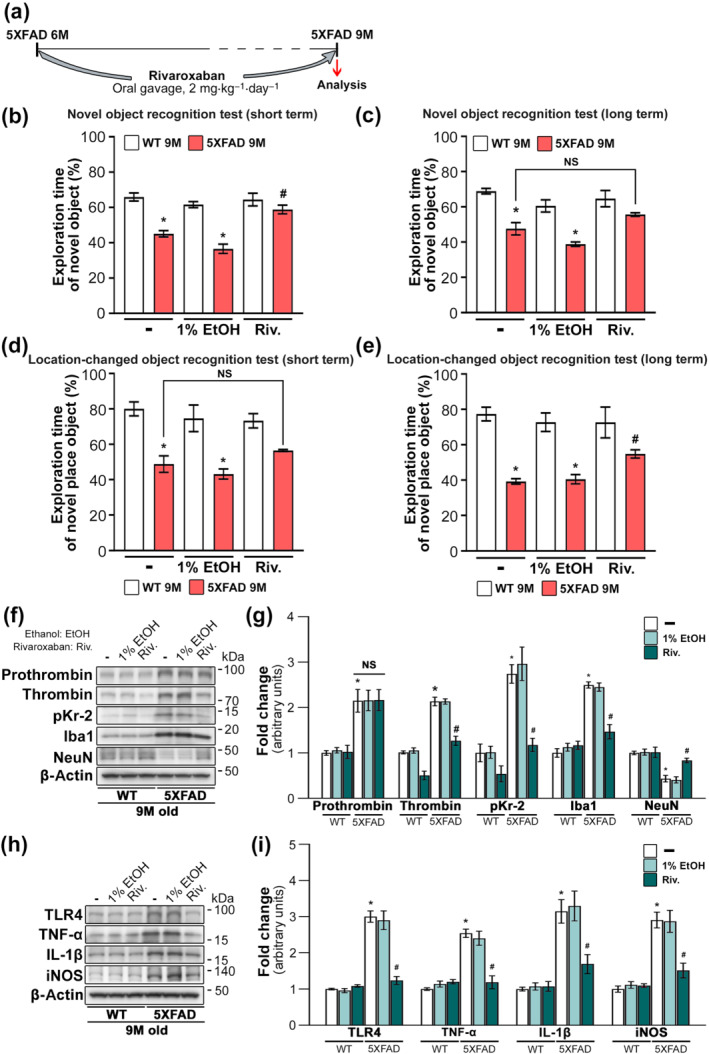
Inhibition of prothrombin kringle‐2 (pKr‐2) generation by rivaroxaban treatment protects hippocampal neurons and diminishes cognitive impairments in five familial Alzheimer's disease (5XFAD) mice. (a) Experimental schematic demonstrating rivaroxaban treatment, which is an inhibitor of factor Xa associated with thrombin production. (b–e) Results of novel object and location‐changed object recognition tests. *P < 0.05 versus nontreated WT mice; # P < 0.05 versus nontreated 5XFAD mice (two‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). (f, g) Western blotting for the protein levels of prothrombin, thrombin, pKr‐2, Iba1 and neuronal nuclei (NeuN) in the hippocampi of 9‐month‐old WT and 5XFAD mice. *P < 0.05, versus nontreated WT mice; # P < 0.05 versus nontreated 5XFAD mice; NS, no significance (two‐way ANOVA with Tukey's post hoc analysis; Friedman test for thrombin and pKr‐2 analysis; n = 5 for each group). (h, i) Western blotting for the protein levels of TLR4, TNF‐α, IL‐1β and iNOS. *P < 0.05 versus nontreated WT mice; # P < 0.05 versus nontreated 5XFAD mice (two‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group). All quantitative values are presented as the mean ± SEM. Note that rivaroxaban treatment significantly inhibited the production of thrombin and pKr‐2 in the hippocampi of 5XFAD mice compared with the levels produced in nontreated 5XFAD mice; however, there was no change in prothrombin expression in 5XFAD mice, suggesting effects of rivaroxaban as an inhibitor of factor Xa
3.5. Caffeine had no effect on the levels of pro‐inflammatory M1 genes in pKr‐2‐treated primary microglial cell cultures and on the levels of amyloid‐β in the hippocampi of 5XFAD mice
To further investigate the potential effects of caffeine alone on the anti‐neuroinflammatory responses observed in 5XFAD mice in vivo, caffeine and pKr‐2 treatments were applied to primary microglial cell cultures in vitro and the levels of pro‐inflammatory M1 genes were assessed. Preliminary quantitative RT‐PCR results demonstrated that 50 μg·ml−1 of pKr‐2, which did not exhibit neurotoxicity in vitro (Kim et al., 2010), increasing the production of pro‐inflammatory M1 genes relative to their production in nontreated microglial cell cultures. Moreover preliminary experiments with the treatment with 1‐μM caffeine, did not show any toxicity to microglial cell viability (Figure 5b) and presumed a higher titre than the expected concentration in the brain based on the pharmacokinetic properties of caffeinated drinking water supply in vivo (Madeira et al., 2017; McLellan et al., 2016; White et al., 2016), had no effect on the up‐regulation of pro‐inflammatory M1 genes after pKr‐2 treatment (Figures 5a and S7). Although it is unclear and not examined in the present preliminary study whether the control of unknown factors, including other endogenous factors such as thrombin and fibrinogen (Benek et al., 2020; Choi et al., 2005; Giannoni et al., 2016; Merlini et al., 2019; Montagne et al., 2015; Sweeney et al., 2018; Vaiserman et al., 2020), following caffeine also contributed to the observed anti‐neuroinflammation in 5XFAD mice, our results suggest that the anti‐neuroinflammatory responses, occurred in 5XFAD mice after maintenance of the BBB with a 0.06‐ or 0.6‐mg·ml−1 caffeine in the drinking water may be mediated by the control of hippocampal pKr‐2 up‐regulation not by caffeine itself. In addition, we found that caffeine had no effect on the levels of amyloid‐β in the hippocampi of 5XFAD mice, as demonstrated by western blotting (Figure 5c).
FIGURE 5.

Caffeine treatment had no effect on the levels of inflammatory molecules increased by prothrombin kringle‐2 (pKr‐2) treatment in primary microglial cultures. (a) Primary microglial cells were pretreated with 1‐μM caffeine, given its pharmacokinetic properties in vivo (Madeira et al., 2017; McLellan et al., 2016; White et al., 2016) for 30 min and subsequently treated with 50 μg·ml−1 of pKr‐2 for 8 h, after which RT‐PCR was performed. Preliminary data shows that the expression levels of pro‐inflammatory M1 genes for proteins such as IL‐1β, TNF‐α, iNOS, CXCL10, LCN2, IL‐23, IL‐12 and IL‐6, were increased in pKr‐2‐treated microglial cells compared with I non‐treated control cells. This was also shown on mRNA transcript levels which are presented as mean ± SD. (b) Preliminary data using a CCK‐8 assay to evaluate the viability of caffeine‐treated primary microglial cells for 1 day indicated that treatment with caffeine alone had no effect on the viability of primary microglial cells. Data are presented as mean ± SD of three independent experiment. (c) Western blotting for the protein levels of amyloid‐β (Aβ) in the hippocampi of 6‐month‐old five familial Alzheimer's disease (5XFAD) mice supplied with noncaffeinated or caffeinated water (0.06 or 0.6 mg·ml−1) for 4 months. *P < 0.05 versus noncaffeinated water‐supplied WT mice (one‐way ANOVA with Tukey's post hoc analysis; n = 5 for each group; NS, no significance). Quantitative values are presented as the mean ± SEM
3.6. pKr‐2 up‐regulation has no effect on amyloid‐ oligomerization and accumulation
In addition to pKr‐2 up‐regulation and microglial activation in the hippocampi of 5XFAD mice, we also examined whether pKr‐2 up‐regulation exacerbates amyloid‐β aggregation, which is one of the characteristics of AD and involved in its pathology, in the hippocampi of WT and 5XFAD mice. Apparent increases in amyloid‐β aggregation and oligomerization were observed in the hippocampi of 5XFAD mice aged 6 months, as demonstrated by thioflavin S and immunohistochemical staining for amyloid‐β and western blotting, respectively (Figure 6a,b). To determine whether pKr‐2 up‐regulation contributed to amyloid‐β aggregation and oligomerization, we administered intrahippocampal injections of pKr‐2 to WT and 5XFAD mice aged 3 months. Administration of pKr‐2 did not increase the levels of amyloid‐β expression and oligomerization in the hippocampi of WT mice, as demonstrated by Western blotting (Figure 6c). Similarly, amyloid‐β oligomerization and expression in the hippocampal tissues of 5XFAD mice, obtained 7 days or 3 months after pKr‐2 administration, did not differ significantly from those in nontreated 5XFAD mice (Figure 6d,e), and these results were confirmed by immunohistochemical staining for amyloid‐β (Figure 6f). However, although there was no change of amyloid‐β expression by pKr‐2 administration in the hippocampi of 5XFAD mice, its treatment significantly decreased neuronal nuclei expression and increased neuroinflammatory molecules in 5XFAD mice compared with nontreated mice (Figure 6g,h). Moreover, pKr‐2 administration in the hippocampi of WT mice induced object recognition impairments compared with untreated WT mice (Figure S8). Taken together, our results suggest that the up‐regulation of pKr‐2 following BBB disruption, which may have no effect on amyloid‐β oligomerization and accumulation, contributes to hippocampal neurodegeneration and object recognition decline through neurotoxic inflammatory responses.
4. DISCUSSION
Accumulation of neurotoxic forms of amyloid‐β and hyperphosphorylated tau proteins is a well‐known pathophysiological hallmark of AD. However, clinical trials conducted with drugs targeting amyloid‐β and tau, including drugs approved by the US Food and Drug Administration for the treatment of AD aiming at neurotransmission, have failed to demonstrate efficacy, provided only modest and temporary relief from symptoms and could not cure the disease; thus, there is a need to explore alternative therapeutic targets (Benek et al., 2020; Vaiserman et al., 2020). A recent accumulation of evidence suggests that systemic inflammation and related vascular dysfunction play important aetiological roles in AD and precede its clinical manifestation (Benek et al., 2020; Choi et al., 2005; Giannoni et al., 2016; Merlini et al., 2019; Montagne et al., 2015; Sweeney et al., 2018; Vaiserman et al., 2020).
Cerebrovascular alteration, which leads to BBB disruption, is a pathological hallmark in the hippocampi of patients with early AD (Giannoni et al., 2016; Sweeney et al., 2018; van de Haar et al., 2016). BBB leakage following disruption results in the infiltration of circulating substances into the brain parenchyma (Merlini et al., 2019; Sweeney et al., 2018; van de Haar et al., 2016). Although the specific endogenous molecules associated with microglial activation, which causes neurotoxic inflammation in the adult brain, require further investigation, for example, major endogenous activators for microglial activation have not yet been elucidated, some studies have shown that hippocampal up‐regulation of blood proteins, such as thrombin and fibrinogen, could contribute to neuroinflammation mediated by microglial cell activation, which results in the induction of neurotoxicity in the hippocampus of the adult murine brain (Choi et al., 2005; Merlini et al., 2019). Taken together, these findings suggest that efforts to develop therapeutic and preventive agents for neurodegenerative diseases, such as AD and Parkinson's disease, must be aimed at the inhibition of microglial cell‐derived neuroinflammation, which would lead to the suppression of microglial activation and its pathogenic mechanisms. Hence, it is essential to examine the endogenous molecules and mechanisms that cause neurotoxic events in the adult brain to develop effective strategies for treating AD.
pKr‐2 is a domain of prothrombin, which can be generated by active thrombin (Kim et al., 2010; Leem et al., 2016; Mann, 1976; Shin et al., 2015; Taneda et al., 1994). Although pKr‐2 levels could be up‐regulated in neurodegenerative diseases, it was largely unknown about the major source and cause for its up‐regulation in the adult brain. In the present study, to determine the source and cause of pKr‐2 up‐regulation in the hippocampus of AD brain, we compared the protein levels of hippocampal pKr‐2 between AD patients and age‐matched controls and examined whether pKr‐2 up‐regulation could be observed in the hippocampi of 5XFAD mice. In addition, we examined whether the up‐regulation of hippocampal pKr‐2 expression could be inhibited by BBB preservations after treatment with caffeine or with rivaroxaban, which is an inhibitor of factor Xa associated with thrombin production (Perzborn et al., 2010; Verma & Brighton, 2009). To investigate the potential role of pKr‐2 as an important pathogenic factor causing neurotoxic inflammation in AD, we examined whether pKr‐2 up‐regulation contributed to neurodegeneration and object cognitive impairments via neuroinflammatory responses in the hippocampi of 5XFAD mice (Eimer & Vassar, 2013; Giannoni et al., 2016; Oakley et al., 2006). Moreover, we determined whether the inhibition of pKr‐2 expression could diminish the pathogenic processes in 5XFAD mice in the present study. In addition to the up‐regulation of pKr‐2 in the hippocampi of patients with AD and 5XFAD mice, our experimental results indicate that the hippocampal pKr‐2, which had no effect on amyloid‐β oligomerization and accumulation in the hippocampi of both WT and 5XFAD mice, can be up‐regulated by BBB disruption, resulting in neurodegeneration and object cognitive impairments through neuroinflammatory responses as shown in 5XFAD mice aged 9 months. Furthermore, we observed that pKr‐2 administration induced the loss of neuronal nuclei expression in 5XFAD mice aged 3 and 6 months and showed no loss of neuronal nuclei expression in nontreated mice at the same age. Consistent with these findings, the increases in the protein levels of neuroinflammatory molecules by pKr‐2 administration were observed in 5XFAD mice.
Previous reports have shown that caffeine treatment can reduce amyloid‐β accumulation and neuroinflammation in neurodegenerative conditions (Cao et al., 2009; Madeira et al., 2017). To examine the role played by caffeine itself on the beneficial effects observed in 5XFAD mice, we assessed whether a 0.06‐ or 0.6‐mg·ml−1 caffeine supply, representing much lower titres in the present study compared with titres used in previous research (Cao et al., 2009), affected amyloid‐β protein levels in the hippocampi of 5XFAD mice and whether treatment with 1‐μM caffeine, presumed as a higher titre than the concentration in the brain according to the pharmacokinetic properties of caffeinated water supply in vivo (Madeira et al., 2017; McLellan et al., 2016; White et al., 2016), could have anti‐inflammatory effects in pKr‐2‐treated primary microglial cell cultures in vitro. A 0.6‐mg·ml−1 caffeine supply, which inhibited hippocampal pKr‐2 expression and exerted anti‐neuroinflammatory effects, did not affect amyloid‐β levels in the hippocampi of 5XFAD mice. Moreover, a 1‐μM caffeine treatment, which showed no toxicity to microglial cell viability, had no effect on the increases in pro‐inflammatory M1 genes after pKr‐2 treatment in microglial cell cultures.
In conclusion, we observed that the hippocampal penetration of prothrombin and pKr‐2 up‐regulation following BBB disruption could induce hippocampal neurodegeneration and object recognition decline through neurotoxic inflammatory responses in 5XFAD mice as depicted in the schema of our results (Figure 7). Furthermore, pKr‐2 up‐regulation had no effect on amyloid‐β oligomerization and accumulation, and the inhibition of hippocampal pKr‐2 expression through BBB preservastions and the control of enzyme activity associated with its production can diminish neurotoxic symptoms in 5XFAD mice. Although it is not possible to rule out the effects of amyloid‐β accumulation (Heppner et al., 2015) and hippocampal penetration of unknown factors, including other endogenous factors such as thrombin (Choi et al., 2005) and fibrinogen (Merlini et al., 2019), can also contribute to neurotoxicity by activated microglia, our results demonstrate that pKr‐2 up‐regulation is one of the major neuroinflammatory mechanisms that cause neurotoxic events in the hippocampus of the adult brain. Thus, inhibiting the generation of pKr‐2 may be useful for preventing and treating AD and related conditions.
FIGURE 7.
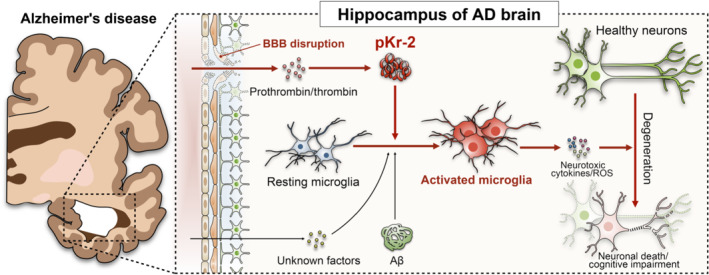
Schematic of neurodegeneration caused by prothrombin kringle‐2 (pKr‐2) up‐regulation in the hippocampus of the adult brain. As demonstrated in the experimental animal model of Alzheimer's Disease (AD) the hippocampal penetration of prothrombin and thrombin after blood‐brain barrier (BBB) disruption may induce the increase in pKr‐2 expression, which consequently leads to microglial activation and hippocampal neurodegeneration. Although the hippocampal penetration of unknown factors, including other endogenous factors such as thrombin and fibrinogen, cannot be ruled out from contributing to microglial activation, our results show that pKr‐2 up‐regulation is one of the major neuroinflammatory mechanisms associated with the microglial activation that causes neurotoxic events
AUTHOR CONTRIBUTIONS
S.K., G.J.M. and S.R.K. conceived and designed the experiments; H.J.K. and J.K. provided the 5XFAD mice; C.M. contributed the human brain samples; S.K., G.J.M., D.‐G.K., K.‐S.K. and S.R.K. conducted the in vivo experiments and generated the figures; H.J.K., S.L. and C.M.H. conducted the in vitro experiments and analysed the data; S.K., J.K., Y.N., E.L. and S.R.K. conducted the experiments for behavioural tests and generated the figures; S.K., Y.N., C.S., H.J.K., S.L. and S.R.K. carried out the revision; W.‐H.S., B.K.J. and S.R.K. supervised the analysis of the data obtained from human brain samples; D.W.K., Y.‐S.O., C.S. and C.‐W.H. contributed to the preparation of references and the schematic of results; S.R.K. supervised the entire project and wrote the paper; and all authors contributed to data analysis and preparation of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1. Protein levels of pKr‐2, NeuN, and neuroinflammatory molecules in the hippocampi of WT mice at 3‐, 6‐, and 9‐months of age. (a‐c) Western blotting for the protein levels of pKr‐2, NeuN, Iba1, TLR4, TNF‐α, IL‐1β and iNOS. Data were analyzed using one‐way ANOVA with Tukey's post hoc analysis and Kruskal‐Wallis test for iNOS analysis (n = 5). Quantitative values are expressed as the mean ± SEM.
Figure S2. Immunohistochemical counterstaining for Nissl (purple) and CD31 (brown) in the hippocampal CA1 regions of WT and 5XFAD mice aged 6 months. CD31 expression, marked by red arrowheads, decreased in the hippocampus of 5XFAD mouse. Scale bar, 50 μm.
Figure S3. Immunohistochemical staining for anti‐ neuronal nuclei (NeuN) in the hippocampal CA1 regions of 9‐month‐old WT and 5XFAD mice. (a) Representative images of immunohistochemical staining for anti‐NeuN in the hippocampal CA1 regions of mice. Scale bar, 50 μm. (b) The number of NeuN‐positive neurons in the hippocampal CA1 regions was expressed quantitatively as a percentage of the number of neurons in 5XFAD mice compared to the number of neurons in WT mice. ***p < 0.05 vs. 9‐month‐old WT mice (Student's t‐test; n = 5 for each group). Quantitative values are expressed as the mean ± SEM.
Figure S4. Total exploration time of object recognition in WT and 5XFAD mice. (a, b) The results of training stage (two identical objects; a) and object recognition tests (one familiar and one novel object; b) were represented as the total exploration time to explore two objects. NS: no significance (t‐test; n = 6 for each group). Quantitative values are expressed as the mean ± SEM.
Figure S5. Pictorial representation of the tracks showing each mouse movement after water or caffeine supply. Novel object (a and b; familiar object: purple square; novel object: blue square) and location‐changed object recognition tests (c and d; familiar object: purple square; location‐changed object: blue square).
Figure S6. Pictorial representation of the tracks showing each mouse movement after rivaroxaban. Novel object (a and b; familiar object: purple square; novel object: blue square) and location‐changed object recognition tests (c and d; familiar object: purple square; location‐changed object: blue square).
Figure S7. Preliminary data on the effects of caffeine treatment on pKr‐2‐induced inflammatory molecules in primary microglial cultures. Primary microglial cells were pretreated with 1 μM caffeine for 30 min, followed by treatment with 50 μg·ml‐1 of pKr‐2 for 8 h. A real‐time PCR analysis using β‐actin as an internal control indicated that the proinflammatory M1 genes were similarly increased in pKr‐2‐treated microglial cells. Caffeine treatment had no significant effect on the pKr‐2‐upregulated M1 genes. The quantification data pertaining to mRNA transcript levels are presented as the mean ± standard deviation (SD) from three independent RT‐PCR experiments.
Figure S8. Intrahippocampal administration of pKr‐2 induces cognitive impairment in WT mice. Behavioural tests for assessing short term recognition were conducted at 7 days after bilateral injections of pKr‐2 in the hippocampal CA1 regions of WT mice. (a, b) The results of the novel object and location‐changed object recognition tests are represented as the time ratio exploring the novel object (novel object/total object exploring time) for short‐term recognition memory. *P < 0.05 vs. nontreated WT mice (one‐way ANOVA with Tukey's post hoc analysis and Kruskal‐Wallis test for novel object recognition analysis; n = 5 for each group). Quantitative values are expressed as the mean ± SEM.
Table S1. Summary of statistical analysis.
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation of Korea (NRF‐2020R1A2C2007954) and the Korea Healthcare Technology R&D (HI14C1135 and HI21C1795) grants funded by the Korean government. We thank the Victorian Brain Bank Network (VBBN) for permission to use human post‐mortem tissues.
Kim, S. , Moon, G. J. , Kim, H. J. , Kim, D.‐G. , Kim, J. , Nam, Y. , Sharma, C. , Leem, E. , Lee, S. , Kim, K.‐S. , Ha, C. M. , McLean, C. , Jin, B. K. , Shin, W.‐H. , Kim, D. W. , Oh, Y.‐S. , Hong, C.‐W. , & Kim, S. R. (2022). Control of hippocampal prothrombin kringle‐2 (pKr‐2) expression reduces neurotoxic symptoms in five familial Alzheimer's disease mice. British Journal of Pharmacology, 179(5), 998–1016. 10.1111/bph.15681
Sehwan Kim, Gyeong Joon Moon, Hyung Jun Kim, Do‐Geun Kim, Jaekwang Kim and Youngpyo Nam contributed equally to this work.
Funding information Korea Healthcare Technology R&D, Grant/Award Number: HI14C1135; HI21C1795; National Research Foundation of Korea, Grant/Award Number: NRF‐2020R1A2C2007954
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Coons, L. , Fuller, P. J. , Korach, K. S. , Young, M. J. , & CGTP collaborators . (2021). The Concise Guide to PHARMACOLOGY 2021/2022: Nuclear hormones receptors. British Journal of Pharmacology, 178, S246–S263. 10.1111/bph.15540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , Cirino, G. , Docherty, J. R. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Mangum, J. , Wonnacott, S. , & Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai, T. , Miklossy, J. , Klegeris, A. , Guo, J. P. , & McGeer, P. L. (2006). Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. Journal of Neuropathology and Experimental Neurology, 65(1), 19–25. 10.1097/01.jnen.0000196133.74087.cb [DOI] [PubMed] [Google Scholar]
- Benek, O. , Korabecny, J. , & Soukup, O. (2020). A perspective on multi‐target drugs for Alzheimer's disease. Trends in Pharmacological Sciences, 41(7), 434–445. 10.1016/j.tips.2020.04.008 [DOI] [PubMed] [Google Scholar]
- Bevins, R. A. , & Besheer, J. (2006). Object recognition in rats and mice: A one‐trial non‐matching‐to‐sample learning task to study ‘recognition memory’. Nature Protocols, 1(3), 1306–1311. 10.1038/nprot.2006.205 [DOI] [PubMed] [Google Scholar]
- Cao, C. , Cirrito, J. R. , Lin, X. , Wang, L. , Verges, D. K. , Dickson, A. , Mamcarz, M. , Zhang, C. , Mori, T. , Arendash, G. W. , Holtzman, D. M. , & Potter, H. (2009). Caffeine suppresses amyloid‐β levels in plasma and brain of Alzheimer's disease transgenic mice. Journal of Alzheimer's Disease, 17(3), 681–697. 10.3233/JAD-2009-1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, O. Y. , Huston, J. P. , Li, J. S. , Wang, A. L. , & de Souza Silva, M. A. (2016). The medial prefrontal cortex–lateral entorhinal cortex circuit is essential for episodic‐like memory and associative object‐recognition. Hippocampus, 26(5), 633–645. 10.1002/hipo.22547 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Gawryluk, J. W. , Wagener, J. F. , Ghribi, O. , & Geiger, J. D. (2008). Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer's disease. Journal of Neuroinflammation, 5, 12. 10.1186/1742-2094-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Ghribi, O. , & Geiger, J. D. (2010). Caffeine protects against disruptions of the blood‐brain barrier in animal models of Alzheimer's and Parkinson's diseases. Journal of Alzheimer's Disease, 20(Suppl 1), S127–S141. 10.3233/JAD-2010-1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Lee, D. Y. , Kim, S. U. , & Jin, B. K. (2005). Thrombin‐induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. Journal of Neuroscience, 25(16), 4082–4090. 10.1523/JNEUROSCI.4306-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, J. L. (2004). Alzheimer's disease. New England Journal of Medicine, 351(1), 56–67. 10.1056/NEJMra040223 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimer, W. A. , & Vassar, R. (2013). Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Aβ42 accumulation and caspase‐3 activation. Molecular Neurodegeneration, 8, 2. 10.1186/1750-1326-8-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorpade, D. S. , Ozcan, L. , Zheng, Z. , Nicoloro, S. M. , Shen, Y. , Chen, E. , Blüher, M. , Czech, M. P. , & Tabas, I. (2018). Hepatocyte‐secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature, 555(7698), 673–677. 10.1038/nature26138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni, P. , Arango‐Lievano, M. , Neves, I. D. , Rousset, M. C. , Baranger, K. , Rivera, S. , Jeanneteau, F. , Claeysen, S. , & Marchi, N. (2016). Cerebrovascular pathology during the progression of experimental Alzheimer's disease. Neurobiology of Disease, 88, 107–117. 10.1016/j.nbd.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Heppner, F. L. , Ransohoff, R. M. , & Becher, B. (2015). Immune attack: The role of inflammation in Alzheimer disease. Nature Reviews. Neuroscience, 16(6), 358–372. 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- Ichikawa, H. , Shimada, M. , Narita, M. , Narita, I. , Kimura, Y. , Tanaka, M. , Osanai, T. , Okumura, K. , & Tomita, H. (2019). Rivaroxaban, a direct factor Xa inhibitor, ameliorates hypertensive renal damage through inhibition of the inflammatory response mediated by protease‐activated receptor pathway. Journal of the American Heart Association, 8(8), e012195. 10.1161/JAHA.119.012195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida, Y. , Nagai, A. , Kobayashi, S. , & Kim, S. U. (2006). Upregulation of protease‐activated receptor‐1 in astrocytes in Parkinson disease: Astrocyte‐mediated neuroprotection through increased levels of glutathione peroxidase. Journal of Neuropathology and Experimental Neurology, 65(1), 66–77. 10.1097/01.jnen.0000195941.48033.eb [DOI] [PubMed] [Google Scholar]
- Jeon, M. T. , Moon, G. J. , Kim, S. , Choi, M. , Oh, Y. S. , Kim, D. W. , Kim, H. J. , Lee, K. J. , Choe, Y. , Ha, C. M. , Jang, I. S. , Nakamura, M. , McLean, C. , Chung, W. S. , Shin, W. H. , Lee, S. G. , & Kim, S. R. (2020). Neurotrophic interactions between neurons and astrocytes following AAV1‐Rheb(S16H) transduction in the hippocampus in vivo. British Journal of Pharmacology, 177(3), 668–686. 10.1111/bph.14882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, M. T. , Nam, J. H. , Shin, W. H. , Leem, E. , Jeong, K. H. , Jung, U. J. , Bae, Y. S. , Jin, Y. H. , Kholodilov, N. , Burke, R. E. , Lee, S. G. , Jin, B. K. , & Kim, S. R. (2015). In vivo AAV1 transduction with hRheb(S16H) protects hippocampal neurons by BDNF production. Molecular Therapy, 23(3), 445–455. 10.1038/mt.2014.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas, C. H. (2003). Clinical practice. Early Alzheimer's disease. New England Journal of Medicine, 349(11), 1056–1063. 10.1056/NEJMcp022295 [DOI] [PubMed] [Google Scholar]
- Kim, S. , Jung, U. J. , Oh, Y. S. , Jeon, M. T. , Kim, H. J. , Shin, W. H. , Hong, J. , & Kim, S. R. (2017). Beneficial effects of silibinin against kainic acid‐induced neurotoxicity in the hippocampus in vivo. Experimental Neurobiology, 26(5), 266–277. 10.5607/en.2017.26.5.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. R. , Chung, E. S. , Bok, E. , Baik, H. H. , Chung, Y. C. , Won, S. Y. , Joe, E. , Kim, T. H. , Kim, S. S. , Jin, M. Y. , Choi, S. H. , & Jin, B. K. (2010). Prothrombin kringle‐2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. Journal of Neuroscience Research, 88(7), 1537–1548. 10.1002/jnr.22318 [DOI] [PubMed] [Google Scholar]
- Kook, S. Y. , Hong, H. S. , Moon, M. , Ha, C. M. , Chang, S. , & Mook‐Jung, I. (2012). Aβ1–42‐RAGE interaction disrupts tight junctions of the blood–brain barrier via Ca2+‐calcineurin signaling. Journal of Neuroscience, 32(26), 8845–8854. 10.1523/JNEUROSCI.6102-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem, E. , Jeong, K. H. , Won, S. Y. , Shin, W. H. , & Kim, S. R. (2016). Prothrombin kringle‐2: A potential inflammatory pathogen in the parkinsonian dopaminergic system. Experimental Neurobiology, 25(4), 147–155. 10.5607/en.2016.25.4.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177, 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25(4), 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Madeira, M. H. , Boia, R. , Ambrosio, A. F. , & Santiago, A. R. (2017). Having a coffee break: The impact of caffeine consumption on microglia‐mediated inflammation in neurodegenerative diseases. Mediators of Inflammation, 2017, 4761081. 10.1155/2017/4761081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, K. G. (1976). Prothrombin. Methods in Enzymology, 45, 123–156. 10.1016/s0076-6879(76)45016-4 [DOI] [PubMed] [Google Scholar]
- McLellan, T. M. , Caldwell, J. A. , & Lieberman, H. R. (2016). A review of caffeine's effects on cognitive, physical and occupational performance. Neuroscience and Biobehavioral Reviews, 71, 294–312. 10.1016/j.neubiorev.2016.09.001 [DOI] [PubMed] [Google Scholar]
- Merlini, M. , Rafalski, V. A. , Rios Coronado, P. E. , Gill, T. M. , Ellisman, M. , Muthukumar, G. , Subramanian, K. S. , Ryu, J. K. , Syme, C. A. , Davalos, D. , Seeley, W. W. , Mucke, L. , Nelson, R. B. , & Akassoglou, K. (2019). Fibrinogen induces microglia‐mediated spine elimination and cognitive impairment in an Alzheimer's disease model. Neuron, 101(6), 1099–1108.e6. 10.1016/j.neuron.2019.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak, Z. , Lebrun, A. , Di Miceli, M. , Rousset, M. C. , Crespel, A. , Coubes, P. , & Rigau, V. (2012). IgG leakage may contribute to neuronal dysfunction in drug‐refractory epilepsies with blood–brain barrier disruption. Journal of Neuropathology and Experimental Neurology, 71(9), 826–838. 10.1097/NEN.0b013e31826809a6 [DOI] [PubMed] [Google Scholar]
- Montagne, A. , Barnes, S. R. , Sweeney, M. D. , Halliday, M. R. , Sagare, A. P. , Zhao, Z. , Toga, A. W. , Jacobs, R. E. , Liu, C. Y. , Amezcua, L. , Harrington, M. G. , Chui, H. C. , Law, M. , & Zlokovic, B. V. (2015). Blood‐brain barrier breakdown in the aging human hippocampus. Neuron, 85(2), 296–302. 10.1016/j.neuron.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, I. , Guzman‐Martinez, L. , Cerda‐Troncoso, C. , Farias, G. A. , & Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Frontiers in Cellular Neuroscience, 8, 112. 10.3389/fncel.2014.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley, H. , Cole, S. L. , Logan, S. , Maus, E. , Shao, P. , Craft, J. , Guillozet‐Bongaarts, A. , Ohno, M. , Disterhoft, J. , van Eldik, L. , Berry, R. , & Vassar, R. (2006). Intraneuronal β‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: Potential factors in amyloid plaque formation. Journal of Neuroscience, 26(40), 10129–10140. 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos, G. , & Franklin, K. B. J. (2004). The mouse brain in stereotaxic coordinates, compact (2nd ed.). Elsevier Academic Press. [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perzborn, E. , Roehrig, S. , Straub, A. , Kubitza, D. , Mueck, W. , & Laux, V. (2010). Rivaroxaban: A new oral factor Xa inhibitor. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(3), 376–381. 10.1161/ATVBAHA.110.202978 [DOI] [PubMed] [Google Scholar]
- Querfurth, H. W. , & LaFerla, F. M. (2010). Alzheimer's disease. New England Journal of Medicine, 362(4), 329–344. 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- Russo, M. V. , Latour, L. L. , & McGavern, D. B. (2018). Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nature Immunology, 19(5), 442–452. 10.1038/s41590-018-0086-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura, J. , Tusell, J. M. , & Serratosa, J. (2003). High‐yield isolation of murine microglia by mild trypsinization. Glia, 44(3), 183–189. 10.1002/glia.10274 [DOI] [PubMed] [Google Scholar]
- Shin, W. H. , Jeon, M. T. , Leem, E. , Won, S. Y. , Jeong, K. H. , Park, S. J. , McLean, C. , Lee, S. J. , Jin, B. K. , Jung, U. J. , & Kim, S. R. (2015). Induction of microglial toll‐like receptor 4 by prothrombin kringle‐2: A potential pathogenic mechanism in Parkinson's disease. Scientific Reports, 5, 14764. 10.1038/srep14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova, E. , & Reiser, G. (2008). Prothrombin/thrombin and the thrombin receptors PAR‐1 and PAR‐4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thrombosis and Haemostasis, 100(4), 576–581. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/18841278 [PubMed] [Google Scholar]
- Song, J. , Choi, S. M. , Whitcomb, D. J. , & Kim, B. C. (2017). Adiponectin controls the apoptosis and the expression of tight junction proteins in brain endothelial cells through AdipoR1 under beta amyloid toxicity. Cell Death & Disease, 8(10), e3102. 10.1038/cddis.2017.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, M. D. , Sagare, A. P. , & Zlokovic, B. V. (2018). Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews. Neurology, 14(3), 133–150. 10.1038/nrneurol.2017.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneda, H. , Andoh, K. , Nishioka, J. , Takeya, H. , & Suzuki, K. (1994). Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. Journal of Biochemistry, 116(3), 589–597. 10.1093/oxfordjournals.jbchem.a124565 [DOI] [PubMed] [Google Scholar]
- Vaiserman, A. , Koliada, A. , & Lushchak, O. (2020). Neuroinflammation in pathogenesis of Alzheimer's disease: Phytochemicals as potential therapeutics. Mechanisms of Ageing and Development, 189, 111259. 10.1016/j.mad.2020.111259 [DOI] [PubMed] [Google Scholar]
- van de Haar, H. J. , Burgmans, S. , Jansen, J. F. , van Osch, M. J. , van Buchem, M. A. , Muller, M. , Hofman, P. A. , Verhey, F. R. , & Backes, W. H. (2016). Blood‐brain barrier leakage in patients with early Alzheimer disease. Radiology, 281(2), 527–535. 10.1148/radiol.2016152244 [DOI] [PubMed] [Google Scholar]
- Verma, A. K. , & Brighton, T. A. (2009). The direct factor Xa inhibitor rivaroxaban. Medical Journal of Australia, 190(7), 379–383. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/19351313 [DOI] [PubMed] [Google Scholar]
- Walter, S. , Letiembre, M. , Liu, Y. , Heine, H. , Penke, B. , Hao, W. , Bode, B. , Manietta, N. , Walter, J. , Schulz‐Schüffer, W. , & Fassbender, K. (2007). Role of the toll‐like receptor 4 in neuroinflammation in Alzheimer's disease. Cellular Physiology and Biochemistry, 20(6), 947–956. 10.1159/000110455 [DOI] [PubMed] [Google Scholar]
- White, J. R. Jr. , Padowski, J. M. , Zhong, Y. , Chen, G. , Luo, S. , Lazarus, P. , Layton, M. E. , & McPherson, S. (2016). Pharmacokinetic analysis and comparison of caffeine administered rapidly or slowly in coffee chilled or hot versus chilled energy drink in healthy young adults. Clinical Toxicology (Philadelphia, Pa.), 54, 308–312. 10.3109/15563650.2016.1146740 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein levels of pKr‐2, NeuN, and neuroinflammatory molecules in the hippocampi of WT mice at 3‐, 6‐, and 9‐months of age. (a‐c) Western blotting for the protein levels of pKr‐2, NeuN, Iba1, TLR4, TNF‐α, IL‐1β and iNOS. Data were analyzed using one‐way ANOVA with Tukey's post hoc analysis and Kruskal‐Wallis test for iNOS analysis (n = 5). Quantitative values are expressed as the mean ± SEM.
Figure S2. Immunohistochemical counterstaining for Nissl (purple) and CD31 (brown) in the hippocampal CA1 regions of WT and 5XFAD mice aged 6 months. CD31 expression, marked by red arrowheads, decreased in the hippocampus of 5XFAD mouse. Scale bar, 50 μm.
Figure S3. Immunohistochemical staining for anti‐ neuronal nuclei (NeuN) in the hippocampal CA1 regions of 9‐month‐old WT and 5XFAD mice. (a) Representative images of immunohistochemical staining for anti‐NeuN in the hippocampal CA1 regions of mice. Scale bar, 50 μm. (b) The number of NeuN‐positive neurons in the hippocampal CA1 regions was expressed quantitatively as a percentage of the number of neurons in 5XFAD mice compared to the number of neurons in WT mice. ***p < 0.05 vs. 9‐month‐old WT mice (Student's t‐test; n = 5 for each group). Quantitative values are expressed as the mean ± SEM.
Figure S4. Total exploration time of object recognition in WT and 5XFAD mice. (a, b) The results of training stage (two identical objects; a) and object recognition tests (one familiar and one novel object; b) were represented as the total exploration time to explore two objects. NS: no significance (t‐test; n = 6 for each group). Quantitative values are expressed as the mean ± SEM.
Figure S5. Pictorial representation of the tracks showing each mouse movement after water or caffeine supply. Novel object (a and b; familiar object: purple square; novel object: blue square) and location‐changed object recognition tests (c and d; familiar object: purple square; location‐changed object: blue square).
Figure S6. Pictorial representation of the tracks showing each mouse movement after rivaroxaban. Novel object (a and b; familiar object: purple square; novel object: blue square) and location‐changed object recognition tests (c and d; familiar object: purple square; location‐changed object: blue square).
Figure S7. Preliminary data on the effects of caffeine treatment on pKr‐2‐induced inflammatory molecules in primary microglial cultures. Primary microglial cells were pretreated with 1 μM caffeine for 30 min, followed by treatment with 50 μg·ml‐1 of pKr‐2 for 8 h. A real‐time PCR analysis using β‐actin as an internal control indicated that the proinflammatory M1 genes were similarly increased in pKr‐2‐treated microglial cells. Caffeine treatment had no significant effect on the pKr‐2‐upregulated M1 genes. The quantification data pertaining to mRNA transcript levels are presented as the mean ± standard deviation (SD) from three independent RT‐PCR experiments.
Figure S8. Intrahippocampal administration of pKr‐2 induces cognitive impairment in WT mice. Behavioural tests for assessing short term recognition were conducted at 7 days after bilateral injections of pKr‐2 in the hippocampal CA1 regions of WT mice. (a, b) The results of the novel object and location‐changed object recognition tests are represented as the time ratio exploring the novel object (novel object/total object exploring time) for short‐term recognition memory. *P < 0.05 vs. nontreated WT mice (one‐way ANOVA with Tukey's post hoc analysis and Kruskal‐Wallis test for novel object recognition analysis; n = 5 for each group). Quantitative values are expressed as the mean ± SEM.
Table S1. Summary of statistical analysis.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.