

Abstract
The relative importance of separation by distance and by environment to population genetic diversity can be conveniently tested in river networks, where these two drivers are often independently distributed over space. To evaluate the importance of dispersal and environmental conditions in shaping microbial population structures, we performed genome‐resolved metagenomic analyses of benthic Microcoleus‐dominated cyanobacterial mats collected in the Eel and Russian River networks (California, USA). The 64 Microcoleus genomes were clustered into three species that shared >96.5% average nucleotide identity (ANI). Most mats were dominated by one strain, but minor alleles within mats were often shared, even over large spatial distances (>300 km). Within the most common Microcoleus species, the ANI between the dominant strains within mats decreased with increasing spatial separation. However, over shorter spatial distances (tens of kilometres), mats from different subwatersheds had lower ANI than mats from the same subwatershed, suggesting that at shorter spatial distances environmental differences between subwatersheds in factors like canopy cover, conductivity, and mean annual temperature decreases ANI. Since mats in smaller creeks had similar levels of nucleotide diversity (π) as mats in larger downstream subwatersheds, within‐mat genetic diversity does not appear to depend on the downstream accumulation of upstream‐derived strains. The four‐gamete test and sequence length bias suggest recombination occurs between almost all strains within each species, even between populations separated by large distances or living in different habitats. Overall, our results show that, despite some isolation by distance and environmental conditions, sufficient gene‐flow occurs among cyanobacterial strains to prevent either driver from producing distinctive population structures across the watershed.
Keywords: benthic cyanobacteria, biogeography, dispersal, metapopulation, population genomics, rivers
1. INTRODUCTION
Natural selection, genetic drift, and recombination are fundamental processes that shape microbial evolution. The relative strengths of these three processes control the frequency of alleles within populations (population evolution) and ultimately speciation. We define a population as a set of co‐occurring cells within a species that are not genetically isolated (Waples & Gaggiotti, 2006). A population is composed of multiple subpopulations with varying degrees of isolation or, conversely, gene flow. Population evolution does not conform to a single model (Hanage, 2016; Shapiro, 2018), and different microbial populations evolve through different processes (Cohan, 2001; Polz et al., 2013). One primary force driving microbial evolution in certain populations is homologous recombination, which can result in promiscuous panmictic populations (Rosen et al., 2015). Other populations are highly clonal, indicating selection of genotypes in populations that evolve through vertical descent and selection on whole genomes (Bendall et al., 2016; Shapiro, 2016). As the genomics of more microbial populations are characterized, we will better understand which evolutionary mechanisms are most common in different bacterial populations.
The potential for genetic drift and selection to differentiate populations is often hampered by dispersal (Martiny et al., 2006; Slatkin, 1987). Microbes disperse through movement of air, flow of water, or transport by organisms, such as insects and birds. Following dispersal, genetic recombination can homogenize populations so that similar strains (variable genomes within a species) and alleles (nucleotide variations at a specific site in the genome) are found in spatially separated populations. In contrast, spatial and environmental barriers throttle dispersal. Once isolated, populations set off on different evolutionary paths (Wright, 1943). Spatial barriers that can limit dispersal include geographic distance, physical obstacles, and directional flows (e.g., air or water). Alternatively, environmental barriers include different abiotic and biotic conditions at a site that prevent the establishment by microbes unfit for the particular environment (Wang & Bradburd, 2014).
The diversity and frequencies of minor alleles within a population provide the genetic background for adaptation to new environmental conditions (Fisher, 1930; Hughes et al., 2008). Strong directional selection can lower the background frequencies of genotypes in the population. An event that selects a minor allele (frequency <50% in the population) may result in either selection on a specific gene or entire genotype, depending on whether or not the gene under selection has recombined into many genomic contexts (high recombination rates) or is found only in one genotypic context (low recombination rates). Genomic analysis methods that only compare assembled sequences are limited because the consensus sequence masks the within‐population sequence variation at any given nucleotide site in the consensus sequence (Garud & Pollard, 2020). Both the dominant genotype within a population (e.g., based on the consensus sequence from a metagenomic assembly of sequencing reads) and the minor alleles present alongside the dominant genotype (single nucleotide variant (SNV) sites within the aligned sequencing reads) must be investigated to characterize the genetic diversity within a population (Olm et al., 2021; Van Rossum et al., 2020).
The dendritic configuration of river habitats makes widely distributed benthic riverine Microcoleus (Cyanobacteria) mats promising targets for genomic analyses investigating how spatial and environmental barriers structure bacterial populations. Microcoleus mats growing in rivers are composed of multiple strains inhabiting the same mat (Bouma‐Gregson et al., 2019; Tee et al., 2020), and can also produce cyanotoxins and pose public health threats (Bouma‐Gregson et al., 2018; McAllister et al., 2016). All rivers are hierarchically branching networks, with many headwater tips converging into fewer tributaries and even fewer mainstems (Benda et al., 2004; Campbell Grant et al., 2007; Power & Dietrich, 2002). At tributary tips (headwaters), similar environments are separated from each other by relatively large distances. At confluences of tributaries with mainstems, contrasting environments are immediately juxtaposed. Environmental distance and spatial distance are, therefore, often independent in river networks (Power & Dietrich, 2002; Winemiller et al., 2010). Comparing natural Microcoleus subpopulations separated by distances of centimetres to populations 100 s of kilometres apart should reveal scales at which environmental and spatial barriers operate in establishing observed patterns of genomic differentiation (Hanson et al., 2012). The degree to which genetic patterns converge under common environmental conditions and/or high dispersal rates, or diverge with spatial or environmental distance may suggest the relative importance of selection, genetic drift, and gene flow in driving genome evolution and population structure of Microcoleus in rivers.
We investigated how the distribution and frequency of Microcoleus within‐species variants are spatially structured across the watershed, at what spatial distances strain diversity overlaps, and if gene flow occurs between subpopulations. We collected Microcoleus mats to sample genetic differentiation at four scales, spanning millimetres to kilometres: (1) within a mat (mm–cm), (2) between mats within a reach (m–km), (3) on tributaries and along adjacent mainstems above and below confluence nodes (m–km), and 4) between subwatersheds (10 s–100 s of km). We hypothesized that (1) Microcoleus mats within the watershed are not a homogenous panmictic population, but have subpopulations structured by environmental conditions and spatial distances; (2) spatial and environmental barriers to gene flow operate over different scales within environmentally diverse habitats to drive strain‐level biogeography; and (3) downstream dispersal will increase genetic diversity at sites with larger upstream subwatersheds.
2. MATERIALS AND METHODS
2.1. Sample collection and DNA sequencing
The Eel River is a 9,547 km2 watershed located in the Coast Range mountains of Northern California (Figure 1). The region has a Mediterranean climate with seasonal summer droughts, and each summer benthic mats of attached algae proliferate (Power et al., 2008). Microcoleus (Oscillatoriales, Cyanobacteria) are common throughout the watershed in summer (Bouma‐Gregson et al., 2018; Kelly et al., 2019).
FIGURE 1.

Sampling locations and field photos of sampling sites. (a) Map of Eel and Russian River watersheds showing which Microcoleus species were recovered from the different sampling sites. (b) Thin (<2 mm) Microcoleus collected from shaded cool‐water creek. (c) Thick (>10 mm) Microcoleus mat over‐growing filamentous Cladophora glomerata in the sunny warm main‐stem. (d) Rattlesnake Creek sampling site (PH2017_40) with riparian vegetation shading the wetted channel (subwatershed drainage area, 50 km2). (e) South Fork Eel sampling site (PH2017_09) with no riparian canopy cover (subwatershed drainage area, 1,392 km2)
Samples were collected from Microcoleus mats over 3 weeks in August 2015 and 3 weeks in July and August 2017 (species and microbial community composition in 2015 samples were reported in Bouma‐Gregson et al., 2019). Microcoleus samples were picked from the cobbles in the river with sterile forceps and immediately frozen in 2 ml cryotubes. Detailed collection methods are described in Bouma‐Gregson et al. (2019). In 2015, 22 samples were collected from 14 sites, and in 2017, 39 samples were collected from 25 sites for a total of 61 samples (Figure 2 and Table S1). In 2017, eight of the 2015 sites were resampled, and 17 new sites were added. At each site, water samples were filtered (0.7 μm, Whatman GF/F) and measured for total dissolved nitrogen, nitrate, ammonium, and phosphorus. Additionally, pH, temperature, alkalinity, conductivity, and dissolved oxygen were measured with handheld probes. Canopy cover was measured with a spherical densiometer. Additional details about environmental measurements are in Bouma‐Gregson et al. (2019). Flow velocity was measured at each collection cobble with an acoustic doppler velocimeter (Sontek FlowMaster) at 80% of the total water depth. For each site, average August water temperature modeling predictions were obtained from the NorWest stream temperature model (Isaak et al., 2017). The upstream watershed area at each sampling point and river network distance between sampling sites was calculated with ArcGIS 10.2 (Esri, Redlands, California, USA). River network distances were calculated with the riverdist R package (https://github.com/mbtyers/riverdist).
FIGURE 2.
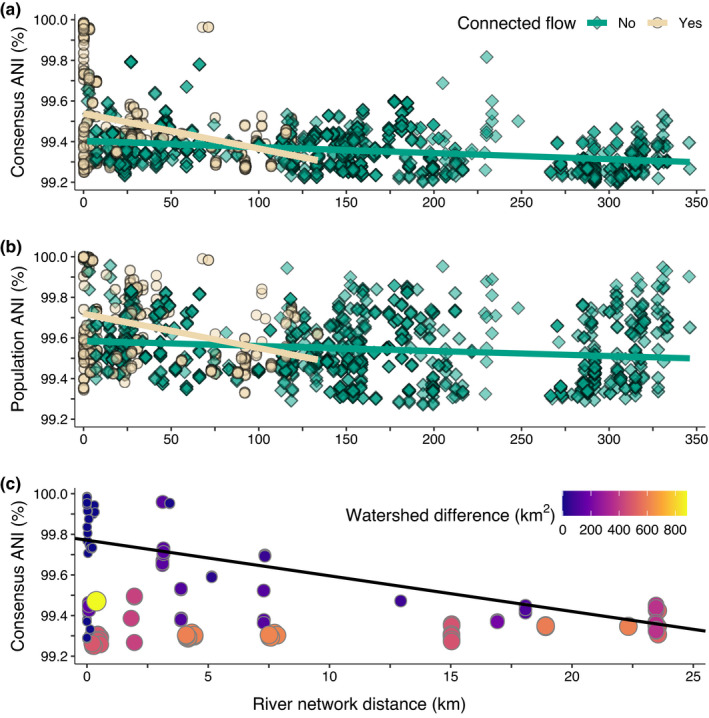
Pairwise average nucleotide identity (ANI) and river network distances between genome pairs in Microcoleus sp. 1. (a) Consensus ANI (ANI of dominant genotype in the mat). (b) Population ANI (ANI of including subdominant genomes in the mat). In a and b, colours show if pairs of sites are connected by downstream flow in the watershed and darker colours signify more overlapping points. (c) Consensus ANI between genome pairs less than 25 km apart and that are connected by downstream flow. Points are coloured and sized according to the difference in their watershed sizes
DNA was extracted from samples using a MoBio DNeasy PowerBiofilm kit. Frozen mat samples were thawed at room temperature for 0.5 h, and ~0.15 g of mat removed for DNA extraction. The DNA extraction followed manufacturer's protocol, except the cell lysis step in the protocol was modified to 5 min of bead beating and submersion for 30 min in a 65°C water bath. DNA was eluted into doubly distilled H2O, and sequenced on an Illumina HiSeq 4000 (San Diego) with 150 bp paired‐end reads at the QB3 Genomics Sequencing Laboratory (http://qb3.berkeley.edu/gsl/, Berkeley, CA, USA).
2.2. Genome assemblies and annotations
Reads were filtered to remove Illumina adapters and contaminants with BBtools, then trimmed with SICKLE (https://github.com/najoshi/sickle) using default parameters. Assembly and scaffolding were performed by IDBA‐UD (Peng et al., 2012). For assembled scaffolds longer than 1 kbp, protein‐coding genes were predicted with Prodigal in the meta‐mode (Hyatt et al., 2010). Predicted genes were then annotated against KEGG (Kanehisa et al., 2014), UniRef100 (Suzek et al., 2007), and UniProt using USEARCH (Edgar, 2010). Genomes were binned manually using coverage, GC content, single copy genes, and taxonomic profile with ggKbase (ggkbase.berkeley.edu), as described in (Raveh‐Sadka et al., 2015). Anatoxin biosynthesis genes were previously identified in Bouma‐Gregson et al. (2019).
2.3. Species diversity and F ST
Microcoleus genome bins were compared with dRep (Olm et al., 2017) to cluster genomes with >96.5% average nucleotide diversity (ANI; see Table S2 for acronym glossary) together into species (Olm et al., 2020; Richter & Rosselló‐Móra, 2009; Varghese et al., 2015). A subset of genes present in most genomes between species was determined by using Roary (Page et al., 2015) to cluster genes at 90% amino acid (AA) identity and align gene clusters with MAFFT (Katoh et al., 2002), which generated 24,826 gene clusters across the genomes. Gene clusters with >50% of the nucleotide sites as gaps or ambiguous bases (Ns) were removed. Then only gene clusters that had sequences from at least 50% of the genomes of each species were kept for analysis. This resulted in 3,744 gene clusters for within species comparisons and 2,244 gene clusters for between species comparisons. For each gene, fixation index (F ST, a measure of the difference in allele frequencies between two populations) was calculated as where Hw is the mean nucleotide diversity (heterozygosity) within a species and Hb is the mean nucleotide diversity between species (Hudson et al., 1992). Calculations were made using the popgenome package (Pfeifer et al., 2014) in R (R Core Team, 2019). The analyses did not calculate population statistics on intergenic regions.
2.4. Strain diversity
To investigate the strain variation within a species at a given site, we mapped sequencing reads to the assembled consensus sequence. A single genome was selected to represent each species based on the quality criteria reported by dRep. On each of the three identified Microcoleus species genomes (Bouma‐Gregson et al., 2019), Prodigal was run with the ‐c flag to ignore any genes that were not closed on a contig. For read mapping, a genome index was built by combining the three Microcoleus species genomes with seven additional genomes of commonly found bacteria in the mats (Bacteroidetes, Betaproteobacteria, Verrucomicrobia, Nostocales, Cytophagales, Oscillatorialles, and Sphingomonadales) to help ensure that reads were not falsely mapped to the Microcoleus genomes. Reads from each sample were mapped with Bowtie 2 (Langmead & Salzberg, 2012) with default parameters.
Population statistics and other metrics were calculated from these mappings with the inStrain program (Olm et al., 2021; Figure S1) https://instrain.readthedocs.io/en/latest/. Reads were filtered with a mapq score >2 to reduce the number of mis‐mapped reads in the analyses. Only read pairs with >96% nucleotide identity were used for the analysis. Nucleotide sites also had to have coverage of >5 for SNV calls and at least 20 paired reads were required for linkage calculations. Population statistics and nucleotide metrics were calculated at both the contig and gene level.
Additional read filtering was done to calculate nucleotide diversity for each gene. Nucleotide diversity (π) is the probability that two reads will have different nucleotides at the same location. First the distribution of coverage was calculated for each genome and genes with coverage values in the lower tenth percentile were removed from further analyses. Of the remaining genes, any gene with breadth <0.9 was also removed from the analysis. Then nucleotide diversity was calculated by summing the squared frequency of each nucleotide across all positions within a gene, (Nei & Li, 1979).
To identify shared SNVs locations from the reference genome, SNV locations in Microcoleus sp. 1 were identified with the inStrain gene_profile module. SNV locations between genome pairs were classified as fixed or biallelic. A fixed site is different from the reference genome and consists of a single nucleotide within the sequencing reads mapped to the site. A biallelic site has two nucleotides mapped to the location, and the dominant nucleotide is the same as the reference genome. Multiallelic sites, with more than two nucleotides mapped to the site, were filtered out of the analysis. Additionally, because sample PH2015_12U was the Microcoleus sp. 1 reference genome, both samples collected at site 12 (PH2015_12U and PH2015_12D) were excluded from the fixed SNV analysis. InStrain identified 142,921 unique fixed sites and 174,320 unique biallelic sites. The unique SNV positions from these two SNV classes were combined into a matrix with columns as the unique SNV position and rows as individual genomes. Cells in the matrix were given a 1 or 0 depending on if a genome contained a particular SNV location. For genomes that did not exceed the coverage threshold at a particular SNV location, a value of 0 for that location was given. Then the percentage of shared SNV sites (Jaccard index) was calculated for each pair of samples using the vegan r package (Oksanen et al., 2019). Genomes with a higher Jaccard index share more SNV positions, while genomes with a lower Jaccard index have more uniquely positioned SNVs between the samples.
SNV frequencies were calculated by inStrain. For each genome, counts of SNV frequencies (rounded to the nearest hundredths) were calculated. Frequencies below 0.05 are not considered by inStrain and are not shown. Then a loess regression was fitted to the SNV frequencies. The smoothed loess curves were visually inspected to identify peaks in SNV frequencies.
2.5. Population ANI
Population and consensus ANI between genomes were calculated with the inStrain program using the compare module. Scaffolds that did not meet coverage thresholds at >25% of sites in both samples compared were removed from the analysis. A consensus ANI SNV site is any site where the consensus nucleotide differs between two samples (Olm et al., 2021; Figure S1). Population ANI sites are when the consensus sequences are different and the minor alleles at the site are also not present in the other sample. For example, if a population has an A to T SNV in the consensus sequence, but the A allele is present in both populations, it is not considered a population SNV (Figure S1). Population ANI is always greater than consensus ANI, because every population SNV is also a consensus SNV, but not vice versa. Therefore, when population ANI and consensus ANI are similar, most SNVs in the genomes do not contain minor alleles present in both populations. In this case, the sample is dominated by a single genome and most SNV sites contain a single allele. When population ANI is close to 100% and consensus ANI is low, then dominant genomes (i.e., consensus sequences) are different, but most SNV sites contain an allele that is present in a minor strain within the sample.
2.6. Recombination
Horizontal gene flow is the sharing of DNA sequences between organisms not related by descent. The four‐gamete test (Garud & Pollard, 2020; Hudson & Kaplan, 1985) was used to predict homologous recombination within species. For a pair of biallelic SNV sites, assuming the infinite‐site model, the presence of all four haplotypes (AB, Ab, aB, ab) within the reads at paired sites can only be explained by at least one recombination event occurring at those sites. The frequency of the different haplotypes was calculated for all linked biallelic sites within genomes from the linkage.tsv output from the inStrain gene‐profile module.
Subpopulation structure based on horizontal gene flow between genomes was investigated by comparing the length of identical DNA sequences in two genomes using the program PopCOGenT (Arevalo et al., 2019; VanInsberghe et al.,2020). The method is based on the observation that when recombination rates between two genomes are high, the length of identical regions shared between genomes increases. The parameter, length bias, was calculated for each pair of genomes as the sum of squares of the difference between observed and expected lengths of identical regions shared by the genome pairs. Length bias increases as recombination rates increase. A genome network of length bias values was then created, estimating horizontal gene flow between genome nodes, and clustered with Infomap (Rosvall et al., 2009) to identify subpopulations with elevated rates of horizontal gene flow within the subpopulation cluster.
2.7. Statistical analyses
Linear regression was used to test for the effect of river network distance (distance traveled within the river network channels and assuming no overland travel) on population and consensus ANI. We used generalized dissimilarity model matrix regressions (Ferrier et al., 2007) implemented with the R package gdm to investigate the relationship of environmental variables with consensus ANI. At sites <25 km we tested for the effect of environmental parameters on ANI. First, to determine if adding sampling‐year as a random effect improved the model, we compared AIC (stepcAIC function; cAIC4 R package) values on models with and without year. Variance associated with year was ~0, so we treated year as a fixed effect. After removing correlated variables, multiple linear regression models were built with year, river distance, watershed size difference, total dissolved phosphorus (TDP), total dissolved nitrogen (TDN), conductivity, canopy cover percent, and NorWest modeled temperature. Model selection (stepAIC function; MASS R package) to select variables that best predicted consensus ANI and population ANI. AIC was then compared between the different model outputs. We also ran ANOVA between model with environmental variables and model with environmental variables, river network distance, and watershed area difference.
Three population diversity metrics – nucleotide diversity, population ANI, and SNV sharing – were used to test the effect of watershed size on population diversity. Watershed area was log10 transformed, and simple linear regression models were built for nucleotide diversity and population ANI data. For SNV sharing, multiple linear regression models to test the main effect and interaction of log10 transformed watershed area and river network distance on SNV site similarity.
Relationships between population age and watershed sizes greater than or less than 500 km2 used a generalized linear binomial model with log10 transformed watershed area with false discovery rate correction (Benjamini & Hochberg, 1995) for the two hypothesis tests. Principal components analysis was performed on scaled and centred environmental variables using the vegan R package. All statistical analyses were conducted in R (R Core Team, 2019). Code for these analyses can be found at https://github.com/keithbg/Microcoleus_Analysis.
3. RESULTS
3.1. Microcoleus species diversity
In this study we analysed metagenomic data from 60 samples collected from sites throughout the Eel River watershed (Northern California) in 2015 (22 samples from 14 sites) and 2017 (38 samples from 24 sites). A single sample was also collected from the Russian River watershed in 2017. Genomes from the 2015 samples were reported in a prior study (Bouma‐Gregson et al., 2019) that investigated microbial community and metabolic diversity in Microcoleus mats.
Combined analyses of all 2015 and 2017 samples led to the recovery of 66 Microcoleus genomes (95% average completeness, Table S3). Genomes were clustered into species‐level groups using a threshold of <96.5% average nucleotide identity (ANI) based on whole genome alignments, resulting in the identification of three species clusters (Table 1, Figure S2). Within each species cluster the average ANI was always >98% (Table 1). In 2017, only Microcoleus sp. 1 and 3 were recovered, whereas all three species were recovered in the 2015 samples. The most common species, Microcoleus sp. 1, with 47 genomes reconstructed was found at 21 sites broadly distributed across the Eel River watershed (Table S1, Figure 1). Only Microcoleus sp. 2 genomes contain predicted anatoxin‐a biosynthesis gene clusters (Bouma‐Gregson et al., 2019), and they were only recovered from the 2015 samples. Microcoleus sp. 3 genomes were recovered in 2015 and 2017 and are most similar to Microcoleus sp. 1 (Table 1). Notably, one of the Microcoleus sp. 3 genomes was recovered from an additional sampling site in the Russian River. This is important because it means that species 3 has dispersed across both the Eel and an adjacent watershed, or it was present in the past when the Russian and Eel Rivers formed a single watershed before they were divided by tectonic activity, about 2 million years ago (Lock et al., 2006).
TABLE 1.
Average nucleotide identity (ANI) and percent alignment coverage (in parentheses) of genomes within and between each Microcoleus species cluster. The number of genomes in each species is given at the top of each column
| Species 1 (47 genomes) | Species 2 (6 genomes) | Species 3 (11 genomes) | |
|---|---|---|---|
| Species 1 | 98.8% (93.5%) | ||
| Species 2 | 86.8% (41.0%) | 99.5% (94.2%) | |
| Species 3 | 92.7% (72.6%) | 86.0% (39.4%) | 99.3% (98.9%) |
Species were not isolated in this study and the assignment to the genus Microcoleus relied on molecular phylogenies in Bouma‐Gregson et al. (2019). Additionally, Conklin et al. (2020) isolated a novel strain with 99.9% 16S rRNA sequence similarity to Microcoleus sp. 2, and assigned it to the genus Microcoleus. Based on the phylogenies and results in these two publications, we placed our genomes within the Microcoleus genus, as revised by (Strunecký et al., 2013).
3.2. Intraspecific diversity across spatial distances
We investigated spatial patterns of strain diversity within Microcoleus sp. 1, the species recovered most often in both sampling campaigns. As the physical distance between mats increased, genome‐wide nucleotide identities decreased (Figure 2), for both consensus ANI (the ANI of the dominant genotype in a sample; conANI, Figure S1, Table S4) and population ANI (a measure that includes consideration of variants in subdominant genomes and minor alleles; popANI, Table S4) (Olm et al., 2021). Sites that were connected through downstream flow had higher overall conANI and popANI (Figure 2a,b). In particular, conANI values higher than 99.82% only occurred at flow‐connected sites. The flow‐connected sites also had a stronger decay relationship with river network distance than sites unconnected by downstream flow (Figure 2a,b).
PopANI values remained higher at large distances compared to conANI values. For example, even at a distances of ~300 km, popANI values between some mats are >99.8%, a similar population ANI value to genomes <25 km apart, while consensus ANI values at long river network distances were much lower than at short river network distances. Sites greater than 150 km were all unconnected by flow, and the distance‐decay between unconnected flow sites was slightly slower for population ANI (Figure 2b; p < .05, slope = −2.5 × 10−4, Table S5) than for consensus ANI (Figure 2a; p < .05, slope = −3.0 × 10−4, Table S5). Additionally, river network distance explained more of the variation in consensus ANI (adj. R 2 = 0.20) than for population ANI (adj. R 2 = 0.12), suggesting these two variables are more independent for population ANI. The slower population ANI distance‐decay and lower R2 for population ANI, together with the highly similar consensus ANI at short distances show that, although highly similar dominant genomes are found spatially close to one another, the alleles within the populations are distributed broadly, indicating that they are not genetically isolated.
Based on the distribution of consensus ANI values in our data (Figure S3), genomes with consensus nucleotide identities >99.6% may be members of the same local subpopulation. Within Microcoleus sp. 1, 67 genome pairs (6% of comparisons) share >99.6% conANI (Figure 2a and S3) and form a long tail in the conANI distribution (Figure S3). These represent more similar genomes than the other 1,014 genome pairs. Most of the genome pairs that share 99.6% conANI are from sites separated by <1 to 1,000 m, with the highest ANI values, >99.8%, often <100 m apart. Additionally, genomes collected from the same cobble (<10 cm) were always >99.9% conANI from one another. However, while the highest conANI values (>99.9%) were primarily separated by river network distances <25 km, some pairs were ~75 km from each other, which may indicate recent dispersal. The 67 genome pairs therefore may represent members from the same subpopulation, as they predominantly inhabit the same river reaches.
Spatial proximity was not always associated with high consensus genetic similarity, however, as many mats <25 km distant had conANI <99.6% (Figure 2c). These lower conANI comparisons often involved genomes from sites in tributaries and mainstem sites, respectively. Despite their proximity, tributary and mainstem sites may have strongly contrasting environmental regimes (Figure 3 and Table S6). In addition to spatial distance, environmental conditions partially explain the conANI patterns across the watershed. For example, conANI was negatively correlated with differences in canopy cover, conductivity, and mean annual temperature between sites (p < .05; Figure 3). Adding watershed difference and river distance to multiple‐linear regression models incorporating environmental parameters significantly reduced (p < .05) unexplained variance in these models (Table S5). In sites <25 km apart (Figure 2c), both increasing differences in subwatershed sizes and increasing river network distances are associated with lower ANI (trend in Figure 2c; p < .05, Table S5). For sites separated by very small river network distances (<1 km), there is a clear relationship involving higher consensus genetic similarity and smaller difference in subwatershed drainage area, which usually indicates more similar environments. Interestingly, however, at distances >7.5 km, differences in subwatershed size do not appear to impact genetic divergence. In fact, almost all genetic comparisons at distances >7.5 km remain below <99.6% conANI, even in subwatersheds of very similar size. We were not able to investigate environmental relationships with ANI using a matrix regression approach because the generalized dissimilarity matrix algorithm could not fit a model to our data because of the highly similar consensus ANI values within Microcoleus sp. 1.
FIGURE 3.
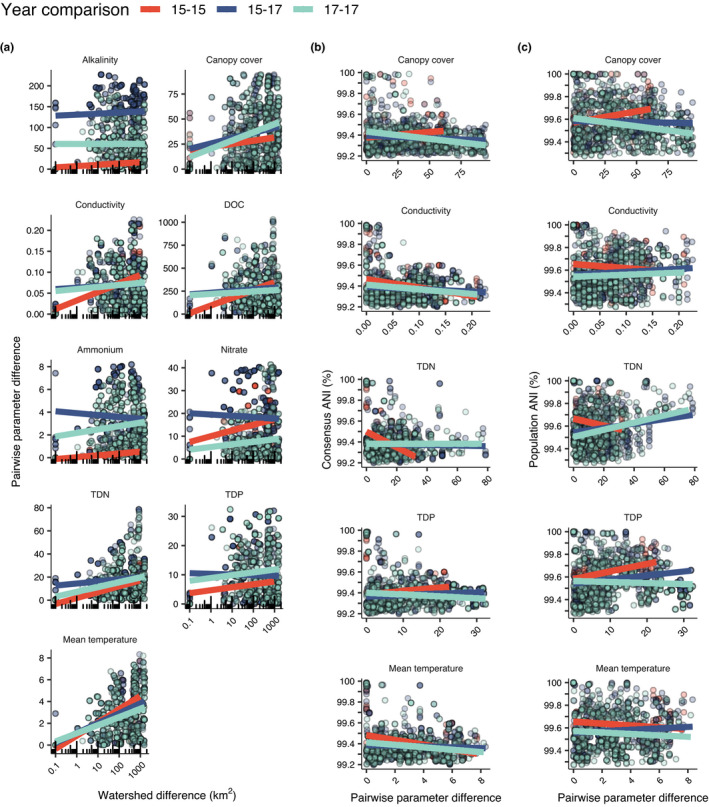
Environmental condition differences and average nucleotide identities (ANI) at different sampling locations. Points are coloured according to the year data was collected. (a) Paired watershed size difference and absolute value of environmental parameter difference. (b) Consensus ANI and absolute value of environmental parameter difference. (c) Population ANI and absolute value of environmental parameter difference. To help interpret trends, simple linear regressions are given for each year comparison; however, not all slopes are statistically significant at p < .05. DOC: dissolved organic carbon, TDN: total dissolved nitrogen, TDP: total dissolved phosphorus, Mean temperature: modelled NorWest mean August temperature
3.3. Intraspecific diversity across different watershed sizes
We hypothesized that mats growing at sites with large upstream subwatersheds (drainage areas) would have higher strain diversity than mats growing in smaller subwatersheds because they would be supplied by more potentially diverse Microcoleus cells by downstream transport. The larger area of subwatershed upstream of a particular site, the greater potential diversity of Microcoleus cells that could be supplied downstream. We tested this hypothesis by evaluating the relationship of subwatershed area with three population diversity metrics calculated from our genomes: nucleotide diversity, single nucleotide variant (SNV) sharing (when two genomes have an SNV at the same genome position), and population ANI. Nucleotide diversity was compared with subwatershed area for Species 1, 2 and 3, while, within Species 1, we investigated SNV sharing and population ANI, which were collected from subwatersheds ranging 2–1,600 km2. We hypothesized nucleotide diversity, population ANI, and SNV sharing would all increase with the size of the upstream subwatershed.
Contrary to our expectations, we did not find higher nucleotide diversity (p > .05; Table S5) in larger subwatersheds; Figure 4a). The highest median nucleotide diversity in genomes was found in mats collected from ~15, 100, and 1,000 km2 subwatersheds. Species 2 and 3 also had lower median nucleotide diversity and were found in larger subwatersheds. However, there were differences in the nucleotide diversity among the three species. Intraspecific nucleotide diversity (π; the probability that two reads will have different nucleotides at the same location) was primarily between 0.0001 and 0.001 for all species (Figure S4). However, the range of nucleotide diversity values among all genes in genomes from the different species spanned several orders of magnitude, with maximum values between 0.02 and 0.48 (Figure S4). Mean population ANI also did not increase (p > .05) with subwatershed areas (Figure 4b). Smaller subwatersheds can contain a similar composition of strains to the overall watershed‐wide composition, explaining the high popANI values between sites.
FIGURE 4.
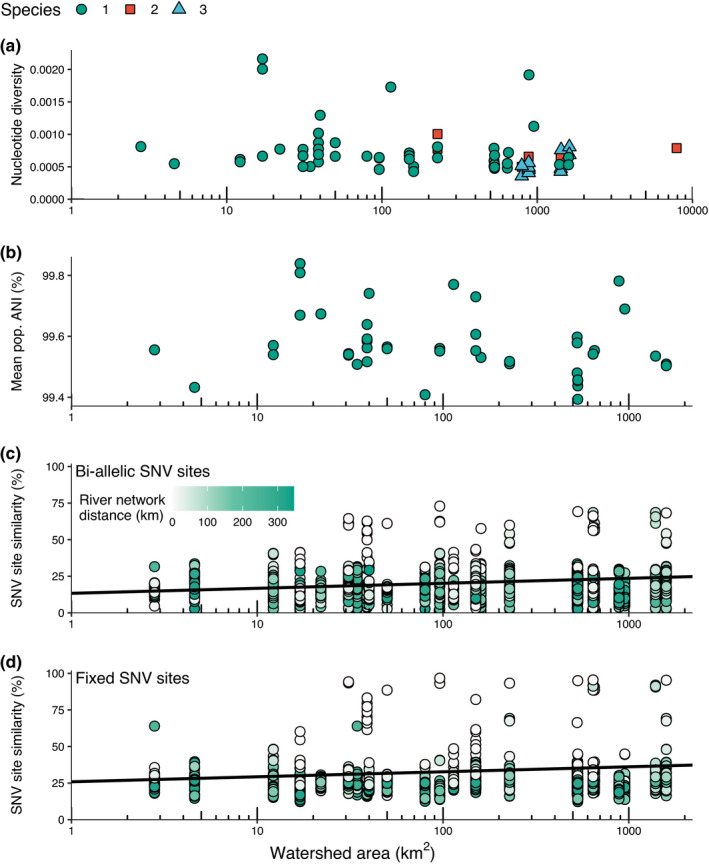
Patterns of intraspecific diversity across watershed sizes. (a) Median nucleotide diversity in each genome and the upstream subwatershed area where each genome was collected. Colours and shapes indicate the ANI species of each genome. (b) Mean population ANI for a Microcoleus sp. 1 mats compared with all other Microcoleus sp. 1 mats and the upstream subwatershed area of where the mat was collected. (c and d) SNV sharing in Microcoleus sp. 1 represented by percentage of shared biallelic (c) or fixed (D) SNV sites between pairs of samples
SNV sharing was higher (p < .05) in larger subwatershed areas for both biallelic and fixed SNV sites (Figure 4c,d). However, this was partially driven by high similarity between samples that were collected near each other (low river network distance) in larger subwatersheds (i.e., significant (p < .05) negative interaction with subwatershed area and river network distance). Samples that were close to one another generally share many SNVs, though one of the highest SNV similarity values occurred between two samples collected 70 km from each other, so high SNV similarity can be present at larger river network distances. In addition to the spatial associations with SNV site similarity, the SNV similarities differed when comparing fixed and biallelic sites. Some samples shared 96% fixed SNV sites and were more similar overall than bi‐allelic sites, for which no samples shared more than 75% biallelic SNV sites. In comparison, some samples shared 96% fixed SNV sites (Figure S5). Taking these results together, the nucleotide diversity, population ANI, and SNV data suggest a limited effect of upstream subwatershed size on Microcoleus strain and allelic diversity within mats.
3.4. Minor allele distribution across the watershed
The percentage of SNV sites where the major allele in one population is present as a minor allele in another population (pMA, percent minor alleles), was calculated as: [1 ‐ (popANI sites / conANI sites)] (Figure S1). Cases where the minor allele in one population is the major allele in another population could arise (1) when cells from a founding population are introduced into another population, (2) through recombination and genetic admixture between populations, or (3) because recurrent mutations are selected for in both locations, resulting in parallel evolution.
Plotting the relationship between conANI and pMA shows the variation in the similarity of both the dominant genomes and the minor strains inhabiting Microcoleus mats (Figure 5). Microcoleus sp. 1 mats exhibit a variety of conANI‐vs‐pMA values, but certain combinations of conANI and pMA do not occur (Figure 5). When the dominant genomes in two mats are similar (conANI >99.9%), pMA in between most mats is relatively high (>75%) and both mats always have relatively low nucleotide diversity (π; <0.001 average nucleotide diversity and <0.001 difference in average nucleotide diversity). There are no samples with similar dominant genotypes (conANI >99.9%) and few shared minor alleles (pMA <35%), a pattern that could indicate a recent clonal sweep within a mat.
FIGURE 5.
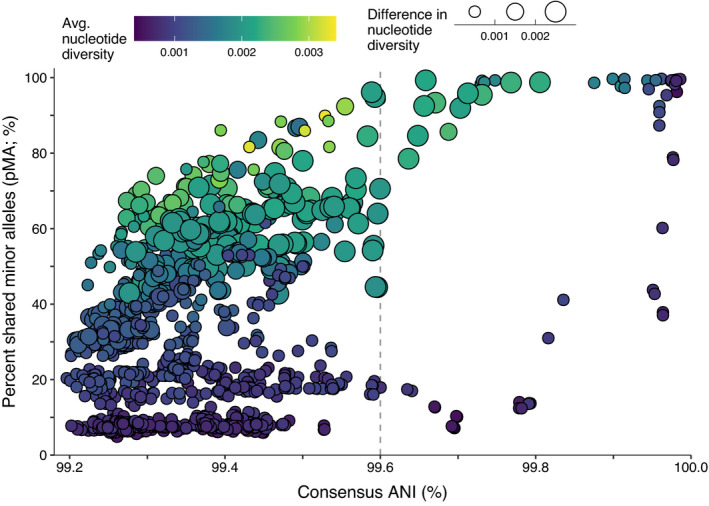
Mean consensus ANI and the percentage of shared minor alleles (pMA) between Microcoleus sp. 1 genomes. Points are coloured by the average nucleotide identity of the two genomes. Points are sized by the absolute difference in nucleotide diversity (π) between the two genomes. Vertical dashed line shows 99.6% consensus ANI
At intermediate conANI levels (99.6%–99.9%), a bi‐modal pattern of pMA and nucleotide diversity is observed. In this conANI range, most pairs of mats either have high pMA (>75%) and high nucleotide diversity >0.002 (indicative of diverse populations that share minor alleles), or low pMA (<20%) and low nucleotide diversity (indicative of relatively clonal, distinct populations that do not share minor alleles). Only two samples in our analysis have intermediate values of pMA (20%–75%) at intermediate values of conANI (99.6%–99.9%).
When conANI is low (99.2%–99.6%), a near continuous range of pMA and nucleotide diversity values are observed, and there is a strong association between increasing pMA and nucleotide diversity. Pairs of mats with high pMA and nucleotide diversity may have acquired new variants without purging minor alleles, and pairs of mats with the same conANI, but low pMA and nucleotide diversity, are pairs in which where either each mat has undergone selective sweeps that have purged diversity, or each mat grew from a relatively clonal source. As conANI increases from 99.2%–99.6%, the maximum pMA values increase as well. Thus, as the dominant genomes become more similar, the population of minor strains in the mat tend to become more similar as well.
Within samples, Microcoleus sp. 1 minor allele frequency distributions show two basic pattern types: either a regular decrease in SNV content with increasing minor allele frequency or a multi‐modal distribution (i.e., one or more peaks with elevated minor allele frequencies; Figure 6 and Figure S6). The peaks in the frequency spectra can be caused by genetically distinct minor strains at higher abundances within the mat. We investigated whether selection is acting differently in populations with and without secondary peaks by calculating the ratio of synonymous to non‐synonymous substitutions (N:S). In mats with secondary peaks, N:S was lower (N:S mean = 0.60, median = 0.59) compared to mats with no secondary peaks (N:S mean = 0.73 median = 0.75). There are fewer nonsynonymous SNVs where mats have secondary peaks (indicating the presence of multiple strains) than in mats with lower abundances of minor strains.
FIGURE 6.
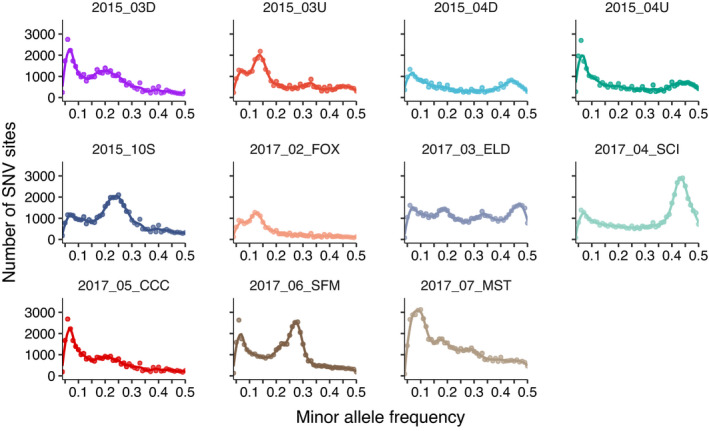
Minor allele frequency distributions for 11 samples that have high abundances of SNV sites at frequencies >0.05 (secondary SNV peaks). The secondary peaks represent minor strains within mats that compose >5% of the mat subpopulation. Points give the number of SNV sites for a 0.02 wide frequency bin. The line is a LOESS smoothing curve to visualize the trend
3.5. Recombination
Both the four‐gamete test and length bias results suggest that recombination occurs within and between mats of the three Microcoleus species. The four‐gamete test postulates that the presence of all four haplotypes (i.e., AB, Ab, aB, ab) of a pair of linked biallelic SNVs occurs through at least one homologous recombination event. All Microcoleus samples had all four alleles (H4 haplotype) present at some linked SNV sites, but the H4 haplotype was rare (<1%–15% of linked SNV sites). The H2 haplotype (AB and ab) was most common at linked SNV sites with ~46% of all linked sites being H2 across all species (Figure 7a). Species 2 had the lowest frequency of H4 sites (median 3.4%, max 5.7%), though there were fewer genomes in that species. Thus recombination occurs within each species, but does not appear to be the dominant mechanism causing nucleotide variation within a given mat.
FIGURE 7.
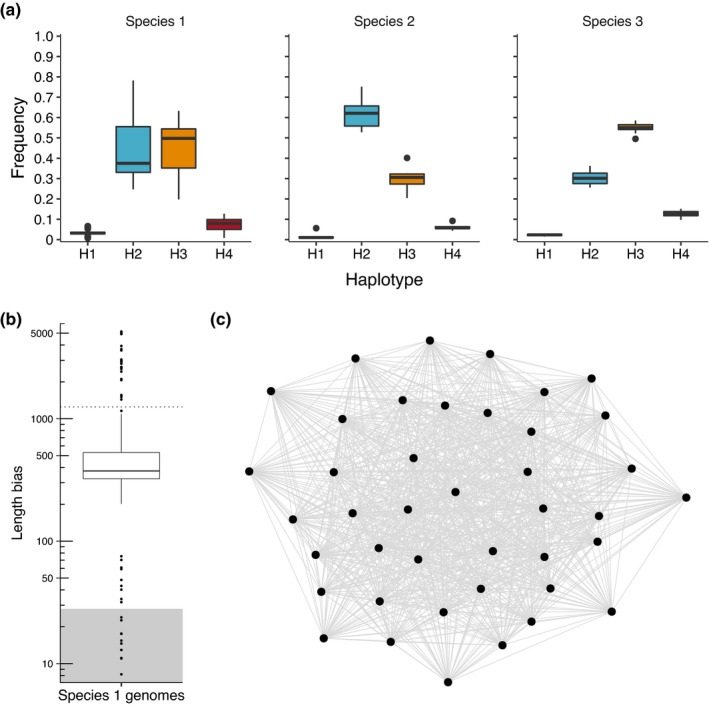
Recombination within Microcoleus species. (a) Haplotype frequencies at linked SNV sites across genomes in each species. Boxplot colours indicate haplotype category. Boxplots boxes range from 25th to 75th quartiles and show median value. Whiskers extend 1.5 times the interquartile range. (b) Boxplot of length bias values of Microcoleus sp. 1 genomes. Dotted horizontal line is at 1,247, the 98th percentile value. Grey shading represents length bias values that could be explained by negative selection (maximum negative selection cutoff = 27.9). (c) Gene flow network built from length bias values showing gene flow among most genomes with no subpopulation clustering. Each node is a Microcoleus sp. 1 genome. Genomes with <0.035% sequence divergence were clustered into a single node
When recombination has occurred, genomes are predicted to share longer sections of identical nucleotide sequences than shared between genomes where no recombination has occurred. Length bias is an estimate of this increased length in identical sequences. Length bias was calculated with the program PopCOGenT by comparing the length distribution of identical sequences to an expected distribution from a null model with only point mutations. In Microcoleus genomes, length bias values suggest recombination between consensus genomes, with median length bias of 374 and an interquartile range of 323–529 (Figure 7b). Most genome pairs have regions of identical DNA sequences longer than expected from a nonrecombinatory null model. However, 18 genome pairs did not have significantly elevated length bias values. Even with some subpopulations not recombining, gene‐flow appears continuous throughout all genomes, as no clusters were identified in the raw network or by the Infomap clustering algorithm (Figure 7c). Recombination occurs between genomes, so we infer that spatial and environmental gene‐flow barriers appear weak and do not substantially reduce recombination rates between different sets of genomes.
Between species, recombination was evaluated by investigating genes with F ST, (fixation index, which is a measure of the genetic differences that can be explained by population structure) the values of which can indicate a recent gene sweep (Figure S7 and Table S7). The lowest F ST values were between Microcoleus sp. 1 and 3, indicating a recent sweep. Particular gene functions were not associated with low F ST values and many genes with low F ST had no functional annotations at all. F ST results suggest that horizontal gene transfer is infrequent between Microcoleus species whose genomes have <96.5% ANI.
4. DISCUSSION
We used the dendritic structure of rivers to compare how spatial distance and environmental factors impact gene flow between genomes and shape population structure. Of the three Microcoleus species we analysed, Microcoleus sp. 1 was most abundant and broadly distributed across the sampling sites and was the subject of our population genomics analyses. First, contrary to our expectation (hypotheses 1 and 2) of strong geographic and environmental population structure, we found little evidence for regional patterning of subpopulation distribution. What spatial structure existed occurred at the local site scale (<1,000 m). We found some evidence that genomes in larger watersheds are more similar to each other (hypothesis 3), but the overall genome and nucleotide diversity in the watershed were not strongly correlated with subwatershed size. At the watershed‐scale, Microcoleus populations are well mixed.
4.1. Microcoleus population structure
Microcoleus sp. 1 genomes form a watershed‐wide population and show no watershed‐wide (>1,000 m) gradients in population structure. The population appears to be primarily structured at the river reach scale (<1,000 m), with organisms with highly similar genomes (>99.6% ANI) at the same site. At local scales, Microcoleus mats are patchy and spatially discontinuous in the river. While visible mats may extend over several tens of metres, there are often hundreds more metres or even kilometres between one large patch and the next. Despite the long distances between mats, there was not a strong spatial decrease in conANI values between 99.2–99.6% across the watershed, and some of the most distant samples (~300 km) had conANI values approaching 99.6%. The rapid decrease with increasing spatial distance of genomes with >99.6% conANI (Figure 2b) suggests that Microcoleus subpopulations are dominated by the same genotype across a few riffle and pool habitats within the river, and that mats outside this local habitat are from different subpopulations dominated by different Microcoleus strains, often with <99.6% ANI.
Dispersal rates across regions within the watershed (i.e., subwatersheds) must exceed mutation or selection rates to maintain a genetically homogenous population (Hanson et al., 2012; Slatkin, 1987). We had hypothesized that upstream or across‐watershed dispersal rates would be low and would isolate mats in different subwatersheds (Morrissey & de Kerckhove, 2009). However, even subwatershed regions up to 250 km apart and those not connected by direct flow share organisms with genomes that share >99.6% ANI (Figure 1). Similarly, within‐species variants of Synechococcus growing in hot springs within 1–1,000 m of each other show strain similarity, and some strains were present at high relative abundances even in hot springs 20–60 km distant (Becraft et al., 2020). Given that Synechococcus in hot spring pools are more severely isolated than sites within a river network, yet have a similar biogeography as Microcoleus mats, dispersal of aquatic cyanobacteria across sites at the 10–100 km scale may be quite frequent. Dispersal upstream or between subwatersheds could occur by wind or hitchhiking on organisms, such as birds, insects, fish, or mammals (Kristiansen, 1996). Cosmopolitan distributions of within‐species variants at the 10–100 km scale could also be generated by low rates of mutation or genetic drift relative to when Microcoleus colonized the watershed. Previous reports do not suggest that Microcoleus have unusually low mutation rates (Dvořák et al., 2012; Segawa et al., 2018). Earliest fluvial deposits of the Eel River are about two million years old (Lock et al., 2006). Although we do not know when Microcoleus arrived within the watershed, given the global distribution of Microcoleus taxa, we have no evidence to suggest the colonization of the Eel was recent, particularly when scaled to the short generation time of actively dividing cells. We conclude that spatial barriers to Microcoleus gene flow are weak within the watershed scale, though barriers probably strengthen at larger regional or continental scales (Becraft et al., 2020; Dvořák et al., 2012; Whitaker et al., 2003).
Like spatial barriers, physical and chemical conditions do not strongly impact the genetic structure of Microcoleus populations. Based on our data, the sites with the strongest potential for environmental selection were at the confluence of Cedar Creek and the South Fork Eel (Cedar Creek samples: PH2015_03D, 03U, 04D, 04U, and PH2017_01; South Fork Eel samples: PH2015_02D, 02U, and PH2017_05). These sites were all separated by <500 m, and environmental data show that the mainstem is warmer and has higher TDN, TDP, and DOC concentrations (Table S6). When comparing across the mainstem and tributary samples, these pairs of Cedar Creek and SF Eel genomes had the lowest consensus ANI values (99.24% – 99.31%) of any of the pairs of samples separated by less than 25 km (Figure 2c). In spite of these lower ANI values, these genomes are still highly similar, and across all samples there was no clear ANI clustering together of tributary or mainstem genomes or strong relationships between ANI and environmental conditions (Figure 3 and Figure S2). Overall, we did not find evidence that selection based on environmental conditions resulted in multiple ecotypes (genetic lineages with ecologically important traits that differentiate the environmental niche used by the lineage; (Cohan, 2001; Rocap et al., 2003). Strong environmental gradients do exist in the Eel River watershed (Finlay et al., 2011), but Microcoleus genomes appear to have sufficient phenotypic plasticity to tolerate this variation.
Microcoleus mats form seasonally in the Eel River. Winter high‐flow events remove biofilms from substrates and can mobilize the riverbed, overturning rocks and scraping surfaces. The patchy local scale (<1,000 m) of Microcoleus subpopulations may be explained by metapopulation theories that posit that transient subpopulations can be reseeded from nearby source populations (Hanski & Gilpin, 1991; Levins, 1969). The frequency of disturbance is a notable difference between seasonally flooding rivers and relatively stable microbial habitats, such as soils and hot springs. High‐flows cause subpopulation bottlenecks when large macroscopic colonies are scoured to microscopic remnants or are removed completely. Notably, substrates are more easily disturbed in channels with larger drainage areas, as river bed sediments decrease in particle size and discharge increases downstream. More stable boulder and bedrock substrates in smaller watersheds could be scour refugia, which might explain the higher than expected within‐site diversity found in many smaller watersheds (Figure 4). In larger subwatersheds where riverbed sediments are more easily mobilized, cobbles with Microcoleus residues could be transported to new sites where variable environmental conditions might determine which strains are able to persist in a site and reinitiate growth. Alternatively, the composition of strains that form mats after scouring could be a random process driven by which cells randomly survived the scour. Riverine processes associated with high‐flow events may, via genetic drift linked to bottlenecks and founder‐events associated with newly introduced strains, strongly influence subpopulation diversity.
While we did not find that environmental gradients at the scale of metres to hundreds of kilometres shaped the biogeography of Microcoleus populations, environmental gradients at centimetre or micrometre scales could still impact the distribution of Microcoleus in the watershed. We still do not know how Microcoleus mats arise out of the microbial community of attached and suspended microorganisms in the watershed (Brasell et al., 2015). Microscale environmental gradients, ecological interactions, and random chance, all play a role in the initial formation of a biofilm community (Battin et al., 2016). Therefore, understanding the assembly process of riverine biofilms and the role of natural selection in controlling the proliferation of Microcoleus mats will benefit from microscale measurements, strain‐resolved genomes, and manipulative experiments to conduct targeted and precise investigations on the evolutionary ecology of Microcoleus genomes.
4.2. Diversity between Microcoleus mats
Our hypothesis that mats with larger upstream catchments would be more similar than mats from more isolated portions of the watershed was only supported by SNV sharing data (Figure 4). Surprisingly, fixed SNV sites are more commonly shared than biallelic SNV sites between mats. Because fixation is harder to achieve, we expected biallelic SNV sites would be more commonly shared between mats. Biallelic SNV sites only require co‐occurrence of two strains and could occur when an upstream cell colonizes and coexists within a downstream mat, while fixed SNV sites require co‐occurrence and recombination or selection to bring an allele, or whole genome, to fixation. Our results show recombination occurs between Microcoleus cells, and previous work has shown recombination to occur in other Oscillatoriales taxa (Lodders et al., 2005; Vos & Didelot, 2009). Furthermore, biofilms facilitate recombination due to the high levels of cell‐to‐cell contact (Cowley et al., 2018; Molin & Tolker‐Nielsen, 2003), therefore, we expect recombination to be integral for SNV sharing between mats.
The different relationships between consensus ANI and minor alleles reveal diversity patterns between mats of both dominant genomes and minor alleles (Figure 6). Notably, certain combinations of consensus ANI (conANI) and percent shared minor alleles (pMA) are absent in our data. For example, most highly similar pairs of dominant genomes (conANI > 99.9%) have very similar minor alleles (pMA > 80%), even though sharing fewer than 80% of alleles is common between less similar pairs of dominant genomes (conANI < 99.9%). Genome pairs with consensus ANI > 99.9% are located close to one another and are members of the same subpopulation. The absence of mats with highly similar consensus genomes (high conANI) but different minor alleles (low pMA) shows that both dominant and minor alleles are highly similar within subpopulations. Dominant genomes are not arising out of diverse gene pools co‐occurring at close distances, nor are strong bottlenecks differentiating mats at close spatial distances. Similarly, Microcystis populations can contain distinct minor strains closely associated with different dominant strains in a bloom (Briand, 2009). The few mat pairs with >99.9% consensus nucleotide identities but <75% shared minor alleles are located 34–71 km away and come from different subpopulations, suggesting spatial and environmental distance accounts for the difference in minor alleles. The minor‐strain similarity in subpopulations builds on work showing highly similar Sulfolobus (Whitaker et al., 2003) and Synechococcus (Becraft et al., 2020) dominant strains within subpopulations, and extends this pattern to minor strains in the subpopulation as well.
The relationship between pMA and conANI is controlled by purging or maintaining diversity through the same mechanisms that control population evolution: gene or genome sweeps, time since colonization, and rates of dispersal and recombination (Hanson et al., 2012; Slatkin, 1987). Notably, in our data when conANI ranges 99.6%–99.9%, there is a bimodal pMA distribution with pMA values either <20% or >75% pMA, and only 2 pMA values between 20% and 75%. Mats with high pMA and high nucleotide diversity may have not undergone a recent selective sweep that removes minor alleles (Bendall et al., 2016). The presence of multiple secondary SNV peaks in mats with high pMA (Figure 6 and Figure S7) provides some evidence that a sweep has not occurred recently, because pMA would decrease if minor strains were purged. In contrast to the bimodal pMA pattern at conANI 99.6%–99.9%, at conANI <99.6%, there is almost continuous pMA variation, and dominant genomes inhabit mats with a range of minor strain diversity. The different patterns above and below the pMA = 99.6% threshold is surprising, as there are few known mechanisms to account for such a rapid change in the distribution of pMA values. Fewer mats with >99.6% conANI were sampled, so the different pattern could result from sampling bias. Alternatively, time since mat formation or purging sweeps could generate low nucleotide diversity and low pMA values, suggesting that most of the mats with >99.6% conANI either experienced a recent sweep or were sampled during a period of mat development when mat diversity is low.
4.3. Diversity within Microcoleus mats
Multiple strains co‐occur within Microcoleus mats, but most mats are strongly dominated by a single strain. Nucleotide diversity was relatively low, up to ten times lower than values reported in soils (Crits‐Christoph et al., 2020), Nitrospira in drinking water filters (Palomo et al., 2020), and planktonic Microcystis blooms (Sabart et al., 2009; Tanabe et al., 2007). Microcoleus mats were thinner in smaller subwatersheds, probably due to light limitation and cooler temperatures, but these had similar nucleotide diversity to thicker mats collected from sunnier warmer sites with larger upstream subwatersheds (Figure 4b). This suggests that biomass accrual is dominated by single clonal lineage within the mat and that the monthly time‐scale of summer growth is not long enough to accumulate substantial diversity through mutations or recombination.
The high abundance of a single Microcoleus genome (Figure S6) within a mat, combined with evidence for recombination among Microcoleus genomes, amounts to an epidemic population structure (Smith et al., 1993), with single clonal genomes sweeping through to dominate over other genomes in an otherwise recombining population. The epidemic population structure has also been shown in planktonic cyanobacterial blooms formed by Microcystis (Tanabe & Watanabe, 2011) and Planktothrix (D’Alelio et al.,2013), and may be a common population structure for cyanobacterial blooms (D’Alelio et al., 2013; Ribeiro et al., 2018).
The low nucleotide diversity and high frequencies of a single genome within mats suggests that dominance of a single genotype is the common process of mat formation. Recombination among Microcoleus genomes does not appear to be strong enough to prevent a dominant strain from arising and forming a mat. Similarly, in hot springs, Synechococcus genomes show that even with frequent recombination among strains (Rosen et al., 2015), population structure exists between mats (Becraft et al., 2020) and highly similar strains (>98% ANI) can possess distinct ecological traits (Olsen et al., 2015). How diversity persists in the face of recombination within Synechococcus populations remains debated (Melendrez et al., 2016; Rosen et al., 2018). However, the final outcome depends on the relative strengths of recombination and selection (Cordero & Polz, 2014; Wiedenbeck & Cohan, 2011). Despite the short (i.e., seasonal) lifespans of Microcoleus mats, recombination is evident, and enables, potentially, adaptation via the sharing of genetic material over larger spatiotemporal scales than the summer growing season and across the watershed.
Although a single strain dominated in most mats, at 12 sites we found multiple co‐occurring strains at higher frequencies (Figure 5 and Figure S6). These samples were from smaller subwatersheds and generally from thinner mats (with the exception of site PH2015_10S which was collected from a subwatershed of 951 km2). If these mats formed very recently, diversity could be in the process of being purged and eventually one strain could dominate. Alternatively, these mats could have stable coexistence of strains within the same mat, at least over the summer growing season, or population size may not be substantially reduced by scouring high‐flows in these smaller subwatersheds with more stable beds. We did not sample many mats over time to track changing strain dominance or diversity. However, mats from one site were sampled in 2015 (PH2015_03D and 03U) and 2017 (PH2017_05_CCC) and had co‐occurring strains in both years (99.7%–99.9% consensus ANI), which may represent a stable co‐existence. Additionally, Microcoleus mats in New Zealand sampled over 20 days contained two co‐occurring Microcoleus species sharing 91% ANI, and the relative abundance of these species was relatively constant over the sampling period (Tee et al., 2020). These observations should motivate further investigation to determine whether mat diversity is stable and that strains are not purged as mats grow and thicken.
Fluctuations in genotype abundance within Microcoleus mats over time have been reported previously. For example, the abundance of anatoxin producing and nonanatoxin producing species can vary over time within mats (Wood & Puddick, 2017). Understanding the population dynamics when anatoxin‐a producing and nontoxic Microcoleus species co‐occur within the same mat could improve predictions about when anatoxin‐a will create public health risks. While our work focused on spatial scales, future temporally‐scaled work focused on within‐mat population diversity over time as mats establish, grow, and senesce (McAllister et al., 2016), will help answer questions about how and when diversity accrues or is purged as mats go through their seasonal summer growth. The processes that enable minor strains to coexist at high frequencies in some samples (Figure S6) remain to be uncovered.
5. CONCLUSION
We conclude that Microcoleus population structures in river networks are well described by metapopulation and epidemic models. Our results show that although river flow is unidirectional downstream, the dendritic river network structure does not strongly isolate Microcoleus subpopulations, and cyanobacterial cells may readily disperse among subwatersheds. More research is needed, however, to determine if other Microcoleus species and bacterial populations inhabiting algal or cyanobacterial dominated biofilms in river networks have similar population structures to Microcoleus sp. 1, or if they are more spatially or environmentally constrained within a river network. Given weak dispersal limitation, our results support hypotheses that emphasize selection to explain biogeographical patterns in bacteria. Bacterial populations in rivers should shift relatively rapidly when changing environmental conditions select for different strains. Selection in rivers may be driven by dynamic flow regimes, as seasonal high‐flow events scour river beds, transform environmental conditions, reduce bacterial population sizes, and transport cells to new locations. Because of these disturbances, the distribution and allele frequencies of cyanobacterial populations may also be partially controlled by genetic drift and founder effects, as local subpopulations expand again after being depleted by scouring floods. Therefore, both selection and variable and random hydrologic processes may shape cyanobacterial population structures river networks.
AUTHOR CONTRIBUTIONS
Conceptualization, Keith Bouma‐Gregson, Mary E. Power, and Jillian F. Banfield; Investigation, Keith Bouma‐Gregson; Formal Analysis, Keith Bouma‐Gregson, Alexander Crits‐Christoph, Mathew R. Olm; Software, Alexander Crits‐Christoph and Mathew R. Olm; Visualization, Keith Bouma‐Gregson; Writing ‐ Original Draft, Keith Bouma‐Gregson and Jillian F. Banfield; Writing ‐ Review and Editing, Mary E. Power, Alexander Crits‐Christoph, Mathew R. Olm; Funding acquisition, Keith Bouma‐Gregson, Mary E. Power, and Jillian F. Banfield.
Supporting information
Fig S1‐S7
Table S1‐S7
ACKNOWLEDGEMENTS
We would like to thank the UC Angelo Coast Range Reserve and Reserve Manager Peter Steel for providing the facilities and resources that enabled this research. Samples could not have been collected and analyzed without the assistance of Keana A. Richmond. We would also like to thank volunteers of the Eel River Recovery Project and staff at the Ranjung Yeshe Gomde for providing access to sampling sites in the Eel River watershed. This work was supported through an NSF Division of Environmental Biology (NSF DEB‐1656009) and the Eel River Critical Zone Observatory (NSF CZO EAR‐1331940), and a US Environmental Protection Agency STAR Graduate Fellowship (91767101‐0). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant.
Bouma‐Gregson, K. , Crits‐Christoph, A. , Olm, M. R. , Power, M. E. , & Banfield, J. F. (2022). Microcoleus (Cyanobacteria) form watershed‐wide populations without strong gradients in population structure. Molecular Ecology, 31, 86–103. 10.1111/mec.16208
DATA AVAILABILITY STATEMENT
Sequencing reads (SRA) and metagenome‐assembled genomes (MAGs) have been deposited at NCBI under BioProject PRJNA448579 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA448579). Output files from bioinformatic programs (dRep, inStrain, popCOGenT, PopGenome and Roary,), as well as all other environmental and sampling data used in this publication are available on Dryad (doi: https://doi.org/10.6078/D1FX44) (Bouma‐Gregson et al., 2021). The analysis code for all figures is available at, https://github.com/keithbg/Microcoleus_Analysis.
REFERENCES
- Arevalo, P. , VanInsberghe, D. , Elsherbini, J. , Gore, J. , & Polz, M. F. (2019). A reverse ecology approach based on a biological definition of microbial populations. Cell, 178(4), 820–834.e14. 10.1016/j.cell.2019.06.033 [DOI] [PubMed] [Google Scholar]
- Battin, T. J. , Besemer, K. , Bengtsson, M. M. , Romani, A. M. , & Packmann, A. I. (2016). The ecology and biogeochemistry of stream biofilms. Nature Reviews Microbiology, 14(4), 251–263. 10.1038/nrmicro.2016.15 [DOI] [PubMed] [Google Scholar]
- Becraft, E. D. , Wood, J. M. , Cohan, F. M. , & Ward, D. M. (2020). Biogeography of American Northwest hot spring A/B′‐lineage Synechococcus populations. Frontiers in Microbiology, 11, 77. 10.3389/fmicb.2020.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda, L. , Poff, N. L. , Miller, D. , Dunne, T. , Reeves, G. , Pess, G. , & Pollock, M. (2004). The network dynamics hypothesis: How channel networks structure riverine habitats. BioScience, 54(5), 413. 10.1641/0006-3568(2004)054[0413:TNDHHC]2.0.CO;2 [DOI] [Google Scholar]
- Bendall, M. L. , Stevens, S. L. R. , Chan, L.‐K. , Malfatti, S. , Schwientek, P. , Tremblay, J. , Schackwitz, W. , Martin, J. , Pati, A. , Bushnell, B. , Froula, J. , Kang, D. , Tringe, S. G. , Bertilsson, S. , Moran, M. A. , Shade, A. , Newton, R. J. , McMahon, K. D. , & Malmstrom, R. R. (2016). Genome‐wide selective sweeps and gene‐specific sweeps in natural bacterial populations. The ISME Journal, 10(7), 1589–1601. 10.1038/ismej.2015.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bouma‐Gregson, K. , Crits‐Christoph, A. , Olm, M. R. , Power, M. E. , & Banfield, J. F. (2021). Microcoleus population genomics data in Eel River (California). Dryad. 10.6078/D1FX44 [DOI]
- Bouma‐Gregson, K. , Kudela, R. M. , & Power, M. E. (2018). Widespread anatoxin‐a detection in benthic cyanobacterial mats throughout a river network. PLoS One, 13(5), e0197669. 10.1371/journal.pone.0197669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma‐Gregson, K. , Olm, M. R. , Probst, A. J. , Anantharaman, K. , Power, M. E. , & Banfield, J. F. (2019). Impacts of microbial assemblage and environmental conditions on the distribution of anatoxin‐a producing cyanobacteria within a river network. The ISME Journal, 13(6), 1618–1634. 10.1038/s41396-019-0374-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasell, K. A. , Heath, M. W. , Ryan, K. G. , & Wood, S. A. (2015). Successional change in microbial communities of benthic Phormidium‐dominated biofilms. Microbial Ecology, 69(2), 254–266. 10.1007/s00248-014-0538-7 [DOI] [PubMed] [Google Scholar]
- Campbell Grant, E. H. , Lowe, W. H. , & Fagan, W. F. (2007). Living in the branches: Population dynamics and ecological processes in dendritic networks. Ecology Letters, 10(2), 165–175. 10.1111/j.1461-0248.2006.01007.x [DOI] [PubMed] [Google Scholar]
- Cohan, F. M. (2001). Bacterial species and speciation. Systematic Biology, 50(4), 513–524. 10.1080/10635150118398 [DOI] [PubMed] [Google Scholar]
- Conklin, K. Y. , Stancheva, R. , Otten, T. G. , Fadness, R. , Boyer, G. L. , Read, B. , Zhang, X. , & Sheath, R. G. (2020). Molecular and morphological characterization of a novel dihydroanatoxin‐a producing Microcoleus species (cyanobacteria) from the Russian River, California, USA. Harmful Algae, 93, 101767. 10.1016/j.hal.2020.101767 [DOI] [PubMed] [Google Scholar]
- Cordero, O. X. , & Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nature Reviews Microbiology, 12(4), 263–273. 10.1038/nrmicro3218 [DOI] [PubMed] [Google Scholar]
- Cowley, L. A. , Petersen, F. C. , Junges, R. , Jimson D. Jimenez, M. , Morrison, D. A. , & Hanage, W. P. (2018). Evolution via recombination: Cell‐to‐cell contact facilitates larger recombination events in Streptococcus pneumoniae. PLOS Genetics, 14(6), e1007410. 10.1371/journal.pgen.1007410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crits‐Christoph, A. , Olm, M. R. , Diamond, S. , Bouma‐Gregson, K. , & Banfield, J. F. (2020). Soil bacterial populations are shaped by recombination and gene‐specific selection across a grassland meadow. The ISME Journal, 14, 1834–1846. 10.1038/s41396-020-0655-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alelio, D. , Salmaso, N. , & Gandolfi, A. (2013). Frequent recombination shapes the epidemic population structure of Planktothrix (Cyanoprokaryota) in Italian subalpine lakes. Journal of Phycology, 49(6), 1107–1117. 10.1111/jpy.12116 [DOI] [PubMed] [Google Scholar]
- Dvořák, P. , Hašler, P. , & Poulíčková, A. (2012). Phylogeography of the Microcoleus vaginatus (Cyanobacteria) from three continents – A spatial and temporal characterization. PLoS One, 7(6), e40153. 10.1371/journal.pone.0040153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19), 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Ferrier, S. , Manion, G. , Elith, J. , & Richardson, K. (2007). Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Diversity and Distributions, 13(3), 252–264. 10.1111/j.1472-4642.2007.00341.x [DOI] [Google Scholar]
- Finlay, J. C. , Hood, J. M. , Limm, M. P. , Power, M. E. , Schade, J. D. , & Welter, J. R. (2011). Light‐mediated thresholds in stream‐water nutrient composition in a river network. Ecology, 92(1), 140–150. 10.1890/09-2243.1 [DOI] [PubMed] [Google Scholar]
- Fisher, R. A. (1930). The genetical theory of natural selection. The Clarendon Press. Retrieved from https://www.worldcat.org/title/genetical‐theory‐of‐natural‐selection/oclc/18500548 [Google Scholar]
- Garud, N. R. , & Pollard, K. S. (2020). Population genetics in the human microbiome. Trends in Genetics, 36(1), 53–67. 10.1016/j.tig.2019.10.010 [DOI] [PubMed] [Google Scholar]
- Hanage, W. P. (2016). Not so simple after all: Bacteria, their population genetics, and recombination. Cold Spring Harbor Perspectives in Biology, 8(7), a018069. 10.1101/cshperspect.a018069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanski, I. , & Gilpin, M. (1991). Metapopulation dynamics: Brief history and conceptual domain. Biological Journal of the Linnean Society, 42(1–2), 3–16. 10.1111/j.1095-8312.1991.tb00548.x [DOI] [Google Scholar]
- Hanson, C. A. , Fuhrman, J. A. , Horner‐Devine, M. C. , & Martiny, J. B. H. (2012). Beyond biogeographic patterns: Processes shaping the microbial landscape. Nature Reviews Microbiology, 10(7), 497–506. 10.1038/nrmicro2795 [DOI] [PubMed] [Google Scholar]
- Hudson, R. R. , & Kaplan, N. L. (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics, 111, 147–164. 10.1093/genetics/111.1.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R. , Slatkin, M. , & Maddison, W. P. (1992). Estimation of levels of gene flow from DNA sequence data. Genetics, 132(2), 583–589. 10.1093/genetics/132.2.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. R. , Inouye, B. D. , Johnson, M. T. J. , Underwood, N. , & Vellend, M. (2008). Ecological consequences of genetic diversity. Ecology Letters, 11(6), 609–623. 10.1111/j.1461-0248.2008.01179.x [DOI] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1). 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaak D. J., Wenger S. J., Peterson E. E., Ver Hoef J. M., Nagel D. E., Luce C. H., Hostetler S. W., Dunham J. B., Roper B. B., Wollrab S. P., Chandler G. L., Horan D. L., Parkes‐Payne Sharon (2017). The NorWeST Summer Stream Temperature Model and Scenarios for the Western U.S.: A Crowd‐Sourced Database and New Geospatial Tools Foster a User Community and Predict Broad Climate Warming of Rivers and Streams. Water Resources Research, 53(11), 9181–9205. 10.1002/2017wr020969 [DOI] [Google Scholar]
- Kanehisa, M. , Goto, S. , Sato, Y. , Kawashima, M. , Furumichi, M. , & Tanabe, M. (2014). Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Research, 42(D1), D199–D205. 10.1093/nar/gkt1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K. , & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, L. T. , Bouma‐Gregson, K. , Puddick, J. , Fadness, R. , Ryan, K. G. , Davis, T. W. , & Wood, S. A. (2019). Multiple cyanotoxin congeners produced by sub‐dominant cyanobacterial taxa in riverine cyanobacterial and algal mats. PLoS One, 14(12), e0220422. 10.1371/journal.pone.0220422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen, J. (1996). 16. Dispersal of freshwater algae—A review. Hydrobiologia, 336(1‐3), 151–157. 10.1007/BF00010829 [DOI] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levins, R. (1969). Some demographic and genetic consequences of environmental heterogeneity for biological control. Bulletin of the Entomological Society of America, 15(3), 237–240. 10.1093/besa/15.3.237 [DOI] [Google Scholar]
- Lock, J. , Kelsey, H. , Furlong, K. , & Woolace, A. (2006). Late Neogene and Quaternary landscape evolution of the northern California Coast Ranges: Evidence for Mendocino triple junction tectonics. GSA Bulletin, 118(9–10), 1232–1246. 10.1130/B25885.1 [DOI] [Google Scholar]
- Lodders, N. , Stackebrandt, E. , & Nubel, U. (2005). Frequent genetic recombination in natural populations of the marine cyanobacterium Microcoleus chthonoplastes . Environmental Microbiology, 7(3), 434–442. 10.1111/j.1462-2920.2005.00730.x [DOI] [PubMed] [Google Scholar]
- Martiny, J. B. H. , Bohannan, B. J. M. , Brown, J. H. , Colwell, R. K. , Fuhrman, J. A. , Green, J. L. , Horner‐Devine, M. C. , Kane, M. , Krumins, J. A. , Kuske, C. R. , Morin, P. J. , Naeem, S. , Øvreås, L. , Reysenbach, A.‐L. , Smith, V. H. , & Staley, J. T. (2006). Microbial biogeography: Putting microorganisms on the map. Nature Reviews Microbiology, 4(2), 102–112. 10.1038/nrmicro1341 [DOI] [PubMed] [Google Scholar]
- McAllister, T. G. , Wood, S. A. , & Hawes, I. (2016). The rise of toxic benthic Phormidium proliferations: A review of their taxonomy, distribution, toxin content and factors regulating prevalence and increased severity. Harmful Algae, 55, 282–294. 10.1016/j.hal.2016.04.002 [DOI] [PubMed] [Google Scholar]
- Melendrez, M. C. , Becraft, E. D. , Wood, J. M. , Olsen, M. T. , Bryant, D. A. , Heidelberg, J. F. , Rusch, D. B. , Cohan, F. M. , & Ward, D. M. (2016). Recombination does not hinder formation or detection of ecological species of Synechococcus inhabiting a hot spring cyanobacterial mat. Frontiers in Microbiology, 6. 10.3389/fmicb.2015.01540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin, S. , & Tolker‐Nielsen, T. (2003). Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Current Opinion in Biotechnology, 14(3), 255–261. 10.1016/S0958-1669(03)00036-3 [DOI] [PubMed] [Google Scholar]
- Morrissey, M. B. , & de Kerckhove, D. T. (2009). The maintenance of genetic variation due to asymmetric gene flow in dendritic metapopulations. The American Naturalist, 174(6), 875–889. 10.1086/648311 [DOI] [PubMed] [Google Scholar]
- Nei, M. , & Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences, 76(10), 5269–5273. 10.1073/pnas.76.10.5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , O'Hara, R. B. , Simpson, G. L. , Solymos, P. , Stevens, M. H. , Szoecs, E. , & Wagner, H. (2019). vegan: Community Ecology Package. Retrieved from https://CRAN.R‐project.org/package=vegan [Google Scholar]
- Olm, M. R. , Brown, C. T. , Brooks, B. , & Banfield, J. F. (2017). dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de‐replication. The ISME Journal, 11(12), 2864–2868. 10.1038/ismej.2017.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm, M. R. , Crits‐Christoph, A. , Bouma‐Gregson, K. , Firek, B. A. , Morowitz, M. J. , & Banfield, J. F. (2021). InStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nature Biotechnology, 39(6), 727–736. 10.1038/s41587-020-00797-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm, M. R. , Crits‐Christoph, A. , Diamond, S. , Lavy, A. , Matheus Carnevali, P. B. , & Banfield, J. F. (2020). Consistent metagenome‐derived metrics verify and delineate bacterial species boundaries. Msystems, 5(1), e00731–19. 10.1128/mSystems.00731-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, M. T. , Nowack, S. , Wood, J. M. , Becraft, E. D. , LaButti, K. , Lipzen, A. , Martin, J. , Schackwitz, W. S. , Rusch, D. B. , Cohan, F. M. , Bryant, D. A. , & Ward, D. M. (2015). The molecular dimension of microbial species: 3. Comparative genomics of Synechococcus strains with different light responses and in situ diel transcription patterns of associated putative ecotypes in the Mushroom Spring microbial mat. Frontiers in Microbiology, 6. 10.3389/fmicb.2015.00604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, A. J. , Cummins, C. A. , Hunt, M. , Wong, V. K. , Reuter, S. , Holden, M. T. G. , Fookes, M. , Falush, D. , Keane, J. A. , & Parkhill, J. (2015). Roary: Rapid large‐scale prokaryote pan genome analysis. Bioinformatics, 31(22), 3691–3693. 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo, A. , Dechesne, A. , Cordero, O. X. , & Smets, B. F. (2020). Evolutionary ecology of natural comammox Nitrospira populations. BioRxiv, 2020.09.24.311399. doi: 10.1101/2020.09.24.311399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y. , Leung, H. C. M. , Yiu, S. M. , & Chin, F. Y. L. (2012). IDBA‐UD: A de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics, 28(11), 1420–1428. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- Pfeifer, B. , Wittelsbürger, U. , Ramos‐Onsins, S. E. , & Lercher, M. J. (2014). Popgenome: An efficient swiss army knife for population genomic analyses in r. Molecular Biology and Evolution, 31(7), 1929–1936. 10.1093/molbev/msu136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polz, M. F. , Alm, E. J. , & Hanage, W. P. (2013). Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends in Genetics, 29(3), 170–175. 10.1016/j.tig.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power, M. E. , & Dietrich, W. E. (2002). Food webs in river networks. Ecological Research, 17(4), 451–471. 10.1046/j.1440-1703.2002.00503.x [DOI] [Google Scholar]
- Power, M. E. , Parker, M. S. , & Dietrich, W. E. (2008). Seasonal reassembly of a river food web: Floods, droughts, and the impacts of fish. Ecological Monographs, 78(2), 263–282. 10.1890/06-0902.1 [DOI] [Google Scholar]
- R Core Team (2019). R: A Language and Environment for Statistical Computing (Version 3.6.2). R Foundation for Statistical Computing. Retrieved from https://www.R‐project.org/ [Google Scholar]
- Raveh‐Sadka, T. , Thomas, B. C. , Singh, A. , Firek, B. , Brooks, B. , Castelle, C. J. , Sharon, I. , Baker, R. , Good, M. , Morowitz, M. J. , & Banfield, J. F. (2015). Gut bacteria are rarely shared by co‐hospitalized premature infants, regardless of necrotizing enterocolitis development. eLife, 4. 10.7554/eLife.05477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro, K. F. , Duarte, L. , & Crossetti, L. O. (2018). Everything is not everywhere: A tale on the biogeography of cyanobacteria. Hydrobiologia, 820(1), 23–48. 10.1007/s10750-018-3669-x [DOI] [Google Scholar]
- Richter, M. , & Rosselló‐Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences, 106(45), 19126–19131. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocap, G. , Larimer, F. W. , Lamerdin, J. , Malfatti, S. , Chain, P. , Ahlgren, N. A. , Arellano, A. , Coleman, M. , Hauser, L. , Hess, W. R. , Johnson, Z. I. , Land, M. , Lindell, D. , Post, A. F. , Regala, W. , Shah, M. , Shaw, S. L. , Steglich, C. , Sullivan, M. B. , … Chisholm, S. W. (2003). Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature, 424(6952), 1042–1047. 10.1038/nature01947 [DOI] [PubMed] [Google Scholar]
- Rosen, M. J. , Davison, M. , Bhaya, D. , & Fisher, D. S. (2015). Fine‐scale diversity and extensive recombination in a quasisexual bacterial population occupying a broad niche. Science, 348(6238), 1019–1023. 10.1126/science.aaa4456 [DOI] [PubMed] [Google Scholar]
- Rosen, M. J. , Davison, M. , Fisher, D. S. , & Bhaya, D. (2018). Probing the ecological and evolutionary history of a thermophilic cyanobacterial population via statistical properties of its microdiversity. PLoS One, 13(11), e0205396. 10.1371/journal.pone.0205396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall, M. , Axelsson, D. , & Bergstrom, C. T. (2009). The map equation. The European Physical Journal Special Topics, 178(1), 13–23. 10.1140/epjst/e2010-01179-1 [DOI] [Google Scholar]
- Sabart, M. , Pobel, D. , Latour, D. , Robin, J. , Salençon, M.‐J. , & Humbert, J.‐F. (2009). Spatiotemporal changes in the genetic diversity in French bloom‐forming populations of the toxic cyanobacterium, Microcystis aeruginosa . Environmental Microbiology Reports, 1(4), 263–272. 10.1111/j.1758-2229.2009.00042.x [DOI] [PubMed] [Google Scholar]
- Segawa, T. , Takeuchi, N. , Fujita, K. , Aizen, V. B. , Willerslev, E. , & Yonezawa, T. (2018). Demographic analysis of cyanobacteria based on the mutation rates estimated from an ancient ice core. Heredity, 120(6), 562–573. 10.1038/s41437-017-0040-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, B. J. (2016). How clonal are bacteria over time? Current Opinion in Microbiology, 31, 116–123. 10.1016/j.mib.2016.03.013 [DOI] [PubMed] [Google Scholar]
- Shapiro, B. J. (2018). What microbial population genomics has taught us about speciation. In Polz M. F., & Rajora O. P. (Eds.), Population genomics: Microorganisms (pp. 31–47). Springer International Publishing. 10.1007/13836_2018_10 [DOI] [Google Scholar]
- Slatkin, M. (1987). Gene flow and the geographic structure of natural populations. Science, 236(4803), 787–792. 10.1126/science.3576198 [DOI] [PubMed] [Google Scholar]
- Smith, J. M. , Smith, N. H. , O’Rourke, M. , & Spratt, B. G. (1993). How clonal are bacteria? Proceedings of the National Academy of Sciences, 90(10), 4384–4388. 10.1073/pnas.90.10.4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunecký, O. , Komárek, J. , Johansen, J. , Lukešová, A. , & Elster, J. (2013). Molecular and morphological criteria for revision of the genus Microcoleus (Oscillatoriales, Cyanobacteria). Journal of Phycology, 49(6), 1167–1180. 10.1111/jpy.12128 [DOI] [PubMed] [Google Scholar]
- Suzek, B. E. , Huang, H. , McGarvey, P. , Mazumder, R. , & Wu, C. H. (2007). UniRef: Comprehensive and non‐redundant UniProt reference clusters. Bioinformatics, 23(10), 1282–1288. 10.1093/bioinformatics/btm098 [DOI] [PubMed] [Google Scholar]
- Tanabe, Y. , Kasai, F. , & Watanabe, M. M. (2007). Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa . Microbiology, 153(11), 3695–3703. 10.1099/mic.0.2007/010645-0 [DOI] [PubMed] [Google Scholar]
- Tanabe, Y. , & Watanabe, M. M. (2011). Local expansion of a panmictic lineage of water bloom‐forming cyanobacterium Microcystis aeruginosa . PLoS One, 6(2), e17085. 10.1371/journal.pone.0017085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee, H. S. , Waite, D. , Payne, L. , Middleditch, M. , Wood, S. , & Handley, K. M. (2020). Tools for successful proliferation: Diverse strategies of nutrient acquisition by a benthic cyanobacterium. The ISME Journal. 10.1038/s41396-020-0676-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum, T. , Ferretti, P. , Maistrenko, O. M. , & Bork, P. (2020). Diversity within species: Interpreting strains in microbiomes. Nature Reviews Microbiology. 10.1038/s41579-020-0368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanInsberghe, D. , Arevalo, P. , Chien, D. , & Polz, M. F. (2020). How can microbial population genomics inform community ecology? Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1798), 20190253. 10.1098/rstb.2019.0253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese, N. J. , Mukherjee, S. , Ivanova, N. , Konstantinidis, K. T. , Mavrommatis, K. , Kyrpides, N. C. , & Pati, A. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Research, 43(14), 6761–6771. 10.1093/nar/gkv657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, M. , & Didelot, X. (2009). A comparison of homologous recombination rates in bacteria and archaea. The ISME Journal, 3(2), 199–208. 10.1038/ismej.2008.93 [DOI] [PubMed] [Google Scholar]
- Wang, I. J. , & Bradburd, G. S. (2014). Isolation by environment. Molecular Ecology, 23(23), 5649–5662. 10.1111/mec.12938 [DOI] [PubMed] [Google Scholar]
- Waples, R. S. , & Gaggiotti, O. (2006). What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular Ecology, 15(6), 1419–1439. 10.1111/j.1365-294X.2006.02890.x [DOI] [PubMed] [Google Scholar]
- Whitaker, R. J. , Grogan, D. W. , & Taylor, J. W. (2003). Geographic barriers isolate endemic populations of hyperthermophilic Archaea. Science, 301(5635), 976–978. 10.1126/science.1086909 [DOI] [PubMed] [Google Scholar]
- Wiedenbeck, J. , & Cohan, F. M. (2011). Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiology Reviews, 35(5), 957–976. 10.1111/j.1574-6976.2011.00292.x [DOI] [PubMed] [Google Scholar]
- Winemiller, K. O. , Flecker, A. S. , & Hoeinghaus, D. J. (2010). Patch dynamics and environmental heterogeneity in lotic ecosystems. Journal of the North American Benthological Society, 29(1), 84–99. 10.1899/08-048.1 [DOI] [Google Scholar]
- Wood, S. , & Puddick, J. (2017). The abundance of toxic genotypes is a key contributor to anatoxin variability in Phormidium‐dominated benthic mats. Marine Drugs, 15(10), 307. 10.3390/md15100307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. (1943). Isolation by distance. Genetics, 28(2), 114–138. 10.1093/genetics/28.2.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Bouma‐Gregson, K. , Crits‐Christoph, A. , Olm, M. R. , Power, M. E. , & Banfield, J. F. (2021). Microcoleus population genomics data in Eel River (California). Dryad. 10.6078/D1FX44 [DOI]
Supplementary Materials
Fig S1‐S7
Table S1‐S7
Data Availability Statement
Sequencing reads (SRA) and metagenome‐assembled genomes (MAGs) have been deposited at NCBI under BioProject PRJNA448579 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA448579). Output files from bioinformatic programs (dRep, inStrain, popCOGenT, PopGenome and Roary,), as well as all other environmental and sampling data used in this publication are available on Dryad (doi: https://doi.org/10.6078/D1FX44) (Bouma‐Gregson et al., 2021). The analysis code for all figures is available at, https://github.com/keithbg/Microcoleus_Analysis.