

Abstract
The Human Epidermal Growth Factor Receptor 2 (HER2) and HER3 form a potent pro-oncogenic heterocomplex upon binding of growth factor neuregulin-1β (NRG1β)1–3. The mechanism by which HER2 and HER3 interact remains unknown in the absence of any structures of the complex. We isolated the NRG1β-bound near full-length HER2/HER3 dimer and obtained a 2.9Å cryo-electron microscopy (cryo-EM) reconstruction of the extracellular domain module which reveals unexpected dynamics at the HER2/HER3 dimerization interface. We show that the dimerization arm of NRG1β-bound HER3 is unresolved because the apo HER2 monomer fails to undergo a ligand-induced conformational change needed to establish a HER3 dimerization arm binding pocket. In a second structure of an oncogenic extracellular domain mutant of HER2, S310F, we observe a compensatory interaction with the HER3 dimerization arm that stabilizes the dimerization interface. Both HER2/HER3 and HER2-S310F/HER3 retain the capacity to bind to the HER2-directed therapeutic antibody, trastuzumab, but the mutant complex does not bind to pertuzumab. Our 3.5Å structure of the HER2-S310F/HER3/NRG1β/trastuzumab Fragment antigen binding (Fab) complex reveals that the receptor dimer undergoes a conformational change to accommodate trastuzumab. Thus, like oncogenic mutations, therapeutics exploit the intrinsic dynamics of the HER2/HER3 heterodimer. The unique features of a singly liganded HER2/HER3 heterodimer underscore the allosteric sensing of ligand occupancy by the dimerization interface and explain why extracellular domains of HER2 do not homo-associate via a canonical active dimer interface.
To form an active complex, HER receptors dimerize upon binding to growth factor ligands. HER2, an orphan receptor, is dependent on heterodimerization with other HER receptors for activation. Its preferred dimerization partner, HER3, binds neuregulin (NRG) ligands1–3. Like HER2, HER3 is an obligate heterodimer partner because it has a catalytically impaired kinase domain (a pseudokinase) and cannot autophosphorylate4,5. In the absence of high-resolution structures of the HER2/HER3 heterocomplex, our current molecular understanding of its activation is inferred from structural studies of the related receptors, EGFR and HER4. Upon activation, HER3 pseudokinase is predicted to allosterically activate the HER2 kinase resulting in phosphorylation of unstructured receptor tails and downstream signaling6,7 (Fig. 1a). The extracellular domains of HER receptors, which are composed of four domains (I-IV), contain a key structural element of the dimerization interface called the dimerization arm (in domain II) (Fig. 1a). In the absence of a ligand, the dimerization arm is obscured in the inactive “tethered” conformation by an intramolecular interaction between domains II and IV8–10. Ligand binding breaks the tether and stabilizes an extended conformation that exposes the dimerization arm (Fig. 1a), which then contacts a pocket formed between domains I and III of the other monomer as seen in the highly symmetric doubly-liganded structures of EGFR and HER411–13.
Fig. 1 |. Overall structure of the HER2/HER3/NRG1β extracellular domain dimer complex.
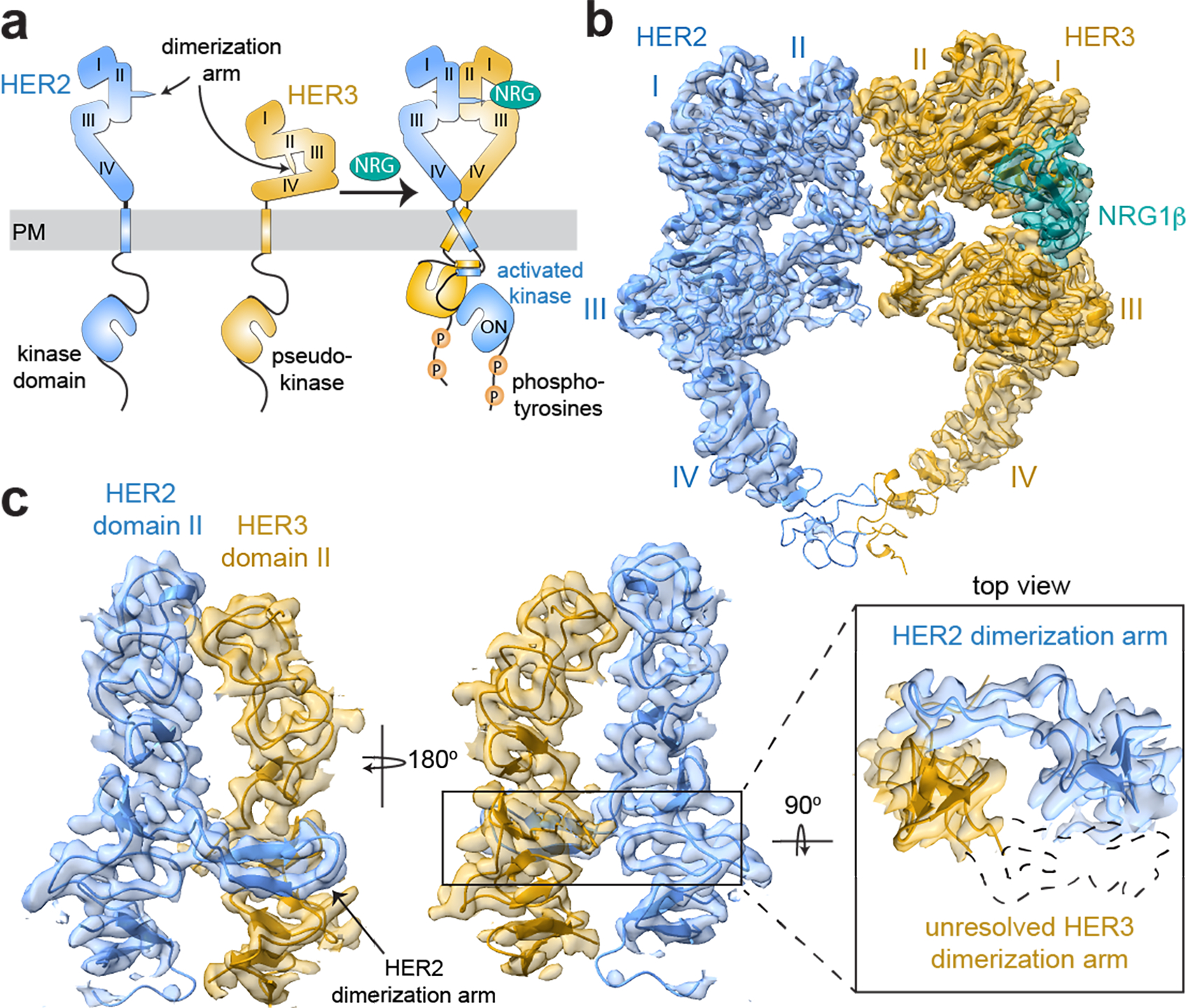
a, Cartoon schematic of the conformational changes that the inactive HER2 and HER3 monomers are predicted to undergo during heterodimerization in the presence of neuregulin (NRG) 1β. PM is plasma membrane. b, Cryo-EM map and the resulting structural model of the HER2/HER3/NRG1β extracellular domain complex, with HER2 shown in light blue, HER3 in gold and NRG1β in teal. Extracellular domains I-IV are marked on the structures. c, Zoomed-in view of the dimerization interface to illustrate lack of density for the HER3 dimerization arm. An outline of the expected location of the HER3 dimerization arm based on previous extracellular domain structures is shown as a dotted path in the top view.
No structures of ligand-bound HER3 have been solved, and, curiously, HER2 is found in an extended conformation in all current structures despite an unoccupied ligand-binding site14,15. In contrast to EGFR and HER4, however, homodimeric interactions mediated by the extended HER2 extracellular domain have not been observed even though its dimerization arm is constitutively exposed. It has been proposed that the existing extended HER2 extracellular domain structures represent a constitutively autoinhibited conformation16. Whether HER2 adopts a different, “active”, conformation when it binds other HER receptors is unknown. In this study, we used cryo-EM to elucidate structure of the active HER2/HER3 complex, which, unlike all structures of liganded EGFR and HER4, is stabilized by the binding of only one growth factor.
Structure of the HER2/HER3/NRG1β dimer
Previous biophysical studies with isolated extracellular domains of HER2 and HER3 did not yield stable heterodimeric complexes in the presence of NRG ligands17. We hypothesized that the transmembrane and intracellular kinase domains help stabilize extracellular domain interactions. Using near-full length HER2 and HER3 receptors that contain cancer-associated mutations in the HER3 pseudokinase to increase dimerization affinity (see Methods, Extended Data Fig. 1a–e)7,18, we obtained homogeneous detergent-solubilized NRG1β-bound HER2/HER3 complexes for cryo-EM (Extended Data Table 1). While the transmembrane and intracellular domains were essential for the stabilization of the HER2/HER3 heterodimer, they do not appear to be rigidly connected to the extracellular domains given the limited resolution of the full-length reconstruction (Extended Data Fig. 1f,g). However, focusing on the extracellular domains yielded a 2.9Å structure of the HER2/HER3/NRG1β complex (Fig. 1b, Extended Data Fig. 2, 3a,b).
In the HER2/HER3/NRG1β structure, the extracellular domains assemble in a “heart-shaped” dimer, resembling ligand-stabilized EGFR and HER4 homodimers11–13,19,20 (Extended Data Fig. 3e,f and Extended Data Fig. 4). EGFR extracellular domains form symmetric dimers when bound to high affinity ligands such as EGF and slightly asymmetric complexes when bound to lower affinity ligands such as epiregulin (EREG)11–13,19,20. The HER2/HER3/NRG1β complex is conformationally distinct and has the highest degree of asymmetry (Extended Data Fig. 3e,f and Extended Data Fig. 4). Membrane-proximal domains IV are visualized at lower resolution for both receptors (Extended Data Fig. 2, 3b) indicating their flexibility, as observed in EGFR and HER412,13,20,21. HER3 extracellular domain is in the previously uncharacterized extended conformation, with NRG1β clearly resolved (Fig. 1b and Extended Data Fig. 5a). Many contacts between NRG1β and HER3 are conserved in the NRG1β-bound HER4 complex (Extended Data Fig. 5a). Like HER3, HER2 adopts an extended conformation in the dimer. This conformation is almost identical to the extended state of HER2 previously seen in structures of its monomeric form (Extended Data Fig. 5b)14,15. Our structure provides evidence that this atypical extended state of HER2 is readily accommodated in the active HER2/HER3 dimer, contradicting the hypothesis that HER2 needs to undergo additional conformational changes in active dimers16.
The most striking feature of the HER2/HER3/NRG1β complex is the lack of resolvable density for the HER3 dimerization arm in domain II (Fig. 1c). In all other known structures, the dimerization arms of both HER receptors provide essential contributions to the dimerization interface. In contrast, only the HER2 dimerization arm is resolved in the HER2/HER3/NRG1β structure making sidechain-backbone and backbone-backbone interactions with HER3 (Fig. 1c and Extended Data Fig. 5c). Additional hydrogen bonds between domains II of HER2 and HER3 stabilize the dimer interface, which while fewer, are similarly positioned to those seen in extracellular crystal structures of other HER receptors (Extended Data Fig. 4 and 5c). Consequently, the total buried surface area at the HER2/HER3 heterodimer interface is reduced compared to that of other HER homodimers (Extended Data Fig. 5d). Our finding that the HER3 dimerization arm need not be engaged with HER2 in the HER2/HER3 heterodimer is supported by the observation that replacing the HER3 dimerization arm with a non-specific linker (HER3-GS-arm) did not change the extent of HER2 and HER3 interactions (Extended Data Fig. 5e,f) and NRG1β−dependent receptor stimulation in cells (Extended Data Fig. 5g,h).
Allosteric control of the dimerization arm
The absence of the HER3 dimerization arm in our structure might be a consequence of the lack of a suitable binding pocket in HER2 that could engage the arm. In the symmetric, EGF-bound EGFR extracellular domain dimers each protomer cradles the dimerization arm of the other protomer via an enclosed binding pocket. In comparison, the domain I/III interface in HER2 does not fully close to form a binding pocket (Fig. 2a). We noticed that a similarly open dimerization arm-binding pocket, albeit to a lesser extent, is present in one of the monomers in the asymmetric structure of the EREG-bound EGFR ectodomain dimer19 (Fig. 2a). While both dimerization arms were resolved in the EGFR/EREG structure, the dimerization arm engaged with the partially open binding pocket was more dynamic (Fig. 2b, Extended Data Fig. 4). Thus, the disengaged dimerization arm in the EGFR/EREG structure represents an intermediate state between the missing dimerization arm of HER3, and the fully engaged dimerization arms in the symmetric EGFR/EGF ectodomain dimer (Fig. 2b).
Fig. 2 |. Analysis of liganded HER receptor states reveals an allosteric mechanism of dimerization arm engagement.
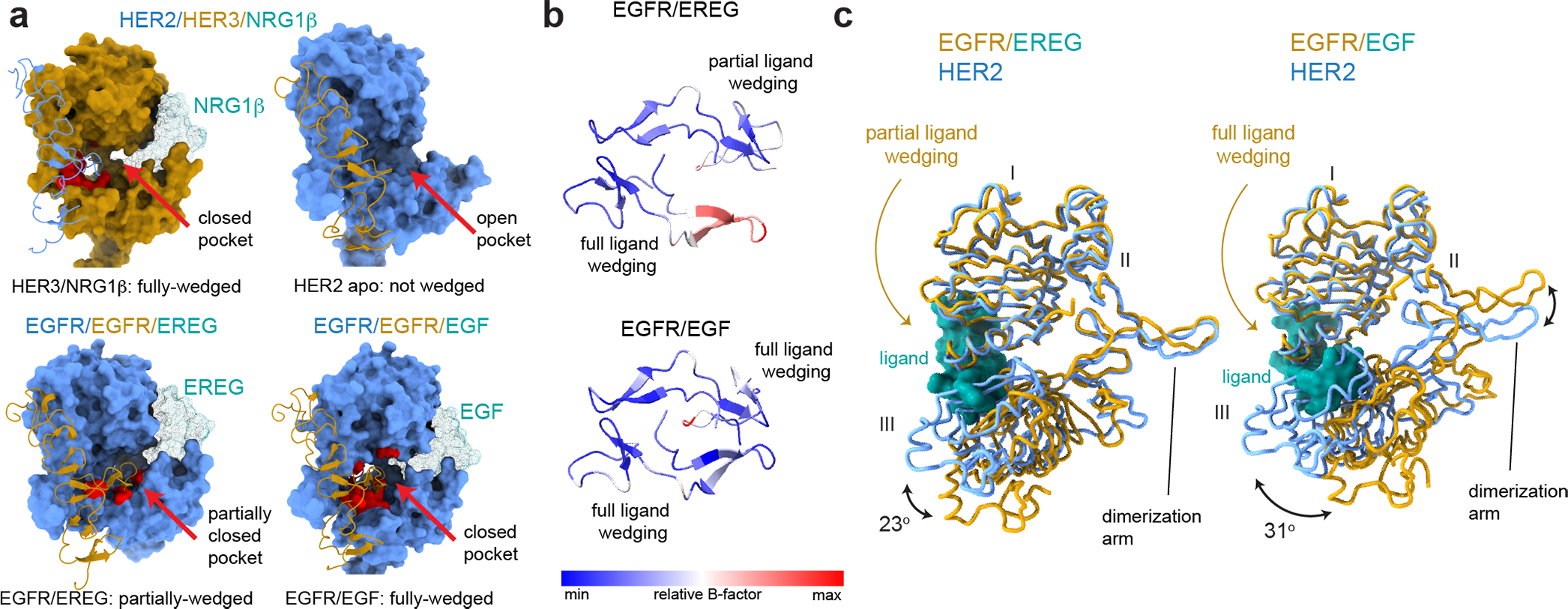
a, Left top panel - closed dimerization arm-binding pocket in HER3 engages the HER2 dimerization arm in the HER2/HER3/NRG1β structure. Right top panel - an open dimerization arm-binding pocket in the ligand-free HER2 does not engage HER3 dimerization arm in the same structure. Left bottom panel – a partially closed dimerization arm-binding pocket in the monomer in the EGFR/EREG structure in which the ligand (EREG) is partially-wedged (PDB ID 5WB7). Right bottom panel – closed binding pocket in EGFR engages a fully-wedged EGF ligand in the EGFR/EGF dimer structure (PDB ID 3NJP). Residues within 4Å of the dimerization arm are shown in red. b, Top view of dimerization arms in the asymmetrically ligand-wedged EGFR/EREG and symmetrically ligand-wedged EGFR/EGF crystal structures indicating different values of B-factors (PDB IDs 5WB7 and 3NJP, respectively). c, Detailed view of domains I-III in the EGFR/EREG or EGFR/EGF crystal structures aligned on HER2 domain I in the HER2/HER3/NRG1β structure. The EGFR monomer in which the EREG ligand is only partially-wedged is shown.
Our analysis points to a previously unappreciated allosteric connection between the dimerization arm-binding pocket and the ligand binding site. Based on the relative rotation between domains I and III induced by ligand, two modes of ligand binding to HER receptors have been described19. The fully-wedged conformation (~31° rotation), seen in the symmetric EGFR/EGF dimer structure, and the partially-wedged conformation (~23° rotation), seen in EGFR/EREG. These differences directly correlate with the state of the dimerization arm-binding pocket (Fig. 2c). A fully-wedged ligand results in a closed, high affinity dimerization arm-binding pocket, and is also seen in the NRG1β-bound HER3 monomer in our structure, providing a high affinity pocket for the HER2 dimerization arm. Partial ligand-wedging results in partial closure of the pocket, increasing dynamics of the dimerization arm, as seen in one monomer of the EGFR/EREG dimer19. In ligand-less HER2, domains I and III do not undergo a relative rotation, and consequently the dimerization arm-binding pocket is fully open and does not engage the HER3 dimerization arm (Fig. 2a, c).
HER2 S310F stabilizes the heterodimer
The most frequent oncogenic missense mutation in HER2 (S310F/Y) found primarily in cancers without HER2 overexpression is localized in the dimerization arm-binding pocket of domain II22,23,24. We reconstituted a nearly full-length HER2-S310F/HER3/NRG1β complex in vitro and observed that it was more stable than heterocomplexes containing wild type HER2 (Extended Data Fig. 6a–c) A cryo-EM reconstruction of the extracellular module of the HER2-S310F/HER3/NRG1β complex at 3.1Å resolution (Fig. 3a, Extended Data Fig. 3c–d, 7, Extended Data Table 1) shows no conformational changes in the HER2 monomer (RMSD: 0.6 Å), but remarkably, an entirely resolved HER3 dimerization arm (Fig. 3a and Extended Data Fig. 6d). The dimerization arm interaction is driven by π-π stacking between the introduced phenylalanine at position 310 (HER2-F310) and HER3-Y265 in addition to a few polar contacts (Fig. 3b). The stabilized HER3 dimerization arm increases the total buried surface area at the HER2/HER3 interface (domains I-III) from 1,977 Å2 in the wild type complex to 3,378 Å2 in the mutant complex, which is even higher than the respective interfaces in structures of the symmetric ligand-bound EGFR and HER4 homodimers (Extended Data Fig. 5d)13,20. We predict that the same mechanism is employed by the HER2 S310Y mutation, which is assumed to form an analogous π-π stacking interaction with HER3 Y265. Thus, the most common HER2 oncogenic mutations act by stabilizing interactions with the HER3 dimerization arm and compensate for the inability of HER2 to undergo a needed rotation between domains I and III.
Fig. 3 |. HER2 oncogenic mutation S310F stabilizes the dimerization arm of HER3.
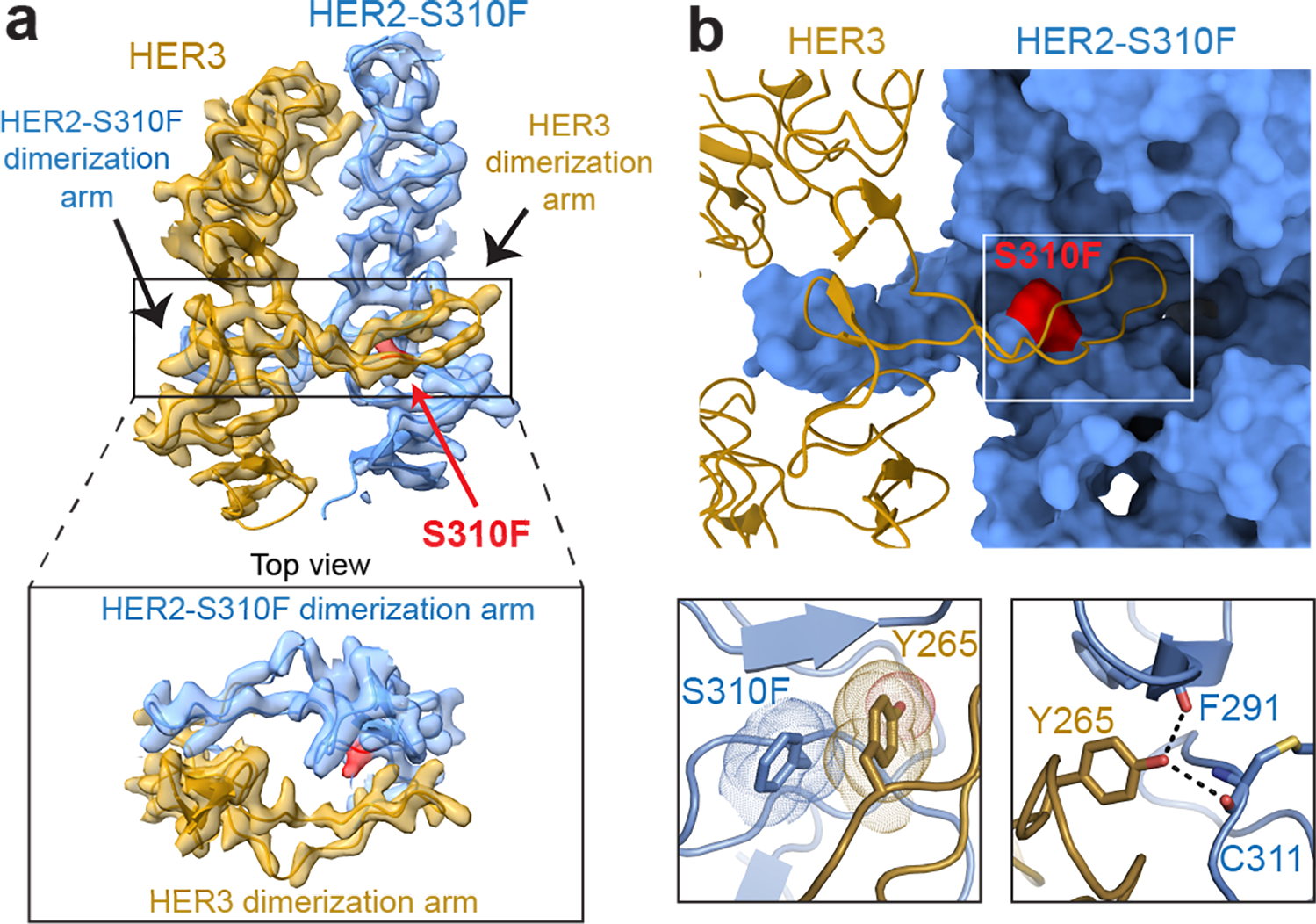
a, Cryo-EM map and model zoomed in on domains II depict a resolved HER3 dimerization arm in the HER2-S310F/HER3/NRG1β complex. Inset shows a top-down view of the HER2 and HER3 dimerization arms. The HER2-S310F mutation is shown in red. b, Top panel, HER2-S310F monomer, shown in surface representation, pins the HER3 dimerization arm, shown as cartoon, in the HER2 dimerization arm-binding pocket despite its inability to close in the ligand-less HER2. Bottom left panel, HER2-S310F forms a π-π interaction with HER3 Y265 that stabilizes the dimerization arm. Bottom right panel, polar contacts (dotted lines) between HER3 Y265 and the backbone residues of HER2 - F291 and C311.
Binding of trastuzumab and pertuzumab
The clinically-approved HER2-targeting monoclonal antibodies, trastuzumab and pertuzumab, target domains IV and II, respectively14,25. Their ability to bind the HER2/HER3 complex has been debated. We found that neither antibody interfered with HER2/HER3 heterodimerization and formed complexes with receptor dimers (Fig. 4a). The cryo-EM reconstruction of the trastuzumab Fab bound to the HER2-S310F/HER3/NRG1β complex at 3.5Å resolution (Fig. 4b, Extended Data Fig. 7, Extended Data Table 1) shows that the flexible domain IV allows the heterodimer to accommodate trastuzumab. Domain IV of HER2 moves away from HER3 as a rigid body with the variable domains of trastuzumab. In addition, HER3 rotates in relation to HER2 to resolve a steric clash between HER3 domain III and the constant domains of the trastuzumab Fab (Extended Data Fig. 8a–b, Fig. 4c).
Fig. 4 |. The HER2/HER3/NRG1β structure accommodates trastuzumab binding.
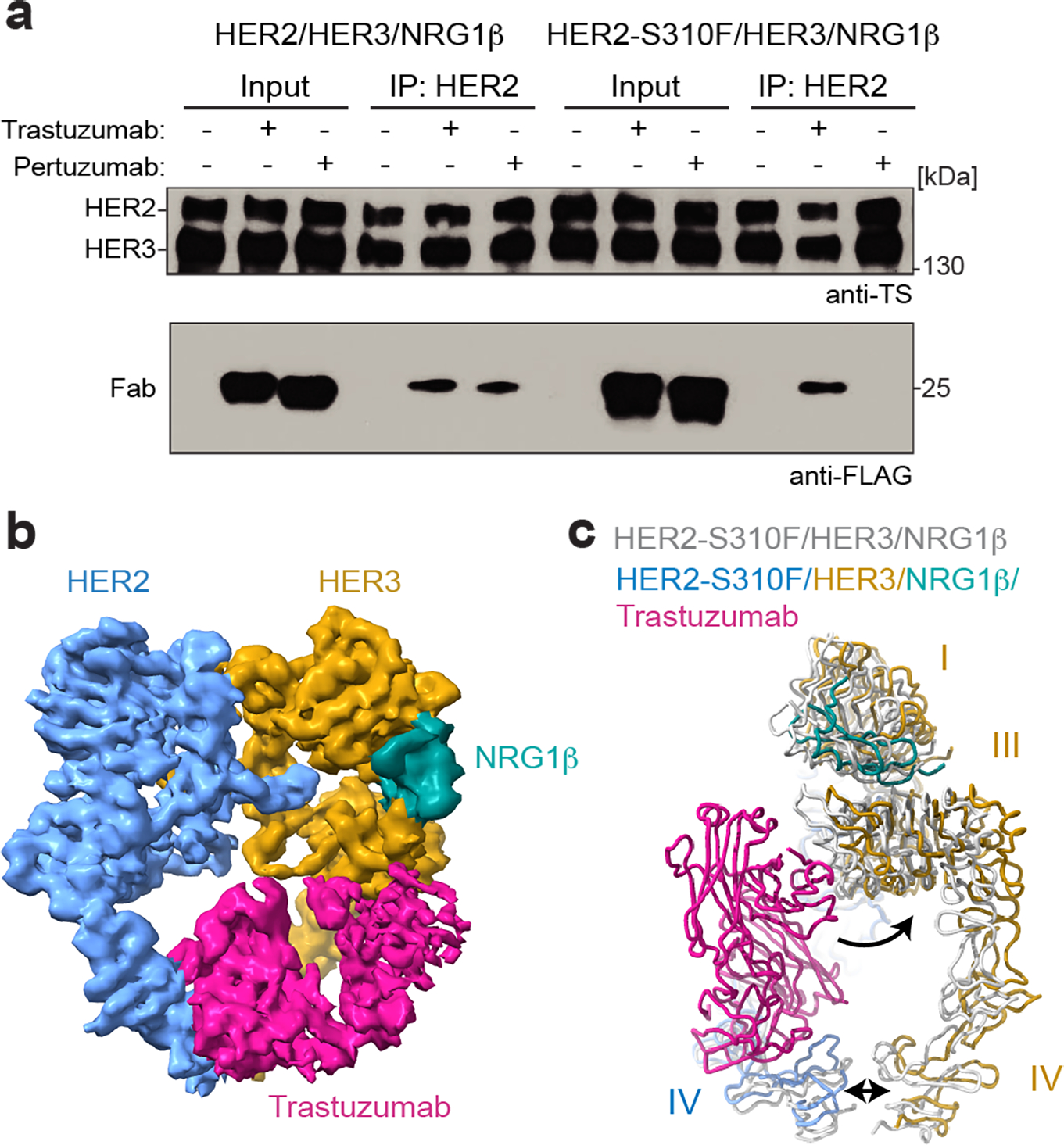
a, Representative Western blot of the purified HER2/HER3 heterodimer pulldowns in the presence of a two-fold molar excess of pertuzumab and trastuzumab FLAG-tagged Fabs. TS – Twin Strep tag (present on both HER2 and HER3) was detected with Strep-Tactin® HRP conjugate. The blot is representative of n=4 independent biological replicates. For gel source data, see Supplementary Figure 1. b, 5Å lowpass filtered density of the HER2-S310F/HER3/NRG1β heterocomplex bound to trastuzumab Fab. c, Ribbon overlay of the HER2-S310F/HER3/NRG1β heterocomplex with (multi-color) and without (light grey) trastuzumab Fab. The Fab pushes HER3 back relative to HER2 (curved arrow) and spreads domains IV further apart (double-headed arrow).
Pertuzumab is positioned to directly block the HER2/HER3 dimerization interface (Extended Data Fig. 8c), and it effectively inhibits NRG1β-dependent HER2/HER3 activation in cells (Extended Data Fig. 8d). Our observation that pertuzumab binds but does not disrupt the purified HER2/HER3/NRG1β complex can be rationalized by the presence of the dimerization-stabilizing intracellular HER3 oncogenic mutations (Extended Data Fig. 1)7,18. Full-length HER2/HER3 heterodimer harboring these mutations is constitutively active and no longer inhibited by pertuzumab in cells (Extended Data Fig. 8d). Our inability to obtain a high-resolution reconstruction of the pertuzumab-bound HER2/HER3/NRG1β complex supports the notion that pertuzumab increases the dynamics of the extracellular domains separated by the bound Fab, while the intracellular domains remain associated.
Neither trastuzumab nor pertuzumab affected the assembly of the mutant HER2-S310F/HER3/NRG1β complex (Fig. 4a) and neither Fab interfered with HER2-S310F/HER3 activation (Extended Data Fig. 8d). Importantly, pertuzumab no longer bound to the HER2-S310F/HER3 complex, possibly due to the direct interference of the mutation with Fab binding (Extended Data Fig. 8e–f), or because the HER2 epitope recognized by pertuzumab is occluded in the mutant complex due to the enhanced dimerization interface (Fig. 3).
Discussion
The HER2/HER3/NRG1β structure captures two obligate heterodimeric HER receptors in an active state stabilized by binding of one ligand, revealing the extended state of HER3. Previously seen only in Drosophila EGFR26, the singly liganded dimer was suggested to be more stable than a doubly liganded one. To the contrary, the HER2/HER3 complex is dynamic, with the HER3 dimerization arm not even engaged at the dimer interface. A destabilized dimerization arm interface might be a general property of the HER2-containing complexes as observed in molecular dynamics simulations of the putative EGFR-HER2 ECD dimer27. This property explains why solution and structural studies of the extracellular domains of HER2 have been unable to identify homodimers despite HER2 being always in an extended conformation with the dimerization arm exposed14,15,28. Because the ligand-free extracellular domain of HER2 does not undergo a necessary rotation between domains I and III, it cannot establish a binding pocket for the partner’s dimerization arm. Therefore, the extracellular domains of HER2 are effectively protected from homo-association via the canonical “heart-shaped” dimer. This intrinsic autoinhibition is likely overridden in cancers where HER2 is hyperactivated due to massive overexpression.
The HER2/HER3 extracellular dimer dynamics likely have important implications for modulating receptor activity. The allosteric coupling between receptor domains across the membrane was described in EGFR homodimers, with recent findings linking the strength of EGFR activity with the degree of separation between domains IV21. We show that the intrinsic dynamics of the HER2/HER3 interface is exploited by the most common HER2 oncogenic mutation in the extracellular domain resulting in aberrant HER2 kinase activation. The interface dynamics likely also accounts for the ability of wild-type HER2/HER3/NRG1β heterocomplex to accommodate both trastuzumab and pertuzumab binding in vitro, although we show that pertuzumab may be less effective than trastuzumab at targeting cancers driven by HER2-S310F/Y. The HER2/HER3 structures presented here provide a platform for the rational design of therapeutics and biomarkers specific to the unique features of the active complex between these two receptors.
MATERIALS AND METHODS
NRG1β expression and purification.
An HRV-3C cleavable Thyrodoxin A (TrxA) was fused to the EGF-like domain of NRG1β (residues 177–236, NRG1 isoform 6 (UniProt: Q02297-6; numbering includes propeptide) with C-terminal Flag and 6x-Histidine tags and subsequently cloned into a p32A vector. The TrxA-3C-NRG1β-Flag-6xHis construct was transformed into Origami E. coli, grown at 37 °C in Terrific Broth in large scale culture until an OD of ~1.0 – 1.5, and induced with 1 mM Isopropyl β-d-1-thiogalactopyranoside (IPTG) overnight at room temperature. Cells were harvested the next day, pelleted, flash frozen, and stored until purification. For the purification, cells were resuspended in NRG lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors (eOComplete, Roche)) and sonicated until thoroughly lysed. Lysate was then clarified through ultracentrifugation, syringe filtered through 0.44 μm filters and incubated with Ni-NTA resin overnight. The beads were washed by gravity through 20 column volumes (CVs) of NRG wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl) containing 20 mM imidazole, then 10 CVs of NRG wash buffer containing 50 mM imidazole, and finally eluted with 3 CVs of NRG wash buffer containing 300 mM imidazole. Imidazole in the eluate was reduced < 30 mM over a 10K concentrator and subsequent dilution with NRG wash buffer. The eluate was cleaved overnight with 3C protease at 4 °C. To remove cleaved TrxA, the elution was again applied on equilibrated Ni-NTA resin, incubated, washed, and eluted as described previously. The elution containing NRG1β was concentrated with a 3K cutoff and applied on an S200 10/300 increase column (GE Healthcare). Protein from the major peak was stored in aliquots at −80 °C for subsequent receptor purifications. Yields ranged from 5–10 mg/L of culture.
Trastuzumab and pertuzumab Fab expression and purification.
The Fragment antigen binding (Fab) heavy chain and light chain sequences encoding trastuzumab and pertuzumab were inserted into the pSVF4 vector. For each Fab, a 1x Flag tag was inserted after the heavy chain constant domain and a 6x-Histidine tag was inserted after the light chain constant domain. Constructs were transformed into BL21 gold E. coli and scaled up to 6L in 2xYT media under Ampicillin antibiotic selection. Cultures were grown at 37 °C until OD of 0.8 – 1.0, induced with 1 mM IPTG for 6 hrs at 37 °C, harvested by centrifugation, and stored at −80 °C. Cells were resuspended in 100 ml of lysis buffer (20 mM sodium phosphate pH 7.4, 500 mM NaCl with DNase I (Roche), 0.5 mM MgCl2 and 1 mM PMSF). Cells were sonicated until fully lysed and resulting lysate was incubated at 65 °C for 30 minutes. The lysate was cooled on ice and spun down at 40,000 rpm for 60 minutes at 4 °C. The clarified lysate was loaded onto a Protein A column equilibrated in Buffer A (20 mM sodium phosphate pH 7.4, 500 mM NaCl), washed with 10 column volumes of Buffer A, and eluted in 100 mM acetic acid by fractionation into neutralizing buffer containing 20 mM Tris-HCl pH 9.0, 150 mM NaCl. Immediately following Protein A purification, eluent was concentrated and loaded onto a Superdex 200 10/300 Increase column (GE Healthcare) equilibrated in a buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl. Fractions corresponding to Fab were pooled and stored at 4 °C until needed.
Near full-length receptor expression.
Human HER2 with a C-terminal tail truncation (Δ1030–1255) followed by maltose binding protein (MBP) and twin-strep tags was cloned into pFastBac with a CMV promoter. A single point mutation in the HER2 kinase domain, G778D, which confers Hsp90 independence29, was introduced to improve yields. Human HER3 with a C-terminal tail truncation (Δ1023 – 1342) followed by a twin-strep tag was cloned in pFastBac with a CMV promoter. Two oncogenic mutations that stabilize the asymmetric kinase domain dimer, Q809R and E928G, were introduced to further improve heterodimer yields7,30. The HER2 and HER3 constructs were each transfected into 60 ml of Expi293 mammalian suspension (Life Technologies) cells cultured to 4×106 cells/ml at 37 °C, 8% CO2 following the standard expression protocol. 10 mM canertinib in DMSO was added 16 – 18 hrs post-transfection to a final concentration of 10 μM along with ExpiFectamine 293 Transfection Kit enahncers 1 and 2. Cells were harvested, flash frozen, and stored at −80 °C 24 hrs after the addition of enhancers. The same procedure was followed for HER2 in the presence and absence of S310F mutation.
Heterodimer purification.
Cells were resuspended with the lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM NaVO3, 1 mM NaF, 1 mM EDTA, protease inhibitors (eOComplete, Roche), DNAse I, and 1% DDM (Inalco)) and lysed for 2 hrs by gentle rocking at 4 °C. Lysate was clarified by centrifugation at 4,000g for 20 min at 4 °C. Purified EGF-like domain of NRG1β was incubated with G1 Flag Resin (Genscript) for 1 hr at 4 °C and serially washed 3x with Buffer A (50 mM Tris-HCl pH 7.4, 150 mM NaCl). Clarified HER2 and HER3 receptor lysates were mixed and incubated O/N in batch mode at 4 °C with NRG1β Flag beads. NRG1β Flag beads were serially 3x washed with Buffer A containing 0.5 mM DDM (Anatrace) and eluted with Buffer A containing 0.5 mM DDM and 250 μg/ml of Flag peptide (SinoBiological). The eluate was then applied to amylose resin in batch mode for 2 hrs, washed serially 3x with Buffer B (50 mM HEPES pH 7.4, 150 mM NaCl) containing 0.5 mM DDM and eluted with amylose elution buffer (Buffer B containing 0.5 mM DDM and 10 mM maltose) O/N at 4 °C. The eluate was concentrated to 0.4 ml with a 100-kDa concentrator (Amicon) and mildly crosslinked in 0.2% glutaraldehyde for 40 min on ice. The sample was loaded on a Superose6 10/300 (GE Healthcare) pre-equilibrated with Buffer A containing 0.5 mM DDM and 0.5 ml fractions were collected. Peak fractions corresponding to heterodimer sample were pooled, concentrated down to ~20 μl with a 100-kDa concentrator, and flash frozen for grid preparation. The same purification protocol was followed for HER2-S310F/HER3 heterocomplex. The HER2-S310F/HER3 + trastuzumab Fab complex sample was generated by incubating a 5x molar excess of Fab with the heterocomplex prior to crosslinking, gel filtration, and imaging.
Electron microscopy sample preparation and imaging.
For negative stain EM, fractions corresponding to heterodimer were applied to negatively glow-discharged carbon coated copper grids, stained with 0.75% uranyl-formate, and imaged on an FEI-Tecnai T12 with an 4k CCD camera (Gatan). The resulting negative stain micrographs were assessed for particle homogeneity and particle density. This analysis was used to determine the target concentration for cryo-EM with graphene oxide grids which typically require 2–5x negative stain concentrations.
Use of graphene oxide coated holey carbon grids enabled solving high-resolution cryo-EM structures at low receptor concentrations31,32. For cryo-EM, 3 μl of purified and concentrated heterodimer sample (as empirically determined by negative stain) was applied to graphene-oxide coated Quantifoil R1.2/1.3 300 mesh Au holey-carbon grids prepared as previously described31, blotted using a Vitrobot Mark IV (FEI) and plunge frozen in liquid ethane (no glow discharge, 30 second wait time, room temperature, 100% humidity, 4–8 seconds blot time, 0 blot force).
Grids were imaged on a 300-keV Titan Krios (FEI) with a K3 direct electron detector (Gatan) and a BioQuantum energy filter (Gatan) using SerialEM v3.8.6 and Digital Micrograph v3.31.2359.033. Data for HER2/HER3/NRG1β and HER2-S310F/HER3/NRG1β were collected in super-resolution mode at a physical pixel size of 0.835Å/pix with a dose rate of 8.0 e− per pixel per second (operated in CDS mode). Images were recorded with a 5.9 s exposure over 118 frames with a dose rate of 0.57 e−/Å2/frame. Data for HER2-S310F/HER3/NRG1β with trastuzumab Fab were collected in super-resolution pixel mode at a physical pixel size of 0.834Å/pix with a dose rate of 8.0 e− per pixel per second. Images were recorded in 6 s exposures over 120 subframes with a dose rate of 0.55 e−/Å2/frame. During data collection the quality of micrographs was estimated in real time using Scipion34 running MotionCor235 and CTFFIND436.
Image processing and 3D reconstruction.
Raw movies were corrected for motion and radiation damage with MotionCor235 and the resulting sums were imported in CryoSPARC237. To account for the reduced GO coverage with detergent sample, all datasets underwent strict micrograph curation with a final yield of ~40–50% of the collected micrograph stack. Micrograph CTF parameters were estimated with the patch CTF job in CryoSPARC2. Particles were initially picked with blob picking, extracted, and 2D-classified. Particles were then template picked with low-pass filtered (to 25Å) 2D class averages, the resulting picks were extracted with 2x Fourier cropping and subjected to iterative rounds of ab initio and heterogeneous refinements (please see processing flow charts). Once reasonable reconstructions were obtained (as judged by the FSC (Fourier Shell Correlation) curves shape), unbinned particles were re-extracted and run through subsequent rounds of heterogeneous and non-uniform refinements to achieve reconstructions with the highest resolution. The final reconstruction of HER2/HER3/NRG1β used for model building included 123,173 particles with C1 symmetry and resulted in an overall resolution of 2.9Å by Gold Standard-Fourier Shell Correlation (GS-FSC) cutoff of 0.143. The final reconstruction of HER2-S310F/HER3/NRG1β used for model building included 99,755 particles with C1 symmetry and attained a GS-FSC resolution of 3.1Å.
HER2-S310F/HER3/NRG1β/Trastruzumab Fab data set was initially processed as above in CryoSPARC2. To address incomplete Fab occupancy, a stack containing 330,000 particles was imported into RELION338 and subclassified through skip-align classification. Particles classified into reconstructions without Fab density were removed from the particle stack. A final particle stack from RELION3 containing 243,376 particles was re-imported into CryoSPARC2 and subjected to non-uniform refinement to produce a reconstruction with a final resolution of 3.45Å.
Each map was assessed for local and directional resolutions through ResMap39 and 3DFSC40 server respectively. For all reconstructions, extracellular domains I-III achieved the highest local resolutions (~3Å) while that of domain IV varied from 4–8Å suggesting that a high degree of flexibility exists closer to the transmembrane domains. All reconstructions achieved a sphericity > 0.9.
To recover micelle and sub-micelle densities, 2x binned particle stacks for HER2/HER3/NRG1β and HER2-S310F/HER3/NRG1β were imported into RELION3 and further 3D-classified. Particles classified into 3D classes with substantial micelle densities were re-extracted with shifted coordinates (PyEM41) on the center of the micelle and refined. Resulting reconstructions featured convincing sub-micelle density with volumes large enough to accommodate transmembrane domains and kinases.
Model refinement and validation.
An initial model was generated by docking HER2 (PDB ID: 1N8Z) with a homology model of liganded HER3 from its closest homolog, HER4, in SwissProt (PDB ID: 3U7U) into the HER2-S310F/HER3/NRG1β map. Given the substantial variation in domain IV local resolution, domain IV was truncated from the model and domains I-III were iteratively rebuilt in Rosetta42. Top scoring models were selected and further edited in Coot43 and ISOLDE44. Domains IV were then placed into the model (HER2 PDB ID: 6OGE, HER3 PDB ID: 1M6B) and fit into the density with a FastRelax Rosetta protocol in torsion space, in Rosetta. For HER2-S310F/HER3/NRG1β + trastuzumab Fab, the Fab (PDB ID: 6OGE) was torsion relaxed with the HER2-S310F/HER3/NRG1β model in Rosetta. Per atom B-factors were assigned in Rosetta indicating the local quality of the map around that atom.
For glycan building, glycans were initially manually placed into the density in Chimera and then were refined with the Rosetta glycan refinement protocol45. Model statistics were routinely assessed in PHENIX MotionCor235,46 and glycan geometries were cross validated in Privateer MKIII47. All structures were deposited into the EMDB and Protein Data Bank (PDB).
Small-scale heterodimer pulldowns and Western Blot.
Tagged HER2 (WT, S310F, S310Y) and HER3 (WT, GS-arm with Q261-E273 replaced with a repeating glycine-serine sequence) expression constructs were co-transfected into 2 ml cultures of Expi293 cells as described above. For this assay, the same expression constructs described above were used, but in a wild type background without stabilizing intracellular mutations. Cell pellets were lysed in 1 ml lysis buffer and clarified lysates were subjected to NRG-pulldown and eluted in 250 μl Flag elution buffer as described above. The extent of heterodimer formation was assessed by Western blot. Samples were boiled in SDS-loading buffer at 95 °C for 5 min, run on 4–15% acrylamide gels and transferred onto PVDF membranes. Membranes were blocked in 3% BSA in TBS with 0.1% Tween (TBST) overnight and incubated with Strep-Tactin® HRP conjugate (IBA, 1:5000) in TBST + 3% BSA for 1 hr at room temperature. Membranes were washed 5x with TBST and signal was detected using ECL Western Blotting detection reagent (GE) or ECL prime (VWR).
Trastuzumab and pertuzumab pulldown assay.
Wild-type HER2 and S310F HER2 heterodimeric complexes with HER3 were expressed and purified as described above until elution from amylose resin with the exception that amylose wash and elution buffers contained 50 mM Tris-HCl pH 7.4 instead of 50 mM HEPES pH 7.4. Eluates were concentrated to 100 μl and maltose was removed via buffer exchange using 7 MWCO Zeba spin desalting columns. 70 nM heterodimer solutions were each incubated with 1 and 10x molar ratios for 30 min and bound to amylose resin overnight. Complexes were eluted as described above and complex formation with trastuzumab and pertuzumab were assessed by Western Blot.
Cell-based assays
Full-length human HER2 tagged with C-terminal 3xFLAG tag and untagged full-length human HER3 in pCDNA4to expression vectors were kindly provided by Mark Moasser. GS-arm mutations encompassing residues (Q261-E273, replaced by GSGSGSGSGSGSG), the S310F, E928G and Q809R mutations were introduced into respective vectors by site-directed mutagenesis. 0.15 million COS-7 cells were seeded into each well of a 6-well plate and transfected with 800 ng total DNA (400 ng per receptor plasmid or 400 ng + 400 ng empty vector for single receptor controls) using the Lipofectime p3000 transfection kit (ThermoFisher Scientific). 24 h post transfection, cells were rinsed in PBS, serum starved for 16 h and, if applicable, stimulated with 10 nM NRG1β (PeproTech) for 10 min at 37° C. For experiments involving Fab fragments, 50 ug/ml trastuzumab or pertuzumab were added 1 h prior to stimulation and were present during NRG1β stimulation. Cells were then cooled on ice for 5 min, washed in ice-cold PBS two times and lysed in 400 ul RIPA buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, Roche Complete protease inhibitors, DNAse, 1 mM sodium orthovanadate, 1 mM sodium fluoride in PBS) per 6-well on ice for 30 min. Lysates were transferred into 1.5 ml microcentrifuge tubes, spun at 15,000 × g for 3 min and supernatants were transferred into fresh tubes with SDS loading dye. HER2, phospho-HER3 (pY1289) and HER3 levels in lysates were determined by Western Blot. Antibodies used for detection were: rabbit anti-HER3 (Cell Signaling, D22C5, 1:1000), rabbit anti-phospho-HER3 recognizing phosphorylated tyrosine position Y1289 (Cell Signaling, 21D3, 1:1000), rabbit anti-HER2 (Cell Signaling, 29D8, 1:1000), anti-rabbit IgG HRP-linked antibody (Cell Signaling, 1:10 000).
Statistical Analysis
Unpaired two tailed t-tests were performed with GraphPad Prism where statistics are reported.
Extended Data
Extended Data Fig. 1 |. Purification, characterization, and reconstruction of the near full-length HER2/HER3/NRG1β heterocomplex.
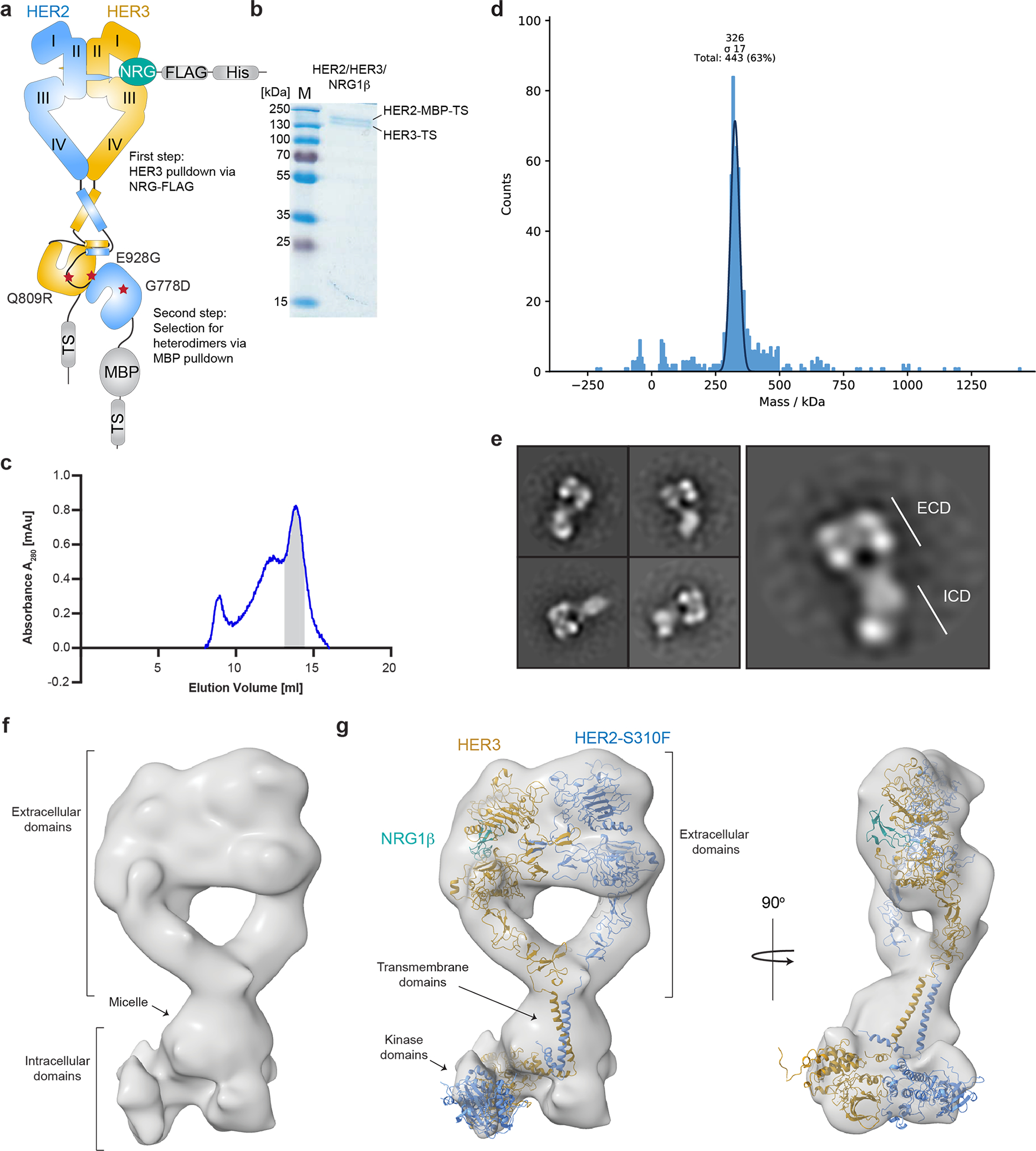
a, Schematic summary of the HER2/HER3/NRG1β complex purification strategy. b, A representative Coomassie-stained SDS-PAGE gel analysis of the HER2/HER3/NRG1β complex after purification showing bands corresponding to the HER2 and HER3 receptors. The gel is representative of three independent experiments. c, Representative size exclusion chromatography profile of the HER2/HER3/NRG1β complex resolved on a Superose6 10/300 Increase column (GE Healthcare). The major peak at ~14 ml elution volume is marked grey and corresponds to the fractions used for structural studies. We routinely obtained a 1–2 mAu peaks for the complex preparations from 120 ml of mammalian culture, which was sufficient for the structural studies. d, Mass photometry of the peak sample indicates that the majority of particles have an average mass of ~326 kDa with a standard deviation of 17 kDa. The mass is consistent with that of the HER2/HER3/NRG1β complex (predicted ~280 kDa without accounting for micelle mass, ~340 kDa assuming a ~60 kDa DDM micelle mass). e, Representative negative stain electron microscopy 2D class averages of sample from HER2/HER3/NRG1β complex fractions (ECDs (extracellular domains), ICDs (intracellular domains)). f, Near full-length reconstruction of HER2-S310F/HER3/NRG1β after particle recentering with center of mass at the micelle in RELION from a ~45,000 particle stack. Rod-shaped density consistent with the asymmetric kinase domain dimer is visible below the micelle. g, The reconstruction accommodates models of the HER2-S310F/HER3/NRG1β extracellular domains from this study, two helical transmembrane domains and juxtamembrane-A (JM-A) segments from HER2 (PDB ID 2N2A), and kinases arranged in the asymmetric dimer (homology model generated based on structures of the EGFR kinase from PDB ID 3KEX and PDB ID 3PP0). HER2 is colored in blue, HER3 in golden yellow, and NRG1β in teal.
Extended Data Fig. 2 |. Resolution estimation, map quality and the workflow for processing of the HER2/HER3/NRG1β dataset.
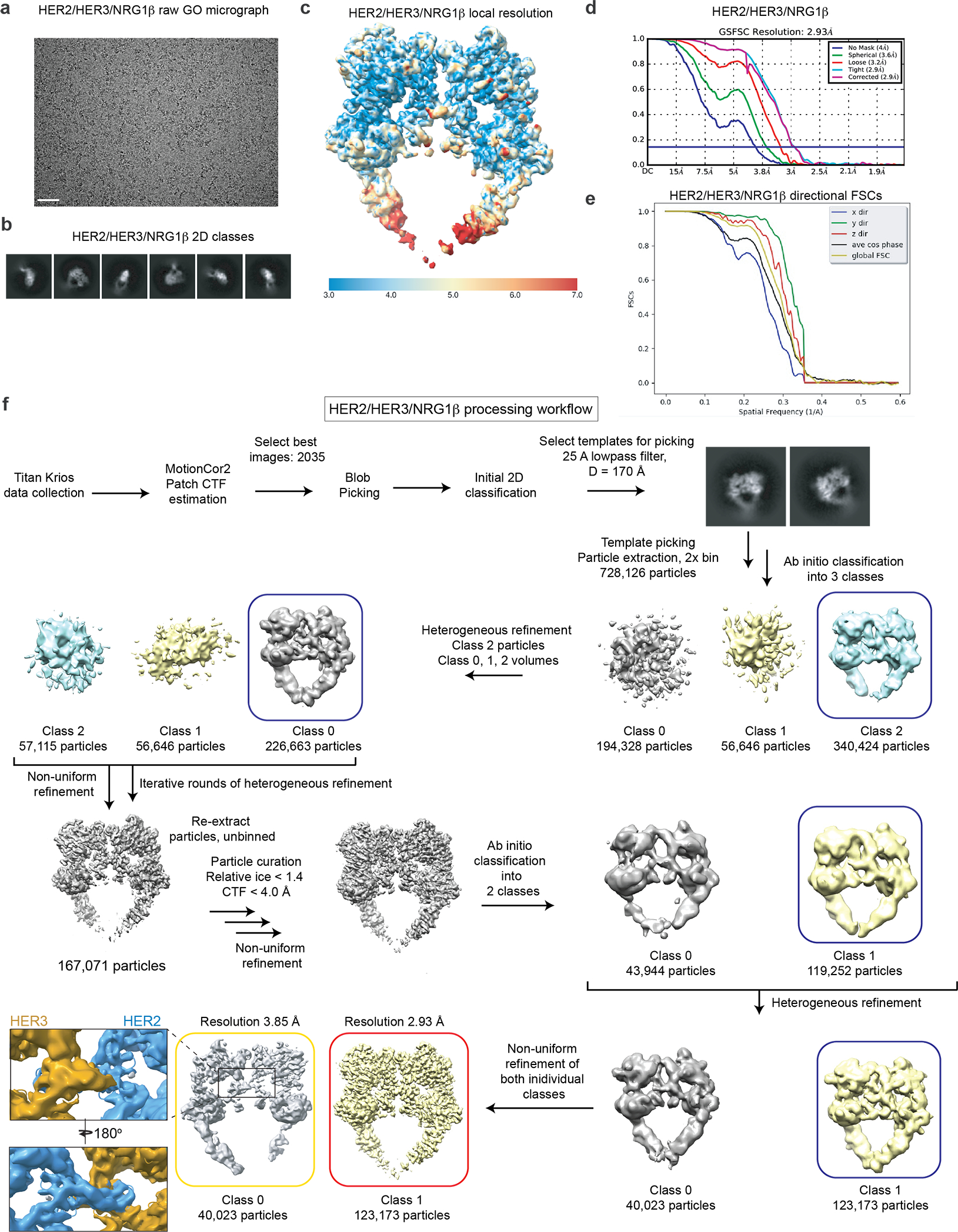
a, Representative micrograph of the HER2/HER3/NRG1β sample on graphene oxide grids, from a dataset with 2035 GO-containing micrographs. The scale bar corresponds to 0.5 μm. b, Example 2D cryo-EM class averages. c, Local resolution estimation from ResMap. d, Gold Standard Fourier Shell Correlation (GSFSC) of the final map used for model building from CryoSPARC2 with a reported resolution of 2.93 Å. e, Directional FSCs calculated by 3DFSC server. Map sphericity was calculated to be 0.927. f, Workflow for processing the HER2/HER3/NRG1β dataset. Blue box indicates model and associated particle stack used for downstream processing. The final model is indicated with a red box. The map in the yellow box represents a slightly different low-occupancy species. The insets show that the HER3 dimerization arm is still poorly resolved in that class.
Extended Data Fig. 3 |. Q-score analysis of the cryo-EM maps and a structural comparison of the HER2/HER3/NRG1β heterocomplex with crystal structures of previously reported HER receptor structures.
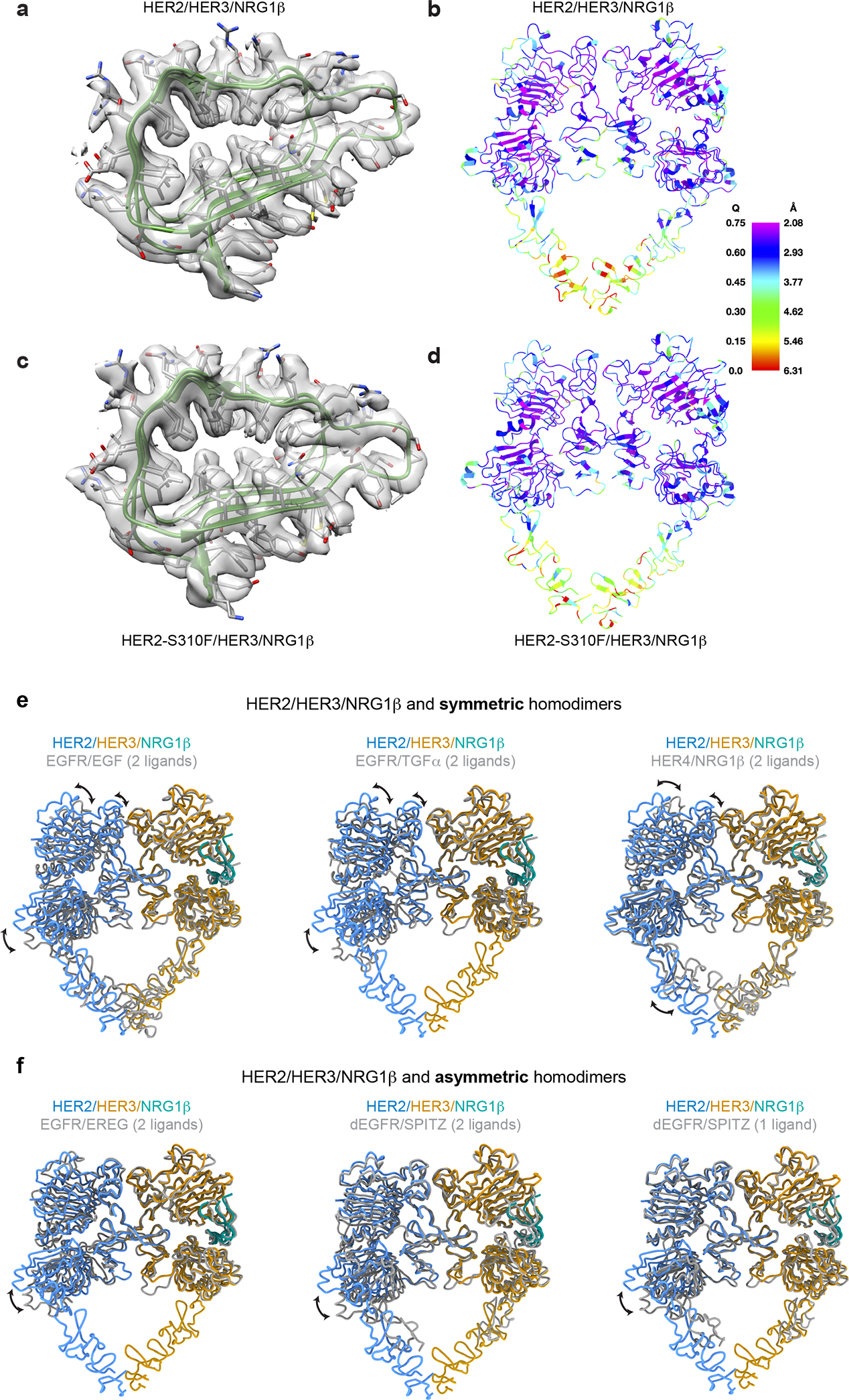
a, Zoomed-in view of the cryo-EM density and model of WT HER2/HER3 (residues 63–150 of HER3) showing features appropriate for the reported resolution. b, WT HER2/HER3 model colored by estimated per residue Q-score ranging from 0 (red) to 0.75 (blue). The color bar shows corresponding estimated resolution in Å for each Q-score. Expected Q-score for 2.9Å map is 0.604. c, Zoomed-in view of the cryo-EM density and model of HER2 S310F/HER3 (residues 63–150 of HER3) showing features appropriate for the reported resolution. d, HER2-S310F/HER3 model colored by estimated Q-score with the same scale as in b. Expected Q-score for 3.1Å structure is 0.569. e, Overlay of the HER2/HER3/NRG1β heterocomplexes with symmetric structures of EGFR/EGF (PDB ID 3NJP), EGFR/TGFα (PDB ID 1MOX) and HER4/NRG1β (PDB ID 3U7U). f, Overlay of the HER2/HER3/NRG1β heterocomplexes with asymmetric structures of EGFR/EREG (PDB ID 5WB7), doubly liganded dEGFR/SPITZ (PDB ID 3LTF) and singly liganded dEGFR/SPITZ (PDB ID 3LTG). All structures were aligned on HER3. Differences between the heterodimer and the homodimers are primarily appreciated in overlays on the HER2 monomer. The heterodimer more closely resembles asymmetric homodimers than symmetric homodimers but reflects a unique conformation that is not seen in previous structures.
Extended Data Fig. 4 |. Comparison of the domain II dimerization interface between HER2/HER3/NRG1β complex domain with crystal structures of previously reported HER receptor homodimers.
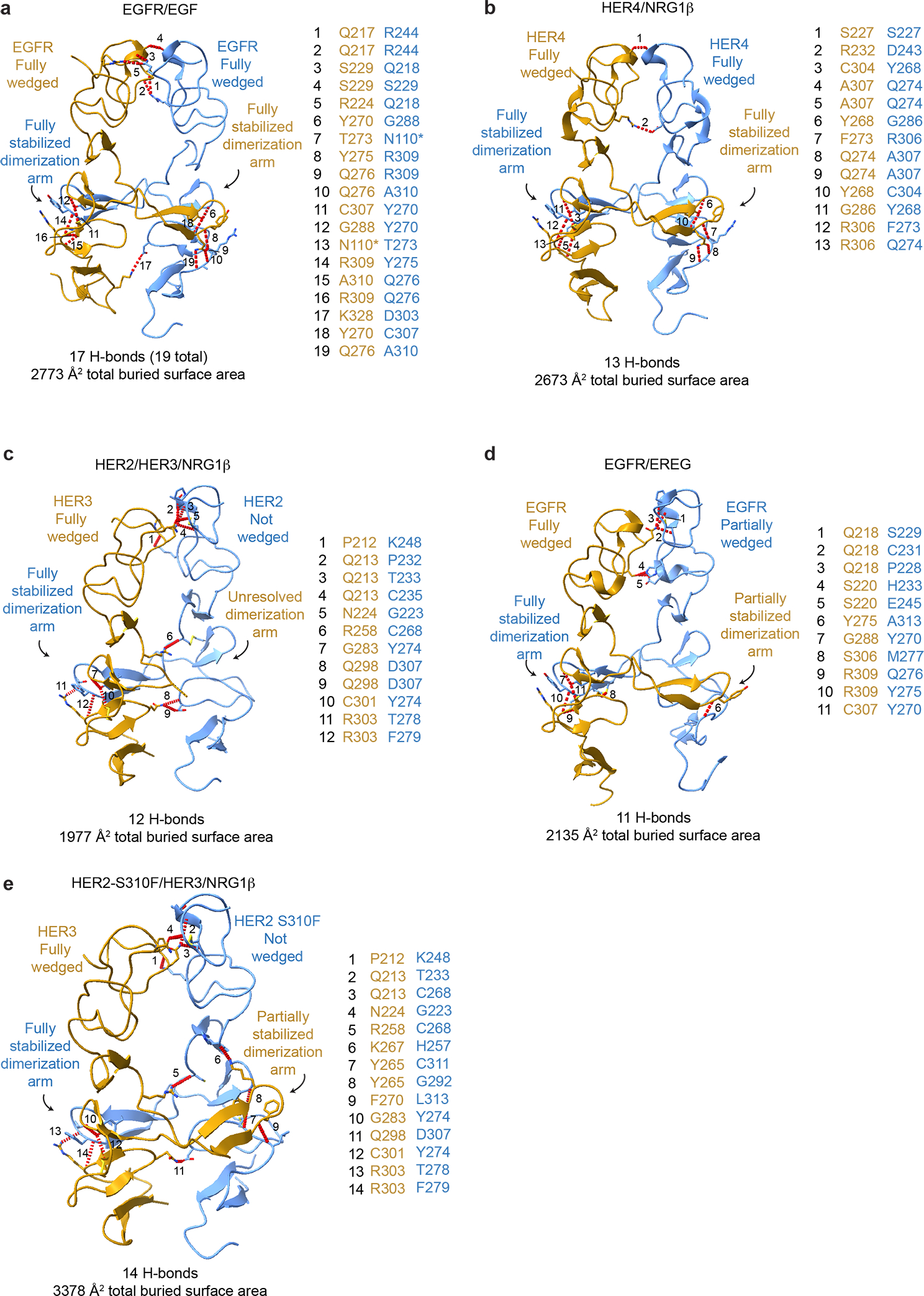
a-e, The domain II interfaces of select HER receptor dimers are shown with the number of hydrogen bonds and the total buried surface area between domains I-III indicated below. Domain IV was excluded from this analysis because it is not resolved in all structures. Hydrogen bonds are shown as red dotted lines, highlighting more substantial interfaces for symmetric homodimers (EGFR/EGF (PDB ID 3NJP), HER4/NRG1β (PDB ID 3U7U) than asymmetric dimers (HER2/HER3/NRG1β, EGFR/EREG (PDB ID 5WB7)), with the exception of the mutant HER2-S310F/HER3/NRG1β heterocomplex in which the mutation stabilizes the domain II interface. All interface hydrogen bonds are formed within domain II, except for an additional hydrogen bond in EGFR with domain III which is not shown here and marked (*) in table.
Extended Data Fig. 5 |. In-depth structural and functional analysis of the NRG1β binding-site and the HER2/HER3 dimerization interface.
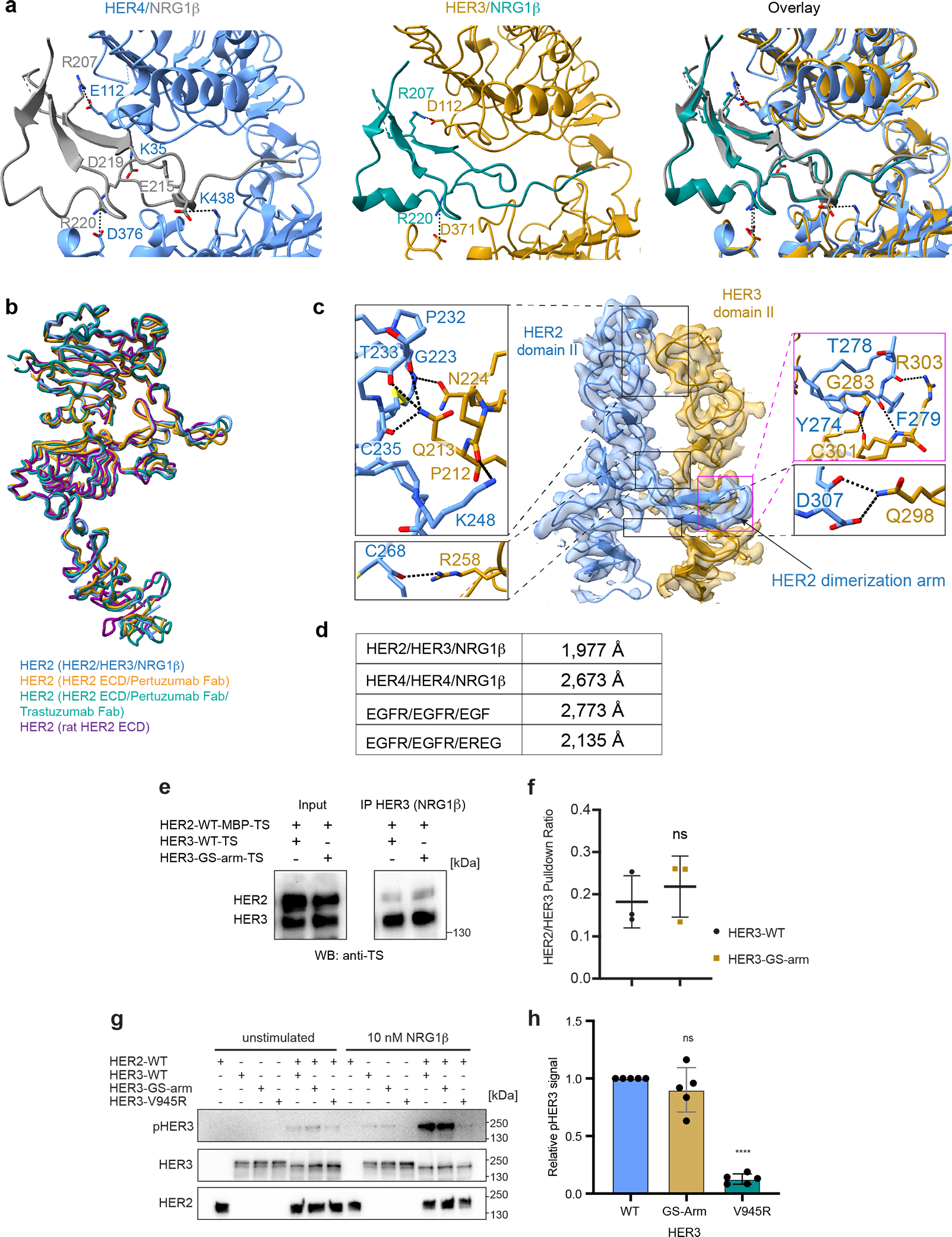
a, Left, HER4 bound to NRG1β (PDB ID 3U7U) with salt-bridge interactions highlighted. Middle, HER3 bound to NRG1β in our structure with salt bridge interactions highlighted. NRG1β engages with HER3 primarily through an extensive interaction network at its C-terminus (total buried surface area: 2,803 Å2) stabilized by salt bridges between R207 (NRG1β) and D112 in HER3 domain I, and R220 (NRG1β) and D371 in HER3 domain III, bringing domains I and III into close proximity. Right, overlay between the two structures shows that the overall orientation of the ligand and some salt-bridge interactions are shared between HER3 and HER4, but overall HER3 forms fewer salt bridges with NRG1β than HER4. b, Structure of HER2 in our HER2/HER3/NRG1β dimer structure is overlayed with the crystal structure of the HER2 extracellular domain bound to trastuzumab Fab (PDB ID 1N8Z), the cryo-EM structure of the HER2 extracellular domain bound to pertuzumab and trastuzumab Fabs (PDB ID 6OGE) and the crystal structure of the rat HER2 extracellular domain (PDB ID 1N8Y). The structures are almost identical with root mean squared deviations (RMSDs) as following: 1.01 Å (1N8Z), 0.74 Å (1N8Y), 0.97 Å (6OGE). Minor conformational changes are observed in the dimerization arm and domain IV. c, Detailed view of the dimerization interface between domains II of HER2 and HER3 in the HER2/HER3/NRG1β structure with all polar contacts between receptors highlighted in the boxes to the left and to the right. d, Calculated buried surface area at the dimerization interface for HER2/HER3/NRG1β complex and the known structures of the dimeric HER extracellular domain complexes. The following structures were used for the analysis: EGFR/EGF (PDB ID 3NJP), EGFR/EREG (PDB ID 5WB7), HER4/NRG1β (PDB ID 3U7U). e, Representative Western Blot analysis of the HER2 and HER3 (wild-type HER3 and HER3 GS-arm) constructs co-transfected in the EXPI293 cells and pulled-down via NRG1β immobilized on FLAG beads. TS – Twin Strep tag (present on both HER2 and HER3). Receptors were detected with the Strep-Tactin® HRP conjugate (anti-TS). For gel source data, see Supplementary Figure 1. f, Quantification of data shown in panel e. Values are presented as mean values +/− SD of mean intensity ratios of HER2 over HER3 for each blot in n=3 independent biological replicates. The HER2/HER3 pulldown ratios are 0.18 +/− 0.06 for HER3-WT and 0.22 +/− 0.07 for HER3-GS-Arm complexes. Significance was determined via unpaired, two-tailed t-test via GraphPad Prism. p=0.5463, t=0.6582, df=4. g, Anti-phospho-HER3 (pHER3, Y1289), anti-HER3 and anti-HER2 Western Blot analysis of lysates from COS7 cells transfected with indicated full-length receptors (wild type or carrying the indicated mutations), serum-starved and stimulated with 10 nM NRG1β for 10 min. HER3-V945R is a negative control due to the presence of mutation in the active kinase dimer interface. For gel source data, see Supplementary Figure 1. h, Quantification of data shown in panel g. Values are presented as mean values +/− SD of pHER3 intensities of the mutant relative to wild-type HER3 for each blot in n=5 independent biological replicates. The mean intensity values are 0.90 +/− 0.19 for the HER3-GS-Arm and 0.13 +/− 0.04 for HER3-V945R relative to the WT with defined values of 1. Significance was determined via unpaired, two-tailed t-test via GraphPad Prism. HER3 GS-arm vs HER3: p=0.2887, t=1.136, df=8, HER3 vs HER3 V945R: p<0.0001, t=43.89, df=8.
Extended Data Fig. 6 |. The oncogenic HER2-S310F/Y mutations increase the yields of the HER2/HER3/NRG1β complex purification.
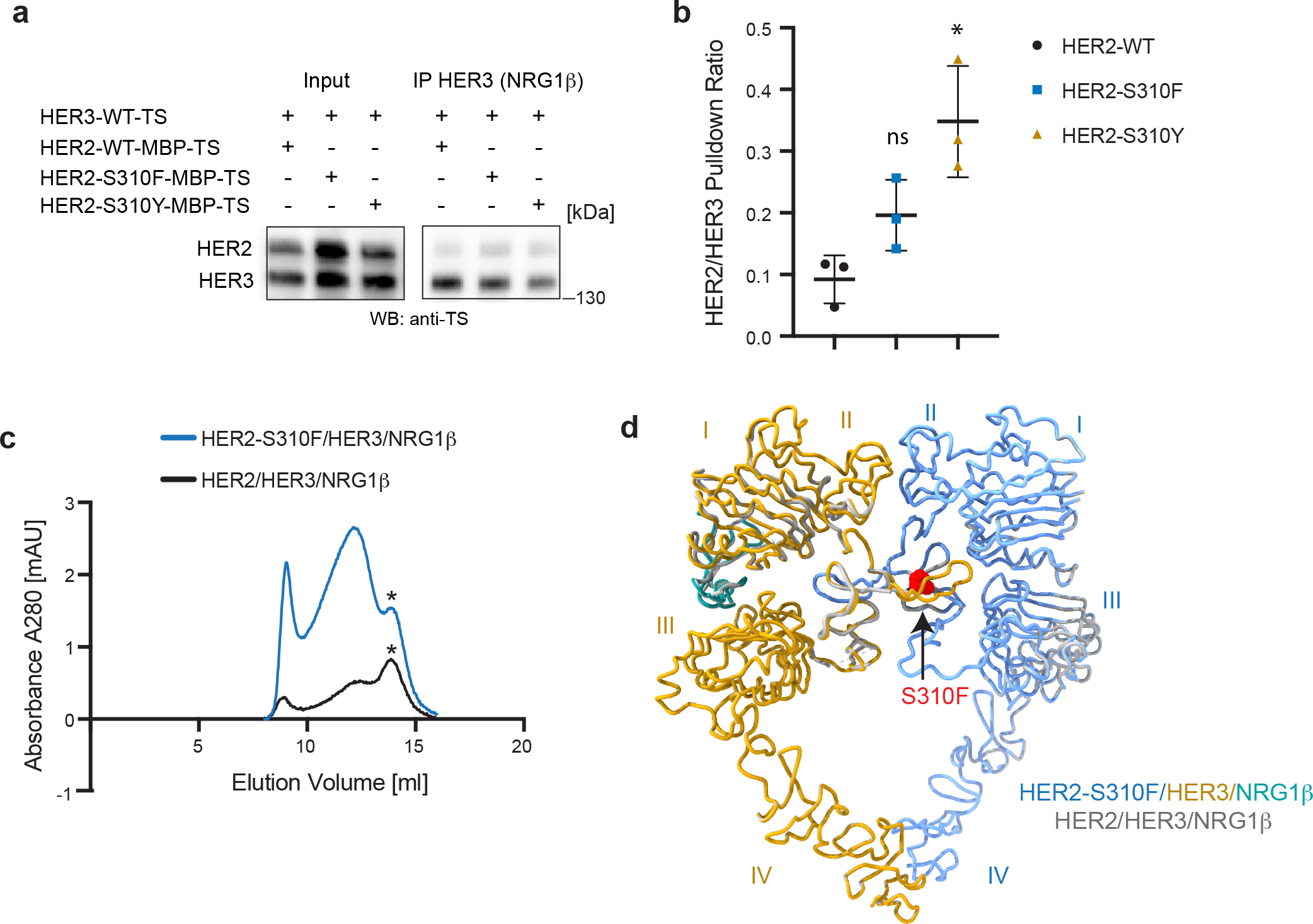
a, Representative Western Blot analysis of the HER2 (wild type, S310F or S310F mutant) and HER3 constructs co-transfected in the EXPI293 cells, and pulled-down via NRG1β immobilized on FLAG beads. TS – Twin Strep tag (present on both HER2 and HER3). Receptors were detected with the Strep-Tactin® HRP conjugate (anti-TS). For gel source data, see Supplementary Figure 1. b, Densitometry analysis of blots for data shown in panel a. Values are presented as mean values +/− SD of mean intensity ratios of HER2 over HER3 for each blot in n=3 independent biological replicates. The HER2/HER3 pulldown ratios are 0.09 +/− 0.04 for HER2-WT, 0.20 +/− 0.06 for HER2-S310F and 0.35 +/− 0.09 for HER2-S310Y complexes. Significance was determined via unpaired, two-tailed t-test via GraphPad Prism. WT vs S310F: p=0.0605, t=2.592, df=4. WT vs. S310Y: p=0.0107, t=4.516, df=4. c, Overlay of representative size exclusion chromatogram profiles from a Superose6 10/300 Increase column (GE Healthcare) of WT and oncogenic HER2-S310F heterocomplexes. Heterodimer peaks are denoted by asterisks. d, Cartoon representation of the cryo-EM structure of HER2-S310F/HER3/NRG1β complex overlayed on HER2/HER3/NRG1β. The HER2-S310F mutation is shown in red.
Extended Data Fig. 7 |. Resolution estimation, map quality and the workflow for processing of the HER2-S310F/HER3/NRG1β and HER2-S310F/HER3/NRG1β/trastuzumab datasets.
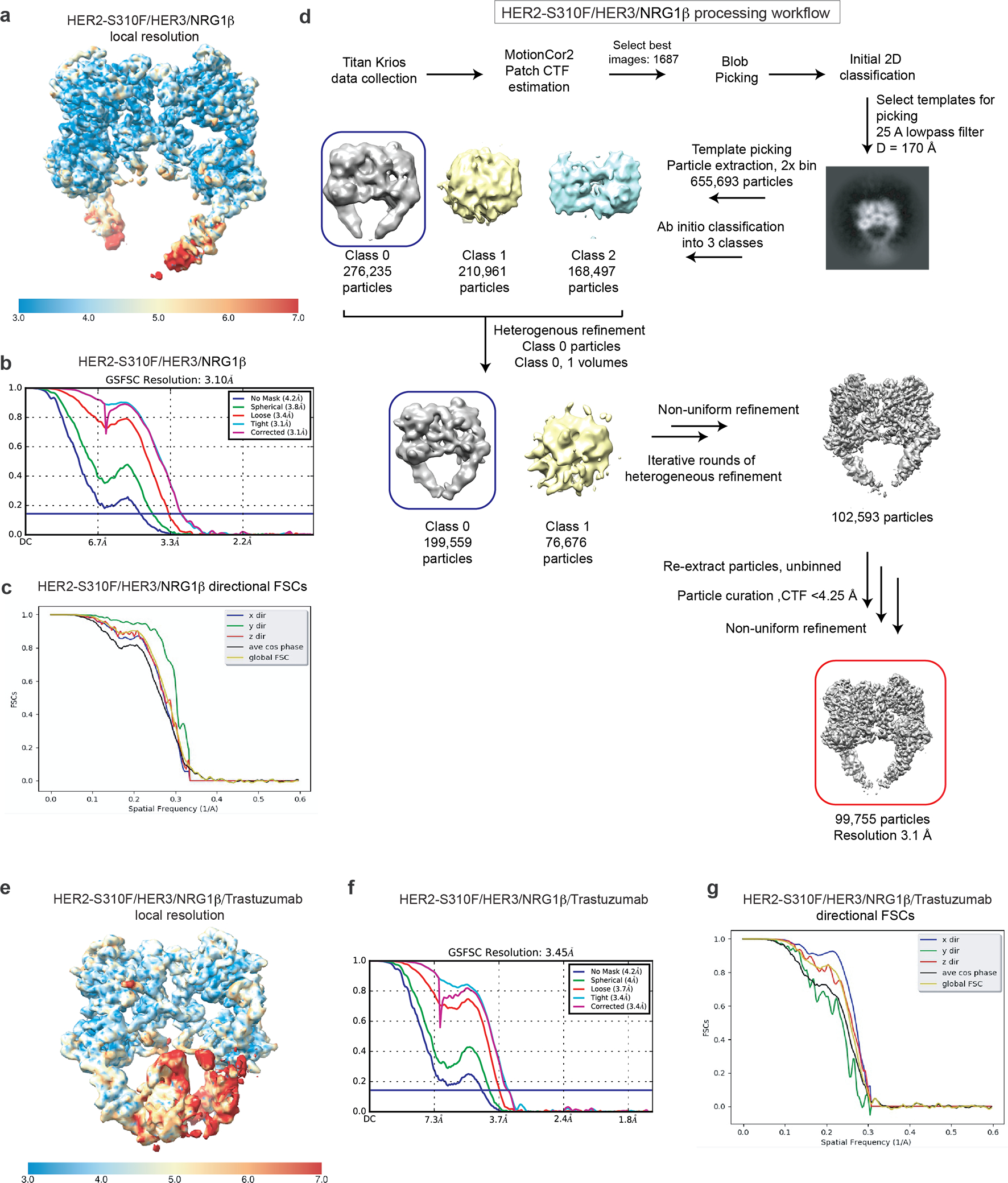
a, Cryo-EM map colored by local resolution as estimated from ResMap. b, Gold Standard Fourier Shell Correlation (GSFSC) of the final map used for model building from CryoSPARC2 with a reported resolution of 3.10 Å. c, Directional FSCs calculated by 3DFSC server. Map sphericity was calculated to be 0.949.d, Workflow for processing the HER2/HER3/NRG1β/trastuzumab dataset. The final model is indicated with a red box. e, Cryo-EM map colored by local resolution as estimated from ResMap. f, Gold Standard Fourier Shell Correlation (GSFSC) of the final map used for model building from CryoSPARC2 with a reported resolution of 3.45Å. g, Directional FSCs calculated by 3DFSC server. Map sphericity was calculated to be 0.941.
Extended Data Fig. 8 |. Functional and structural analysis of interactions between the therapeutic Fabs and HER receptors in the HER2/HER3/NRG1β complex.
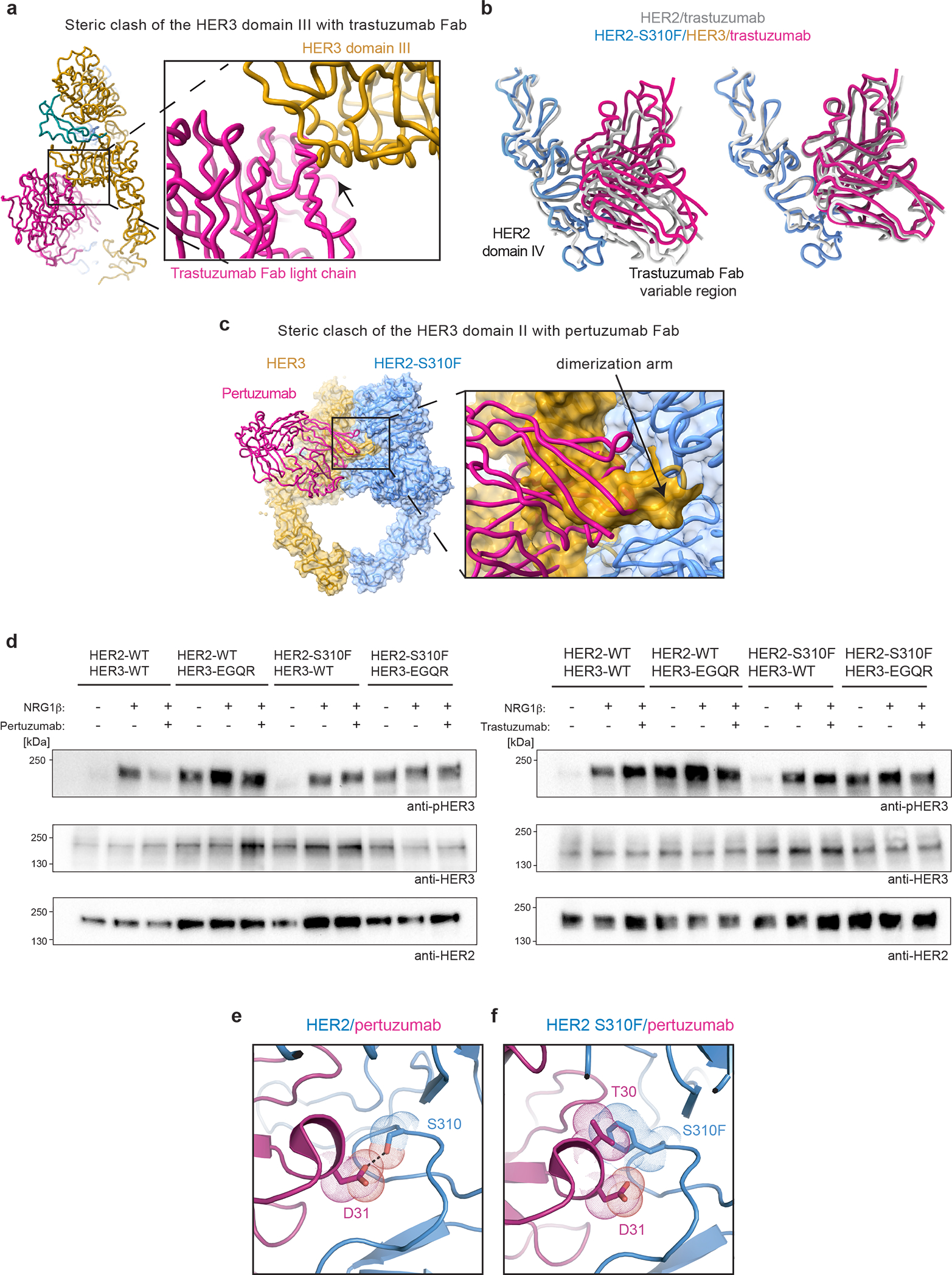
a, Overlay of the HER2/trastuzumab structure (PDB ID 6OGE) with the HER2-S310F/HER3/NRG1β structure reveals a steric clash between the Fab light chain constant domain and HER3 domain III. b, Left, overlay of the trastuzumab Fab variable domains in the HER2/trastuzumab structure (PDB ID 1N8Z) with the trastuzumab Fab in the HER2/HER3/NRG1β complex (HER2 domain IV in blue, trastuzumab Fab variable domains in magenta). The structures were aligned on HER2 domains I-III. Right, the HER2 domain IV bound to Fab variable domain from each structure was overlayed (RMSD: 1.569 Å) demonstrating that a Fab-binding epitope on HER2 is the same. Trastuzumab therefore avoids a steric clash with HER3 by inducing a rigid body rotation of HER2 domain IV relative to HER3. c, Overlay of the HER2/pertuzumab structure (PDB: 6OGE) with the HER2-S310F/HER3/NRG1β structure reveals a steric clash between the Fab variable domains, the HER3 dimerization arm, and HER3 domain II. d, Anti-phospho-HER3 (pHER3, Y1289), anti-HER3 and anti-HER2 Western Blot analysis of lysates from COS7 cells transfected with indicated full-length wild-type or mutant receptors. Cells were serum-starved, pre-incubated with pertuzumab or trastuzumab for 1h, as indicated, and then stimulated with 10 nM NRG1β. HER3-EGQR is HER3-E928G/Q809R. Representative Western Blot is shown, n=3. For gel source data, see Supplementary Figure 1. e, D31 of the pertuzumab variable light chain forms a polar contact with HER2 S310. f, HER2-S310F removes the polar contact with D31 and introduces a steric clash with T30 of the pertuzumab variable light chain. The predicted net effect of the oncogenic HER2-S310F mutation would be a decrease in pertuzumab affinity for HER2 (PDB ID 6OGE).
Extended Data Table 1.
Cryo-EM data collection, refinement and validation statistics.
| HER2/HER3/NRG1β (EMDB-23916) (PDB 7MN5) | HER2-S310F/HER3/NRG1β (EMDB-23917) (PDB 7MN6) | HER2-S310F/HER3/NRG1β + Herceptin Fab (EMDB-23918) (PDB 7MN8) | |
|---|---|---|---|
|
| |||
| Data collection and processing | |||
| Magnification | 105000x | 105000x | 105000x |
| Voltage (kV) | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 67 | 67 | 66 |
| Defocus range (μm) | 0.9–2.0 | 0.9–2.0 | 0.9–2.0 |
| Pixel size (Å) | 0.835 | 0.835 | 0.834 |
| Symmetry imposed | C1 | C1 | C1 |
| Initial particle images (no.) | 800000 | 650000 | 1500000 |
| Final particle images (no.) | 123173 | 99755 | 243376 |
| Map resolution (Å) | 2.9 | 3.1 | 3.4 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 3–7 | 3–7 | 3–9 |
| Refinement | |||
| Initial model used (PDB code) | 60GE, 1M6B, 3U7U | 60GE, 1M6B, 3U7U | 60GE, 1M6B, 3U7U |
| Model resolution (Å) | 3.2 | 3.3 | 3.6 |
| FSC threshold | 0.5 | 0.5 | 0.5 |
| Model resolution range (Å) | |||
| Map sharpening B factor (Å2) | −94.3 | −89.8 | −100.3 |
| Model composition | |||
| Non-hydrogen atoms | 9527 | 9652 | 12949 |
| Protein residues | 1213 | 1227 | 1661 |
| Ligands | 13 | 13 | 13 |
| B factors (Å2) | |||
| Protein | 116.53 | 69.28 | 239.91 |
| Ligand | 74.42 | 123.59 | 131.77 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.012 | 0.012 | 0.013 |
| Bond angles (°) | 1.572 | 1.616 | 1.048 |
| Validation | |||
| MolProbity score | 0.85 | 0.84 | 0.92 |
| Clashscore | 0.38 | 0.27 | 1.70 |
| Poor rotamers (%) | 0 | 0 | 0.14 |
| Ramachandran plot | |||
| Favored (%) | 96.65 | 96.37 | 98.11 |
| Allowed (%) | 3.35 | 3.47 | 1.17 |
| Disallowed (%) | 0 | 0.17 | 0.18 |
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Manglik for advice on receptor expression, and D. Bulkley, G. Gilbert, E. Tse, and Z. Yu from the UCSF EM facility. We also thank members of the Verba and Jura labs for their helpful discussions, and E. Linossi and H. Torosyan for critical comments on the manuscript. We would also like to acknowledge D. Suveges for his experimental contributions to the generation of our first HER expression constructs. This work was funded through UCSF Program for Breakthrough Biomedical Research to K.V. and N.J., NIH/NIGMS R35-GM139636 to N.J., Genentech research grant to N.J., DFG German Research Foundation GZ: TR 1668/1-1 to R.T. and NIH/NCI 1F30CA247147 to D.D.
Footnotes
COMPETING INTERESTS
N.J. is a member of the SAB and a shareholder of Turning Point Therapeutics, SUDO Biosciences and Type6 Therapeutics. The Jura laboratory has received sponsored research support from Genentech. Other authors do not declare competing interests.
Note
The original publication is available at https://www.nature.com/articles/s41586–021-04084-z
DATA AVAILABILITY STATEMENT
3D cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under the accession numbers: EMD-23916 (HER2/HER3/NRG1b), EMD-23917 (HER2-S310F/HER3/NRG1b), and EMD-23918 (HER2-S310F/HER3/NRG1b/Herceptin Fab). Atomic coordinates for the atomic models have been deposited in the RCSB Protein Data Bank under the accession numbers: PDB ID 7MN5 (HER2/HER3/NRG1b), PDB ID 7MN6 (HER2-S310F/HER3/NRG1b), and PDB ID 7MN8 (HER2-S310F/HER3/NRG1b/Herceptin Fab).
REFERENCES
- 1.Sliwkowski MX et al. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem 269, 14661–14665 (1994). [PubMed] [Google Scholar]
- 2.Wallasch C et al. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. The EMBO Journal 14, 4267–4275 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moasser MM The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487, doi: 10.1038/sj.onc.1210477 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sierke SL, Cheng K, Kim H & Koland JG Biochemical characterization of the protein tyrosine kianse domain of the ErbB3 (HER3) receptor protein. Biochem J 322, 757–763 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jura N, Shan Y, Cao X, Shaw DE & Kuriyan J Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A 106, 21608–21613, doi: 10.1073/pnas.0912101106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Gureasko J, Shen K, Cole PA & Kuriyan J An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149, doi: 10.1016/j.cell.2006.05.013 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Littlefield P et al. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Science Signaling 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 8.Ferguson K et al. EGF Activates Its Receptor by Removing Interactions with Autoinhibit Ectodomain Dimerization. Molecular Cell 11, 507–517 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Bouyain S, Longo PA, Li S, Ferguson KM & Leahy DJ The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci U S A 102, 15024–15029, doi: 10.1073/pnas.0507591102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho H-S & Leahy DJ Structure of the Extracellular Region of HER3 Reveals an Interdomain Tether. Science 297, 1330–1333 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Garrett JT et al. Crystal Structure of a Truncated Epidermal Growth Factor Receptor Extracellular Domain Bound to Transforming Growth Factor alpha. Cell 110, 763–773 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Ogiso H et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 110, 775–787 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Liu P et al. A single ligand is sufficient to activate EGFR dimers. Proc Natl Acad Sci U S A 109, 10861–10866, doi: 10.1073/pnas.1201114109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho H-S et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760, doi: 10.1038/nature01423 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Hao Y, Yu X, Bai Y, McBride HJ & Huang X Cryo-EM Structure of HER2-trastuzumab-pertuzumab complex. PLoS One 14, e0216095, doi: 10.1371/journal.pone.0216095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarado D, Klein DE & Lemmon MA ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 461, 287–291, doi: 10.1038/nature08297 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferguson KM, Darling PJ, Mohan MJ, Macatee TL & Lemmon MA Extracellular domains drive homo- but not hetero- dimerization of erbB receptors. The EMBO Journal 19, 4632–4643 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanker AB et al. Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell, doi: 10.1016/j.ccell.2021.06.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freed DM et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 171, 683–695 e618, doi: 10.1016/j.cell.2017.09.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol Cell Biol 30, 5432–5443, doi: 10.1128/MCB.00742-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y et al. A molecular mechanism for the generation of ligand-dependent differential outputs by the epidermal growth factor receptor. bioRxiv, 2020.2012.2008.417006, doi: 10.1101/2020.12.08.417006 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greulich H et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A 109, 14476–14481, doi: 10.1073/pnas.1203201109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T et al. HER2 somatic mutations are associated with poor survival in HER2-negative breast cancers. Cancer Sci 108, 671–677, doi: 10.1111/cas.13182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Network TCGA Comprehensive Molecular Portraits fo Human Breast Tumors. Nature 490, 61–70, doi: 10.1038/nature11412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franklin MC et al. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Alvarado D, Klein DE & Lemmon MA Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 142, 568–579, doi: 10.1016/j.cell.2010.07.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arkhipov A, Shan Y, Kim ET, Dror RO & Shaw DE Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. Elife 2, e00708, doi: 10.7554/eLife.00708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrett JT et al. The Crystal Structure of a Truncated ErbB2 Ectodomain Reveals an Active Conformation, Poised to Interact with Other ErbB Receptors. Molecular Cell 11, 495–505 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Xu W et al. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol 12, 120–126, doi: 10.1038/nsmb885 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Jaiswal BS et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617, doi: 10.1016/j.ccr.2013.04.012 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Palovcak E et al. A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J Struct Biol 204, 80–84, doi: 10.1016/j.jsb.2018.07.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang F et al. General and robust covalently linked graphene oxide affinity grids for high-resolution cryo-EM. Proc Natl Acad Sci U S A 117, 24269–24273, doi: 10.1073/pnas.2009707117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51, doi: 10.1016/j.jsb.2005.07.007 (2005). [DOI] [PubMed] [Google Scholar]
- 34.de la Rosa-Trevin JM et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99, doi: 10.1016/j.jsb.2016.04.010 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221, doi: 10.1016/j.jsb.2015.08.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kucukelbir A, Sigworth F & Tagare H Quantifying the local resolution of cryo-EM density maps. Nature Methods 11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan YZ et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796, doi: 10.1038/nmeth.4347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asarnow D, Palovcak E & Cheng Y UCSF pyem v0.5. Zenodo; (2019). [Google Scholar]
- 42.DiMaio F et al. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nat Methods 12, 361–365, doi: 10.1038/nmeth.3286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol 74, 519–530, doi: 10.1107/S2059798318002425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frenz B et al. Automatically Fixing Errors in Glycoprotein Structures with Rosetta. Structure 27, 134–139 e133, doi: 10.1016/j.str.2018.09.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agirre J et al. Privateer: software for the conformational validation of carbohydrate structures. Nat Struct Mol Biol 22, 833–834, doi: 10.1038/nsmb.3115 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
3D cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under the accession numbers: EMD-23916 (HER2/HER3/NRG1b), EMD-23917 (HER2-S310F/HER3/NRG1b), and EMD-23918 (HER2-S310F/HER3/NRG1b/Herceptin Fab). Atomic coordinates for the atomic models have been deposited in the RCSB Protein Data Bank under the accession numbers: PDB ID 7MN5 (HER2/HER3/NRG1b), PDB ID 7MN6 (HER2-S310F/HER3/NRG1b), and PDB ID 7MN8 (HER2-S310F/HER3/NRG1b/Herceptin Fab).