

Abstract
α‐Diimine ligands have significantly shaped the coordination chemistry of most transition metal complexes. Among them, bis(imino)acenaphthene ligands (BIANs) have recently been matured to great versatility and applicability to catalytic reactions. Besides variations of the ligand periphery, the great versatility of BIAN ligands resides within their ability to undergo facile electronic manipulations. This review highlights key aspects of BIAN ligands in metal complexes and summarizes recent contributions of metal‐BIAN catalysts to syntheses of small and functionalized organic molecules.
Keywords: imines, redox-active ligands, redox-non-innocent ligands, BIAN ligands, diimines
Redox‐non‐innocent ligands: Bis(imino)acenaphthenes (BIANs) are redox‐active α‐diimine ligands that display a multi‐facetted coordination and redox chemistry. Recent reports of metal‐BIAN complexes in unusual coordination modes, redox and spin states have greatly expanded the range of catalytic applications of metal/BIAN complexes to organic transformations. This review summarizes key aspects of their synthesis, properties, and recent applications to metal catalysis.
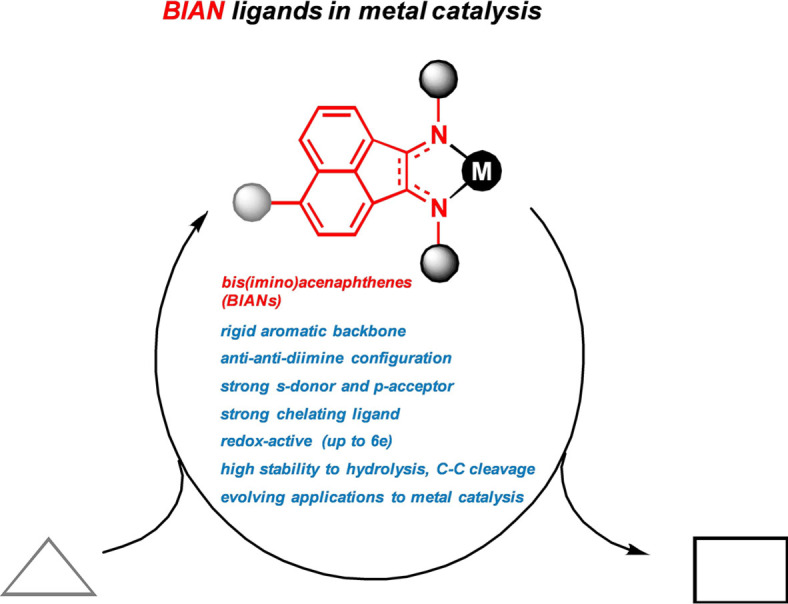
1. Introduction
Since the pioneering studies of Alfred Werner in 1893 on the coordination of ammonia to platinum ions, N‐donor ligands have been abundantly employed as ligands in the coordination chemistry of virtually all metal ions with widespread applications to inorganic materials, bioinorganic complexes, and many fields of catalysis including the synthesis of polymers, pharmaceuticals, fine chemicals, and agrochemicals, electro‐ and photo‐catalysis.[ 1 , 2 , 3 , 4 ] With the advent of new C−N bond forming reactions in the past decades, the availability of diverse and functional N‐ligands has been greatly expanded. In comparison with phosphines, N‐ligands are mostly less expensive, lower in molecular weight, highly modular, and easily tunable. While ligands containing sp3‐hybridized nitrogen atoms serve as σ‐donors, often as multi‐dentate chelates, more diverse modes of coordination can be derived from sp2‐hybridized nitrogen ligands due to the possibility of additional π‐acceptor interactions. Today, sp2‐ and sp3‐hybridized nitrogen‐based ligands constitute key players in organometallic chemistry. The occurrence of sp‐hybridized N‐ligands is mainly limited to the coordination of solvent molecules of acetonitrile as labile placeholders (Scheme 1, top). Ligands (that are not being consumed in a chemical reaction within the coordination sphere of a metal) are often used to modulate the stereoelectronic properties of a central metal ion. The development of modern coordination chemistry over the past 100 years has exploited various stereoelectronic roles of ligands in the stabilization of ground states or activated states, the modulation of thermodynamic or kinetic properties, the accessibility of neighboring sites, the direction and activation of substrates, the control of selectivities (chemo, regio, stereo), and the geometrical control over size and shape of molecules. The ability of ligands to actively interfere in the course of chemical bond cleavage and formation events at the metal center of coordination compounds has only recently been utilized to control the activity and selectivity of metal complexes.[ 5 , 6 ] These framework conditions laid the foundation for the development of new ligands that enabled wide electronic manipulations of metal complexes by ligand‐centered redox activity. Especially fruitful applications of such new ligand design principles were discovered in studies of 3d‐transition metal complexes that display distinct reactivities from the heavier noble metals. [7] The electronic participation of ligands in the bond‐breaking and bond‐making events at the metal center is differentiated into the roles of a spectator or an actor ligand. An actor ligand will electronically engage directly with the substrate, while the spectator ligand exerts its effect on the metal center (Scheme 1, bottom). Among the first non‐innocent ligands were α‐diimines such as pyridyl‐(di)imines that enabled new chemical reactivities through their facile redox activities.[ 8 , 9 ] These pioneering manifestations of reaction development by catalyst design principles involving redox‐active N‐ligands led to a revival of many imine ligand families. [10] Among them, BIAN ligands (bis(imino)‐acenaphthenes) were demonstrated to harbor especially facile redox and rich coordination chemistry with many metal ions (Scheme 1, center).[ 11 , 12 ] Structurally, BIANs can be considered as a central 1,4‐diazabutadiene merged with a naphthalene backbone. This union provides several advantages in chemical applications. The exocyclic imines are expected to lead to better σ‐donating as well as π‐accepting properties to comparable chelating N‐ligands such as 2,2’‐bipyridine (bpy) and 1,10‐phenanthroline (phen). This ensures stabilization of metal ions in both higher and lower oxidation states.[ 13 , 14 , 15 ] Additionally, BIAN derivatives are more rigid than related acyclic diimine ligands, which imparts a high chemical stability towards both hydrolysis and rupture of the central C−C‐bond. [16] Moreover, the aromatic backbone enforces the anti‐anti conformation on the α‐diimine moiety, thus encouraging strong chelation to a metal center. [13]
Scheme 1.

The diversity of N‐ligands including redox‐active α‐diimines. The BIAN ligand family displays high rigidity, stability, yet facile redox chemistry to facilitate electron transfer events at coordinated metal ions. [5]
BIAN ligands have been reported to effectively coordinate to almost all main group elements[ 17 , 18 , 19 , 20 , 21 ] and transition metals.[ 22 , 23 , 24 , 25 , 26 , 27 ] The resultant stereoelectronic properties including coordination modes, redox states, magnetic properties as well as the applications to chemical transformations have been varied to a great extent. Although N‐aryl substituted BIAN (ArBIAN) compounds were first described in the 1960s,[ 28 , 29 ] it was not until the 1990s that they were first employed in catalytic applications by the group of Elsevier. [30] A few years later, Brookhart introduced BIAN ligands to the field of olefin polymerizations (Scheme 2).[ 22 , 31 ]
Scheme 2.

ArBIAN ligands in metal‐catalyzed ethylene polymerizations. [31]
By virtue of their high modularity and stereoelectronic tunability, α‐diimine metal complexes have developed into a powerful class of non‐metallocene polymerization catalysts with wide applications ranging from polyethylene to highly functional polyesters and polycarbonates.[ 22 , 31 ] The different stereoelectronic properties of BIAN ligands, including their oxidation states, have enabled the modulation of catalyst properties, polyethylene branching and polymer micro‐structure.[ 32 , 33 , 34 ] Very recently, Garcia and coworkers discussed syntheses, properties, and catalytic behavior of p‐block element complexes with BIAN ligands. [35] Liu and Szostak reviewed applications of BIAN/N‐heterocyclic carbene ligands in transition metal‐catalyzed reactions. [36] Such applications of BIAN ligands are outside of the scope of this review which highlights the benefit of using BIAN ligands in catalytic reactions for the synthesis of small and functionalized molecules.
2. Synthesis of BIAN ligands
The main building block for all BIAN syntheses is acenaphthene quinone, an intermediate for many dyes, pharmaceuticals, and pesticides, which is obtained from the oxidation of the coal tar component acenaphthene (Scheme 3). [37] Most ArBIANs are synthesized via acetic acid catalyzed imine formation. However, Ragaini reported in 2002 that this equilibrium lies well on the side of the unwanted N‐acetanilide by condensation with the catalyst. [38] Key to a successful and high‐yielding synthesis of N‐aryl BIAN compounds is the shift of the reaction to the desired diimines by precipitation (Scheme 3). This precipitation strategy was mostly realized by the formation of the Zn or Ni complexes. [17] These complexes are mostly insoluble in acetic acid and easily filtered off; the free ligand is obtained by subsequent treatment with bases. This understanding of the thermodynamic conditions enabled the preparations of hitherto capricious ArBIANs. Anilines containing strongly electron‐withdrawing CF3 groups were hardly accessible before, but then could be obtained by a solvent change that led to the precipitation of the otherwise soluble Zn complexes of these ligands. [38] Similar optimizations of solubility behaviors were successful in the preparation of unsymmetrical ArAr'BIAN ligands, [16] which are generally challenging. Early attempts by Ragaini in 2004 documented that syntheses via monoimination of acenaphthene quinone were problematic. [16] Unsymmetrical ArAr'BIANs could be prepared but the key monoimine intermediates could only be accessed from sterically extremely bulky anilines (Scheme 4). [39] A different approach used a transimination step, in which one electron‐deficient substituent is replaced by an electron‐rich aniline. This transimination procedure required the formation of the Zn‐BIAN complexes to ensure good yields.
Scheme 3.

Acenaphthene quinone as precursor to BIAN syntheses; suppression of unwanted amidation by precipitation of ArBIANs.
Scheme 4.

Unsymmetrical ArAr’BIAN synthesis via transimination.
Early efforts to expand the BIAN library to N‐alkyl‐substituted derivatives were met with failure. [40] The oxidizing power of the acenaphthene quinone precursor effected oxidations of the aliphatic amines. The alkyl substituents also underwent rapid tautomerization to give mixture of undesired amines and imines (Scheme 5, top).[ 40 , 41 ] These imine degradations were presumably driven by the relief of ring strain, [41] as demonstrated by reactions employing cycloalkyl amines bearing even higher strain (Scheme 5, center). The use of chiral cyclopropylamines enabled the preparation of asymmetric alkyl BIAN derivatives.[ 42 , 43 ] Tertiary amines – which lack the problematic α‐H atom that results in unwanted tautomerization – were still not compatible with the available procedures (e. g. t‐butyl, 1‐adamantyl), most likely due to the steric bulk building in the transamination step. Cowley et al. reported in 2011 a new procedure that addressed the synthesis of tert‐alkyl BIANs via the reaction of aminoalanes with acenaphthene quinone (Scheme 5, bottom). Crystal structure analyses of the resultant alkyl BIAN mixtures revealed the predominant formation of the syn‐anti stereoisomers. [44]
Scheme 5.

i) Tautomerization and transamination of alkyl BIANs. ii) Synthesis of cyclopropyl‐BIAN ligands. iii) Synthesis of alkyl‐BIAN using aminoalanes.
The installation of substituents at the acenaphthene unit of BIAN ligands, such as halide, nitro, and carboxylate functions, has been effected by aromatic substitution reactions.[ 45 , 46 , 47 ] Besides structural and steric variations of the ligand periphery, the great versatility of BIAN ligands also resides within their ability to undergo facile electronic manipulations. Reductions of the central diamine motif lead to radical anions, enediamido, and diamido ligand states. Reductions can also involve the aromatic backbone. Under standard conditions with common reductants such as LiAlH4 or Na, BIANs can accept up to four electrons. For example, the diamine BIAN‐H4 can be accessed from hydride addition with lithium aluminium hydride; the enediamine BIAN‐H2 is derived from sequential one‐electron reduction with Na and protolysis (Scheme 6).[ 12 , 48 ]
Scheme 6.

The rich reduction chemistry of the α‐diimine motif within ArBIANs.
3. Applications to metal catalysis
BIAN ligands display an especially rich coordination and redox chemistry in metal complexes of many main group and transition elements.[ 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 ] Despite numerous literature reports on metal/BIAN‐catalyzed reactions, this review will be limited to successful implementations that have documented a distinct behavior of BIAN ligands. Therefore, investigations in which various ligand families have been employed and the use of BIANs showed no beneficial effect over other (diimine) ligands are not specifically addressed. The selection of the most significant and distinctive applications of BIAN ligands to metal‐catalyzed reactions is intended to provide a deeper insight into catalyst properties, method developments, and mechanistic rationales. The binding strength of BIAN ligands to metal ions was reported to be somewhat lower than with the related phenanthrolines and bipyridines, however, the modular structure of BIANs enable wide stereoelectronic variations by proper choice of the pendant amine. Investigations by Ragaini revealed a linear correlation between the coordination strength of aryl‐BIANs to Pd complexes and the Hammett σ‐constants of the N‐aryl substituents (Scheme 7). [49] ArBIAN‐Pd complexes bearing electron‐donating N‐aryl substituents led to stronger coordination with Pd(0)‐ and Pd(II)‐complexes. [49] Similar Hammett correlations were observed by Zysman‐Colman et al. from the UV‐Vis lowest energy absorption bands of the free ligands that displayed red shifts with electron rich substituents. [50]
Scheme 7.

Equilibrium constants of ligand exchange and Hammett‐σ‐constants for the N‐aryl substituents of ArBIAN‐Pd(II) complexes. [49]
3.1. Hydrogenations and hydrofunctionalizations
Metal‐catalyzed hydrogenation reactions constitute one of the key chemical transformations for bulk chemicals, functionalized building blocks, pharmaceuticals, and materials.[ 51 , 52 ] The first concise studies of metal‐catalyzed alkene hydrogenations employing BIAN ligands were reported by the group of Elsevier. Trivalent Pd‐BIAN‐olefin complexes were shown to be active hydrogenation catalysts for electron‐deficient alkenes (Scheme 8, top). [30] It was postulated that the enhanced reactivity of (BIAN)Pd(alkene) complexes over bis(phosphine)Pd(alkene) complexes was a consequence of an effective overlap of π‐ligand orbitals with metal‐d‐ and the σ*‐H2‐orbital in the overall‐HOMO of the transition state (Scheme 8). [30] These insights enabled a further development of stereoselective semihydrogenations of alkynes to give (Z)‐alkenes. [53] Based on kinetic, spectroscopic, and theoretical studies, a catalytic mechanism was proposed that involves ligand‐assisted heterolytic activation of H2 across the Pd−N bond (Scheme 8, center).[ 54 , 55 ] Stereoelectronic variations of the ligand properties showed lower (Z)‐stereocontrol with electron‐deficient N‐aryl‐BIANs (Table 1). Enhanced alkane formation was observed with bulky ligands due to better (Z)‐alkene coordination. [53] With the same catalyst and identical conditions, the regioselective 2,3‐semihydrogenation of substituted allenes was effected (Scheme 8, bottom). [56]
Scheme 8.

i) Palladium catalysts and the relevant HOMO‐σ*(H2) interaction. [30] ii) Semi‐hydrogenation of alkynes.[ 53 , 54 , 55 ] iii) Semihydrogenation of allenes. [56]
Table 1.
Pd‐catalyzed semihydrogenation of phenylpropyne. [53] Pre‐catalyst structures are shown in Scheme 8, top.
|
Entry |
Pre‐catalyst |
Z‐Alkene [%] |
E‐Alkene [%] |
Alkane [%] |
|---|---|---|---|---|
|
1 |
[Pd]a |
92 |
2 |
6 |
|
2 |
[Pd]b |
85 |
5 |
10 |
|
3 |
[Pd]c |
80 |
7 |
13 |
|
4 |
[Pd]d |
62 |
3 |
35 |
The activity of iron‐BIAN complexes in catalytic hydrogenations of alkenes was recently investigated. The coordination of less bulky aryl‐BIANs to iron(II) ions resulted in octahedral homoleptic complexes that proved inactive. Use of the bulky 2,6‐diiso‐propylphenyl derivative (dippBIAN) in combination with a co‐catalytic reductant (LiBHEt3 or nBuLi) gave a highly active pre‐catalyst that even allowed hydrogenations of very challenging tri‐ and tetra‐substituted olefins. [25] Highest catalyst activities were observed from pre‐catalyst mixtures that involved BIAN‐Fe(II) complexes and three reduction equivalents which may indicate ligand‐ and iron‐centered reduction to an active BIAN‐ferrate species (Scheme 9). The 2e‐reduced neutral BIAN‐Fe(0) complex [57] exhibited no catalytic activity under the reaction conditions. These observations illustrated the potential of BIAN ligands to act as electron reservoirs (in their radical anion or dianion states) that facilitate metal‐catalyzed redox mechanisms. [25]
Scheme 9.

dippBIANFeCl2 as pre‐catalyst in the hydrogenation of alkenes. [25]
Jacobi von Wangelin and coworkers reported a detailed synthetic, spectroscopic, and mechanistic study of structurally novel cobalt complexes. [58] Reactions of the pre‐catalyst dippBIANCoBr2 with the co‐catalyst LiBHEt3 afforded three distinct reduced species that document the redox non‐innocence of BIAN ligands. (Scheme 10, top). The formal 2e‐reduced complex (BIAN)Co(η6‐benzene) ([Co]a) bearing a formal radical anion of BIAN showed no catalytic activity in alkene hydrogenations. The formal 3e‐reduced cobaltate complex [Co]b bearing BIAN in its dianionic enediamido state proved to be highly active. A most unusual dimeric species was isolated from the pre‐catalyst mixture in diethylether. This formal 2e‐reduced trihydrido cobaltate ([Co]c] constitutes the first reported anionic cobalt complex containing μ‐hydride ligands and showed high activity in catalytic alkene hydrogenations. It is very likely that such anionic BIAN complexes are catalytic intermediates of Co‐catalyzed hydrogenation and dehydrogenation reactions. [58] A broad variety of substrates, including tri‐ and tetra‐substituted alkenes as well as imines and quinolines were cleanly hydrogenated (Scheme 10, top). The monomeric cobaltate catalyzed the dehydrogenation of ammine‐boranes. In the presence of alkenes, a new transfer hydrogenation protocol was realized (Scheme 10, bottom). [59]
Scheme 10.

i) dippBIANCoBr2 as pre‐catalyst in the hydrogenation of alkenes, imines, and quinolines. [58] ii) dippBIAN‐H2‐cobaltate‐catalyzed ammine‐borane dehydrogenation and transfer‐hydrogenation. [59]
While proceeding under similar conditions via similar mechanisms as hydrogenation reactions, the operational simplicity of using easily available and liquid hydride reagents makes hydro‐functionalizations especially attractive reduction methods. Findlater and coworkers explored the catalytic activity of 2e‐reduced (arene)Fe(0)‐BIAN complexes in catalytic hydro‐functionalizations (Schemes 9 and 11). [57] With 1 mol % catalyst, clean hydrosilylations of aldehydes, ketones, and imines were achieved, without excess silane. [60] Ketimines afforded lower yields. With the same complex, Findlater and coworkers performed catalytic hydrosilylation of esters with excess amounts of silane. The reaction showed broad functional group tolerance and good to excellent yields. [61] The same group reported hydroborations of alkynes and alkenes with DippBIAN‐FeCl2 (Scheme 11, center). [62] High regioselectivities and stereo‐selectivities were obtained (up to 98 % for alkyl boronates; exclusive formation of trans‐vinyl boronates). The anionic BIAN‐ferrate complex [DippBIANFe(cod)]K catalyzed the hydroboration of ketones under very mild conditions (Scheme 11, bottom). [63] With 0.1 mol % catalyst, a turnover frequency of 220,000 h−1 was recorded.
Scheme 11.

Fe(BIAN)‐catalyzed hydrofunctionalizations: i) Hydrosilylation of aldehydes, ketones, imines;[ 57 , 60 ] ii) hydroboration of alkynes, alkenes; [62] iii) hydroboration of ketones. [63]
3.2. C−C Bond formations
Metal‐catalyzed cross‐couplings and closely related C−C bond forming reactions have significantly shaped the art of modern organic synthesis in the past decade. [64] Applications of the BIAN ligand family to cross‐coupling protocols were first investigated by Elsevier and coworkers. [65] The same trivalent (BIAN)Pd‐olefin complexes showed good activity in Kumada‐, Negishi‐, and Stille‐type reactions of various electrophiles (alkyl, aryl, acyl, allyl). Variations of the BIAN substitution patterns had only little effect on catalyst activities and selectivities. Despite slightly lower conversions than with the commonly used (phosphine)palladium catalysts, the employment of BIAN ligands in palladium‐catalyzed cross‐coupling led to lower amounts of unwanted homo‐coupling products. [66] The suppression of homo‐coupling with (BIAN)Pd catalysts was proposed to be a consequence of slower trans‐metallation steps between organopalladium and organometal reagents, possibly due to the reduced basicity of the Pd ions in the coordination of π‐accepting BIAN ligands. Formation of a Pd(IV) complex was excluded (Scheme 12). [67] Elsevier and coworkers documented that effective stabilization of Pd in the oxidation states 0, II and IV can be realized with BIAN ligands. An example of the occurrence of these species is the palladium‐catalyzed alkyne‐inserting cross‐coupling reaction shown in Scheme 13. [68] This mechanistic combination of a cross‐coupling reaction with an alkyne dimerization was reported to proceed with (BIAN)Pd catalysts (Scheme 13).[ 69 , 70 ] The three‐component synthesis of conjugated dienes from two alkynes, an organic halide and tetramethyl tin most likely involves the intermediacy of Pd(IV) states which are highly sensitive to sterics. [71] Consequently, bulky N‐aryl substituents of the BIAN ligands inhibit the second oxidative addition event. Interestingly, the direct cross‐coupling between the organic halide and the organotin reagent was not operative. [71]
Scheme 12.

Scheme 13.

Pd/BIAN catalyzed alkyne‐inserting cross‐coupling by Elsevier. [71]
Pd(BIAN) complexes were employed as pre‐catalysts in the Suzuki‐Miyaura coupling of aryl halides with arylboronic acids in water. The authors proposed a heterogeneous reaction mechanism where the bulky BIAN derivative enabled stabilization of the catalyst and facile catalyst recovery (Scheme 14). [72] Palladium‐catalyzed oxidative Heck reactions between arylboronic acids and α,β‐unsaturated carbonyl derivatives were reported by Minnaard et al. in 2011 (Scheme 15). The commonly observed catalyst deactivation, catalyst dimerization, and ligand oxidation pathways of alternative Pd catalysts under the oxidative conditions were effectively suppressed with bulky BIAN ligands. The active Pd catalysts and mild conditions allowed coupling reactions with various arylboronic acids and otherwise capricious Michael acceptors, including conjugated enals. [73] A mechanism involving Heck‐type olefination at a Pd(II) center with a terminal β‐hydride elimination and subsequent oxidation to regenerate the active Pd(II) species was proposed earlier. [74]
Scheme 14.

Suzuki‐Miyaura coupling supported by bulky BIAN ligand. [72]
Scheme 15.

Metal‐catalyzed olefin polymerizations have been studied extensively in the presence of various diimine ligands, including many reports where BIAN ligands induce high polymer yields and selectivities. [31] The iterative Kumada cross‐coupling of 1‐bromo‐phenyl‐4‐magnesium bromides was effectively catalyzed by BIAN‐nickel bromide complexes (Scheme 16). [75] Such living polymerizations are typically very challenging when electron‐deficient monomers are being employed. Electron‐rich N‐aryl‐BIANs gave very high selectivities that even enabled controlled block‐copolymerization upon addition of further monomers. [76] Alternating additions of electron‐rich and electron‐deficient para‐halo aryl magnesium halides were used for the synthesis of novel donor‐acceptor block copolymers. [77]
Scheme 16.

Nickel/BIAN‐catalyzed cross‐coupling polymerization. [76]
3.3. Nitrobenzene reductions
A field of metal‐catalyzed reduction reactions that has been widely explored with BIAN‐coordinated complexes is the reduction of aromatic nitro compounds with CO as reductant. Upon combination with trapping reactions of the reduced intermediates (nitroso, hydroxylamine, amine derivatives), a diverse set of N‐containing molecules can be accessed in straight‐forward manner. Key to the reduction of the nitro function is the ability of coordinated CO‐ligands to act as deoxygenation reagent in the presence of water. Such scenarios have been applied to metal/BIAN‐catalyzed syntheses of anilines, indoles, pyrroles, oxazines and allylic amines.[ 78 , 79 , 80 ] Combinations of N‐aryl‐BIAN ligands with [Ru3(CO)12] have been shown to be especially reactive. [78] The postulated catalytically active complex [Ru(CO)3(ArBIAN)] is generated at elevated temperature. Nitro‐benzene reductions to anilines displayed the reactivity order H>4‐Me>4‐Cl>4‐OMe of the N‐phenyl substituents. This trend is most likely a compromise of higher activity of electron‐rich BIAN ligands and faster BIAN dissociation and catalyst deactivation to [Ru(CO)5] with the same ligands. [79] Attempts to recover and recycle the air‐sensitive Ru(0) catalysts were studied with alkyl‐substituted BIAN‐Ru complexes immobilized on a PEEK‐WC (modified polyether ether ketone) polymer membrane. [83] The catalytic membrane showed good thermal and chemical stability and was utilized for repetitive reduction cycles of nitrobenzene. However, dissociation of the BIAN ligands under the reaction conditions was observed. From spectroscopic studies and stoichiometric control experiments, a dual catalytic mechanism was postulated: the catalyst [Ru3(CO)12] is capable of reducing ArBIAN to ArBIAN‐H2 in aqueous solution. This 2e‐reduced BIAN derivative with an enediamino motif undergoes rapid reduction of nitrobenzene, possibly facilitated by π‐π‐interactions (Scheme 17). This report supports the notion of the versatile redox chemistry of BIANs that may facilitate metal‐centered redox reactions but engage as electron (and proton) shuttle in metal‐free electron transfer processes. [83]
Scheme 17.

i) Ru‐catalyzed reduction of nitrobenzene to aniline. ii) Catalyst formation and deactivation pathways. [82] ii) Dual catalysis involving BIAN as electron/proton shuttle.
A sequential combination of nitroarene reduction and allylic CH‐amination was reported by Ragaini and coworkers. [84] The fine balance of effective alkene coordination and sufficiently fast nitro‐benzene reduction was optimal for electron‐deficient nitroarenes and moderately electron‐rich N‐aryl‐BIAN ligands. Very electron‐rich BIANs accelerate gave high activities but low selectivities, steric BIANs resulted in low conversions (Scheme 18). [85] When employing 2,3‐dimethylbutadiene, a similar reaction mechanism can be followed toward the formation of oxazines and – after thermal elimination of water – pyrroles (Scheme 19). [86] A similar C,O‐bound hydroxylamine intermediate is believed to form that undergoes reductive elimination to release the N‐aryl oxazine. [86]
Scheme 18.

i) Allylic aminations of unactivated olefins with nitroarenes. ii) Mechanism of the [Ru(CO)3(ArBIAN)]‐catalyzed reductive CH‐amination.
Scheme 19.

Synthesis of N‐aryl oxazines and N‐aryl pyrroles from nitroarene reductions.
3.4. 1,2‐Additions
Hydroaminations of alkenes are one of the most straight‐forward and atom‐economic processes for the synthesis of valuable substituted amines and N‐heterocycles from inexpensive and readily available starting materials. A severe challenge for this reaction, however, is the poor reactivity of internal olefins. BIAN complexes of alkaline earth metals and early transition metals that bear highly basic alkyl ligands have been successfully employed as catalysts in such hydroamination reactions. Hill and coworkers reported a new series of Mg, Ca, and Sr complexes derived from BIAN precursors. [87] Upon treatment with strong organometallics, a formal 1,6‐carbometallation of DippBIAN led to the formation of dearomatized ligands with an amidoimine bonding motif at the metal centers (Scheme 20). [87] The calcium complex showed superior catalytic activity in intramolecular hydroaminations of internal alkenes (Ca>Sr≫Mg). The Mg analogue also effected 7‐membered ring formation. [20]
Scheme 20.

Hydroaminations with alkaline earth metal complexes derived from DippBIAN. [20]
Under very similar conditions, intramolecular hydroaminations were performed in the presence of Zr and Y complexes. Catalyst formation from organometallic alkyl metallates and various N‐aryl BIAN resulted in the formation of the 2e‐reduced enediamido complexes of Zr and Y, respectively. The anionic [(BIAN2−)YR2] afforded excellent yields in the formations of 5‐ and 6‐membered N‐heterocycles from both, terminal and internal alkenes (Scheme 21). [88] The authors proposed that the BIAN ligand effectively prevents catalyst deactivation under the strongly basic conditions.
Scheme 21.

Hydroaminations of internal alkenes catalyzed by reduced Y‐BIAN catalysts. [88]
Sterically demanding N‐aryl‐BIANs gave highest catalyst activities. [89] Recently, Fedushkin and coworkers complemented the set of BIAN‐metal catalysts for hydroaminations by the related Ca and Mg complexes. Very high yields of intramolecular hydroamination of terminal alkenes was observed. In the absence of other basic reagents, the amido functions of the BIAN ligand act as intramolecular proton shuttle which commences with amine by deprotonation. [88]
A rare example of a copper‐catalyzed reaction with BIAN ligands was reported by Aviles and Gomes.[ 90 , 91 ] The azide‐alkyne cycloaddition proceeded smoothly with only 1 mol % of catalyst (Scheme 22). A wide survey of copper‐BIAN catalyst was performed, including monomeric and dimeric, neutral and cationic complexes with various BIAN ligands and co‐ligands such as phosphines. Cationic copper complexes bearing bulky and electron‐rich BIAN ligands were most reactive. The authors postulated that the stabilization of soluble copper(I) species and the prevention of coordinative saturation were key factors of high catalyst activities.[ 90 , 91 ]
Scheme 22.

Copper/BIAN‐catalyzed azide‐alkyne cycloaddition.
The application of photoactive BIAN‐copper complexes to visible light‐driven radical additions was reported by Soo and coworkers. A bromo‐substituted [(ArBIAN)2Cu]+ initiated a radical chain reaction that enabled the synthesis of various bromopropyl benzene derivatives. [45] The homoleptic copper(I) complex acts as 1e‐photoreductant in the presence of tetrabromomethane to form the intermediate tribromomethyl radical that engages in an atom transfer radical addition (ATRA) to styrenes with yields up to 75 % (Scheme 23). The introduction of the bromide substituent in the BIAN backbone led to increased lifetime of the photoexcited state of the catalyst. [45]
Scheme 23.

ATRA reaction photocatalyzed by [(Ar(CO)OMeBIANBr)2Cu]PF6. [45]
3.5. Oxidations
Oxidative transformations with metal‐BIAN complexes are much rarer than reductive processes, possibly due to the electron‐accepting properties of the ligands. However, many of the reported examples of metal‐BIAN‐catalyzed reactions operate in the presence of oxidizing reagents or even under aerobic conditions (see for example Scheme 15, 18, 19). A biomimetic catalyst that mimics vanadium‐based enzymatic reactivities in oxidation reactions with hydroperoxides was reported by the group of Calhorda in 2015. The square‐pyramidal V(III)‐BIAN complex shown in Scheme 24 efficiently catalyzed the epoxidation of terminal and internal olefins with tert‐butyl‐hydroperoxide or hydrogen peroxide. [92] A significant correlation of substrate reactivities with the stereoelectronic properties of the BIAN ligands were observed. Electrochemical studies corroborated that BIAN ligands effectively stabilize the intermediate V(IV) redox state, so that only 1 mol % catalyst were sufficient for high catalytic activities. [92] Following this work, the same authors developed a related vanadium(IV) complex that enabled facile CH‐oxidations of alkenes and alkanes. Only 0.5 mol % vanadium‐BIAN catalyst were employed with stoichiometric H2O2 at 50 °C (Scheme 24, bottom). [93]
Scheme 24.

BIAN/vanadium‐catalyzed oxidations of alkenes and alkanes.
4. Conclusions
The multi‐facetted coordination and redox chemistry of bis(imino)‐acenaphthene (BIAN) ligands has been acknowledged in numerous reports of metal complex syntheses with virtually all stable elements. Diverse topologies, nuclearities, and substitution patterns have been realized in coordination compounds of main group elements, transition metals, and rare earth elements. Most emphasis has been laid on complexes in lower oxidation states that benefit from the facile and reversible ligand‐centered reduction of BIAN ligands. However, applications to metal catalysis have remained scarce, with hydrogenations, reductions of nitrobenzenes, and hydroaminations being the most prominent examples. (In comparison with such small‐molecule syntheses, far more attention has been directed at olefin polymerizations in the presence of metal/BIAN catalysts.) Recent reports of metal complexes in unusual coordination modes, redox and spin states, as well as the isolation of hitherto unknown reactive intermediates may contribute to a wider perception of the untapped potential of BIAN ligands. Future applications may very likely include photocatalytic and electrocatalytic reactions. Transfer of knowledge from the versatile coordination chemistry with main group elements to catalytic reactions are very likely to complement the current arsenal of methods. With the advent of novel alkyl‐BIAN derivatives and chiral BIAN ligands, further grounds will be broken. The recent reports of new BIAN‐metal complexes exhibiting highly reduced oxidation states, redox‐non‐innocence, rare coordination motifs, and catalytic activities document that BIAN ligands have become a trusted member in the family of powerful and versatile ligand architectures.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Josef Bernauer hails from the beautiful Danube riverbanks in Bavaria. He studied Chemistry at the University of Regensburg and obtained his Ph.D. from the University of Hamburg on metal‐catalyzed cross‐couplings and hydrogenations. Josef recently returned to Bavaria for an R&D position in desalination technologies.

Biographical Information
Jennifer Pölker calls the Münsterland her home. She studied Chemistry at the University of Münster where she graduated with an M.Sc. thesis in the group of Fabian Dielmann. As a doctoral fellow of the Evonik Foundation, Jenny currently investigates earth‐abundant metals in reduction and coupling reactions.

Biographical Information
Axel Jacobi von Wangelin was born in Berlin and saw the graffiti‐free side of the wall collapsing. He later enjoyed vibrant college lives in Erlangen, Utah, Cardiff, Rostock, Stanford, Cologne, and Regensburg. Axel is Professor of Molecular Chemistry at the University of Hamburg. His group's research spans all aspects of catalysis.

Acknowledgements
We gratefully acknowledge generous support from the European Research Council (ERC, CoG 683150). J.P. is a doctoral fellow of the Evonik Foundation. Open Access funding enabled and organized by Projekt DEAL.
J. Bernauer, J. Pölker, A. Jacobi von Wangelin, ChemCatChem 2022, 14, e202101182.
Dedicated to Kilian Muniz
BIAN=bis(imino)acenaphthene
References
- 1. Werner A., Z. Anorg. Chem. 1893, 3, 267–330. [Google Scholar]
- 2. Togni A., Venanzi L. M., Angew. Chem. Int. Ed. Engl. 1994, 33, 497–526. [Google Scholar]
- 3. Fache F., Schulz E., Tommasino M. L., Lemaire M., Chem. Rev. 2000, 100, 2159–2232. [DOI] [PubMed] [Google Scholar]
- 4. Pombeiro A. J. L., Dalton Trans. 2019, 48, 13904–13906. [DOI] [PubMed] [Google Scholar]
- 5. Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279. [Google Scholar]
- 6. Luca O. R., Crabtree R. H., Chem. Soc. Rev. 2013, 42, 1440–1459. [DOI] [PubMed] [Google Scholar]
- 7. Chirik P. J., Wieghardt K., Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- 8. Bouwkamp M. W., Bowman A. C., Lobkovsky E., Chirik P. J., J. Am. Chem. Soc. 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]
- 9. Bart S. C., Chłopek K., Bill E., Bouwkamp M. W., Lobkovsky E., Neese F., Wieghardt K., Chirik P. J., J. Am. Chem. Soc. 2006, 128, 13901–13912. [DOI] [PubMed] [Google Scholar]
- 10. Mashima K., Bull. Chem. Soc. Jpn. 2020, 93, 799–820. [Google Scholar]
- 11. Kaim W., Dalton Trans. 2019, 48, 8521–8529. [DOI] [PubMed] [Google Scholar]
- 12. Fedushkin I. L., Skatova A. A., Chudakova V. A., Fukin G. K., Angew. Chem. Int. Ed. 2003, 42, 3294–3298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3416–3420. [Google Scholar]
- 13. van Asselt R., Elsevier C. J., Smeets W. J. J., Spek A. L., Benedix R., Recl. Trav. Chim. Pays-Bas 1994, 113, 88–98. [Google Scholar]
- 14. Cavell K. J., Stufkens D. J., Vrieze K., Inorg. Chim. Acta 1981, 47, 67–76. [Google Scholar]
- 15. Reinhold J., Benedix R., Birner P., Hennig H., Inorg. Chim. Acta 1979, 33, 209–213. [Google Scholar]
- 16. Gasperini M., Ragaini F., Gazzola E., Caselli A., Macchi P., Dalton Trans. 2004, 33, 3376–3382. [DOI] [PubMed] [Google Scholar]
- 17. Hill N. J., Vargas-Baca I., Cowley A. H., Dalton Trans. 2009, 38, 240–253. [DOI] [PubMed] [Google Scholar]
- 18. Fedushkin I. L., Moskalev M. V., Lukoyanov A. N., Tishkina A. N., Baranov E. V., Abakumov G. A., Chem. Eur. J. 2012, 18, 11264–11276. [DOI] [PubMed] [Google Scholar]
- 19. Fedushkin I. L., Skatova A. A., Bazyakina N. L., Chudakova V. A., Khvoinova N. M., Nikipelov A. S., Eremenko O. V., Piskunov A. V., Fukin G. K., Lyssenko K. A., Russ. Chem. Bull. 2013, 62, 1815–1828. [Google Scholar]
- 20. Arrowsmith M., Hill M. S., Kociok-Köhn G., Organometallics 2014, 33, 206–216. [Google Scholar]
- 21. Wang J., Ganguly R., Yongxin L., Díaz J., Soo H. S., García F., Inorg. Chem. 2017, 56, 7811–7820. [DOI] [PubMed] [Google Scholar]
- 22. Johnson L. K., Killian C. M., Brookhart M., J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar]
- 23. Rosa V., Santos C. I. M., Welter R., Aullón G., Lodeiro C., Avilés T., Inorg. Chem. 2010, 49, 8699–8708. [DOI] [PubMed] [Google Scholar]
- 24. Fedushkin I. L., Makarov V. M., Sokolov V. G., Fukin G. K., Maslov M. O., Ketkov S. Y., Russ. Chem. Bull. 2014, 63, 870–882. [Google Scholar]
- 25. Villa M., Miesel D., Hildebrandt A., Ragaini F., Schaarschmidt D., Jacobi von Wangelin A., ChemCatChem 2017, 9, 3203–3209. [Google Scholar]
- 26. Quintal S., Pires da Silva M. J., Martins S. R. M., Sales R., Félix V., Drew M. G. B., Meireles M., Mourato A. C., Nunes C. D., Saraiva M. S., Machuqueiro M., Calhorda M. J., Dalton Trans. 2019, 48, 8449–8463. [DOI] [PubMed] [Google Scholar]
- 27. Tanahashi H., Ikeda H., Tsurugi H., Mashima K., Inorg. Chem. 2016, 55, 1446–1452. [DOI] [PubMed] [Google Scholar]
- 28. Matei I., Lixandru T., Bul. Inst. Politeh. Iasi 1967, 13, 245–255. [Google Scholar]
- 29. Dvolaitzky M., C. R. Acad. Sci. Ser. IIc 1969, 268, 1811–1813. [Google Scholar]
- 30. van Asselt R., Elsevier C. J., J. Mol. Catal. 1991, 65, L13-L19. [Google Scholar]
- 31. Ittel S. D., Johnson L. K., Brookhart M., Chem. Rev. 2000, 100, 1169–1204. [DOI] [PubMed] [Google Scholar]
- 32. Zhao M., Chen C. L., ACS Catal. 2017, 7, 7490–7494. [Google Scholar]
- 33. Anderson W. C., Rhinehardt J. L., Tennyson A. G., Long B. K., J. Am. Chem. Soc. 2016, 138, 774–777. [DOI] [PubMed] [Google Scholar]
- 34. Kaiser J. M., Anderson W. C., Long B. K., Polym. Chem. 2018, 9, 1567–1570. [Google Scholar]
- 35. Wang J., Soo H. S., Garcia F., Commun. Chem. 2020, 3, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen C., Liu F.-S., Szostak M., Chem. Eur. J. 2021, 27, 4478–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmidt R., Griesbaum K., Behr A., Biedenkapp D., Voges H.-W., Garbe D., Paetz C., Collin G., Mayer D., Höke H., In: Ullmann's Encyclopedia of Industrial Chemistry (Eds.: Gerhartz W., Yamamoto Y. S.), VCH, Weinheim, 1985. –1996. [Google Scholar]
- 38. Gasperini M., Ragaini F., Cenini S., Organometallics 2002, 21, 2950–2957. [Google Scholar]
- 39. Liu H., Zhao W., Hao X., Redshaw C., Huang W., Sun W.-H., Organometallics 2011, 30, 2418–2424. [Google Scholar]
- 40. Ragaini F., Gasperini M., Parma P., Gallo E., Casati N., Macchi P., New J. Chem. 2006, 30, 1046. [Google Scholar]
- 41. Ragaini F., Gasperini M., Gallo E., Macchi P., Chem. Commun. 2005, 1031–1033. [DOI] [PubMed] [Google Scholar]
- 42. Hagar M., Ragaini F., Monticelli E., Caselli A., Macchi P., Casati N., Chem. Commun. 2010, 46, 6153–6155. [DOI] [PubMed] [Google Scholar]
- 43. Viganò M., Ferretti F., Ragaini F., Macchi P., Inorg. Chim. Acta 2018, 483, 305–309. [Google Scholar]
- 44. Moore J. A., Vasudevan K., Hill N. J., Reeske G., Cowley A. H., Chem. Commun. 2006, 2913–2915. [DOI] [PubMed] [Google Scholar]
- 45. Ng Y. Y., Tan L. J., Ng S. M., Chai Y. T., Ganguly R., Du Y., Yeow E. K. L., Soo H. S., ACS Catal. 2018, 8, 11277–11286. [Google Scholar]
- 46. Outis M., Rosa V., Laia C. A. T., Lima J. C., Barroso S., Carvalho A. L., Calhorda M. J., Avilés T., Eur. J. Inorg. Chem. 2020, 2900–2911. [Google Scholar]
- 47. Hasan K., Wang J., Pal A. K., Hierlinger C., Guerchais V., Soo H. S., Garcia F., Zysman-Colman E., Sci. Rep. 2017, 7, 15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Viganò M., Ferretti F., Caselli A., Ragaini F., Rossi M., Mussini P., Macchi P., Chem. Eur. J. 2014, 20, 14451–14464. [DOI] [PubMed] [Google Scholar]
- 49. Gasperini M., Ragaini F., Organometallics 2004, 23, 995–1001. [Google Scholar]
- 50. Hasan K., Zysman-Colman E., J. Phys. Org. Chem. 2013, 26, 274–279. [Google Scholar]
- 51. Nishimura S., Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001. [Google Scholar]
- 52. de Vries J. G., Elsevier C. J., The Handbook of Homogeneous Hydrogenation, Wiley, 2006. [Google Scholar]
- 53. van Laren M. W., Elsevier C. J., Angew. Chem. Int. Ed. 1999, 38, 3715–3717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 3926–3929. [Google Scholar]
- 54. Dedieu A., Humbel S., Elsevier C., Grauffel C., Theor. Chem. Acc. 2004, 112. [Google Scholar]
- 55. Kluwer A. M., Koblenz T. S., Jonischkeit T., Woelk K., Elsevier C. J., J. Am. Chem. Soc. 2005, 127, 15470–15480. [DOI] [PubMed] [Google Scholar]
- 56. Guo H., Zheng Z., Yu F., Ma S., Holuigue A., Tromp D. S., Elsevier C. J., Yu Y., Angew. Chem. Int. Ed. 2006, 45, 4997–5000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5119–5122. [Google Scholar]
- 57. Wekesa F. S., Arias-Ugarte R., Kong L., Sumner Z., McGovern G. P., Findlater M., Organometallics 2015, 34, 5051–5056. [Google Scholar]
- 58. Sandl S., Maier T. M., van Leest N. P., Kröncke S., Chakraborty U., Demeshko S., Koszinowski K., de Bruin B., Meyer F., Bodensteiner M., Herrmann C., Wolf R., Jacobi von Wangelin A., ACS Catal. 2019, 9, 7596–7606. [Google Scholar]
- 59. Maier T. M., Sandl S., Shenderovich I. G., Jacobi von Wangelin A., Weigand J. J., Wolf R., Chem. Eur. J. 2019, 25, 238–245. [DOI] [PubMed] [Google Scholar]
- 60. Saini A., Smith C. R., Wekesa F. S., Helms A. K., Findlater M., Org. Biomol. Chem. 2018, 16, 9368–9372. [DOI] [PubMed] [Google Scholar]
- 61. Raj Tamang S., Cozzolino A. F., Findlater M., Org. Biomol. Chem. 2019, 17, 1834–1838. [DOI] [PubMed] [Google Scholar]
- 62. Singh A., Shafiei-Haghighi S., Smith C. R., Unruh D. K., Findlater M., Asian J. Org. Chem. 2020, 9, 416–420. [Google Scholar]
- 63. Maier T. M., Gawron M., Coburger P., Bodensteiner M., Wolf R., van Leest N. P., de Bruin B., Demeshko S., Meyer F., Inorg. Chem. 2020, 59, 16035–16052. [DOI] [PubMed] [Google Scholar]
- 64. Biffis A., Centomo P., Del Zotto A., Zecca M., Chem. Rev. 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- 65. van Asselt R., Elsevier C. J., Organometallics 1992, 11, 1999–2001. [Google Scholar]
- 66. van Asselt R., Elsevier C. J., Tetrahedron 1994, 50, 323–334. [Google Scholar]
- 67. van Asselt R., Elsevier C. J., Organometallics 1994, 13, 1972–1980. [Google Scholar]
- 68. van Asselt R., Rijnberg E., Elsevier C. J., Organometallics 1994, 13, 706–720. [Google Scholar]
- 69. Elsevier C. J., Coord. Chem. Rev. 1999, 185–186, 809–822. [Google Scholar]
- 70. van Asselt R., Vrieze K., Elsevier C. J., J. Organomet. Chem. 1994, 480, 27–40. [Google Scholar]
- 71. van Belzen R., Hoffmann H., Elsevier C. J., Angew. Chem. Int. Ed. Engl. 1997, 36, 1743–1745. [Google Scholar]
- 72. Gholinejad M., Karimkhani V., Kim I., Appl. Organomet. Chem. 2014, 28, 221–224. [Google Scholar]
- 73. Gottumukkala A. L., Teichert J. F., Heijnen D., Eisink N., van Dijk S., Ferrer C., van den Hoogenband A., Minnaard A. J., J. Org. Chem. 2011, 76, 3498–3501. [DOI] [PubMed] [Google Scholar]
- 74. Enquist P.-A., Nilsson P., Sjöberg P., Larhed M., J. Org. Chem. 2006, 71, 8779–8786. [DOI] [PubMed] [Google Scholar]
- 75. Magurudeniya H. D., Sista P., Westbrook J. K., Ourso T. E., Nguyen K., Maher M. C., Alemseghed M. G., Biewer M. C., Stefan M. C., Macromol. Rapid Commun. 2011, 32, 1748–1752. [DOI] [PubMed] [Google Scholar]
- 76. Bridges C. R., McCormick T. M., Gibson G. L., Hollinger J., Seferos D. S., J. Am. Chem. Soc. 2013, 135, 13212–13219. [DOI] [PubMed] [Google Scholar]
- 77. Bridges C. R., Yan H., Pollit A. A., Seferos D. S., ACS Macro Lett. 2014, 3, 671–674. [DOI] [PubMed] [Google Scholar]
- 78. Ragaini F., Cenini S., Gallo E., Caselli A., Fantauzzi S., Curr. Org. Chem. 2006, 10, 1479–1510. [Google Scholar]
- 79. Ferretti F., Formenti D., Ragaini F., Rend. Fis. Acc. Lincei 2017, 28, 97–115. [Google Scholar]
- 80. Ferretti F., Ramadan D. R., Ragaini F., ChemCatChem 2019, 11, 4450–4488. [Google Scholar]
- 81. Ragaini F., Cenini S., Tollari S., J. Mol. Catal. 1993, 85, L1-L5. [Google Scholar]
- 82. Ragaini F., Cenini S., Gasperini M., J. Mol. Catal. A 2001, 174, 51–57. [Google Scholar]
- 83. Viganò M., Ragaini F., Buonomenna M. G., Lariccia R., Caselli A., Gallo E., Cenini S., Jansen J. C., Drioli E., ChemCatChem 2010, 2, 1150–1164. [Google Scholar]
- 84. Cenini S., Ragaini F., Tollari S., Paone D., J. Am. Chem. Soc. 1996, 118, 11964–11965. [Google Scholar]
- 85. Ragaini F., Cenini S., Tollari S., Tummolillo G., Beltrami R., Organometallics 1999, 18, 928–942. [Google Scholar]
- 86. Ragaini F., Cenini S., Borsani E., Dompé M., Gallo E., Moret M., Organometallics 2001, 20, 3390–3398. [Google Scholar]
- 87. Arrowsmith M., Hill M. S., Kociok-Köhn G., Organometallics 2011, 30, 1291–1294. [Google Scholar]
- 88. Yakub A. M., Moskalev M. V., Bazyakina N. L., Fedushkin I. L., Russ. Chem. Bull. 2018, 67, 473–478. [Google Scholar]
- 89. Cimino A., Moscatelli F., Ferretti F., Ragaini F., Germain S., Hannedouche J., Schulz E., Luconi L., Rossin A., Giambastiani G., New J. Chem. 2016, 40, 10285–10293. [Google Scholar]
- 90. Li L., Lopes P. S., Figueira C. A., Gomes C. S. B., Duarte M. T., Rosa V., Fliedel C., Avilés T., Gomes P. T., Eur. J. Inorg. Chem. 2013, 1404–1417. [Google Scholar]
- 91. Li L., Lopes P. S., Rosa V., Figueira C. A., Lemos M. A. N. D. A., Duarte M. T., Avilés T., Gomes P. T., Dalton Trans. 2012, 41, 5144–5154. [DOI] [PubMed] [Google Scholar]
- 92. Nunes C. D., Vaz P. D., Félix V., Veiros L. F., Moniz T., Rangel M., Realista S., Mourato A. C., Calhorda M. J., Dalton Trans. 2015, 44, 5125–5138. [DOI] [PubMed] [Google Scholar]
- 93. Fomenko I. S., Gushchin A. L., Shul'pina L. S., Ikonnikov N. S., Abramov P. A., Romashev N. F., Poryvaev A. S., Sheveleva A. M., Bogomyakov A. S., Shmelev N. Y., Fedin M. V., Shul'pin G. B., Sokolov M. N., New J. Chem. 2018, 42, 16200–16210. [Google Scholar]