

Abstract
The climate fluctuations of the Quaternary shaped the movement of species in and out of glacial refugia. In Europe, the majority of species followed one of the described traditional postglacial recolonization routes from the southern peninsulas towards the north. Like most organisms, barn owls are assumed to have colonized the British Isles by crossing over Doggerland, a land bridge that connected Britain to northern Europe. However, while they are dark rufous in northern Europe, barn owls in the British Isles are conspicuously white, a contrast that could suggest selective forces are at play on the islands. Yet, our analysis of known candidate genes involved in coloration found no signature of selection. Instead, using whole genome sequences and species distribution modelling, we found that owls colonised the British Isles soon after the last glaciation, directly from a white coloured refugium in the Iberian Peninsula, before colonising northern Europe. They would have followed a hitherto unknown post‐glacial colonization route to the Isles over a westwards path of suitable habitat in now submerged land in the Bay of Biscay, thus not crossing Doggerland. As such, they inherited the white colour of their Iberian founders and maintained it through low gene flow with the mainland that prevents the import of rufous alleles. Thus, we contend that neutral processes probably explain this contrasting white colour compared to continental owls. With the barn owl being a top predator, we expect future research will show this unanticipated route was used by other species from its paleo community.
Keywords: demographic inference, MC1R, plumage coloration, reference genome, species distribution modelling, whole‐genome resequencing
1. INTRODUCTION
The dramatic climate fluctuations of the Quaternary were key in shaping the global distribution of species and communities observed today (Ficetola et al., 2017; Hewitt, 2000). During the last glaciation, northern Europe was largely covered by ice caps, and the resulting lower sea levels unveiled an expanded coastline widely different from that of today. The inhospitable conditions throughout the continent forced many temperate species into warmer refugia, most commonly the southern peninsulas of Iberia, Italy and Balkans (Hewitt, 1999, 2011). Once temperatures started increasing about 18 thousand years BP, ice sheets melted, the sea rose and these species re‐expanded northwards into central and northern Europe, a key step in determining their modern distribution and genetic structure across the continent. Early comparative phylogeography studies described differences in the route and timing of colonisation from each refuge population and identified the main post‐glacial recolonization patterns from the south (Hewitt, 1999, 2000; Taberlet et al., 1998). However, advances in sequencing technology and the consequent increase in studies with high representation molecular markers have since provided numerous examples of alternative routes and cryptic refugia for different taxa in mainland Europe as well as on islands (Bilton et al., 1998; Deffontaine et al., 2005; García‐Vázquez et al., 2019; Herman et al., 2017; Stewart & Lister, 2001).
The colonisation of the British Isles by terrestrial organisms has often been described in the context of the main phylogeographic patterns, with mainland north‐western Europe as its origin (Hewitt, 1999, 2000; Montgomery et al.,2014). Such a route would have been facilitated by Doggerland, a large land bridge of alluvial plains that connected Great Britain (GB) to mainland northern Europe (from today's Belgium to Denmark) before submerging under the North Sea 8000 years BP (Coles, 1998; Ward et al., 2006). Most terrestrial vertebrates of GB do appear to have arrived via Doggerland, as evidenced by the similarity between its mammal fauna and that of northern rather than southern Europe (Montgomery et al., 2014). Nonetheless, some species believed to have followed this path were found to have had glacial refugia on the islands themselves (Stewart & Lister, 2001), including plants (Kelly et al., 2010), amphibians (Snell et al., 2005; Teacher et al., 2009) and mammals (Boston et al., 2015; Lister, 1984). Some taxa revealed other surprising post‐glacial patterns such as colonization of the British Isles from multiple refugia in independent waves (badger: O’Meara et al., 2012; water vole: Brace et al., 2016) and even separate colonisation of Ireland and GB (stoat: Martínková et al., 2007).
Barn owls (Tyto alba) recolonised western Europe following the last glaciation from a refugium in the Iberian Peninsula (Antoniazza et al., 2014; Burri et al., 2016). On the mainland, barn owl ventral plumage colouration follows a latitudinal cline ranging from mostly white in the southern populations to dark rufous in the north (Antoniazza et al., 2010, 2014). Despite their post‐glacial expansion route, the clinal variation in colour was not a neutral byproduct of range expansion, but was rather created and maintained by an independent post‐glacial selective process (Antoniazza et al., 2014). The genetic basis of this pheomelanin‐based trait is not fully understood, but a specific nonsynonymous variant (V126I) in the melanocortin‐1 receptor (MC1R) gene has been found to explain roughly 30% of its variation in Europe (San‐Jose et al., 2015). The derived MC1R rufous allele produces the darkest owl phenotypes and follows the European colour cline of increasing frequency with latitude (Burri et al., 2016).
It is hypothesised that, given their aversion to crossing large water bodies, barn owls recolonized Great Britain following the traditional north‐western route by crossing over Doggerland (Martin, 2017). However, barn owls from the British Isles are famously white (Martin, 2017; Roulin & Randin, 2016) in stark contrast to their darker mainland counterparts at similar latitudes. Over‐land expansion from a north‐western European population, inhabited mostly by rufous owls with 10%–45% rufous MC1R allele, would be at odds with the whiteness of the GB population. This disparity is especially startling, given that rufous individuals disperse further than white ones (van den Brink et al.,2012; Roulin, 2013), and would thus be more likely to colonise the islands in the first place. Finally, with GB being a recently isolated island, its avifauna is very similar to that of continental Europe (albeit less species rich), and examples of such phenotypic divergence from the mainland are rare; the barn owl is thus an intriguing exception. Being sensitive to extreme cold (Altwegg et al., 2006), a northern refugium seems unlikely. However, such phenotypic disparity suggests that, unless strong selective pressure is involved, the colonisation timing and route of barn owls of the British Isles might have been less straightforward than has been assumed.
Here, we address the post‐glacial colonisation history of barn owls in the British Isles in light of the puzzling whiteness of their plumage. First, with a new broad sampling of 147 individuals from western Europe, we confirm that owls from the British Isles do not fit into the expected colouration and MC1R pattern of the mainland, with darker individuals at higher latitudes. Taking advantage of a highly contiguous newly‐assembled reference genome and using the whole‐genome sequences of 61 individuals, we use the neutral genetic structure to model the demographic history of barn owl colonisation of the northern part of Europe and the British Isles from a glacial refugium in Iberia. Then, we use ringing data to support estimations of current gene flow. Lastly, we investigate the potential role of other colour‐linked genes in maintaining the phenotypic disparity in plumage colour between the British Isles and mainland Europe.
2. MATERIALS AND METHODS
2.1. Tissue sampling, MC1R genotyping and colour measurement
In total, 147 individual barn owls were sampled for this study from six European populations (Table S1): Ireland (IR), Great Britain (GB), France (FR), Switzerland (CH), Denmark (DK) and Portugal (PT). This scheme allowed the sampling along the continental latitudinal colour cline, from the refugium white population of PT up to the northern dark rufous DK. Central and western continental Europe were colonised from the Iberia Peninsula (Antoniazza et al., 2014; Burri et al., 2016) and therefore we did not sample any populations further east as they would be highly unlikely to contribute to the colonisation of the British Isles. A denser sampling was performed in the Great Britain and Ireland (total n = 113) as this was the first time these populations were studied, while for the mainland populations data was already available (Burri et al., 2016). Genomic DNA was extracted from blood, feathers or soft tissue using the DNeasy Blood & Tissue kit (Qiagen) following the manufacturer's instructions, including RNA digestion with RNase A. A previously established allelic discrimination (AD) assay (San‐Jose et al., 2015) was used to molecularly determine individual genotypes at the amino acid position 126 of the Melanocortin 1 receptor (MC1R) gene of the 147 individuals (Table S1). Briefly, the region surrounding the MC1R mutation was pre‐amplified and its product was diluted 100 times for the AD assay using allele‐specific fluorescent‐labelled probes in an ABI 7500 qPCR machine (Life Technologies). On each plate, positive controls of each genotype and a negative control were also run. Additional allelic frequencies at this locus published in Burri et al. (2016) from the mainland populations of interest to this study were used for context (n = 247 individuals; Appendix S1).
For all individuals with available breast feathers (n = 145), pheomelanin‐based colour, which is homogenous on barn owl breast feathers, was estimated as the brown chroma of the reflectance spectra (for detailed description see Antoniazza et al., 2010). Briefly, the brown chroma represents the ratio of the red part of the spectrum (600–700 nm) to the complete visible spectrum (300–700 nm), with higher values indicating larger amounts of reddish pigments on the feathers. The reflectance of four points of the top of three overlapping breast feathers was measured using a S2000 spectrophotometer (Ocean Optics) and a dual deuterium and halogen 2000 light source (Mikropackan, Mikropack). An individual's brown chroma score was obtained as the average of these four points. This method correlates well with observational assessments using colour chips (r = –0.78, p < .0001, Dreiss & Roulin, 2010) and has high repeatability (97.6%; Antoniazza et al., 2010). Brown chroma data from Burri et al. (2016) were used to complete the data set, using the same individuals as for the MC1R analysis (Appendix S1). Given the marked non‐normality of the data, a nonparametric Kruskal‐Wallis test was performed to detect differences in coloration between the six populations. Further, a pairwise Wilcoxon rank sum test was used to identify significant differences between pairs of populations using a Bonferroni correction.
2.2. New reference genome
As the available reference genome for the European Tyto alba was fragmented (Ducrest et al., 2020), a new reference was produced in order to achieve a near chromosome‐level assembly. A full description of the process and its detailed results are given in Appendix S2. Briefly, a long‐read PacBio library was produced from a blood sample of a Swiss individual at an expected coverage of 100x for the barn owl's 1.3 Gb genome. falcon and falcon‐unzip v.3 (Chin et al., 2016) were used to assemble PacBio reads. Then, a high molecular weight DNA Bionano optical mapping library was used to assemble pacbio contigs into scaffolds. Finally, repeated regions were identified using repeatmodeler v.1.0.11 (Smit & Hubley, 2008–2015) and masked with repeatmasker v.4.0.7 (Smit, et al., 2013–2015). Coding regions were identified using the braker2 pipeline v.2.0.1 (Brůna et al.,2020; Hoff et al., 2016, 2019; Stanke et al., 2006, 2008).
2.3. Whole‐genome resequencing and SNP calling
For the population genomics analyses of this study, the whole genomes of 61 out of the 147 individual barn owls were sequenced (Table S2). In addition, one eastern (T. javanica from Singapore) and one American barn owl (T. furcata from California, USA) were used as outgroups. See Methods S1 for a complete description of the library preparation, sequencing, SNP calling and filtering. Briefly, individual 100 bp TruSeq DNA PCR‐free libraries (Illumina) were sequenced with Illumina HiSeq 2500 high‐throughput paired‐end sequencing technology at the Lausanne Genomic Technologies Facility (GTF, University of Lausanne, Switzerland). The bioinformatics pipeline used to obtain analysis‐ready SNPs was adapted from the Genome Analysis Toolkit (GATK) Best Practices (van der Auwera et al., 2013) to a non‐model organism following the developers’ recommendations, producing a full data set of 6,721,999 SNP for the 61 European individuals with an average coverage of 21.1x (3.36 SD).
2.4. Population structure and genetic diversity
To investigate population structure among our samples, sNMF v.1.2 (Frichot et al., 2014) was run for K 2 to 6 in 25 replicates to infer individual clustering and admixture proportions. For this analysis, singletons were excluded and the remaining SNPs were pruned for linkage disequilibrium (LD) with PLINK v1.946 (Purcell et al., 2007; parameters ‐indep‐pairwise 50 10 0.1) as recommended by the authors, yielding 319,801 SNP. The same data set was used to perform a principal component analysis (PCA) with the R package SNPRelate (Zheng et al., 2012). Treemix (Pickrell & Pritchard, 2012) was used to infer population splits in our data, using the LD‐pruned data set further filtered to include no missing data (180,764 SNP). To detect meaningful admixture between populations, 10 replicates were run for 0 to 8 migration events, with the tree rooted on the PT population, representative of the glacial refugium. An extra run without migration events was conducted with a North American owl as an outgroup in the data set to verify that the root did not affect the topology of the tree.
To estimate population statistics, individuals found to be mis‐assigned to their given population based on genetic structure analyses (PCA and sNMF) were removed so as not to bias allelic frequencies (n = 3 individuals from Ireland). Individual expected and observed heterozygosity and population‐specific private alleles were estimated from the full data set (6,721,999 SNP) using custom R scripts for each genetic lineage identified by sNMF with K = 4 (Figure 1c). To account for differences in sample sizes, private alleles were calculated by randomly sampling 9 individuals from the larger populations (GB and western Europe) 10 times in a bootstrap‐fashion and estimating the mean. Individual‐based relatedness (β; Weir & Goudet, 2017), inbreeding coefficient for SNP data, overall and population pairwise F ST (Weir & Cockerham, 1984) were calculated from the full data set with SNPRelate.
FIGURE 1.
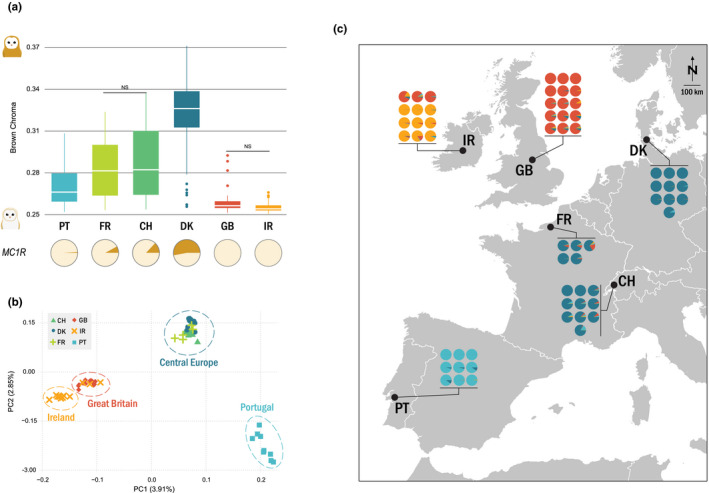
Coloration and genetic structure of barn owl populations in western Europe. (a) Brown chroma distribution and MC1R allelic frequencies of each studied population (total n = 145). Higher brown chroma indicates redder owls. NS denotes the nonsignificant pairwise comparisons. The pies below the plot illustrate the populations’ MC1R allelic frequencies: the rufous allele in brown and the white in beige. (b) PCA based on the pruned data set (319,801 SNP) of the 61 individuals whose whole genome was resequenced. Point shape and colour denote populations according to the legend. Dashed circles enclose sample clusters identified in sNMF. Values in parenthesis indicate the percentage of variance explained by each axis. (c) Population structure. Small pie charts denote the individual proportion of each of k = 4 lineages as determined by sNMF. Black dots are located at the approximate centroid of each sampled population
2.5. Gene flow and migration analyses
2.5.1. Migration surface estimate
The estimated effective migration surface (EEMS) v.0.0.9 software (Petkova et al., 2016) was used to visualize geographic regions with higher or lower than average levels of gene flow within our data set. The provided tool bed2diff was used to compute the matrix of genetic dissimilarities, from the data set pruned for LD produced above. The free google maps api v.3 tool (http://www.birdtheme.org/useful/v3tool.html) was used to draw the polygon outlining the study area in western Europe (Figure 2a). EEMS was run with 750 demes in three independent chains of 5 million MCMC iterations with a 1 million iterations burnin. Results were checked for MCMC chain convergence visually and through the linear relation between the observed and fitted values for within‐ and between‐demes estimates using the accompanying r package reemsplots v.0.0.1 (Petkova et al., 2016). The three MCMC chains were combined to produce maps of effective migration and diversity surfaces with the provided functions in reemsplots.
FIGURE 2.
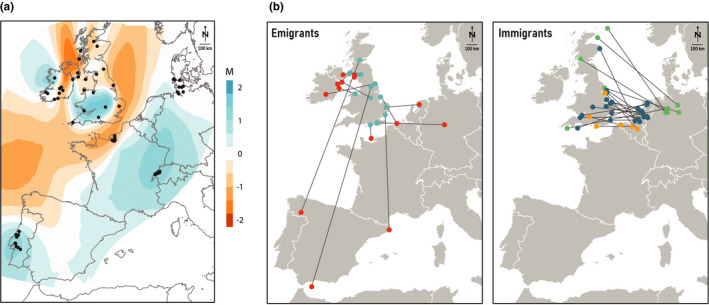
Barn owl gene flow and dispersal between the British Isles and mainland Western Europe. (a) Estimated effective migration surface (EEMS) based on whole‐genome data. Blue and orange shading denote regions of higher and lower than average gene flow, respectively. Black dots indicate individual sampling location. (b) Ringing and recapture locations of barn owls known to have flown out of (Emigrants) or into (Immigrants) Great Britain from 1910 to 2019, based on data courtesy of EURING. Lines simply connect two capture points and do not represent the actual path travelled by birds. Emigrant ringing locations in GB are coloured in blue, and recaptures in red. Immigrants into GB are coloured according to country of origin (orange – Belgium; green – Germany; blue – The Netherlands)
2.5.2. Treatment and analyses of capture‐recapture data
In addition to genomic data, recapture data of ringed barn owls across Europe were obtained from the EURING database (obtained in March 2020; du Feu et al., 2016; Speek et al., 2001). Specifically, we estimated the frequency of crossings over open water (flights between two land masses over a water body) between GB and central and western Europe, as well as between GB and Ireland. To do so, we kept records of birds that had been recaptured at least once after ringing (n = 94,797 recaptures, n = 80,083 individuals, from 1910 to 2019) and filtered the accuracy of the “time of capture” parameter to a period of within 6 weeks of the reported date to exclude potentially unreliable data points. We extracted the number of birds ringed and recaptured in GB and Ireland, as well as in the countries that produced or received migrant birds from these islands and western Europe (Belgium, Denmark, France, Spain, Germany, Switzerland and The Netherlands). Crossings between GB and Ireland and to/from the mainland are reported and include birds that were found dead in the sea (n = 8). All counts and percentages reported are relative to the number of individual birds recaptured (rather than number of recapture events, as a single bird can be recaptured multiple times).
2.6. Post‐glacial species distribution
To support the demographic scenarios tested in the following section, we modelled the past spatial distribution of barn owls in western Europe, in order to identify the regions of high habitat suitability at the Last Glacial Maximum (LGM, 20,000 years BP). A complete description of the models can be found in Methods S1. Briefly, using maximum entropy modelling (MaxEnt), a presence‐only based modelling tool, we built species distribution models (SDM) for the Western Palearctic (Figure S1) based on climatic variables extracted from the WorldClim database (Figure S2; Hijmans et al., 2005) at 5 arc min resolution. The best combination of feature and regularization multiplier based on the corrected AIC (as recommended by Warren & Seifert, 2011) was achieved with a quadratic model with 1 as regularization multiplier (Table S6). Then, the output of the models was transformed into a binary map of suitability in which only cells suitable in 90% of the models are presented as such in the map. All models were then projected to the mid‐Holocene (6000 years BP) and LGM (20,000 years BP) conditions extracted from WorldClim at the same resolution as current data. For each timepoint, the results of the models were merged and transformed into a binary map as for the current data.
2.7. Maximum‐likelihood demographic inference
2.7.1. Data preparation
To discriminate between different demographic scenarios for the colonisation of the British Isles by barn owls we used the software fastsimcoal2 (Excoffier et al., 2013; Excoffier & Foll, 2011). Individuals and variants in the data set used here as input went through additional filtering steps in an attempt to ensure neutrality and homogeneity between samples (Methods S1). Given their similarity (Figure 1b,c), the original populations of France, Denmark and Switzerland were combined into a western European population (EU) with eight individuals. The remaining populations were Portugal (PT), Great Britain (GB) and Ireland (IR), also with eight individuals each (Table S1, Appendix S1). Population pairwise site frequency spectrum (SFS) were produced from the filtered data set of 739,168 SNP.
2.7.2. Demographic scenarios and parameters
Three different scenarios of colonization of western Europe and the British Isles from the Iberian Peninsula were simulated (Figure 3), distinguishable by the difference in timing and origin of the insular populations: north‐western (NW) European origin, Iberian origin and insular refugium. Each scenario was further split in two versions (A and B) to accommodate small changes in topology. For all scenarios, wide search ranges for initial simulation parameters were allowed for population sizes, divergence times and migration rates while accounting for census and geological data (Table S8). Splits were preceded by instantaneous bottlenecks, in which the founding population size was drawn from a log‐uniform distribution between 0.01 and 0.5 of current population sizes. Splits represent the time at which two populations coalesce into the past or, in a forward‐in‐time interpretation, when two populations are differentiated enough to be considered as independent evolutionary units. Due to the model simplification with instantaneous bottlenecks, splits occur in a near‐discrete fashion in one single generation. All times were relative to the end of the last glaciation (18,000 years BP, rounded to 6000 generations ago), bounded between the present and the previous demographic event in the model.
FIGURE 3.
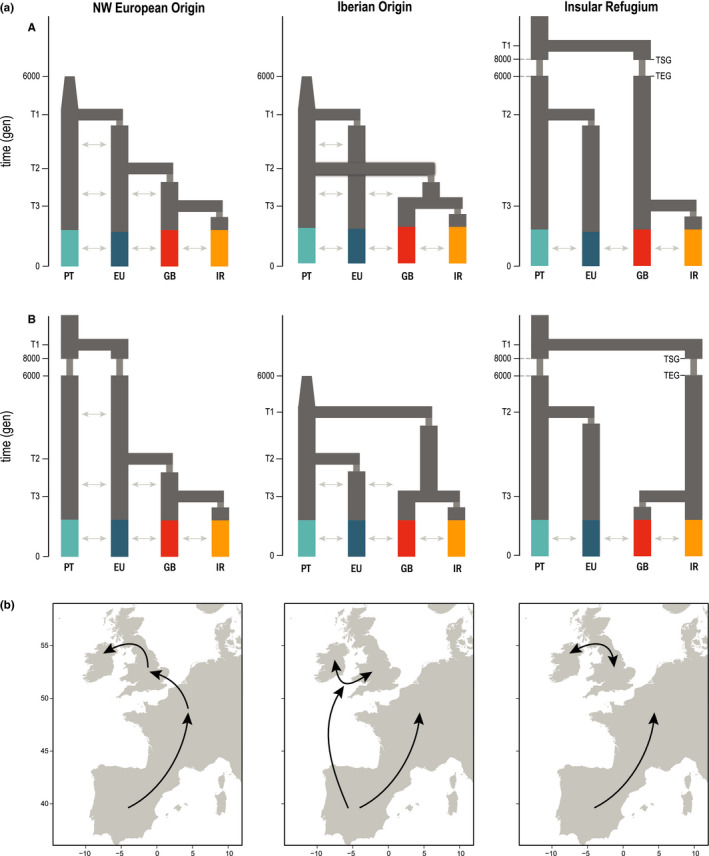
Hypothesized demographic scenarios for the colonization of the British Isles by barn owls. (a) Tested demographic scenarios for the colonization of the British Isles by barn owls. There are three main topologies – NW European Origin, Iberian Origin and Insular Refugium – each with two version (A & B; first and second line, respectively). The four main genetic clusters in our data set were used: Portugal (PT), Western Europe (EU), Great Britain (GB) and Ireland (IR). Population EU in this analysis is composed of individuals from FR and DK. Indicated times were fixed in the models (6000 and 8000 generations ago), and the remaining time parameters were inferred relative to them or to the event immediately before (e.g., T3 was bound between the present and T2). Cones depict post‐glacial size increase and arrows gene flow between adjacent populations. In Insular Refugium topologies, TSG, time of start of glaciation in the insular lineage; TEG, time of end of glaciation in the insular lineage. (b) Schematic representation of the colonisation route to the British Isles for each scenario
In scenario NW European origin A, after an initial post‐glaciation size expansion, the ancestral PT population colonized western Europe. From here, barn owls sequentially reached Great Britain and Ireland, potentially across the Doggerland land bridge. In version B, a smaller second glacial refugium is hypothesized to have existed in southern France, above the Pyrenees, as the founder of the western European population after the glaciation. In both versions, barn owls reached the British Isles from western Europe. In the Iberian origin scenarios, the insular populations originated directly from PT. Spatially, this could have taken place across now submerged land in the Bay of Biscay, west of current‐day France and north of Spain. Genetically, the insular birds would have been derived from the initial genetic pool in Iberia rather than from the subset in western Europe. Versions A and B of this scenario differ in the timing of colonization, with Europe being colonized before the islands in A and after in B. Lastly, the insular refugium scenarios hypothesize a separate and smaller glacial refugium in the south of the British Isles that would have been the origin of today's populations on the islands. Such refugia have been described for some terrestrial organisms albeit not birds (Kelly et al., 2010; Ravinet et al., 2014; Stewart & Lister, 2001; Teacher et al., 2009). Western Europe would be colonized post‐glacially from PT. In version A and B of this scenario, the second glacial refugium would be part of an ancestral GB or IR population, respectively.
In summary, the NW European origin scenario reflects the shortest overland path based on current geography, whereas the remaining scenarios attempt to address the whiteness in the British Isles by avoiding shared ancestry with darker‐coloured populations at different time scales, as well as the changes in the coastline during and after the last glaciation. For all scenarios, migration was allowed between neighbouring populations (Figure 3; Table S8).
2.7.3. Demographic inference
Demographic simulations and parameter inference were performed under a composite‐likelihood approach based on the joint SFS as implemented in fastsimcoal2 (Excoffier et al., 2013; Excoffier & Foll, 2011). For each scenario, 100 independent estimations with different initial values were run (Methods S1). The best‐fitting scenario was determined based on Akaike's information criterion (AIC; Akaike, 1974) and confirmed through the examination of the likelihood ranges of each scenario as proposed in Kocher et al. (1989). For the best‐fitting scenario, nonparametric bootstrapping was performed to estimate 95% confidence intervals (CI) of the inferred parameters. For each block‐bootstrapped SFS, 50 independent parameter inferences were run for the best‐fitting scenario (see Methods S1 for a detailed explanation).
2.8. Genome scans of colour‐linked genes
Genome‐wide scans were used to compare patterns of divergence and diversity between populations. SNPs were filtered to a minimum derived allelic frequency of 5%, and vcftools was used to calculate nucleotide diversity (π) for each population and to estimate F ST (Weir & Cockerham, 1984) between pairs of populations in 20 kb sliding windows with 5kb steps across the whole genome. For our comparisons, Great Britain and Ireland were combined as British Isles; France and Switzerland as western Europe. Denmark was not included in the latter due to its markedly darker phenotype (Figure 1a). The British Isles were compared to all other groups of individuals: white in Portugal, intermediate in western Europe and dark rufous in Denmark. Further, Portugal and Denmark were also compared.
In our genomic data set, owls from the British Isles and Portugal carried the same genotypes at the MC1R mutation (100% V allele) despite there being considerably more variation in colour among Portuguese individuals (Figure 1a). As such, we first investigated whether insular individuals showed particular diversity or divergence at the surrounding positions within the MC1R gene that could relate to their pure white colour. Since the MC1R gene in barn owls is particularly GC rich (San‐Jose et al., 2015) and is embedded in a region with a lot of homopolymeric sequences, the sequencing in this region has a considerably lower coverage than the average of the genome. To account for this, the scaffold containing this gene was extracted from the raw SNP set and re‐filtered with similar site thresholds as described above, except for allowing 25% overall missing data (instead of 5%), limiting the minimum individual DP to 5 (instead of 10) and the minimum minor allelic count to 3. VCFtools was used to calculate nucleotide diversity for each population and to estimate F ST (Weir & Cockerham, 1984) between pairs of populations in 5 kb sliding windows with 1kb steps along this scaffold.
Second, to widen our search to other colour‐linked genes besides MC1R, we mapped 22 autosomal candidate genes (Appendix S3) onto the reference genome using blast v.2.9.0 (Zhang et al., 2000). Windows including the candidate genes were plotted onto genomic scans (5 kb windows with 1 kb step) to check for overlap with peaks or drops in diversity and/or differentiation.
3. RESULTS
3.1. MC1R genotyping and colour measurements
Plumage colour comparisons showed that the British Isles have the whitest owls of all measured European populations (Figure 1a; X 2 = 243.28, p <.001). Most pairwise comparisons were significantly different after correction, with the exception of between GB and IR owls, and between CH and FR. As for MC1R genotyping, notably no rufous allele was found among the 113 genotyped individuals of the British Isles indicating it is absent from these populations or at very low frequency.
3.2. New reference genome
The new reference genome produced for European barn owl was a near chromosome level assembly, and has been deposited at DDBJ/ENA/GenBank under the accession JAEUGV000000000. Sequencing of the new reference genome's PacBio library yielded 7.3 million long reads with a total sum length of unique single molecules of 135 Gbp (N50 > 31Kb) yielding a realized coverage of 108x. Its assembly with FALCON and FALCON‐Unzip resulted in 478 primary contigs partially phased, and 1736 fully phased haplotigs which represented divergent haplotypes. Optical mapping with Bionano produced a final assembly of 70 scaffolds, slightly more than the barn owl's karyotype of 46 chromosomes (Ducrest et al., 2020). The final assembly was 1.25 Gbp long, with an N50 of 36 Mbp and BUSCO score of 96.9% (see Appendix S2: Table S1 for full assembly metrics). In comparison, the previous reference assembly (Ducrest et al., 2020) had 21,509 scaffolds, with an N50 of 4.6 Mbp.
3.3. Population structure and genetic diversity
Our data set was composed of four main genetic clusters identified by individual ancestry analyses (sNMF) and PCA clustering. Individuals from Portugal (PT), Great Britain (GB) and Ireland (IR) generally belonged to their specific population ancestry, while individuals from France (FR), Denmark (DK) and Switzerland (CH) formed a single western European cluster (Figure 1b,c; Figure S3). Consistently, the first axis of the PCA (Figure 1b) opposed PT to GB & IR, as seen with sNMF K = 2 (Figure S3). The second axis clustered the western European individuals together and opposed them to PT (Figure 1b). GB and IR segregate in both the first and second axes. Three barn owls sampled in Ireland showed a clear genetic signal of belonging to the Great Britain genetic cluster (Figure 1b,c; Figure S3). To avoid their interference in estimating allelic frequencies, they were omitted when estimating diversity and differentiation statistics.
The overall F ST was 0.035. Population pairwise F ST were the highest between Ireland and western Europe (Table S4). Overall, populations within western Europe showed the smallest differentiation (F ST below 0.02) and the British Isles had the highest values in comparison to all mainland populations (Table S4). Diversity estimates showed higher levels in PT than in any other population and the British Isles had the lowest (Table S3). Individual relatedness was highest within IR, followed by GB (Figure S4). On the opposite end, PT had the lowest within‐population relatedness as well as with the other populations, consistent with its higher diversity.
Analyses of genetic drift with Treemix yielded a population tree with two branches splitting from PT. The first is a long branch of drift that divides into GB and IR, while the second, shorter branch, diversified into the three western European populations (Figure 4a). Plotting the likelihood of runs and the standard error (SE) of each tree showed that including one migration event from PT to CH (migration edge weight =0.27) considerably increased the fit of the tree to the data (Figure S5).
FIGURE 4.
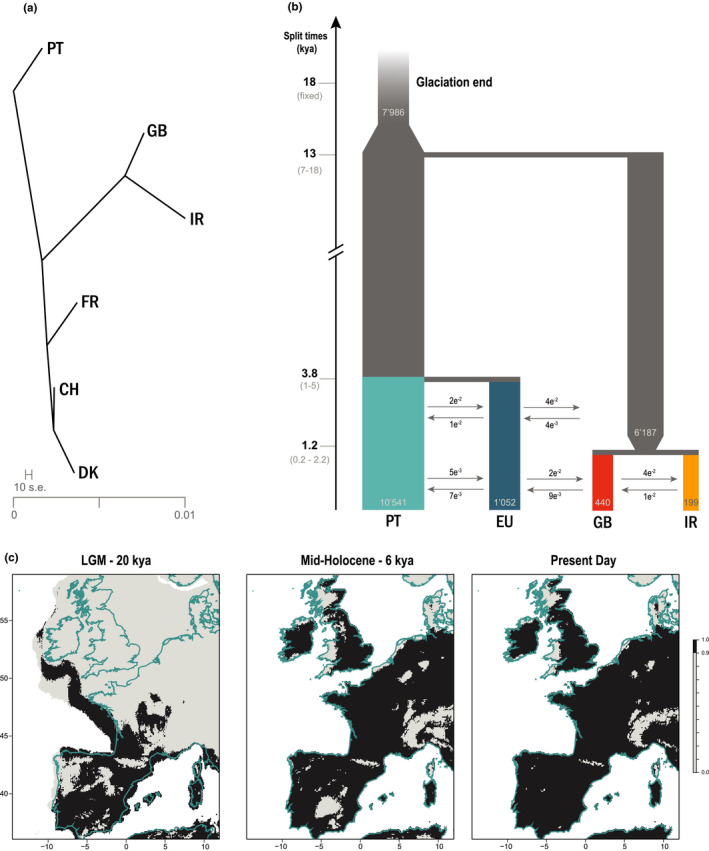
Demographic history of barn owls of the British Isles. (a) Treemix analysis with zero migration events. (b) Best supported demographic model for the colonisation of the British Isles as determined by fastsimcoal2. Time is indicated in thousands of years, with a 3‐year generation time, confidence intervals at 95% are given between brackets. Population sizes (haploid) are shown inside each population bar; arrows indicate forward‐in‐time migration rate and direction. Population EU in this analysis is composed of individuals from FR and DK. (c) Species distribution model of barn owls projected into past conditions – Last Glacial Maximum (20,000 years BP) and mid‐Holocene (6000 years BP) – compared to today's distribution. Only locations with high suitability in at least 90% model averaging are coloured in dark grey. Below that threshold cells were considered as unsuitable (lightest grey shade on the graph). Modern coastline is shown in blue
3.4. Migration and gene flow
The English Channel – including the Straits of Dover and the southernmost part of the North Sea – was identified by estimated effective migration surface (EEMS) as a region with lower than average gene flow between populations (Figure 2a). This corridor extended west to the Atlantic. Furthermore, this analysis highlighted a region of low gene flow between the British and Irish populations. It put a barrier in Ireland by separating the north from the rest of the island, effectively isolating the three individuals sampled in Ireland that genetically resemble the British and clustering them with GB, while the remaining individuals from further south in Ireland formed a separate Irish cluster.
Analyses of capture‐recapture data of ringed owls (n = 80,083 individuals, from 1910 to 2019) revealed that all individuals ringed in Ireland (n = 81 individuals) were recaptured in Ireland. As for GB, the vast majority (99.92%) of its ringed individuals (n = 17,903) were also recaptured in GB and only 14 migrated out of the island: seven to Ireland (100% of this island's immigrants) and seven to mainland Europe (Figure 2b – Emigrants; Table S5a). In the opposite direction, GB received 21 individuals from the mainland (Figure 2b – Immigrants), specifically from Belgium, the Netherlands and northern Germany (Table S5b). Of the immigrant birds, 19 were found dead, one severely injured with unknown fate, and one breeding. The latter was a female from the Netherlands, but the fate of its brood is not known. In the mainland, western European countries show considerably higher exchanges of individuals with each other (Table S5c) than with GB (Table S5b).
3.5. Post‐glacial species distribution
Habitat suitability projections for barn owls in the past showed that, at the time of the last glaciation, there was suitable land for barn owls outside of the known refugium of Iberia from a climatic perspective (Figure 4c). Specifically, south of today's British Isles there was a corridor of suitable land currently submerged, as well as along the south and western coasts of France, and a small cluster in inland southern France. At the mid‐Holocene (6000 years BP), the coastline resembled that of the present day, and the distribution of suitable habitat for barn owls resembled that of nowadays (Figure 4c).
3.6. Demographic inference
AIC and raw likelihood comparisons showed the Iberian origin B model to be the best at explaining the SFS of our data set (Table S7; Figure 4b). In this model, an ancestral insular lineage split from the mainland refugium lineage in Iberia fairly soon after the end of the glaciation, estimated at approximately 13,000 years ago (95% CI: 7000–17,000 years BP; calculated with 3‐year generation time). Only much later, the model predicted the split of the central EU population from PT at 4000 years BP (95% CI: 1000–5000 years BP) and the separation between GB and IR at 1200 years BP (95% CI: 220–2200 years BP). Estimated effective population size was the largest in the PT population, followed by EU, GB and IR (Figure 4b). Migration between populations was higher before these split than in recent times (Table S9; Ancestral vs. Recent migration). Highest recent gene flow was observed from PT to EU, agreeing with Treemix's first migration event (Figure S5). Migration levels between the two islands (GB and IR) and with the European mainland were of a similar order of magnitude and less than half of that between mainland populations (PT and EU), consistent with the two barriers to gene flow identified by EEMS (Figure 2a). Point estimates with 95% confidence intervals for all parameters of the best model are provided (Table S9), as well as single point estimates for the rest of the models (Table S8).
3.7. Genome scans of colour‐linked genes
Genome‐wide scans revealed some high peaks of differentiation between populations, but none overlapped with the colour‐linked candidate genes tested (Appendix S3). Windows containing the MC1R mutation were the most differentiated between light and dark populations (Figure 5a). However, the MC1R region around the mutation showed no particular sign of increased differentiation between pairs of populations, nor drop in diversity, with the exception of the known causal SNP between populations with different genotypes (Figure 5b; Appendix S3).
FIGURE 5.
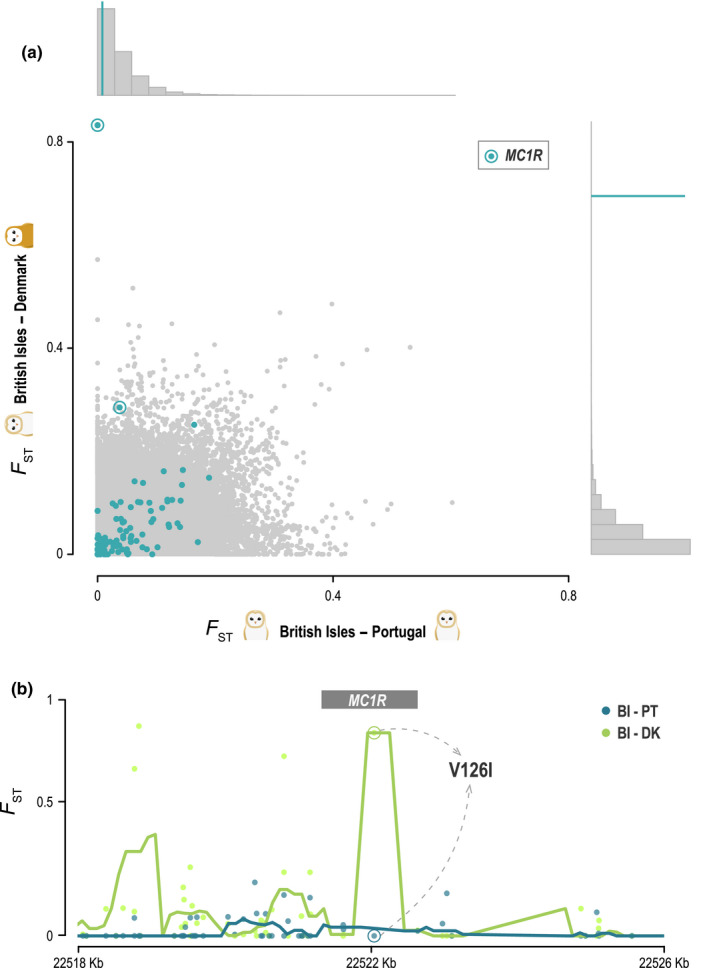
Differentiation at the colour‐linked locus V126I of the MC1R gene between differently coloured barn owl populations in Europe. (a) Genome‐wide F ST values per window (in grey, 20 Kbp windows with 5Kbp steps), between two white barn owl populations on the horizontal axis – British Isles (BI) and Portugal (PT) – and between one white and one rufous on the vertical axis – BI and Denmark (DK). The distribution of each axis is shown on the histograms. Blue dots indicate the F ST at windows containing the tested colour‐linked genes. Windows containing the MC1R are encircled, and their mean is shown with the blue line on the histograms. (b) F ST per site (dots) around the MC1R gene (grey box). Lines show the mean over sliding windows (500bp with 100bp step), for the same comparisons as above: BI and PT in blue; BI and DK in green. Circled dots indicate the V126I locus in both comparisons
4. DISCUSSION
Like most terrestrial species, barn owls are assumed to have colonized the British Isles after the last glaciation by crossing over Doggerland, a land bridge that connected GB to northern Europe. In continental Europe, barn owls display a marked latitudinal colour cline maintained through local adaptation (Antoniazza et al., 2010). However, in the British Isles they are conspicuously white in comparison to their nearest mainland counterparts questioning whether this is their source population. The currently held interpretation for their whiteness is a strong selection on this trait after colonisation. Here we provide evidence for a simpler explanation that does not require selection. Using whole‐genome sequences and demographic simulations, we show that the colour disparity can be explained by the patterns in neutral genetic differentiation, resulting from an unexpected colonization route to the British Isles. We provide evidence for an early split of the insular lineage and low levels of gene flow with the mainland. Having found no evidence of selection on colour in the British Isles, it is plausible that this population has simply remained the white colour of its founders.
4.1. Genetic isolation from the mainland
Our results based on whole genomes revealed genetic structure among western European barn owls despite shallow differentiation for a cosmopolitan bird (overall F ST 0.035) and showed genome wide genetic isolation between the islands and the mainland, accompanied by low levels of gene flow and migration. On the mainland, Portugal displayed the highest levels of genetic diversity (Table S3) and the largest estimated population size (Figure 4b; Table S9), in accordance with its known role as a glacial refugium (Antoniazza et al., 2014). While forming its own population cluster (Figure 1b,c), we found evidence of considerable gene flow towards western Europe (Figures 2a, 4a,b; Table S9), consistent with a recent split between the two populations (<5000 years BP; Figure 4a) and the relatively low differentiation between them (Table S4). This suggests that the Pyrenees are permeable to barn owl migration, unlike other higher and larger mountain ranges (Machado et al., 2018). In western Europe, barn owl populations appear to be remarkably homogenous genetically, despite covering a large geographical and colour range (Figure 1, Table S4), in accordance with previous studies of continental Europe with traditional markers (Antoniazza et al., 2010), and supported by capture‐recapture data that revealed high amounts of exchanges in western Europe (Table S5c).
Ireland and GB showed the lowest diversity and estimated effective population sizes in our study (Figure 4; Tables S3, S9). Barn owl populations of each island are genetically distinct from each other as well as from the mainland (Figure 1; Table S4). Further, they are genetically homogenous within each island, with increased individual relatedness (Figure S4) thus appearing to have a single origin (Figures 1, 4a) unlike other species that show patterns of population replacement or swamping (e.g., Marková et al., 2020; Searle et al., 2009). Genomic differentiation (Figures 1, 2a, 4a,b; Table S4) suggests gene flow between the islands and the mainland is low, with faint signals of admixture (less than 5% of individual ancestry; Figure S3) limited geographically to narrow areas on each side of the English Channel. Accordingly, capture‐recapture data showed crosses over the sea are rare in contrast to the frequent exchanges observed overland in continental Europe across considerably larger distances (Figure 2b; Table S5a–c). Specific analyses highlighted a barrier to gene flow extending from the Celtic Sea, through the English Channel to the North Sea (Figure 2a), effectively isolating the British Isles from the mainland. Between the two islands, isolation appears to be recent (<2230 years BP; Figure 4a,b; Table S9), despite relatively high genetic differentiation (Table S4) probably exacerbated by an important effect of genetic drift in such small populations. There is little sign of current pervasive admixture in either direction (Figure 1c), consistent with the role of the Irish Sea as a strong barrier. However, there are records of owls from GB migrating into northern parts of Ireland (Figure 2b – Emigrants), the most easily accessible part of the island, while avoiding major water bodies by island‐hopping from Scotland. Curiously, three of the individuals we sampled in Ireland for whole‐genome sequencing (all sampled from found carcasses in the northern part of the island) appeared to be genetically from GB (Figure 1b,c), driving EEMS to place a gene flow barrier nearly along the political border between the two countries of Ireland instead of the sea (Figure 2a). Northern Ireland appears to be inhospitable for barn owls, at least in modern times, with only 1 to 3 pairs recorded per year in the whole country (Barn Owl Report, 2019). It could be acting as an extension of the sea barrier with the birds that fly in from GB being unable to find mates and thus not contributing to the genetic pool of the southern population, accentuating the differentiation between the two islands.
4.2. Disparity in plumage coloration
Plumage colouration in barn owls, and the linked MC1R locus, follow a clinal distribution in continental Europe maintained by local adaptation (Antoniazza et al., 2010; Burri et al., 2016). Here, we formally establish that barn owls from the British Isles do not follow the continental latitudinal cline and are whiter than any continental population in Europe, including even Portugal (Figure 1a), confirming what was previously untested common knowledge among ornithologists. The rufous MC1R allele appears to be virtually absent in these populations in contrast to its close to 50% frequency at similar latitudes on the mainland, where dark morphs are positively selected (Figure 1a; Antoniazza et al., 2014; Burri et al., 2016). While genome‐wide scans confirmed the important role of the known MC1R mutation in determining rufous coloration (Figure 5a), it appears to be restricted to the SNP variant itself and not the adjacent genomic regions (Figure 5b). Our results are consistent with previous studies that showed that carrying a single copy of the rufous allele is sufficient to ensure a darker phenotype, while individuals homozygous for the white allele can have a wide range of colouration (Burri et al., 2016; San‐Jose et al., 2015).
This colour trait is probably polygenic, given that the known MC1R mutation explains only 30% of its variation (Burri et al., 2016; San‐Jose et al., 2015) and its high heritability (Roulin & Dijkstra, 2003). Other loci could act in conjunction with a homozygous white MC1R to either produce whiter birds in GB or slightly darker morphs in Iberia. However, none of the other known colour‐linked genes tested here explain how white owls homozygous for the white MC1R allele from Portugal reach darker phenotypes than those of the British Isles (Figures 1a, 5a; Appendix S3). Alternatively, it is conceivable that the phenotype we observe – colouration – simply reflects the pleiotropic effect of insular local adaptation on other linked cryptic traits. The melanocortin system regulates behaviour and physiology alongside the production of melanin, and associations between these traits are common among vertebrates (Ducrest et al., 2008; Roulin & Ducrest, 2011). Further work, potentially focusing on colour‐varied populations to avoid the confounding factor of population structure could help elucidate the genetic basis of barn owl plumage colouration. If such other loci are found, it would be fascinating to investigate their distributions and interaction with MC1R along the continental colour cline as well as on the British Isles.
4.3. Colonisation of the British Isles
Demographic simulations based on neutral sites showed that the British Isles were colonized from the glacial refugium in the Iberian Peninsula soon after the end of the glaciation (Figure 3b). This would have occurred while the British Isles were still connected to the mainland and the landmass extended considerably further south than today's islands, following a corridor of suitable climatic conditions along the coast leading west (Figure 4c). This contrasts with the suspected route over Doggerland which, despite being part of the same large European landmass at the time, was climatically unsuitable for barn owls (Figure 4c). It is also possible that the corridor on the Bay of Biscay was already occupied by barn owls in a continuous population with Iberia before becoming isolated, as this species easily maintains high over‐land gene flow (Figures 1b,c, 2a; Table S5c). Our wide confidence intervals make it hard to pinpoint exactly the time of the actual split between the insular lineage from that of Iberia, but with the fast rise of sea levels and opening of the delta in the English Channel, the southern route to the islands would have been closed by 10,000 years BP (Lambeck et al., 2014; Leorri et al., 2012). Crucially, at this time prey would already be available in the form of voles, shrews, lemmings and bats (Montgomery et al., 2014). Once separated, the insular lineage underwent a long period of genetic drift, isolated from the mainland population in Iberia but homogenous within itself before splitting between the two islands (Figure 4a,b).
On the mainland, western European populations split genetically from the Iberian refugium much later (<5000 years BP; Figure 4b). This inference of such strikingly recent split probably reflects the large population sizes and high overland gene flow (Figure 4b; Table S9) that would have maintained a relatively homogenous population from Iberia to western Europe. Only over a long period of time could they accumulate differentiation and become two genetically distinct populations, and be recognised as such by the model. This is consistent with the still low divergence currently observed between them (Table S4; Figure S3) and with a separate estimate in a new paper describing the complete history of barn owls in continental Europe (Cumer et al., 2021). Additionally, it appears that climatic conditions north of the Pyrenees took longer to become favourable for barn owls (Figure 4c), which might have delayed the north‐eastern expansion from Iberia. In contrast, the climate in the south of the British Isles was already suitable at the end of the glaciation. This could render the traditional view of Doggerland as the overland point of arrival into the islands further unlikely, as they might not have yet reached such high latitudes before Doggerland submerged 8000 years BP. Intriguingly, our demographic model predicts high migration from GB into western Europe between the splits of the latter with Iberia and between the two islands (Figure 4b), which appears unlikely with all land bridges submerged at this point (<5000 years BP). It is possible that the migration rate was inflated as the model did not allow for gene flow between the ancestral insular and mainland populations before the first split and thus forced all migration to occur in a short time interval (Figure 3).
In light of the inferred demographic history, barn owls of the British Isles would have inherited their whiteness from their source mainland population, the refugium in the Iberian Peninsula, and kept it through small population size, genetic drift and low gene flow. Although it is conceivable that some copies of the rufous MC1R allele were present in the founding insular population, similar to its frequency in Iberia (1%; Figure 1a), in the absence of strong positive selection in the insular environment, it could have disappeared through genetic drift given the small effective sizes (Figure 4b; Table S9). Thus, the selective pressure that renders the rufous colour and allele adaptive in northern continental Europe (Antoniazza et al., 2010; Burri et al., 2016), may be absent in the British Isles. Still, we cannot rule out that gene flow with the mainland is too weak and over too short a period of time to offer selection sufficient variation in the British Isles to increase the frequency of imported rufous alleles, especially considering that they occur at a maximum frequency of about 50% on the mainland (in DK; Figure 1a) and therefore very few alleles are probably imported. If, conversely, the white morph was positively selected on the islands – potentially explaining its purer shade – we could have expected to find extended haplotypic differentiation when comparing it to the white mainland birds, which we did not (Figure 5; Appendix S3). This could suggest that the whiter shade was the ancestral phenotype and that Iberian birds got darker via the high gene flow they maintain with northern mainland populations. Therefore, it appears the white insular morph can be parsimoniously explained by relaxation or absence of selective pressure in contrast to the mainland. Such a pattern is actually common among insular birds which, due to relaxed selection, tend to display less colourful plumage than their mainland counterparts (Doutrelant et al., 2016; Grant, 1965), as also observed in the barn owl worldwide (Romano et al., 2021).
This early history of colonisation of the British Isles inferred here from whole‐genome sequences and supported by SDM projections on past climatic features is apparently unique among terrestrial vertebrates thus far, but it is far from the first to deviate from the most common colonisation route directly over Doggerland (e.g., Boston et al., 2015; Kelly et al., 2010; Snell et al., 2005; Stewart & Lister, 2001; Teacher et al., 2009) or to indicate an earlier colonisation than generally assumed (Martínková et al., 2007; McDevitt et al., 2020). Actually, it has often been conjectured that some organisms of the British Isles had an Iberian origin though this remained unconfirmed (e.g., Arntzen, 2019; Corbet, 1961; McDevitt et al., 2011). The case of the stoat (Mustela erminea) is particularly interesting as it was found to have had an isolated glacial refugium also in now submerged land southwest of today's French coastline on the Bay of Biscay (Figure 4c – LGM; Martínková et al., 2007). From there, they reached Ireland very early as the temperatures started rising but, as the Celtic Sea opened 15,000 years BP, only colonized GB much later over Doggerland (Martínková et al., 2007). The key difference between the two cases lies in the fact that barn owls maintained a homogenous population between GB and Ireland through flight.
5. CONCLUSION
Our study demonstrates that barn owls followed a highly uncommon post‐glacial colonisation route to the British Isles. Probably taking advantage of the since submerged suitable habitat on the Bay of Biscay, barn owls reached the islands much earlier than expected from this southern point. The inferred demographic history could explain the whiteness of these populations through a combination of founder effect and low gene flow, and without the need to invoke selective pressures. We contend high quality population genomic data associated with species distribution hindcasting will reveal an unusual demographic history and post‐glacial colonization for many non‐model species. We speculate how often an intuitive selective explanation for a conspicuous phenotype could turn out to be the result of purely neutral processes.
AUTHOR CONTRIBUTIONS
Ana Paula Machado, Tristan Cumer, Alexandre Roulin, and Jérôme Goudet designed the study; Ana Paula Machado produced whole‐genome resequencing libraries; Ana Paula Machado and Tristan Cumer conducted the analyses; Anne‐Lyse Ducrest and Melanie Dupasquier produced the new reference genome; Christian Iseli, Emmanuel Beaudoing, and Nicolas Guex assembled it; Tristan Cumer identified coding regions; Klaus Dichmann, Rui Lourenço, John Lusby, Hans‐Dieter Martens, Laure Prévost and David Ramsden provided samples for the study; Ana Paula Machado led the writing of the manuscript with input from all authors.
Supporting information
Appendix S1
Appendix S2
Appendix S3
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the following institutions and individuals that provided samples or aided in sampling to our study: Barn Owl Trust (Devon, UK), BirdWatch Ireland (Kilcoole, IE), National University of Ireland (Galway, IE), The European Barn Owl Network, CHENE Association (Seine‐Maritime, FR), Burke Museum of Natural History and Culture (Washington, USA), San Diego Natural History Museum (California, USA), the late Richard F. Shore, Emy Guibault, Sylvain Antoniazza and Reto Burri. We thank Céline Simon, Guillaume Dumont, Luis San‐José, Valérie Ducret and Kathleen Salin for their valuable assistance with molecular work. Finally, we thank Laurent Excoffier and Nina Marchi for their assistance with demographic inference in Fastsimcoal2, Olivier Broennimann for his advice and review of the SDM, and John Pannell for insightful review of the manuscript. This study was funded by the Swiss National Science Foundation with grants 31003A_138180 & 31003A_179358 to JG and 31003A_173178 to AR. Open Access Funding provided by Universite de Lausanne.
Machado, A. P. , Cumer, T. , Iseli, C. , Beaudoing, E. , Ducrest, A.‐L. , Dupasquier, M. , Guex, N. , Dichmann, K. , Lourenço, R. , Lusby, J. , Martens, H.‐D. , Prévost, L. , Ramsden, D. , Roulin, A. , & Goudet, J. (2022). Unexpected post‐glacial colonisation route explains the white colour of barn owls (Tyto alba) from the British Isles. Molecular Ecology, 31, 482–497. 10.1111/mec.16250
Alexandre Roulin and Jérôme Goudet are Co‐senior authors.
DATA AVAILABILITY STATEMENT
The new refence genome for European barn owl (Tyto alba) has been deposited at DDBJ/ENA/GenBank under the accession JAEUGV000000000, and the corresponding PacBio reads in the BioProject PRJNA694553. The raw Illumina reads for the whole‐genome sequenced individuals are available in BioProject PRJNA700797. Colour and MC1R data are provided in Appendix S1.
REFERENCES
- Akaike, H. (1974). A new look at the statistical model identification. IEEE Transactions on Automatic Control, 19(6), 716–723. 10.1109/TAC.1974.1100705 [DOI] [Google Scholar]
- Altwegg, R. , Roulin, A. , Kestenholz, M. , & Jenni, L. (2006). Demographic effects of extreme winter weather in the barn owl. Oecologia, 149(1), 44–51. 10.1007/s00442-006-0430-3 [DOI] [PubMed] [Google Scholar]
- Antoniazza, S. , Burri, R. , Fumagalli, L. , Goudet, J. , & Roulin, A. (2010). Local adaptation maintains clinal variation in melanin‐based coloration of European barn owls (Tyto alba). Evolution, 64(7), 1944–1954. [DOI] [PubMed] [Google Scholar]
- Antoniazza, S. , Kanitz, R. , Neuenschwander, S. , Burri, R. , Gaigher, A. , Roulin, A. , & Goudet, J. (2014). Natural selection in a postglacial range expansion: The case of the colour cline in the European barn owl. Molecular Ecology, 23(22), 5508–5523. 10.1111/mec.12957 [DOI] [PubMed] [Google Scholar]
- Arntzen, J. W. (2019). An amphibian species pushed out of Britain by a moving hybrid zone. Molecular Ecology, 28(23), 5145–5154. 10.1111/mec.15285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , del Angel, G. , Levy‐Moonshine, A. , Jordan, T. , Shakir, K. , Roazen, D. , Thibault, J. , Banks, E. , Garimella, K. V. , Altshuler, D. , Gabriel, S. , & DePristo, M. A. (2013). From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics, 43(Suppl. 43), 11.10.1‐11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilton, D. T. , Mirol, P. M. , Mascheretti, S. , Fredga, K. , Zima, J. , & Searle, J. B. (1998). Mediterranean Europe as an area of endemism for small mammals rather than a source for northwards postglacial colonization. Proceedings of the Royal Society B: Biological Sciences, 265(1402), 1219–1226. 10.1098/rspb.1998.0423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boston, E. S. M. , Montgomery, W. I. , Hynes, R. , & Prodöhl, P. A. (2015). New insights on postglacial colonization in western Europe: The phylogeography of the Leisler’s bat (Nyctalus leisleri). Proceedings of the Royal Society B: Biological Sciences, 282(1804), 10.1098/rspb.2014.2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace, S. , Ruddy, M. , Miller, R. , Schreve, D. C. , Stewart, J. R. , & Barnes, I. (2016). The colonization history of British water vole (Arvicola amphibius (Linnaeus, 1758)): Origins and development of the Celtic fringe. Proceedings of the Royal Society B: Biological Sciences, 283(1829). 10.1098/rspb.2016.0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna, T. , Hoff, K. J. , Lomsadze, A. , Stanke, M. , & Borodovsky, M. (2020). BRAKER2: Automatic Eukaryotic Genome Annotation with GeneMark‐EP+ and AUGUSTUS Supported by a Protein Database. BioRxiv, 2020.08.10.245134. 10.1101/2020.08.10.245134 [DOI] [PMC free article] [PubMed]
- Burri, R. , Antoniazza, S. , Gaigher, A. , Ducrest, A.‐L. , Simon, C. , Fumagalli, L. , Goudet, J. , & Roulin, A. (2016). The genetic basis of color‐related local adaptation in a ring‐like colonization around the Mediterranean. Evolution, 70(1), 140–153. 10.1111/evo.12824 [DOI] [PubMed] [Google Scholar]
- Chin, C.‐S. , Peluso, P. , Sedlazeck, F. J. , Nattestad, M. , Concepcion, G. T. , Clum, A. , Dunn, C. , O'Malley, R. , Figueroa‐Balderas, R. , Morales‐Cruz, A. , Cramer, G. R. , Delledonne, M. , Luo, C. , Ecker, J. R. , Cantu, D. , Rank, D. R. , & Schatz, M. C. (2016). Phased diploid genome assembly with single‐molecule real‐time sequencing. Nature Methods, 13(12), 1050–1054. 10.1038/nmeth.4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles, B. J. (1998). Doggerland: A Speculative Survey. Proceedings of the Prehistoric Society, 64(January), 45–81. 10.1017/S0079497X00002176 [DOI] [Google Scholar]
- Corbet, G. B. (1961). Origin of the British insular races of small mammals and of the “Lusitanian” fauna. Nature, 191(4793), 1037–1040. 10.1038/1911037a0 [DOI] [Google Scholar]
- Cumer, T. , Machado, A. P. , Dumont, G. , Bontzorlos, V. A. , Ceccherelli, R. , Charter, M. , Dichmann, K. , Martens, H.‐D. , Kassinis, N. , Lourenço, R. , Manzia, F. , Prévost, L. , Rakovic, M. , Siverio, F. , Roulin, A., & Goudet, J. (2021). Landscape and climatic variations of the Quaternary shaped multiple secondary contacts among barn owls (Tyto alba) of the Western Palearctic. BioRxiv, 10.1101/2021.06.09.447652 [DOI] [PMC free article] [PubMed]
- Deffontaine, V. , Libois, R. , Kotlik, P. , Sommer, R. , Nieberding, C. , Paradis, E. , Searle, J. B. , & Michaux, J. R. (2005). Beyond the Mediterranean peninsulas: Evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Molecular Ecology, 14(6), 1727–1739. 10.1111/j.1365-294X.2005.02506.x [DOI] [PubMed] [Google Scholar]
- Doutrelant, C. , Paquet, M. , Renoult, J. P. , Grégoire, A. , Crochet, P. A. , & Covas, R. (2016). Worldwide patterns of bird colouration on islands. Ecology Letters, 19(5), 537–545. 10.1111/ele.12588 [DOI] [PubMed] [Google Scholar]
- Dreiss, A. N. , & Roulin, A. (2010). Age‐related change in melanin‐based coloration of Barn owls (Tyto alba): Females that become more female‐like and males that become more male‐like perform better. Biological Journal of the Linnean Society, 101(3), 689–704. 10.1111/j.1095-8312.2010.01503.x [DOI] [Google Scholar]
- du Feu, C. R. , Clark, J. A. , Schaub, M. , Fiedler, W. , & Baillie, S. R. (2016). The EURING Data Bank – a critical tool for continental‐scale studies of marked birds. Ringing and Migration, 31(1), 1–18. 10.1080/03078698.2016.1195205 [DOI] [Google Scholar]
- Ducrest, A.‐L. , Keller, L. , & Roulin, A. (2008). Pleiotropy in the melanocortin system, coloration and behavioural syndromes. Trends in Ecology and Evolution, 23(9), 502–510. 10.1016/j.tree.2008.06.001 [DOI] [PubMed] [Google Scholar]
- Ducrest, A.‐L. , Neuenschwander, S. , Schmid‐Siegert, E. , Pagni, M. , Train, C. , Dylus, D. , Nevers, Y. , Vesztrocy, A. W. , San‐Jose, L. M. , Dupasquier, M. , Dessimoz, C. , Xenarios, I. , Roulin, A. , & Goudet, J. (2020). New genome assembly of the barn owl (Tyto alba alba). Ecology and Evolution, 10(5), 2284–2298. 10.1002/ece3.5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , Dupanloup, I. , Huerta‐Sánchez, E. , Sousa, V. C. , & Foll, M. (2013). Robust demographic inference from genomic and SNP data. PLoS Genetics, 9(10), 1003905. 10.1371/journal.pgen.1003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , & Foll, M. (2011). fastsimcoal: A continuous‐time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics, 27(9), 1332–1334. 10.1093/bioinformatics/btr124 [DOI] [PubMed] [Google Scholar]
- Ficetola, G. F. , Mazel, F. , & Thuiller, W. (2017). Global determinants of zoogeographical boundaries. Nature Ecology & Evolution, 1(4), 89. 10.1038/s41559-017-0089 [DOI] [PubMed] [Google Scholar]
- Frichot, E. , Mathieu, F. , Trouillon, T. , Bouchard, G. , & François, O. (2014). Fast and efficient estimation of individual ancestry coefficients. Genetics, 196(4), 973–983. 10.1534/genetics.113.160572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Vázquez, A. , Pinto Llona, A. C. , & Grandal‐d’Anglade, A. (2019). Post‐glacial colonization of Western Europe brown bears from a cryptic Atlantic refugium out of the Iberian Peninsula. Historical Biology, 31(5), 618–630. 10.1080/08912963.2017.1384473 [DOI] [Google Scholar]
- Grant, P. R. (1965). Plumage and the evolution of birds on Islands. R. Grant Source: Systematic Zoology, 14(1), 47–52. 10.2307/2411902 [DOI] [Google Scholar]
- Herman, J. S. , Jóhannesdóttir, F. , Jones, E. P. , Mcdevitt, A. D. , Michaux, J. R. , White, T. A. , Wójcik, J. M. & Searle, J. B. (2017). Post‐glacial colonization of Europe by the wood mouse, Apodemus sylvaticus: Evidence of a northern refugium and dispersal with humans. Biological Journal of the Linnean Society, 120(2), 313–332. 10.1111/bij.12882 [DOI] [Google Scholar]
- Hewitt, G. M. (1999). Postglacial re‐colonisation of European biota. Biological Journal of the Linnean Society, 68, 87–112. 10.1111/j.1095-8312.1999.tb01160.x [DOI] [Google Scholar]
- Hewitt, G. M. (2000). The genetic legacy of the Quaternary ice ages. Nature, 405(6789), 907–913. 10.1038/35016000 [DOI] [PubMed] [Google Scholar]
- Hewitt, G. M. (2011). Mediterranean Peninsulas: The evolution of hotspots. In Zachos F. E., & Habel J. C. (Eds.), Biodiversity hotspots (pp. 123–147). Springer Berlin Heidelberg. 10.1007/978-3-642-20992-5_7 [DOI] [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25(15), 1965–1978. 10.1002/joc.1276 [DOI] [Google Scholar]
- Hoff, K. J. , Lange, S. , Lomsadze, A. , Borodovsky, M. , & Stanke, M. (2016). BRAKER1: Unsupervised RNA‐Seq‐based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32(5), 767–769. 10.1093/bioinformatics/btv661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K. J. , Lomsadze, A. , Borodovsky, M. , & Stanke, M. (2019). Whole‐genome annotation with BRAKER. In Kollmar M. (Ed.), Gene prediction: Methods and protocols (pp. 65–95). Springer New York. 10.1007/978-1-4939-9173-0_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, A. , Charman, D. J. , & Newnham, R. M. (2010). A last glacial maximum pollen record from Bodmin Moor showing a possible cryptic northern refugium in southwest England. Journal of Quaternary Science, 25(3), 296–308. 10.1002/jqs.1309 [DOI] [Google Scholar]
- Kocher, T. , Thomas, W. , Meyer, A. , Edwards, S. , Pääbo, S. , Villablanca, F. , & Wilson, A. (1989). Dynamics of mitochondrial DNA evolution in animals : Amplification and sequencing with conserved primers. Proceedings of the National Academy of Sciences of the United States of America, 86, 6196–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeck, K. , Rouby, H. , Purcell, A. , Sun, Y. , & Sambridge, M. (2014). Sea level and global ice volumes from the Last Glacial Maximum to the Holocene. Proceedings of the National Academy of Sciences of the United States of America, 111(43), 15296–15303. 10.1073/pnas.1411762111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leorri, E. , Cearreta, A. , & Milne, G. (2012). Field observations and modelling of Holocene sea‐level changes in the southern Bay of Biscay: Implication for understanding current rates of relative sea‐level change and vertical land motion along the Atlantic coast of SW Europe. Quaternary Science Reviews, 42, 59–73. 10.1016/j.quascirev.2012.03.014 [DOI] [Google Scholar]
- Lister, A. M. (1984). Evolutionary and ecological origins of British deer. Proceedings of the Royal Society of Edinburgh. Section B. Biological Sciences, 82(4), 205–229. 10.1017/S0269727000003754 [DOI] [Google Scholar]
- Machado, A. P. , Clément, L. , Uva, V. , Goudet, J. , & Roulin, A. (2018). The Rocky Mountains as a dispersal barrier between barn owl (Tyto alba) populations in North America. Journal of Biogeography, 45, 1288–1300. 10.1111/jbi.13219 [DOI] [Google Scholar]
- Marková, S. , Horníková, M. , Lanier, H. C. , Henttonen, H. , Searle, J. B. , Weider, L. J. , & Kotlík, P. (2020). High genomic diversity in the bank vole at the northern apex of a range expansion: The role of multiple colonizations and end‐glacial refugia. Molecular Ecology, 29(9), 1730–1744. 10.1111/mec.15427 [DOI] [PubMed] [Google Scholar]
- Martin, J. R. (2017). The Barn Owl: Guardian of the Countryside. Whittet Books. Retrieved from http://library1.nida.ac.th/termpaper6/sd/2554/19755.pdf [Google Scholar]
- Martínková, N. , McDonald, R. A. , & Searle, J. B. (2007). Stoats (Mustela erminea) provide evidence of natural overland colonization of Ireland. Proceedings of the Royal Society B: Biological Sciences, 274(1616), 1387–1393. 10.1098/rspb.2007.0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt, A. , Coscia, I. , Browett, S. , Ruiz‐González, A. , Statham, M. , Ruczyńska, I. , Statham, M. J. Ruczyńska, I. , Roberts, L. , Stojak, J. , Frantz, A. C. , Norén, K. , Ågren, E. O. , Learmount, J. , Basto, M. , Fernandes, C. , Stuart, P. , Tosh, D. G. , Sindicic, M. , … Wójcik, J. (2020). Next‐generation phylogeography resolves post‐glacial colonization patterns in a widespread carnivore, the red fox (Vulpes vulpes), in Europe. BioRxiv, 10.1101/2020.02.21.954966 [DOI] [PubMed] [Google Scholar]
- McDevitt, A. D. , Vega, R. , Rambau, R. V. , Yannic, G. , Herman, J. S. , Hayden, T. J. , & Searle, J. B. (2011). Colonization of Ireland: Revisiting the pygmy shrew syndrome using mitochondrial. Y chromosomal and microsatellite markers. Heredity, 107(6), 548–557. 10.1038/hdy.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, W. I. , Provan, J. , McCabe, A. M. , & Yalden, D. W. (2014). Origin of British and Irish mammals: Disparate post‐glacial colonisation and species introductions. Quaternary Science Reviews, 98, 144–165. 10.1016/j.quascirev.2014.05.026 [DOI] [Google Scholar]
- O'Meara, D. B. , Edwards, C. J. , Sleeman, D. P. , Cross, T. F. , Statham, M. J. , Mcdowell, J. R. , Dillane, E. , Coughlan, J. P. , O'leary, D. , O'reilly, C. , Bradley, D. G. , & Carlsson, J. (2012). Genetic structure of Eurasian badgers Meles meles (Carnivora: Mustelidae) and the colonization history of Ireland. Biological Journal of the Linnean Society, 106(4), 893–909. 10.1111/j.1095-8312.2012.01927.x [DOI] [Google Scholar]
- Petkova, D. , Novembre, J. , & Stephens, M. (2016). Visualizing spatial population structure with estimated effective migration surfaces. Nature Genetics, 48(1), 94–100. 10.1038/ng.3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell, J. , & Pritchard, J. (2012). Inference of population splits and mixtures from genome‐wide allele frequency data. Nature Preceding’s, 10.1038/npre.2012.6956.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. R. , Bender, D. , Maller, J. , Sklar, P. , de Bakker, P. I. W. , Daly, M. J. , & Sham, P. C. (2007). PLINK: A tool set for whole‐genome association and population‐based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet, M. , Harrod, C. , Eizaguirre, C. , & Prodöhl, P. A. (2014). Unique mitochondrial DNA lineages in Irish stickleback populations: Cryptic refugium or rapid recolonization? Ecology and Evolution, 4(12), 2488–2504. 10.1002/ece3.853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, A. , Séchaud, R. , & Roulin, A. (2021). Evolution of wing length and melanin‐based coloration in insular populations of a cosmopolitan raptor. Journal of Biogeography, 48(4), 961–973. 10.1111/jbi.14053 [DOI] [Google Scholar]
- Roulin, A. (2013). Ring recoveries of dead birds confirm that darker pheomelanic Barn Owls disperse longer distances. Journal of Ornithology, 154(3), 871–874. 10.1007/s10336-013-0949-0 [DOI] [Google Scholar]
- Roulin, A. , & Dijkstra, C. (2003). Genetic and environmental components of variation in eumelanin and phaeomelanin sex‐traits in the barn owl. Heredity, 90(5), 359–364. 10.1038/sj.hdy.6800260 [DOI] [PubMed] [Google Scholar]
- Roulin, A. , & Ducrest, A.‐L. (2011). Association between melanism, physiology and behaviour: A role for the melanocortin system. European Journal of Pharmacology, 660(1), 226–233. 10.1016/j.ejphar.2011.01.036 [DOI] [PubMed] [Google Scholar]
- Roulin, A. , & Randin, C. F. (2016). Barn owls display larger black feather spots in cooler regions of the British Isles. Biological Journal of the Linnean Society, 119(2), 445–454. 10.1111/bij.12814 [DOI] [Google Scholar]
- San‐Jose, L. M. , Ducrest, A.‐L. , Ducret, V. , Béziers, P. , Simon, C. , Wakamatsu, K. , & Roulin, A. (2015). Effect of the MC1R gene on sexual dimorphism in melanin‐based colorations. Molecular Ecology, 24(11), 2794–2808. 10.1111/mec.13193 [DOI] [PubMed] [Google Scholar]
- Searle, J. B. , Kotlík, P. , Rambau, R. V. , Marková, S. , Herman, J. S. , & McDevitt, A. D. (2009). The Celtic fringe of Britain: Insights from small mammal phylogeography. Proceedings of the Royal Society B: Biological Sciences, 276(1677), 4287–4294. 10.1098/rspb.2009.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A. F. , & Hubley, R. (2008‐2015). RepeatModeler Open‐1.0. Retrieved from http://www.repeatmasker.org
- Smit, A. F. , Hubley, R. , & Green, P. (2013‐2015). RepeatMasker Open‐4.0. Retrieved from http://www.repeatmasker.org
- Snell, C. , Tetteh, J. , & Evans, I. H. (2005). Phylogeography of the pool frog (Rana lessonae Camerano) in Europe: Evidence for native status in Great Britain and for an unusual postglacial colonization route. Biological Journal of the Linnean Society, 85(1), 41–51. 10.1111/j.1095-8312.2005.00471.x [DOI] [Google Scholar]
- Speek, G. , Clark, J. A. , Rohde, Z. , Wassenaar, R. , & Van Noordwijk, A. J. (2001). The EURING exchange‐code 2000. Heteren. [Google Scholar]
- Stanke, M. , Diekhans, M. , Baertsch, R. , & Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics (Oxford, England), 24(5), 637–644. 10.1093/bioinformatics/btn013 [DOI] [PubMed] [Google Scholar]
- Stanke, M. , Schöffmann, O. , Morgenstern, B. , & Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 7(1), 62. 10.1186/1471-2105-7-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, J. R. , & Lister, A. M. (2001). Cryptic northern refugia and the origins of the modern biota. Trends in Ecology and Evolution, 16(11), 608–613. 10.1016/S0169-5347(01)02338-2 [DOI] [Google Scholar]
- Taberlet, P. , Fumagalli, L. , Wust‐Saucy, A. G. , & Cosson, J. F. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Molecular Ecology, 7(4), 453–464. 10.1046/j.1365-294x.1998.00289.x [DOI] [PubMed] [Google Scholar]
- Teacher, A. G. F. , Garner, T. W. J. , & Nichols, R. A. (2009). European phylogeography of the common frog (Rana temporaria): Routes of postglacial colonization into the British Isles, and evidence for an Irish glacial refugium. Heredity, 102(5), 490–496. 10.1038/hdy.2008.133 [DOI] [PubMed] [Google Scholar]
- Ulster Wildlife (2019). Barn Owl Report. https://www.ulsterwildlife.org/sites/default/files/2020‐09/Ulster%20Wildlife%20Barn%20Owl%20Report%202019_0.pdf
- van den Brink, V. , Dreiss, A. N. , & Roulin, A. (2012). Melanin‐based coloration predicts natal dispersal in the barn owl. Tyto Alba. Animal Behaviour, 84(4), 805–812. 10.1016/j.anbehav.2012.07.001 [DOI] [Google Scholar]
- Ward, I. , Larcombe, P. , & Lillie, M. (2006). The dating of Doggerland – post‐glacial geochronology of the southern North Sea. Environmental Archaeology, 11(2), 207–218. 10.1179/174963106x123214 [DOI] [Google Scholar]
- Warren, D. L. , & Seifert, S. N. (2011). Ecological niche modeling in Maxent: The importance of model complexity and the performance of model selection criteria. Ecological Applications, 21(2), 335–342. 10.1890/10-1171.1 [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.2307/2408641 [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Goudet, J. (2017). A unified characterization of population structure and relatedness. Genetics, 206(4), 2085–2103. 10.1534/genetics.116.198424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Schwartz, S. , Wagner, L. , & Miller, W. (2000). A greedy algorithm for aligning DNA sequences. Journal of Computational Biology, 7(1‐2), 203–214. 10.1089/10665270050081478 [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28(24), 3326–3328. 10.1093/bioinformatics/bts606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Appendix S3
Supplementary Material
Data Availability Statement
The new refence genome for European barn owl (Tyto alba) has been deposited at DDBJ/ENA/GenBank under the accession JAEUGV000000000, and the corresponding PacBio reads in the BioProject PRJNA694553. The raw Illumina reads for the whole‐genome sequenced individuals are available in BioProject PRJNA700797. Colour and MC1R data are provided in Appendix S1.