

Abstract
A metabolome‐guided screening approach in the novel myxobacterium Corallococcus sp. MCy9072 resulted in the isolation of the unprecedented natural product myxofacycline A, which features a rare isoxazole substructure. Identification and genomic investigation of additional producers alongside targeted gene inactivation experiments and heterologous expression of the corresponding biosynthetic gene cluster in the host Myxococcus xanthus DK1622 confirmed a noncanonical megaenzyme complex as the biosynthetic origin of myxofacycline A. Induced expression of the respective genes led to significantly increased production titers enabling the identification of six further members of the myxofacycline natural product family. Whereas myxofacyclines A–D display an isoxazole substructure, intriguingly myxofacyclines E and F were found to contain 4‐pyrimidinole, a heterocycle unprecedented in natural products. Lastly, myxofacycline G features another rare 1,2‐dihydropyrol‐3‐one moiety. In addition to a full structure elucidation, we report the underlying biosynthetic machinery and present a rationale for the formation of all myxofacyclines. Unexpectedly, an extraordinary polyketide synthase‐nonribosomal peptide synthetase hybrid was found to produce all three types of heterocycle in these natural products.
Keywords: 4-pyrimidinole, biosynthesis, isoxazole, myxobacteria, secondary metabolites
Discovering the natural route to “synthetic” heterocycles: Myxofacyclines are natural products isolated from myxobacteria. Myxofacyclines A–D display an isoxazole substructure, myxofacyclines E and F contain 4‐pyrimidinole, and myxofacycline G features another rare 1,2‐dihydropyrol‐3‐one moiety. The isoxazole, 4‐pyrimidinole or 1,2‐dihydropyrrol‐3‐one heterocycle in the myxofacyclines and the underlying biosynthesis are rarely described in natural products. Isolation, structure elucidation, mutagenesis of the producer strain and heterologous expression of the identified biosynthetic genes are presented.
Introduction
Natural products, featuring unique and specific biological activities, are indispensable for drug discovery. [1] These activities are often driven by the wide range of unique and intriguing chemical scaffolds afforded by specialized biosynthesis. [2] However, the discovery of new chemical scaffolds has become increasingly more challenging, since numerous secondary metabolites considered to be “low‐hanging fruits” [3] have already been characterized in‐depth, and typical prolific producers of natural products seem to be rather exploited. [4] Encouragingly, underexploited sources of natural products have entered the stage such as the myxobacteria, with the propitious prospect to provide new chemical entities [5] and avoid rediscovery of known natural products. [6]
Several heterocycles such as 1,3‐oxazoles are frequently found in secondary metabolites. [7] In contrast to 1,3‐oxazoles, the 1,2‐oxazole motif (simply termed isoxazole) is rarely present in natural products and the biosynthesis for these compounds remains largely elusive.[ 8 , 9 ] Among the limited number of natural products featuring an isoxazole, [8] only isoxazolidinone formation of d‐cycloserine (8) has been characterized through in vitro reconstitution of the biosynthesis with the respective recombinantly produced biosynthetic enzymes. [10] The amino acid l‐serine first undergoes O‐acetylation, followed by reaction with hydroxyurea and racemization before heterocyclization catalyzed by DcsG, an ATP‐grasp fold family protein, [11] affords the final 8 . [10]
The isoxazoline‐containing bacterial natural products acivicin (9) and 4‐hydroxyacivicin represent another class of 1,2‐oxazole compounds from Streptomycetes. However, despite the availability of the producer strain and its genome sequence, no genetic correlation to a corresponding biosynthetic gene cluster (BGC) has yet been made.
Paralleling the lack of biosynthetic knowledge corresponding to 9, little was known concerning the biosynthesis of ibotenic acid and the isoxazole muscimol (10) from Amanita muscaria [12] until recently. Recombinant production of the glutamate hydroxylase IboH from this fungus, [13] revealed that the biosynthesis of 10 starts from 3‐hydroxyglutamate. [14] In contrast to the uncommon occurrence of isoxazoles in Nature, numerous isoxazoles have been chemically synthesized and described as pharmaceuticals [15] or as chemical crop protection agents. [16]
Similar to the isoxazole type heterocycle, the 4‐pyrimidinole scaffold is accessible by chemical synthesis and has been described in synthetic tankyrase inhibitors. [17] Despite some structural similarity to pyrimidine nucleobases, 2,5‐diketopiperazines or 4‐aminopyrimidines – which is found in bleomycin and originates from two asparagines [18] – no natural product featuring a 4‐pyrimidinole heterocycle has been discovered to date. However, some secondary metabolites featuring a 4‐quinazolinone scaffold such as farinamycin (11) [19] or terremide B (12) [20] have been described and the biosynthesis of 11 was shown to start with the formation of the heterocycle through condensation of 3‐hydroxyanthranilic acid and 3,4‐aminohydroxybenzamide.
Heterocycles are commonly found in numerous myxobacterial natural products such as the oxazole rings in ajudazols [21] and disorazols, [22] thiazole ring formation during the biosynthesis of epothilone and myxothiazol, [23] and six‐membered ring formations during the biosynthesis of ambruticin [24] and pyrronazols. [25] Those heterocycles are often formed by cyclization domains within a nonribosomal peptide synthetase (NRPS) module that performs the amide‐bond‐forming condensation with l‐serine or l‐cysteine followed by subsequent heterocyclization to yield an oxazoline/thiazoline intermediate. Further oxidation by oxidase domains yield the unsaturated heterocycles oxazole and thiazole, respectively. [26]
Although the modular nature of biosynthetic megasynthetases like NRPSs, polyketide synthases (PKSs) or hybrids thereof greatly facilitate in silico identification and characterization of genes encoding biosynthetic pathways, the formation of several alternative heterocyclic scaffolds in natural products remains elusive, such as the uncommon 1,2‐dihydropyrrol‐3‐one ring found in hyapyrroline A (13). [27] The heterocyclic structure presented in discoipyrrole A (14) – which roughly resembles the heterocycle in 13 – highlights an interesting biosynthetic case, where the heterocycle is formed by specific and concerted non‐enzymatic reactions in aqueous media from the biosynthetic substrates anthranilic acid, 4‐hydroxybenzaldehyde and 4‐hydroxysattabacin. [28] Nevertheless it seems likely, that the 1,2‐dihydropyrrol‐3‐one ring found in 13 is formed differently, since its chemical scaffold suggest a PKS‐derived biosynthetic pathway.
In this study we identified an unprecedented class of natural products from the novel myxobacterial isolate Corallococcus sp. MCy9072. Isolation and structure elucidation revealed myxofacycline congeners to bear either isoxazole (1–4), a 4‐pyrimidinole (5, 6) or 1,2‐dihydropyrrol‐3‐one (7) ring structures (Figure 1). The corresponding biosynthetic gene cluster was identified and heterologous expression showed that one noncanonical PKS‐NRPS system can produce all seven myxofacyclines in parallel. Inspired by the new biosynthetic logic, a hypothesis for the heterocyclization in myxofacyclines is proposed that does not involve the classical NRPS‐based extension with an amino acid.
Figure 1.

A) Chemical structure and numbering of isolated myxofacyclines A–D (1–4), which feature an isoxazole substructure, myxofacyclines E and F (5, 6), featuring a 4‐pyrimidinole heterocycle, and one derivative termed myxofacycline G, which features a pyrrolinone ring (7). B) Natural products featuring an isoxazole(‐related) ring, d‐cycloserine (8), acivicin (9), muscimol (10); natural products partially resembling the 4‐pyrimidinole heterocycle; farinamycin (11), terremide B (12), which feature a 4‐quinazolinone scaffold; hyapyrroline A (13) exhibits a 1,2‐dihydropyrrol‐3‐one heterocycle, whereas discoipyrrole A (14) features a 3H‐benzo[d]pyrrolo[1,3]‐oxazine‐3,5‐dione core.
Results and Discussion
Isolation and structure elucidation of myxofacycline A
In the course of a metabolome‐guided analysis of the crude extract of Corallococcus sp. MCy9072 we identified 1 as target mass, that displayed an ion peak at m/z 268.1919 [M+H]+ (calcd. for C15H25N1O3). Fermentation of Corallococcus sp. MCy9072 permitted its isolation for full structure elucidation.
The 1H spectrum of 1 in [D6]DMSO exhibited a signal at δ H2‐10 6.15 characteristic for a double bond. Six methylene signals were identified at δ H2‐8 2.68, δ H2‐14 2.57, δ H2‐16 2.24, δ H2‐15 1.79, δ H2‐7 1.60 and δ H2‐4 1.13 typical for alkyl chains. In addition, one methine at δ H‐2 1.49 was identified together with an intense signal at δ H3‐1/3 0.84 that implied the presence of two methyl groups. Examination of the HSQC spectrum revealed the presence of two additional methylene groups (δ C‐6 29.7, δ H2‐6 1.32, δ C‐5 27.5, δ H2‐5 1.31). A sequential spin system deduced from COSY correlations implied the presence of an iso‐branched alkyl chain and a butyric acid chain (Figure 2).
Figure 2.

Observed correlations of 1.
The HMBC spectrum was examined to establish connectivity between the alkyl chain and butyric acid chain. The methylene protons H2‐8 showed HMBC correlations to a carbon resonance at δ C‐9 173.0 and to the carbon of a sp2 hybridized methine resonance at δ C‐10 100.7. This methine proton at δ H‐10 6.15 also showed HMBC correlations to carbon resonances at δ C‐11 163.0 and δ C‐9 173.0. In addition, from H2‐14, HMBC correlations to C‐11 and C‐10 were observed, suggesting both, alkyl chain and butyric acid chain are connected by a heteroaromatic system. The chemical shift values of C‐9,10 and 11 are characteristic for an isoxazole heterocycle. [29] The determined 15N chemical shift of δ N‐12 361 is in the range of reported values for an isoxazole heterocycle. [30] Furthermore, a 1,1‐ADEQUATE NMR experiment [31] could confirm this connectivity (Figure 2).
Biosynthetic origin and heterologous expression
According to the retro‐biosynthetic analysis of its structure we propose 1 to be biosynthesized by a modular PKS pathway, featuring an aminotransferase and an extraordinary cyclization reaction. As the genome sequence of Corallococcus sp. MCy9072 was not available, we conducted a survey of our in‐house myxobacterial metabolome collection – which contains LC‐MS datasets from around 2300 myxobacterial strains [32] – in order to identify alternative producers with available genome sequences. This search identified M. xanthus Mx x48 and Stigmatella aurantiaca Sg a32 as alternative producers of 1 (Table S1 in the Supporting Information). AntiSMASH analysis of both genome sequences led to a candidate PKS‐NRPS hybrid BGC that might indeed be responsible for the biosynthesis of 1.
The putative PKS‐NRPS BGC contains nine open reading frames (iso1–9; iso=isoxazole‐containing natural product) (Figure 3A) encoding four modules; three of these are PKS modules (Iso2, Iso5, Iso9), whereas one module is NRPS derived (Iso6). M. xanthus Mx x48 was selected to establish a genetic manipulation procedure for gene disruption experiments on the proposed BGC, since it offers beneficial growth characteristics [33] and is phylogenetically closely related to the myxobacterial model strain M. xanthus DK1622. [34] The essential role of the biosynthetic genes iso1‐6 and iso8–9 in the biosynthesis of 1 was confirmed by targeted single crossover based gene inactivation (Figure S18) leading to the loss of myxofacycline production. This result connected the in silico identified BGC to 1. Importantly, comparative metabolome analysis of mutants versus wild‐type revealed further novel secondary metabolites produced by this biosynthetic pathway.
Figure 3.

A) Genetic BGC operon iso1–9. Insertion of a vanillate‐inducible promoter (PVan) in front of iso1 (not shown) or iso2 in M. xanthus Mx x48 led to increased production of secondary metabolites associated with the iso BGC. B) High‐performance liquid chromatography–mass spectrometry base peak chromatogram (HPLC‐MS BPC) of M. xanthus Mx x48 Pvan_iso2 with and without supplementation of 2 mM vanillate. The isolated myxofacyclines and their corresponding peaks are shown.
To achieve stable and improved production, the vanillate‐inducible promoter system, which was used in numerous studies describing genetic engineering of myxobacterial pathways, [23] was inserted by single‐crossover recombination in front of iso1 or iso2 in M. xanthus Mx x48. To unambiguously show that the BGC is sufficient for myxofacycline biosynthesis, the corresponding BGC from S. aurantiaca Sg a32 (mobilized on a cosmid), was used for heterologous expression in M. xanthus DK1622 also enabling investigation of the biosynthesis of myxofacyclines.
After introducing a vanillate‐inducible promoter upstream of iso1 or iso2 (as in the later construct the gene iso1 was removed), both generated mutant strains were capable of producing all previously identified myxofacyclines; the respective peaks in the chromatogram showed approximately the same intensity as in the genetically engineered M. xanthus Mx x48 mutants.
Gratifyingly, the previously unreliable production of most myxofacyclines was stabilized and increased upon this genetic modification and chemical induction of the BGC. (Figures 3B and S18).
Isolation and structure elucidation of myxofacycline B–G
Based on the conducted genetic experiments as outlined above, we isolated six additional metabolites from M. xanthus Mx x48, which are directly linked to the identified biosynthetic origin of 1. Structure elucidation showed that 2–4 were congeners of 1; myxofacycline B (2) features a linear alkyl chain indicated, whereas myxofacycline C (3) has a keto group at position 7, which is reduced to an alcohol in myxofacycline D (4).
Myxofacycline E (5) contains an iso‐branched alkyl chain and a butyric acid moiety like 1, as shown by NMR data, but the sum formula of C15H25N2O3 and N‐HMBC correlations from δ H‐11 5.92 and δ H2‐7 2.48 to two nitrogen resonances at δ N‐9175 and δ N‐13 242, indicate the presence of a different heterocycle.
Inspection of HMBC showed correlations from sp2 hybridized H‐11 to two carbon resonances at δ C‐10 162.2 and δ C‐12 167.1. In addition, HMBC correlations from δ H2‐7 2.48 to a third fully substituted resonance at δ C‐8 161.9 were observed. In order to establish C‐C connectivities, we acquired a 1,1‐ADEQUATE NMR spectrum, that displayed the methine group at pos‐11 neighbors both C‐10 and C‐12. In addition, the 1,1‐ADEQUATE showed that other fully substituted carbon, C‐8 neighbors the iso‐branched alkyl. Considering the determined connectivities and the chemical shift values, we conclude a 4‐pyrimidinole heterocycle between the iso‐branched alkyl chain and butanoic acid chain (Figure 4). Myxofacycline F (6) has a linear and shorter alkyl chain, comparable to 2. The observed 5 J HMBC correlation between H‐11 and C‐7 does not contradict this structure, as these type of correlations can be found in other heterocycles. [35]
Figure 4.

Observed correlations of the pyrimidinole ring in 5.
Although myxofacycline G (7) has the same sum formula (C15H26NO4) as 4 and also features an sp2 hybridized methine group in the HSQC spectrum (δ C‐10 93.0, δ H‐10 4.62), distinct NMR signals of 7 suggest a third heterocycle. In contrast to 1–6, a nitrogen proton at δ H‐12 8.06 was observed in the 1H spectrum.
Starting from δ H‐12 8.06, HMBC correlations to three fully substituted carbons at δ C‐8 87.5, δ C‐9 201.0, δ C‐11 179.8 and the sp2 hybridized methine group at δ C‐10 93.0 were found. The shift value of C‐9 strongly implies this carbon to be a keto group while the shift value of C‐8 is characteristic for carbons with multiple heteroatoms as substituents.
Based on the shift value of C‐11, this carbon shows properties of an enol or enamine. Since the same correlations were observed from methine proton H‐10, we deduced that methine group, nitrogen and the three fully substituted carbons form a five membered heterocycle. The alkyl chain shows HMBC correlations to C‐8 and C‐9 from δ H2‐7 1.49 while the butyric acid chain shows HMBC correlations from δ H2‐13 2.36 to C‐11 and methine carbon C‐10. Based on these observations, we propose a 1,2‐dihydropyrrol‐3‐one type heterocycle in 7 (Figure 5).
Figure 5.

Observed correlations of the 1,2‐dihydropyrrol‐3‐one ring in 7.
While 1 showed weak antifungal and cytotoxic activity towards Mucor hiemalis DSM 2656 (MIC: 32 μg/mL) and Cellosaurus KB‐3‐1 cell line (MIC: 24.06 μg/mL), the absence of activity of the myxofacyclines 2–7 in the performed biological assays indicates that this new natural product family is likely to have a distinctive biological function that is currently unknown (pages S52 and S53 in the Supporting Information).
Proposed biosynthesis of the myxofacyclines
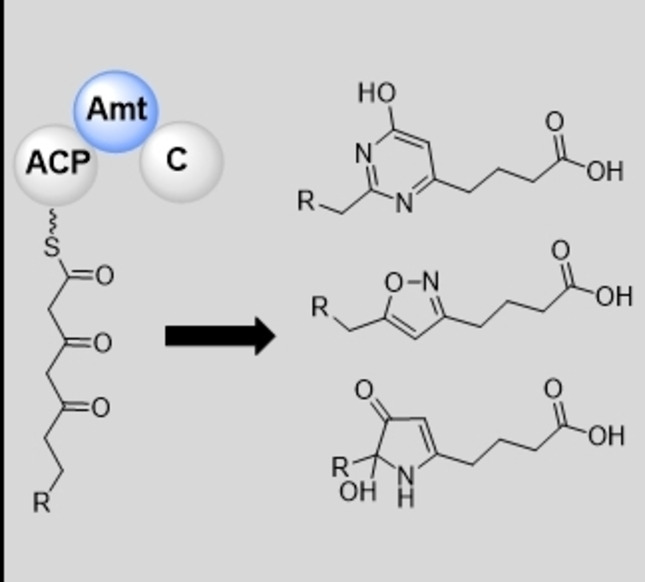
The most intriguing part of myxofacycline biosynthesis is the formation of three exceptional heterocycles through the catalysis of one biosynthetic assembly line. According to our hypothesis the biosynthesis of the heterocycles featured in the different myxofacyclines starts either with the activation of 3‐oxodecanoic acid or 9‐methyl‐3‐oxodecanoic acid to its respective acid‐adenosine monophosphate (AMP) activated derivative by the fatty acyl‐AMP ligase domain on Iso2 (Figure 6A, B). The first chain extension occurs with the condensation of the decanoic acid backbone with malonyl‐coenzyme A (CoA), catalyzed by the PKS module Iso5. The acyl carrier protein (ACP) from Iso6 acts in this case as the swinging arm for tethering of the biosynthetic intermediates. Iso2 harbors a putatively functional acyl‐CoA dehydrogenase which might play a role during the formation of different myxofacycline derivatives (page S38 in the Supporting Information).
Figure 6.

A) BGC operon iso1–9 and proposed biosynthetic route leading to the different myxofacyclines (B–D). Proposed reactions catalyzed by module 3 and further tailoring enzymes leading to the heterocycles found in the different myxofacyclines. B) Formation of the isoxazole ring in 1–4, C) hydroxylated 1,2‐dihydropyrrol‐3‐one ring formation in (7) and D) biosynthesis of the 4‐pyrimidinole ring. E) Proposed mechanism of the Beckmann rearrangement during the biosynthesis of 5 and 6.The catalytic domains for each biosynthetic step are colored in blue.
As 3‐oxodecanoic acid precursors are rarely described for the biosynthesis of natural products, the first chain extension might alternatively be initiated through the condensation of octanoic acid or 7‐methyloctanoic acid with malonyl‐CoA, catalyzed by the PKS module Iso5. Iterative use of module 2 would logically explain the incorporation of two ketide units onto the growing polyketide backbone, which provides the fundamental requirements for the subsequent modifications on module 3.
The downstream noncanonical NRPS module Iso6 does not contain an adenylation (A) domain and presumably accepts the acyl chain as a substrate, either directly or after transacylation of the nascent intermediate to the ACP of module 3. The aminotransferase domain of the same module can next perform a reductive transamination on either the second or the third carbonyl of the tethered backbone, eventually resulting in the formation of different heterocycles (Figure 6B–D).
Potential biosynthetic pathways to all three extraordinary heterocycles are outlined below and are shown in Figure 6. In order to obtain an isoxazole, the generated amino group undergoes hydroxylation by Iso4 to yield an oxime, and subsequent cyclization of the tethered backbone leads to a proposed hemiacetal intermediate. The condensation (C) domain from Iso6 is phylogenetically closely related to heterocyclization domains such as those described for the biosynthesis of epothilone and myxothiazol [23] (Figure S15), and might thus perform the cyclization of the generated oximes to yield the hemiacetal scaffold. This hemiacetal intermediate might then be spontaneously dehydrated to yield the oxidized derivative myxofacycline 1 or 2 as the growing intermediate (Figure 6B).
The formation of the hydroxylated pyrrolinone ring found in 7 does not require the formation of an oxime on the growing polyketide backbone, but rather a ϵ‐ketone. The hydroxylation of the ϵ‐position on the third ketone might be catalyzed by Iso4, which could also explain formation of 3 and 4; for the formation of 3 and 7, the respective hydroxy group would be oxidized to yield a ketone at this position (Figure 6C). The question if the required oxidation to form the ϵ‐ketone is catalyzed by Iso4 or a freestanding FAD‐dependent enzyme encoded elsewhere in the genome, remains elusive. The reaction cascade to obtain the 4‐pyrimidinole ring starts with two reductive transaminations on the second and the third carbonyl of the tethered backbone. After biosynthetic formation of the ϵ‐ketone – similar to the formation of 3 and 7 – Iso4 generates an oxime on the δ‐amine. The remaining secondary amine of the tethered backbone might perform a nucleophilic attack on the ϵ‐ketone to correspondingly generate a cyclized five‐membered scaffold in the nascent intermediate. After spontaneous dehydration, the oxime of this five‐membered ring undergoes a Beckmann rearrangement possibly also catalyzed by the C domain (such as the cyclizations) on Iso6 to yield a cyclized dihydropyrimidinole scaffold in the nascent intermediate (Figure 6E); such rearrangement reactions are proposed for the biosynthesis of marine oxazole‐containing natural products, [36] since a mechanistically related dehydration of an oxime to nitrile was achieved with a bacterial dehydratase. [37] Nevertheless no Beckmann rearrangement‐based mechanism in the biosynthesis of natural products has been confirmed yet. Subsequent oxidation by Iso3 would yield the oxidized derivative myxofacycline 5 or 6 (Figure 6D).
The standalone ACP domain encoded by iso7 might alternatively play a role as the tethering arm for the NRPS module 3 encoded by iso6 and might be necessary for the transfer of the biosynthetic intermediate to Iso9. The proposed heterocyclization mechanisms for the myxofacyclines, which seem to exclusively use the C domain from the NRPS module are unprecedented in the biosynthetic logic of PKS‐NRPS hybrids. [38]
The last step of myxofacycline biosynthesis involves extension by a malonyl‐CoA unit by the PKS module 4 encoded by iso9. In accordance with the reduction state observed in 1–7, Iso9 performs a full reduction loop of the loaded extender unit. The linear PKS‐NRPS chain is then released by the action of the thioesterase (TE) domain in module 4 yielding the various products of the pathway.
Gene disruption of iso9 resulted in abolished production of 1–7 and the associated identified metabolites (Figure S18). Interestingly, a significant increase of several metabolites corresponding to derivatives truncated by C2H4 supports the proposed function of Iso9. Accordingly, the proposed myxofacycline biosynthetic pathway combined with the sum formula and marginally earlier retention time of the identified metabolites enabled the clear assignment of the direct precursors 1 a–7 a (Table S19). This finding underlines that myxofacycline biosynthesis can function independently of Iso9, and chain release can occur without catalysis by the TE domain.
Similar to the genetic disruption of iso9, genetic inactivation of iso8 abolishes the production of 1–7 and the associated identified metabolites, whereas the production of 1 a–7 a is increased in the secondary metabolome of M. xanthus Mx x48. This finding raises the question about the biosynthetic role of the standalone A domain encoded by iso8, as its genetic disruption does not abolish the production of 1 a–7 a. In addition, the inactivation could only be done by single crossover mutagenesis and is thus expected to result in polar effects also inactivating iso9. Therefore, we cannot conclude that Iso8 plays any role in the formation of myxofacyclines. However, Iso8 might feature a biosynthetic purpose which could be complemented by some other A domain encoded in the genome.
The proposed biosynthetic pathway consequently explains the order of biosynthetic steps to generate the isoxazole moiety in 1–4, the 4‐pyrimidinole scaffold in 5 and 6, and the hydroxylated pyrrolinone ring found in 7. Furthermore, the herein proposed and genetically characterized biosynthetic pathway of the Myxofacyclines is supported by the fact, that after preparation of this manuscript, [39] the discovery of another family of natural products termed Myxadazoles, which feature a similar building block to the structure of 1–4, was published. [40]
Conclusion
The myxofacyclines are a new natural product class featuring heterocycles rarely, if ever, produced by other biosynthetic pathways. In this study, we report the identification through targeted gene inactivation and heterologous expression of the biosynthetic gene cluster encoding the biosynthetic machinery required to produce the myxofacyclines. These efforts led to a proposed biosynthetic pathway that differs from those forming acivicin, ibotenic acid or d‐cycloserine in the biosynthesis of the isoxazole scaffold. In addition, we provide the first report of the isolation and biosynthesis of a 4‐pyrimidinole scaffold in natural products. These heterocycles have, to date, been described exclusively for synthetic compounds. Further in‐depth investigation of the biosynthetic pathway by in vitro biochemical characterization of the enzymes involved is required to characterize the intriguing steps during the formation of the different myxofacyclines. In particular the unique interplay of PKS and NRPS functionality in myxofacycline biosynthesis exemplifies the notable biosynthetic capabilities of myxobacteria.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank Dr. Chantal D. Bader for her assistance in structure elucidation, Dr. Bastien Schnell, Dr. Fabian Panter, Dr. Chengzhang Fu, Dr. Domen Pogorevc and Jan Dastbaz for biosynthetic discussions, Dr. Daniel Krug and Dr. Jake Haeckl for proofreading the manuscript, Stefanie Neuber, Irene Kochems, and Victoria George for performing bioactivity assays and Dr. Nestor Zaburannyi for bioinformatics support. J.J.H. acknowledges funding by a PhD fellowship of the Boehringer Ingelheim Fonds. Open Access funding enabled and organized by Projekt DEAL.
A. Popoff, J. J. Hug, S. Walesch, R. Garcia, L. Keller, R. Müller, Chem. Eur. J. 2021, 27, 16654.
A previous version of this manuscript has been deposited on a preprint server (https://chemrxiv.org/engage/chemrxiv/article‐details/60f147e7f1a54f9bc0504d79).
References
- 1. Newman D. J., Cragg G. M., J. Nat. Prod. 2020, 83, 770. [DOI] [PubMed] [Google Scholar]
- 2. Chen Y., Garcia de Lomana M., Friedrich N.-O., Kirchmair J., J. Chem. Inf. Model. 2018, 58, 1518. [DOI] [PubMed] [Google Scholar]
- 3. Flemming A., Nat. Chem. Drug. Discov. 2013, 12, 826. [DOI] [PubMed] [Google Scholar]
- 4. Pye C. R., Bertin M. J., Lokey R. S., Gerwick W. H., Linington R. G., Proc. Natl. Acad. Sci. USA 2017, 114, 5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Skinnider M. A., Magarvey N. A., Proc. Natl. Acad. Sci. USA 2017, 114, E6271-E6272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Herrmann J., Fayad A. A., Müller R., Nat. Prod. Rep. 2017, 34, 135. [DOI] [PubMed] [Google Scholar]
- 6. Hug J. J., Bader C. D., Remškar M., Cirnski K., Müller R., Antibiotics 2018, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin Z., Nat. Prod. Rep. 2016, 33, 1268. [DOI] [PubMed] [Google Scholar]
- 8. Rahbœk L., Christophersen C. in The Alkaloids: Chemistry and Biology (Eds.: Alper K. R., Brossi A., Cordell G. A., Glick S. D., Holmes H. L., Manske R. H. F., Suffness M., Rodrigo R. G. A.), Elsevier, 1983, pp. 185–233. [Google Scholar]
- 9.
- 9a. Kochetkov N. K., Sokolov S. D. in Advances in Heterocyclic Chemistry, Vol. 2 (Ed.: Katritzky A. R.), Academic Press, New York, 1963, pp. 365–422; [DOI] [PubMed] [Google Scholar]
- 9b. Kaur K., Kumar V., Sharma A. K., Gupta G. K., Eur. J. Med. Chem. 2014, 77, 121. [DOI] [PubMed] [Google Scholar]
- 10. Uda N., Matoba Y., Kumagai T., Oda K., Noda M., Sugiyama M., Antimicrob. Agents Chemother. 2013, 57, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matoba Y., Uda N., Kudo M., Sugiyama M., FEBS J. 2020, 287, 2763. [DOI] [PubMed] [Google Scholar]
- 12. Bowden K., Drysdale A. C., Mogey G. A., Nature 1965, 206, 1359. [DOI] [PubMed] [Google Scholar]
- 13. Müller M., Obermaier S., Angew. Chem. Int. Ed. 2020, 59, 12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsumoto T., Trueb W., Gwinner R., Eugster C. H., HCA 1969, 52, 716. [Google Scholar]
- 15. Hu Feng, Szostak Michal, Adv. Synth. Catal. 2015, 357, 2583. [Google Scholar]
- 16. Lamberth C., J. Heterocycl. Chem. 2018, 55, 2035. [Google Scholar]
- 17.
- 17a. Kim M. K., Oncol. Lett. 2018, 16, 6895; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b.H.-P. Buchstaller, D. Dorsch (Merck), WO/2015/169421, 2015.
- 18. Shen B., Du L., Sanchez C., Edwards D. J., Chen M., Murrell J. M., J. Ind. Microbiol. Biotechnol. 2001, 27, 378. [DOI] [PubMed] [Google Scholar]
- 19. Nett M., Hertweck C., J. Nat. Prod. 2011, 74, 2265. [DOI] [PubMed] [Google Scholar]
- 20. Wang Y., Zheng J., Liu P., Wang W., Zhu W., Mar. Drugs 2011, 9, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Essig S., Bretzke S., Müller R., Menche D., J. Am. Chem. Soc. 2012, 134, 19362. [DOI] [PubMed] [Google Scholar]
- 22. Kopp M., Irschik H., Pradella S., Müller R., ChemBioChem 2005, 6, 1277. [DOI] [PubMed] [Google Scholar]
- 23. Hug J. J., Müller R. in Comprehensive Natural Products III (Eds.: (B. H.-W.) Liu, T. P. Begley), Elsevier, Oxford, 2020, pp. 149–216. [Google Scholar]
- 24. Julien B., Tian Z. Q., Reid R., Reeves C. D., Chem. Biol. 2006, 13, 1277. [DOI] [PubMed] [Google Scholar]
- 25. Witte S. N. R., Hug J. J., Géraldy M., Müller R., Kalesse M., Chem. Eur. J. 2017, 23, 15917. [DOI] [PubMed] [Google Scholar]
- 26. Bloudoff K., Schmeing T. M., Biochim. Biophys. Acta 2017, 1865, 1587. [DOI] [PubMed] [Google Scholar]
- 27. Okanya P. W., Mohr K. I., Gerth K., Kessler W., Jansen R., Stadler M., Müller R., J. Nat. Prod. 2014, 77, 1420. [DOI] [PubMed] [Google Scholar]
- 28. Colosimo D. A., MacMillan J. B., J. Am. Chem. Soc. 2016, 138, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pinto D. C. G. A., Santos C. M. M., Silva A. M. S. in Recent Research Developments in Heterocyclic Chemistry, 2007, pp. 397–475. [Google Scholar]
- 30. Thorn K. A., PLoS One 2019, 14, e0224112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin G. E. in Annual Reports on NMR Spectroscopy (Ed.: Webb G. A.), Academic Press, 2011, pp. 215–291. [Google Scholar]
- 32. Hoffmann T., Krug D., Bozkurt N., Duddela S., Jansen R., Garcia R., Gerth K., Steinmetz H., Müller R., Nat. Commun. 2018, 9, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Freese A., Reichenbach H., Lünsdorf H., J. Bacteriol. 1997, 179, 1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Krug D., Garcia R., Müller R., BIOspektrum 2020, 26, 32; [Google Scholar]
- 34b. Yang Y.-J., Singh R. P., Lan X., Zhang C.-S., Li Y.-Z., Li Y.-Q., Sheng D.-H., Biomol. Eng. 2018, 8, 137. [Google Scholar]
- 35. Araya-Maturana R., Pessoa-Mahana H., Weiss-López B., Nat. Prod. Commun. 2008, 3, 1934578X0800300. [Google Scholar]
- 36. Ichino T., Arimoto H., Uemura D., Chem. Commun. 2006, 1742. [DOI] [PubMed] [Google Scholar]
- 37. Kato Y., Ooi R., Asano Y., J. Mol. Catal. B 1999, 6, 249. [Google Scholar]
- 38. Miyanaga A., Kudo F., Eguchi T., Nat. Prod. Rep. 2018, 35, 1185–1209. [DOI] [PubMed] [Google Scholar]
- 39. Popoff A., Hug J., Walesch S., Garcia R., Keller L., Müller R., ChemRxiv. 2021, preprint DOI: 10.33774/chemrxiv-2021-qklc0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y., Zhuo L., Li X., Zhu Y., Wu S., Hu W., Shen T., Li Y.-Z., Wu C., Angew. Chem. 2021, 60, 21679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information