

Abstract
Fine‐tuning of G protein‐coupled receptor (GPCR) signaling is important to maintain cellular homeostasis. Recent studies demonstrated that lateral GPCR interactions in the cell membrane can impact signaling profiles. Here, we report on a one‐step labeling method of multiple membrane‐embedded GPCRs. Based on short peptide tags, complementary probes transfer the cargo (e. g. a fluorescent dye) by an acyl transfer reaction with high spatial and temporal resolution within 5 min. We applied this approach to four receptors of the cardiovascular system: the endothelin receptor A and B (ETAR and ETBR), angiotensin II receptor type 1, and apelin. Wild type‐like G protein activation after N‐terminal modification was demonstrated for all receptor species. Using FRET‐competent dyes, a constitutive proximity between hetero‐receptors was limited to ETAR/ETBR. Further, we demonstrate, that ETAR expression regulates the signaling of co‐expressed ETBR. Our orthogonal peptide‐templated labeling of different GPCRs provides novel insight into the regulation of GPCR signaling.
Keywords: bioorganic chemistry, membrane proteins, protein modification, receptors, signal transduction
GPCR‐GPCR interactions can influence signaling events but are difficult to investigate in live cell setups. Here, we report on an orthogonal labeling approach, which excludes intracellular background signals and allows the installment of different dyes. Using this technique, the interaction of homo‐receptors demonstrated for different GPCRs, but hetero‐interaction was limited to ETAR/ETBR. For the latter, impaired ETBR signaling was shown.
Introduction
G protein‐coupled receptors (GPCR) interact with several different protein species for efficient signal transduction. [1] GPCR interactions with downstream effectors like G proteins and arrestins have been widely described to play an important role in (patho‐)physiology.[ 2 , 3 ] In the cardiovascular system GPCRs like the endothelin receptor A and B (ETAR, ETBR) are known to be potent regulators of the vascular tone. [4] ETAR activation by endothelin 1 (ET‐1) leads to long‐lasting vasoconstriction. [5] In contrast, the ETBR is involved in ET‐1 clearance, and contributes to the basal vascular tone, balancing ETAR‐mediated vasoconstriction.[ 6 , 7 , 8 , 9 , 10 ] Other important GPCRs include the angiotensin II receptor type 1 (AT1R), which conveys vasoconstriction, and the apelin receptor (APJ), which acts as a vasodilator.[ 11 , 12 ] Counteracting effects between the physiological roles of these GPCR species have been described in the context of cardiovascular homeostasis.[ 13 , 14 ] In recent years, the local compartmentalization of hetero‐ and homo‐GPCR complexes has been shown to regulate signaling.[ 15 , 16 , 17 , 18 , 19 ]
Bio‐orthogonal fluorescence labeling techniques are widely applied to study GPCR‐GPCR interactions and the formation of receptor complexes in living cells.[ 20 , 21 ] Commonly, resonance energy transfer (RET) techniques like fluorescence and bioluminescence RET (FRET/BRET) are used to study the proximity of membrane‐embedded GPCRs.[ 22 , 23 , 24 ] Often, RET‐based methodologies rely on auto‐fluorescent proteins, fused to the C‐terminus of the protein of interest (POI), which prevents discrimination between membrane‐embedded proteins from intracellular POI subpopulations. Recently, we reported on a peptide‐templated acyl transfer reaction using short N‐terminal peptide tags, carrying a coiled‐coil motif, to introduce fluorescent reporters at distinct time points to monitor GPCR trafficking in mammalian cells.[ 25 , 26 ] Coiled‐coil motifs provide an interface, which displays a defined pattern of ionic and hydrophobic amino acids, allowing the selective interaction with a complementary peptide sequence. For covalent peptide‐templated labeling, the acceptor peptide is genetically fused to the extracellularly exposed N‐terminus of a GPCR, equipped with an N‐terminal cysteine residue, and the complementary peptide probe carries a reporter by thioester linkage.[ 25 , 27 ] Parallel alignment of both peptides triggers an acyl transfer reaction, which leads to covalent modification of the respective GPCR.
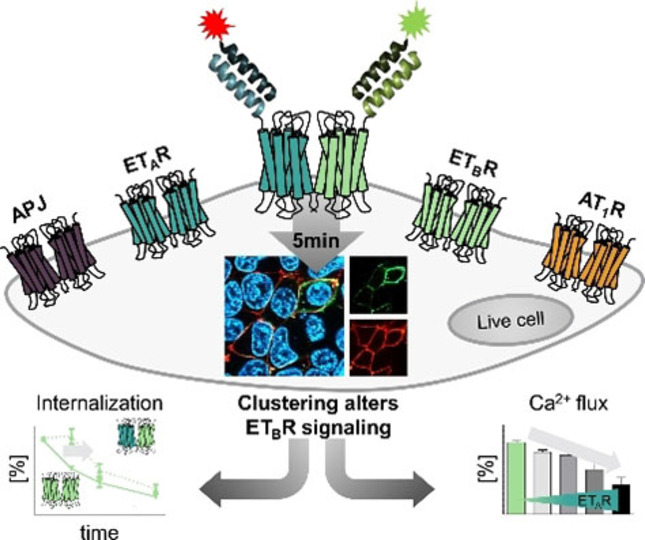
Here, we report on the establishment of an orthogonal labeling platform for two GPCR species. For multiplexed labeling, the previously reported orthogonal coiled‐coil peptide sequences P1/P2 and P3/P4 were applied.[ 28 , 29 ] By introduction of the required cysteine residue into the P1‐ and P3 acceptor sequences (Cys‐P1‐tag: C‐EIQALEE ENAQLEQ ENAALEE EIAQLEY; Cys‐P3‐tag: C‐EIQQLEE EIAQLEQ KNAALKE KNQALKY), we generated POI tags, accessible for labeling by the complementary peptide probes (P2: KIAQLKE KNAALKE KNQQLKE KIQALKY; P4: KIAQLKQ KIQALKQ ENQQLEE ENAALEY), carrying the respective reporter. This technique was applied to four different GPCRs – ETAR, ETBR, AT1R, and APJ – and enables the transfer of different fluorescent dyes to two membrane‐embedded POIs in a one‐pot reaction within 5 min. The transfer of different dyes allows the investigation of GPCR proximity in the plasma membrane of live mammalian cells by FRET. We were able to confirm the ETAR/ETBR interaction, while we did not find evidence for a specific proximity of both endothelin receptor subtypes with either AT1R or APJ, respectively. By using on‐surface labeling, we were able to elucidate a delayed onset of the constitutive ETBR internalization in the presence of co‐expressed ETAR, which we did not detect in co‐expression analyses with AT1R or APJ. Furthermore, initial G protein activation by the ETBR is suppressed by ETAR expression, adding a novel level of signal regulation to the endothelin system.
Results
Set‐up of the orthogonal labeling system
First, we investigated the applicability of the orthogonal coiled‐coil peptide pairs P1/P2 and P3/P4 to GPCR labeling by using both ETAR and ETBR, carrying a C‐terminal GFP. The sequences encoding the Cys‐P1‐ and Cys‐P3‐tag were positioned at the GPCR N‐terminus behind the endogenous signal peptides (SP) of the ETAR (ETAR1−20) and the ETBR (ETBR1−26).[ 30 , 31 , 32 ] The impact of the tag insertion on receptor activation was assessed in signal transduction assays using the Gq signaling of both ETAR and ETBR. For both GPCR subtypes, wild type (wt)‐like activation of the phospholipase C signaling pathway was detected (Supporting Information Figure S1 and Table S2). The ETAR variants, carrying the Cys‐P1‐ and Cys‐P3‐tag, displayed subnanomolar EC50 values (EC50 range: 0.5 to 0.6 nM), whereas the ETBR variants exhibited low nanomolar EC50 values (EC50 range: 1.4 to 2.4 nM). Using the C‐terminal GFP as control we further investigated the membrane localization of the tagged receptor constructs in live mammalian cells. Both wt ETAR‐GFP and ETBR‐GFP display localization to the cell membrane with only minor intracellular fluorescence (Supporting Information Figure S1). Similar intracellular distribution and membrane localization was observed for both the Cys‐P1‐ and Cys‐P3‐tagged receptor constructs. We then removed the C‐terminal fluorophore to limit protein visualization to membrane‐embedded GPCRs and exclude intracellular background. Signal transduction assays using the Gq pathway confirmed the activation of the C‐terminally untagged GPCRs. Characteristic EC50 values similar to the wt ETAR (EC50: 0.6 nM) and ETBR (EC50: 2.4 nM) were obtained for the N‐terminally tagged GPCRs, indicating their full functionality based on the endogenous Gq signaling pathway (Table 1 and Figure 1A).
Table 1.
Gq activation profiles of (SP)‐Cys‐P1/P3‐tagged ETAR, ETBR, AT1R, and APJ. Application of the respective agonist (ETAR/ETBR: ET‐1, AT1R: AngII, APJ: Ap13) was carried out in a concentration‐dependent range from 10−6 to 10−12 M in inositol monophosphate accumulation assays (performed in transiently transfected COS‐7 cells; n≥3 in triplicates). Signal transduction data were normalized to the N‐terminally untagged receptors (wt) and represent the mean over all assay repetitions.
|
|
wt |
(SP‐)Cys‐P1‐tag |
(SP‐)Cys‐P3‐tag |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
GPCR |
EC50 [nM] |
pEC50 ±SEM |
Emax [%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
Emax [%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
Emax [%] ±SEM |
|
ETAR |
0.6 |
9.2±0.03 |
100±1 |
0.9 |
9.0±0.07 |
89±3 |
0.8 |
9.1±0.08 |
83±4 |
|
ETBR |
2.4 |
8.6±0.03 |
99±2 |
2.0 |
8.7±0.07 |
97±3 |
2.4 |
8.6±0.09 |
93±5 |
|
AT1R |
0.4 |
9.3±0.02 |
100±1 |
0.4 |
9.4±0.06 |
118±3 |
0.4 |
9.4±0.09 |
94±3 |
|
APJ |
1.7 |
8.8±0.02 |
100±1 |
1.5 |
8.8±0.10 |
91±5 |
1.3 |
8.9±0.07 |
105±3 |
Figure 1.

Activation profile and peptide‐templated labeling of membrane embedded and N‐terminally modified ETAR, ETBR, AT1R, and APJ in live cells. (A) Receptor activation was investigated by inositol monophosphate accumulation using N‐terminally unmodified receptors (wt) as control in transiently transfected COS‐7 cells (n≥3, performed in triplicates). GPCR activation was facilitated by agonist application (ETAR/ETBR: ET‐1, AT1R: AngII, APJ: Ap13) in a concentration range from 10−6 to 10−12 M. Signal transduction data represent the mean over all assay repetitions. (B) Reaction scheme of the peptide‐templated acyl transfer reaction, which relies on a membrane‐embedded protein, which extracellularly exposes the Cys‐P1/P3‐tag. The exposed tag is addressed by the complementary P2 or P4 peptide, respectively, equipped with a cargo moiety (red star). (C) Application of peptide‐template labeling to visualize membrane‐embedded ETAR, ETBR, AT1R, and APJ (from left to right) by fluorescence microscopy (n=3), using Atto488‐P2 (P1‐tagged GPCRs, green) or Atto565/TAMRA‐P4 (P3‐tagged GPCRs, red) in transiently transfected HEK293 cells. Scale bar: 10 μm.
To fuse the Cys‐P1/P3‐tag to the AT1R and APJ, the SP, derived from the N‐terminus of the ETBR (ETBR1−26 single‐letter amino acid sequence: MQPPPSLCGRALVALVLACGLSRIWG) was added additionally to the Cys‐P1‐ and Cys‐P3‐tag (SP‐Cys‐P1/P3) since both the AT1R and APJ do not contain an endogenously encoded SP. The modified AT1R and APJ displayed EC50 values similar to the wt receptors (EC50(AT1R): 0.4 nM; EC50(APJ): 1.7 nM) in Gq‐based signal transduction assays. Additionally, the N‐terminally modified receptors displayed Emax values comparable to the wt receptors (Table 1; Figure 1A), demonstrating the compatibility of the SP‐Cys‐P1/P3 tags with AT1R and APJ activation and signaling.
For proof‐of‐principle labeling experiments, we applied the peptide‐templated acyl transfer (reaction scheme in Figure 1B) to live cells expressing either Cys‐P1‐ETAR/ETBR‐GFP or Cys‐P3‐ETAR/ETBR‐GFP, using 6‐carboxytetramethyl‐rhodamine (TAMRA)‐P2 (for P1‐tagged GPCRs) or TAMRA‐P4 (for P3‐tagged GPCRs) (Supporting Information Figure S1). Successful TAMRA‐staining was limited to GFP‐positive cells and no off‐target labeling was detected in these experiments. To validate the specificity of the coiled‐coil labeling reaction, we administered a dye‐thioester‐peptide conjugate (control peptide TAMRA‐MPAA‐GSGSG), lacking the coiled‐coil interaction motif of the P1/P2 and P3/P4 peptides (Supporting Information Figure S2). Cells expressing Cys‐P1‐ETAR‐GFP or Cys‐P3‐ETAR‐GFP were successfully labeled with the TAMRA fluorophore by TAMRA‐P2 and TAMRA‐P4, respectively, but no labeling of GFP‐positive cells was detectable after incubation with the control peptide. The failed labeling by the control peptide indicates the need for the proximity of the thiol/thioester moieties for efficient acyl transfer on live cells.
Selective receptor labeling in living cells
Next, we broadened the range of dyes for GPCR labeling on cell surfaces. Applying the labeling probes P2 and P4, equipped with either Atto488/TAMRA (for the P2 probe) or Atto565/TAMRA (for the P4 probes) facilitated staining of membrane‐embedded receptor subpopulation (Figure 1C) of Cys‐P1‐ and Cys‐P3‐tagged ETAR, ETBR, AT1R, and APJ without the addition of C‐terminal auto‐fluorescent proteins. All four GPCR species were successfully visualized without background signals from intracellular receptor populations. In addition, efficient cargo transfer was observed for both coiled‐coil pairs (P1/P2 and P3/P4), independent of the receptor species, emphasizing the general applicability of this visualization technique.
Since the labeling protocol includes an incubation step under reducing conditions using the mild reducing agent tris(2‐carboxyethyl)phosphine (TCEP) at low concentration (100 μM), we questioned whether this step subsequently impairs the signal transduction of labeled GPCR species after the acyl transfer reaction. Therefore, Gq protein signaling profiles of both endothelin receptor subtypes were determined pre‐ and post‐labeling in Ca2+ flux analyses (Table 2; Supporting Information Figure S3). For wt receptors without N‐terminal modification, low nanomolar EC50 values were obtained prior to TCEP addition (ETARuntreated: 2.5 nM; ETBRuntreated: 1.1 nM). Post‐treatment, EC50 values and Emax values similar to the untreated receptors were obtained (EC50(ETARlabeled): 2.0 nM; EC50(ETBRlabeled): 0.8 nM), demonstrating the robustness of the GPCRs towards reducing conditions. For the Cys‐P1‐ and Cys‐P3‐tagged GPCRs, the EC50 and Emax values remained wt‐like independent of the TCEP‐containing buffer and the transfer of a TAMRA fluorophore onto the tagged GPCRs. In comparison to the untagged receptors, ligand potencies for Cys‐P1‐ETAR (EC50(untreated): 2.6 nM), Cys‐P3‐ETAR (EC50(untreated): 2.0 nM), Cys‐P1‐ETBR (EC50(untreated): 0.9 nM), and Cys‐P3‐ETBR (EC50(untreated): 0.7 nM) were similar to the respective wt GPCR. Additionally, the observed Emax values remained wt‐like even after transfer of the additional TAMRA group onto the N‐terminus of the Cys‐P1/P3‐carrying GPCRs, demonstrating the low impact of the peptide‐templated acyl transfer labeling approach and its high compatibility with GPCR signaling.
Table 2.
Receptor activation profiles after receptor labeling by peptide‐templated acyl transfer. Wild type (wt) ETAR/ETBR‐GFP, Cys‐P1/P3‐ETAR‐GFP or Cys‐P1/P3‐ETBR‐GFP activation was assessed by Ca2+ flux analyses before (untreated) and after fluorescent labeling (labeled; n≥3 performed in duplicates). Signal transduction data were normalized to the wt receptors and represent the mean over all assay repetitions.
|
Receptor |
ETAR |
ETBR |
|||||
|---|---|---|---|---|---|---|---|
|
N‐terminal tag |
EC50 [nM] |
pEC50 ±SEM |
Emax [%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
Emax [%] ±SEM |
|
|
wt |
untreated |
2.5 |
8.6±0.07 |
105±3 |
1.1 |
8.9±0.07 |
99±2 |
|
labeled |
2.0 |
8.7±0.07 |
106±3 |
0.8 |
9.1±0.07 |
106±3 |
|
|
Cys‐P1 |
untreated |
2.6 |
8.6±0.15 |
89±6 |
0.9 |
9.1±0.17 |
108±7 |
|
labeled |
2.5 |
8.6±0.11 |
84±4 |
0.7 |
9.2±0.17 |
119±7 |
|
|
Cys‐P3 |
untreated |
2.0 |
8.7±0.13 |
113±6 |
0.7 |
9.1±0.10 |
112±4 |
|
labeled |
1.7 |
8.8±0.10 |
118±5 |
0.5 |
9.3±0.18 |
126±6 |
|
Distinct labeling of co‐receptor expression and proximity studies
Next, we envisioned the simultaneous labeling of co‐expressed Cys‐P1‐ and Cys‐P3‐tagged receptors in a one‐pot reaction. Therefore, the labeling reaction requires a high selectivity and specificity of the tag/probe pairs Cys‐P1/P2 and Cys‐P3/P4. The selectivity of the thioester peptide probes towards their complementary POI tag (Supporting Information Figure S4) was investigated in a live cell setup. Cells, expressing either Cys‐P3‐ETAR‐GFP or Cys‐P1‐ETBR‐GFP were incubated with either TAMRA‐P2 or TAMRA‐P4. The C‐terminal GFP was used as a control element to discriminate between successful receptor labeling and unspecific background staining. When incubating the P1‐tagged receptor with the P4 probe and the P3‐tagged receptor with the P2 peptide, no successful TAMRA labeling was detectable (Supporting Information Figure S4). Rapid TAMRA staining within 5 min was only observable for the Cys‐P1‐GPCR treated with TAMRA‐P2 and the Cys‐P3‐GPCR treated with TAMRA‐P4. No cross‐reactivity was observable by fluorescence microscopy since the cargo transfer was limited to the probes, which are complementary to the respective acceptor tag. Additionally, the TAMRA/GFP ratio of labeled receptors was used to estimate labeling efficiencies on live cells and to exclude labeling bias between the coiled‐coil peptide pairs. Similar labeling rates were determined for the ETAR and ETBR, equipped with either the Cys‐P1‐ or Cys‐P3‐tag, using TAMRA‐carrying peptide probes (Supporting Information Figure S5). No labeling bias was detectable for the endothelin receptor subtypes and peptide‐templated labeling achieved similar TAMRA/GFP ratios between all tested receptor setups.
Since the selectivity and specificity of the coiled‐coil peptide probes towards the respective tag (P1/P2 and P3/P4) and their transfer versatility was demonstrated, we simultaneously targeted two POIs expressed in the same cell by transferring different dyes to study GPCR interactions by FRET. The dyes Atto488 (attached to the P2 probe) and Atto565 (attached to the P4 probe) were used because of their overlapping spectral properties suitable for FRET (Figure 2A). Receptor titration experiments were performed by co‐transfecting constant amounts of the P1‐tagged GPCR with increasing amounts of a P3‐tagged GPCR, which were then simultaneously labeled in a one‐pot reaction (Figure 2B). Receptor expression was estimated by labeling either P1‐ or P3‐tagged GPCR. First, we applied this approach to homo‐receptor setups. A saturation of the FRET signal with increasing A/D ratio was achieved for all four GPCR species, allowing a hyperbolic fit (Supporting Information Table S3). However, varying FRETmax values (maximum FRET signal in saturation) were observed for the different homo‐receptor setups. Higher FRETmax values were observed for the ETAR (0.76) and the AT1R (0.72), followed by the APJ (0.58) and ETBR (0.57). Using the calculated FRETmax derived from the hyperbolic fit (resembling the theoretic signal at complete saturation), both AT1R and ETAR displayed higher FRET values compared to the observed FRETmax (0.98 and 0.96, respectively), whereas the APJ and ETBR showed lower values (0.75 and 0.57, respectively). For the latter receptor, the calculated FRETmax value correlates to the observed FRETmax. FRET50 values, corresponding to an observed (pseudo‐)affinity, were then determined from the hyperbolic fit, which were lower for both endothelin receptor subtypes (ETAR: 0.15; ETBR: 0.25) than for the APJ (0.63) and AT1R (0.85).
Figure 2.

Determination of constitutive proximity between GPCRs in homo‐receptor clusters by FRET analyses. (A) Close proximity of two GPCR protomers, equipped with either Atto488 (green star) or Atto565 (red star) by peptide‐templated acyl transfer enables Förster resonance energy transfer (FRET). Activation of labeled GPCRs by agonist application can trigger changes in FRET fluorescence due to conformational rearrangement of the receptor. (B) To assess local association of membrane‐embedded GPCRs from the same species (left to right: ETAR, ETBR, AT1R, APJ), receptor titration experiments were performed by transfecting HEK293 cells with constant amounts of P1‐tagged GPCR (subsequently addressed with the FRET donor) and increasing amounts of P3‐tagged GPCR (subsequently labeled with the FRET acceptor). Labeling was simultaneously performed with Atto488‐P2 (FRET donor, green) and Atto565‐P4 (FRET acceptor, red). To determine the acceptor/donor fluorescence ratio at the cell membrane (x axis), either acceptor or donor labeling was performed to determine the respective fluorescence signal of membrane‐embedded GPCRs and exclude intracellularly retained receptor subpopulations. FRET measurement was performed in quadruplicates (n≥2). NetFRET values were determined by subtraction of fluorescence values derived from cells, expressing only the P1‐GPCR (donor). Data represent the mean over all assay repetitions. GPCR proximity is indicated by signal saturation. (C, D) The influence of receptor activation on the proximity‐induced FRET was determined by transfecting P1‐tagged donor GPCR and P3‐tagged acceptor GPCR using equal amounts (1 : 1 ratio). After receptor labeling, the baseline (basal FRET) was monitored before addition of different ligand concentrations (C: 10 nM ligand; D: 500 nM ligand) at t=0 s. ET‐1 was used for activation of ETAR and ETBR, AngII for activation of AT1R, and Ap13 for activation of APJ (left to right: ETAR, ETBR, AT1R, APJ). Ligand‐induced effects on the FRET signal were observed for 10 min after ligand addition. Curves represent the deviation of the FRET signal after ligand addition to the basal FRET (prior to ligand addition) relative to the respective buffer control. (kinetic analysis n=3, each performed in quadruplicates; black: buffer, light blue: 10 nM ligand, blue: 500 nM ligand; representative kinetic data shown).
Agonist‐induced proximity of receptors
Next, we investigated the effect of agonist administration and receptor activation on modulating the N‐terminal FRET signal by means of kinetic analyses. Based on the receptor titration experiments and saturation studies, a 1 : 1 transfection ratio of the GPCR species was chosen. By keeping the equal transfection ratio and overall transfection rates low, potential crowding effects due to protein overexpression were minimized. To ensure efficient receptor activation and exclude cross‐reactivity between the different ligands and the GPCRs in the kinetic experiments, we validated ligand potencies and the respective ligand/receptor selectivity in signal transduction assays (Supporting Information Figure S6 and Table S4). Concentration dependent activation of ETAR, ETBR, AT1R, and APJ was performed with either angiotensin II (AngII), apelin‐13 (Ap13) or ET‐1. For the respective ligand/receptor systems (AngII/AT1R, Ap13/APJ, ET‐1/ETAR, and ET‐1/ETBR) low nanomolar or subnanomolar EC50 values were determined for all GPCRs (EC50(APJ): 1.7 nM; (EC50(AT1R): 0.4 nM; EC50(ETAR): 0.6 nM; EC50(ETBR): 2.4 nM). No inter‐system cross‐activation was observable between the applied peptide agonists and the different GPCR species. Based on the signal transduction assays, we applied low nanomolar ligand concentrations (10 nM, correlating to an at least >4‐fold higher ligand concentration relative to the respective EC50 value) to facilitate efficient receptor activation for kinetic studies without cellular overstimulation. Generally, the circulating plasma levels of these peptide hormones are in the picomolar range.[ 33 , 34 , 35 ] Applying 10 nM peptide ligand (Figure 2C) to the co‐expressed and simultaneously labeled GPCRs did not induce fluorescence changes for ETAR, AT1R, and APJ compared to the control experiment without ligand (buffer control). The observed signals for these three GPCR species did not deviate from the buffer condition for 10 min after ligand addition. Contrary, the homomeric expressed ETBR displayed high sensitivity towards receptor activation even at low ligand concentrations. 10 nM ET‐1 induced a decline of the FRET signal (relative to the buffer control). To exclude assay artefacts due to slow diffusion of the peptide after addition to the GPCR‐expressing cells, an increased ligand amount was applied to facilitate immediate activation of all membrane‐embedded GPCRs (500 nM agonist, correlating to an at least >100‐fold higher ligand concentration relative to the respective EC50 value). After application of the high ligand concentration an immediate decrease in FRET was observed for the homo‐receptor expression of AT1R and APJ in comparison to the respective buffer control (Figure 2D). The observed changes were similar for both AT1R and APJ. Contrary to these two GPCR species, the ETAR displayed a continuous decay of the FRET signal upon addition of 500 nM ET‐1, in comparison to the buffer control. Interestingly, whereas 10 nM ET‐1 initiated a continuous FRET decay for the ETBR, 500 nM ET‐1 induced an immediate loss in observed FRET signal, emphasizing the N‐terminal sensitivity of the ETBR towards receptor activation.
Membrane residence time and constitutive internalization
We used the peptide‐templated labeling of membrane‐embedded GPCR subpopulations to investigate the membrane residence time and constitutive internalization of the homo‐receptor GPCRs in mammalian cells by fluorescence labeling. We monitored ligand‐independent internalization of the four GPCR species up to 60 min after fluorescence labeling (Figure 3). Fluorescently labeled ETAR did not translocate into the cytoplasm in the absence of agonist and remained at the cell surface. The membrane fluorescence remained unchanged and no intracellular vesicles were detectable. Similarly, the membrane fluorescence for both AT1R and APJ remained constant over 30 min post‐labeling (AT1R: 86±7 %; APJ: 96±1 %). However, after 1 h, a decrease in membrane fluorescence was observed for both receptor species (AT1R: 75±5 %; APJ: 81±9 %), indicating a minor internalization rate in the absence of added agonist. Contrary to these GPCRs, the ETBR showed a distinct different membrane residence time. The receptor quickly internalizes without agonist‐mediated activation, characterized by a vanishing membrane fluorescence, which significantly deviates from the ETAR, AT1R, and APJ (Supporting Information Figure S9). Receptor internalization was detectable as early as 15 min (membrane fluorescence: 60±3 %) after ETBR visualization by the acyl transfer reaction and continuously decreased over the observation time period (30 min: 42±6 %; 60 min: 23±3 %). We further verified the impact of receptor activation on GPCR internalization after peptide‐templated labeling (Supporting Information Figure S7). In contrast to the limited constitutive internalization in the absence of agonist, complete internalization of membrane‐embedded receptors was observable for ETAR, ETBR, AT1R, and APJ after activation by the respective endogenous ligand. Further, the synthetic ligand [4Ala1,3,11,15, Nle7]‐ET‐1 (linear ET‐1) facilitated efficient internalization of the ETBR, but not the ETAR.
Figure 3.

Membrane residence time of N‐terminally labeled GPCRs in the absence of agonist administration. Membrane‐embedded Cys‐P3‐ETAR (A), Cys‐P1‐ETBR (B) SP‐Cys‐P3‐AT1R (C) or SP‐Cys‐P3‐APJ (D) were stained using the Atto488‐P2 (green) or Atto565‐P4 (red) peptide, respectively. Image acquisition was performed at distinct time point (0, 15, 30 and 60 min) post‐labeling. The membrane fluorescence was quantified for each time point and normalized to 0 min (100 %) and background fluorescence (0 %) (ETAR – blue, ETBR – light green, AT1R – orange, APJ – purple; ETAR/ETBR n=3, AT1R/APJ n=2; quantitative data represent the average over all assay repetitions and 10–15 cells were analyzed per time point and experiment; representative image shown). Scale bar: 10 μm.
Next, the orthogonal labeling was applied to study GPCRs in a hetero‐receptor co‐expression setup by receptor titration experiments similar to the homo‐receptor studies. We verified efficient peptide‐templated labeling for each hetero‐GPCR combination by fluorescence microscopy (Figure 4). All receptors were successfully labeled in the co‐expression setups depending on the N‐terminal tag (P1‐GPCR: Atto488, P3‐GPCR: Atto565). For co‐expressed ETAR/ETBR, a FRET signal saturation was observed (Supporting Information Table S3). However, signal saturation was limited to the endothelin receptor hetero‐expression. A linear FRET to A/D ratio‐correlation was observed for the GPCR co‐expressions containing either AT1R or APJ, which showed a comparable quality of the linear fit (R2 AT1R/ETAR: 0.92; R2 AT1R/ETBR: 0.90; R2 APJ/ETAR: 0.99; R2 APJ/ETBR: 0.91). Additionally, no FRET saturation was detectable for the AT1R/APJ co‐expression.
Figure 4.

Determination of constitutive proximity between GPCRs in hetero‐receptor clusters by proximity‐dependent FRET analyses. For constitutive interactions of membrane‐embedded GPCRs from different species, receptor titration experiments were performed by transfecting HEK293 cells with constant amounts of P1‐tagged GPCR (subsequently addressed with the FRET donor) and increasing amounts of P3‐tagged GPCR (subsequently labeled with the FRET acceptor). Labeling was simultaneously performed with Atto488‐P2 (FRET donor, green) and Atto565‐P4 (FRET acceptor, red). Formation of specific interaction is indicated by hyperbolic fitting of the ETAR/ETBR proximity‐dependent FRET data (A). A linear correlation was observed for ETAR/AT1R (B), ETAR/APJ (C), ETBR/AT1R (D), ETBR/APJ (E), and AT1R/APJ (F) in the absence of receptor activation. Successful peptide‐templated labeling was validated for all investigated GPCR combinations (fluorescence microscopy n=3; representative image shown). (G) For ligand‐driven effects on constitutive ETAR/ETBR proximity‐dependent FRET, FRET was observed without ligand post labeling (basal). Upon stimulation (10 nM ET‐1: light blue; 500 nM ET‐1: dark blue), the FRET fluorescence was observed for 10 min (n≥2 each performed in quadruplicates, representative kinetic data shown). ETAR: blue, ETBR: light green, AT1R: orange, APJ: purple. Scale bar: 10 μm.
Since we only detected a signal saturation for the ETAR and ETBR, we investigated the impact of receptor activation on co‐expressed ETAR/ETBR (Figure 4G). Both, high (500 nM) and low (10 nM) ligand concentrations induced a decrease in the observed FRET signal. Whereas 10 nM ET‐1 led to a slow, but continuous FRET decay over several minutes, 500 nM ET‐1 induced an immediate loss of FRET. Both time courses of FRET decay showed similarities to the homo‐ETBR FRET kinetics.
Next, we investigated the role of the ETAR/ETBR interaction on cellular signaling and downstream effects. As no FRET interaction was determinable for ETBR co‐transfected AT1R or APJ, these receptors were used for control. First, the influence of the GPCR co‐expression on ETBR membrane residence time and constitutive internalization was determined by fluorescence microscopy (Figure 5). The co‐expression of ETAR/ETBR significantly prolonged the membrane residence time of the ETBR (Supporting Information Figure S9). The constitutive internalization of the latter was delayed (membrane fluorescence after 15 min: 102±10 %, relative to 0 min), but not abolished by the cellular expression of the ETAR (ETBR membrane fluorescence after 60 min: 33±6 %). Despite the constitutive translocation of the ETBR from the cell membrane into intracellular vesicles, membrane‐embedded ETAR remain at the cell surface without co‐internalization after 60 min post labeling (ETAR: 91±7 %). Interestingly, co‐expression of the AT1R did not delay the constitutive internalization of the ETBR, but this receptor displayed a potential co‐trafficking. This led to a decreased membrane‐residence time (after 60 min post labeling) of the AT1R (44±8 %) despite no ligand‐independent FRET between ETBR and AT1R was detectable. Co‐expression of the APJ did not interfere with the ligand‐independent behavior of the ETBR. APJ remained at the cell membrane for 30 min (86±5 %) but displays minor internalization at 1 h post labeling (membrane fluorescence after 60 min: 71±4 %), which is similar to the APJ membrane residence time without ETBR expression. The ETBR translocation into intracellular vesicles for the APJ/ETBR setup was comparable to the constitutive ETBR internalization without co‐expressed APJ (Supporting Information Figure S9).
Figure 5.

Membrane residence time of ETBR co‐expressed with different GPCRs. Membrane‐embedded Cys‐P1‐ETBR and Cys‐P3‐ETAR (A), SP‐Cys‐P3‐AT1R (B) or SP‐Cys‐P3‐APJ (C) were labeled using the Atto488‐P2 (green) and Atto565‐P4 (red) peptide probe. Picture acquisition was performed at distinct time point (0, 15, 30 and 60 min) post‐labeling without agonist application. Membrane fluorescence was quantified for each time point and normalized to 0 min (100 %) and background fluorescence (0 %) (ETAR: blue, ETBR: light green, AT1R: orange, APJ: purple). Representative images are shown, and quantitative data represent the average over all assay repetitions (n≥2 with 10–15 cells analyzed per time point and experiment). Scale bar: 10 μm.
Downstream signaling in cellular systems with heteromeric endothelin receptor expression
We assessed the agonist‐driven internalization of the ETBR co‐expressed with either ETAR, AT1R, or APJ by addition of the respective peptide agonists (Supporting Information Figure S8). For cells, expressing both ETAR and ETBR, the dual agonist ET‐1 induces efficient internalization of both GPCRs. Contrary, the ETBR‐selective linear ET‐1 induces efficient and complete internalization of the ETBR, but not the ETAR. For the ETBR co‐transfected with either AT1R or APJ, ET‐1 stimulated cells show intracellular uptake of both AT1R and APJ in the presence of ETBR similar to the constitutive internalization experiments. Complete intracellular localization of ETBR was observed due to constitutive internalization even when the cells were stimulated with AngII (AT1R activation) and Ap13 (APJ activation), which do not induce ETBR activation.
Since the co‐expression of ETAR and ETBR affected the signaling of this receptor in an unstimulated state, we probed whether the ETAR/ETBR co‐expression influences the signaling events of activated ETBR, which was characterized by Ca2+ flux analyses. To ensure similar ETBR expression throughout all investigations, an auto‐fluorescent GFP was fused to the C‐terminus of the ETBR, enabling the correlation of GFP fluorescence with ETBR expression. We demonstrated by fluorescence microscopy of the wt ETBR‐GFP, that most of this protein is transported to the cell membrane (Supporting Information Figure S1) and only small fractions of this receptor are intracellularly retained (e. g. in the Golgi apparatus and endoplasmic reticulum) or intracellularly localized due to constitutive internalization. To test the impact of GPCR stoichiometry on ETBR signaling, constant amounts of ETBR‐GFP and increasing amounts of non‐fluorescent ETAR were co‐transfected in titration experiments. Using these studies, no impact on intracellular Ca2+ release was detectable with the non‐selective endothelin receptor agonist ET‐1 (Figure 6). The EC50 and Emax values, derived from concentration‐dependent analyses, were comparable to the mock control (no co‐transfection of ETAR; EC50: 1.3 nM; Emax: 100±5 %) (Table 3). The EC50 and Emax values were independent of the amount of co‐transfected ETAR after ET‐1 administration. Interestingly, the GFP fluorescence, used to determine the total ETBR expression, positively correlated with increasing amounts of ETAR and, thus, increased ETAR/ETBR ratios. At a 1 : 1 transfection ratio, the ETBR‐GFP expression was increased by ∼30 %. Since ET‐1 activates both endothelin receptor subpopulations, stimulation with this peptide results in an integrated signal response from both ETAR and ETBR. To selectively activate ETBR in the presence of ETAR, we used a linear ETBR‐selective analog [4Ala1,3,11,15, Nle7]‐ET‐1. When cells, expressing both ETAR and ETBR, were activated with the linear ETBR‐selective agonist, the intracellular Ca2+ response was influenced by the ETAR/ETBR ratio compared to the mock control. In the presence of co‐transfected ETAR, the signaling of selectively activated ETBR displayed a decreased maximum signaling. ETBR‐specific signaling was reduced by ∼15 % for a 1 : 0.25 ETBR/ETAR ratio and by ∼50 % for a 1 : 1 transfection ratio. By adding ETAR in excess the loss of ETBR signaling was increased to ∼60 % compared to the mock control (Supporting Information Figure S10 and Table S6). In all cases of decreased ETBR‐mediated Ca2+ release, ligand potency remained similar to the ETBR/mock control (EC50: 2.0 nM). To exclude ligand scavenging by the ETAR, we investigated the effect of ET‐1 on ETBR signaling by blocking the ETAR with sitaxentan. This competitive antagonist displays a low‐nanomolar IC50 value at the ETAR (∼1 nM), but requires micromolar concentrations to efficiently inhibit ETBR (IC50∼10 μM). [36] No differences in both ligand efficacy and potency were detectable between the mock+vehicle (no ETAR expression, DMSO used as vehicle) and mock+sitaxentan condition (Figure 6B). By co‐expression of ETAR and addition of sitaxentan, the ETBR response after ET‐1 activation was reduced to ∼50 % (Table 4), which is comparable to the data obtained for the ETBR‐selective peptide. Similar results were obtained, when the ETBR/ETAR ratio was increased further (Supporting Information Figure S10 and Table S6). ETBR activation by the ETBR‐selective peptide induced only a ∼40 % signal compared to the mock control, whereas the non‐selective agonist ET‐1 displayed only minor Emax reduction. Pre‐incubation with the competitive and ETAR‐selective antagonist sitaxentan allowed the detection of decreased ETBR signaling after ET‐1 application at low ETBR transfection levels (Supporting Information Figure S10 and Table S7). In contrast to the ETAR, ETBR signaling was not altered in co‐expression studies with either AT1R (Figure 6C) or APJ (Figure 6D). EC50 and Emax values derived from ETBR/AT1R or ETBR/APJ co‐expressions were comparable to the respective mock controls (EC50: 1.8 nM (ETBR/AT1R) and 1.9 nM (ETBR/APJ); Table 3). To probe the specificity of the impaired ETBR signaling by ETAR expression, the Gq‐coupled AT1R‐YFP was co‐transfected with the ETAR and similar analyses of Gq activation were performed (Figure 6E, Table 5). ETAR co‐expression did not alter AT1R‐mediated Ca2+ release after AngII administration. Since the aforementioned studies were performed in transiently transfected COS‐7 cells, we additionally verified the signaling impact in a human‐derived cellular system (Supporting Information Figure S11, Supporting Information Tables S8 and S9).
Figure 6.

Investigation of ETBR signaling by co‐expression with other GPCRs. COS‐7 cells were transfected with constant amounts of ETBR‐GFP and increasing amounts of Cys‐P3‐tagged GPCRs (transfection ratio ETBR/GPCR: 1 : 0–1 : 1). Cells, expressing both ETBR and ETAR stimulated with either the non‐selective ET‐1 in the absence (A upper panel) or presence (B) of the ETAR‐selective antagonist sitaxentan (sita, red) or the ETBR‐selective agonist [4Ala1,3,11,15, Nle7]‐ET‐1 (linear ET‐1, A lower panel). Cells, expressing ETBR‐GFP and SP‐Cys‐P3‐AT1R (C), or SP‐Cys‐P3‐APJ (D) were stimulated with the ET‐1 for selective ETBR activation. Co‐expression of ETBR with ETAR, but not with AT1R and APJ, reduced the Ca2+ release upon ETBR activation. Co‐transfection of AT1R‐YFP and Cys‐P3‐ETAR (E) was used to validate the specificity of the impaired ETBR signaling by ETAR expression. Ca2+ flux was normalized to the respective receptor activation without receptor co‐transfection (mock control, 1 : 0 transfection ratio). ETBR‐GFP and AT1R‐YFP expression was monitored by GFP/YFP fluorescence. Ca2+ flux mediated by Gq protein (turquoise) activation was monitored in COS‐7 cells in duplicates and concentration‐response curved represent the average over all assay repetitions (n≥3; significance was determined by one‐way ANOVA and Tukey's post‐test, n.s.: not significant, **: P<0.01; ***: P<0.001).
Table 3.
Impact of ETBR/GPCR co‐expression on ETBR‐mediated Gq signaling. COS‐7 cells were transfected with constant amounts of ETBR‐GFP and increasing amounts of Cys‐P3‐tagged ETAR, AT1R, and APJ. Ca2+ response was monitored after administration of ET‐1 or [4Ala1,3,11,15, Nle7]‐ET‐1 (linear ET‐1). Ca2+ flux was normalized to the respective receptor activation without receptor co‐transfection (mock control, 1 : 0 transfection ratio). ETBR‐GFP and AT1R‐YFP expression was monitored by GFP/YFP fluorescence. Ca2+ flux was monitored in duplicates and concentration‐response curved represent the average over all assay repetitions (n≥3).
|
Co‐transfection ratio |
Relative Ca2+ release of ETBR |
|||||||
|---|---|---|---|---|---|---|---|---|
|
|
ETBR expression |
ET‐1 |
Linear ET‐1 |
|||||
|
|
[%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
[%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
[%] ±SEM |
|
|
ETBR/ETAR |
1 : 0 |
102±2 |
1.3 |
8.9±0.09 |
100±5 |
2.0 |
8.7±0.11 |
100±5 |
|
|
1 : 0.25 |
100±1 |
0.9 |
9.0±0.11 |
100±6 |
1.0 |
9.0±0.12 |
85±5 |
|
|
1 : 0.5 |
110±3 |
0.8 |
9.1±0.11 |
100±5 |
1.0 |
9.0±0.09 |
76±3 |
|
|
1 : 0.75 |
114±4 |
0.7 |
9.2±0.12 |
95±5 |
0.6 |
9.2±0.16 |
60±4 |
|
|
1 : 1 |
135±4 |
0.9 |
9.0±0.10 |
85±4 |
0.7 |
9.1±0.14 |
52±3 |
|
ETBR/AT1R |
1 : 0 |
95±2 |
1.8 |
8.7±0.12 |
99±6 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.25 |
90±7 |
1.6 |
8.8±0.13 |
99±7 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.5 |
114±7 |
1.8 |
8.7±0.11 |
109±6 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.75 |
91±14 |
1.2 |
8.9±0.14 |
93±6 |
n.d. |
n.d. |
n.d. |
|
|
1 : 1 |
81±3 |
1.3 |
8.9±0.09 |
83±4 |
n.d. |
n.d. |
n.d. |
|
ETBR/APJ |
1 : 0 |
104±4 |
1.9 |
8.7±0.12 |
96±6 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.25 |
102±8 |
1.1 |
8.9±0.10 |
92±4 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.5 |
93±4 |
0.9 |
9.0±0.11 |
97±5 |
n.d. |
n.d. |
n.d. |
|
|
1 : 0.75 |
93±3 |
0.5 |
9.3±0.13 |
92±5 |
n.d. |
n.d. |
n.d. |
|
|
1 : 1 |
103±1 |
0.9 |
9.0±0.11 |
83±4 |
n.d. |
n.d. |
n.d. |
n.d.=not determined.
Table 4.
ETBR signaling in response to ET‐1 and antagonist‐blocked ETAR. COS‐7 cells, expressing both ETBR and ETAR stimulated with either the non‐selective ET‐1 in the absence or presence of the ETAR‐selective antagonist sitaxentan. Ca2+ flux was monitored in duplicates and concentration‐response curved represent the average over all assay repetitions (n≥3).
|
Co‐transfection ratio |
|
Relative Ca2+ flux initiated by ETBR‐GFP |
||||||
|---|---|---|---|---|---|---|---|---|
|
|
ETBR expression |
ET‐1+vehicle |
ET‐1+sitaxentan |
|||||
|
|
[%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
[%] ±SEM |
EC50 [nM] |
pEC50 ±SEM |
[%] ±SEM |
|
|
ETBR/ETAR |
1 : 0 |
100±1 |
0.7 |
9.2±0.06 |
100±3 |
0.5 |
9.3±0.09 |
89±4 |
|
|
1 : 1 |
149±8 |
0.7 |
9.1±0.14 |
73±4 |
0.4 |
9.4±0.12 |
49±3 |
Table 5.
AngII‐induced and AT1R‐mediated Ca2+ response presence of co‐transfected ETAR. COS‐7 cells were transiently transfected with AT1R‐YFP and Cys‐P3‐ETAR. Ca2+ flux was normalized to the respective receptor activation without receptor co‐transfection (mock control, 1 : 0 transfection ratio). AT1R‐YFP expression was monitored by YFP fluorescence. Ca2+ flux was monitored in COS‐7 cells in duplicates and concentration‐response curved represent the average over all assay repetitions (n=3).
|
Co‐transfection ratio |
Relative Ca2+ release of AT1R |
||||
|---|---|---|---|---|---|
|
|
AT1R expression |
AngII |
|||
|
|
[%] ±SEM |
EC50 [nM] |
[%] ±SEM |
EC50 [nM] |
|
|
AT1R/ETAR |
1 : 0 |
100±1 |
0.2 |
9.8±0.07 |
100±2 |
|
|
1 : 0.25 |
122±4 |
0.3 |
9.6±0.08 |
98±3 |
|
|
1 : 0.5 |
135±21 |
0.3 |
9.6±0.12 |
89±4 |
|
|
1 : 0.75 |
171±21 |
0.3 |
9.6±0.09 |
83±3 |
|
|
1 : 1 |
153±44 |
0.4 |
9.4±0.15 |
96±5 |
Similar to the COS‐7 experiments, co‐expression of the ETAR with ETBR reduced the ETBR‐mediated Ca2+ release but did not affect AT1R‐mediated Gq activation, excluding cell culture artefacts. To determine the temporal frame of impaired ETBR signaling by ETAR co‐expression, we investigated the recruitment of arrestin 3 (arr3) as additional intracellular effector. For arr3 recruitment experiments, the ETBR‐GFP receptor was co‐transfected with either Cys‐P3‐ETAR, SP‐Cys‐P3‐AT1R or SP‐Cys‐P3‐APJ and a Nluc‐arr3 construct. Upon addition of the non‐selective ET‐1 or the ETBR‐selective [4Ala1,3,11,15, Nle7]‐ET‐1 to cells, the arr3 recruitment kinetics as well as the concentration‐dependent analyses of arr3 recruitment were comparable to the mock control for all experimental setups (Supporting Information Figure S12 and Table S10). The ETAR/ETBR‐co‐expressing cells display an arr3 recruitment similar to ETBR‐expressing cells, indicating a short‐term regulation of ETBR signal transduction by the ETAR.
Discussion
In recent years, it has become clear, that the local compartmentalization of GPCRs in the cell membrane is directly involved in the fine‐tuning of receptor signaling.[ 37 , 38 , 39 , 40 , 41 ] GPCR complexes can alter protein localization and intracellular signaling pathways.[ 42 , 43 ] Hints towards GPCR interactions have been found in crystal structures of model GPCRs like the C−X−C chemokine receptor 4, κ‐ and μ‐opioid receptors, and the β1 adrenergic receptor.[ 44 , 45 , 46 , 47 ] Since tight regulation of GPCR signaling is required for vascular homeostasis, GPCR interactions play an important role in the regulation of the cardiovascular system. The proximity of ETAR and ETBR was demonstrated by C‐terminal fluorophores in heterologous expression systems.[ 15 , 16 ] The AT1R forms constitutive oligomeric complexes and interacts with GPCRs like the angiotensin II receptor type 2 and the Mas1 oncogene receptor.[ 17 , 18 , 48 , 49 ] The APJ forms oligomers and interacts with e. g., the bradykinin type 1 receptor and the neurotensin receptor1.[ 19 , 50 , 51 ] However, lateral GPCR interactions in the cell membrane are difficult to investigate in complex cellular environments due to intracellular background noise due to GPCR crowding. Therefore, the exclusion of intracellular signals has been demonstrated to be superior in differentiating the behavior of membrane‐embedded GPCRs from the intracellular bulk. [52] Based on our previously reported peptide‐templated labeling, which targeted one GPCR species and enabled pulse‐chase studies, we expand here this approach to the visualization of two GPCR species in a one‐pot reaction.[ 25 , 26 , 27 ] This approach uses the proximity‐triggered acyl transfer reaction, which was demonstrated to covalently transfer an organic fluorophore onto membrane‐embedded GPCRs.[ 25 , 27 ] Here, the simultaneous labeling of different proteins allows the assessment of GPCR‐GPCR proximities with high spatial and temporal control. N‐terminal labeling has been recently reviewed, highlighting advantages like fast reaction kinetics and versatility of coiled‐coil‐based systems compared to e. g. enzyme‐based protein modification. [21] Our novel approach based on the Cys‐P1/P3‐tags adds only a small molecular weight increase (∼3 kDa) to the POI. Since the typical molecular weight of a GPCR is ∼45 kDa, this is a minor modification compared to other common protein labeling techniques like the SNAP‐tag (20 kDa) or Halo‐tag (33 kDa), which minimizes the danger of impaired POI function and localization.[ 53 , 54 ] Commonly applied for investigation of GPCRs are auto‐fluorescent reporters like GFP. Especially cyan or yellow fluorescent protein have been widely applied as FRET sensors to study GPCR‐GPCR interactions. However, they can alter protein mobility and no discrimination of membrane populations from the intracellular bulk is possible due to background signals.[ 55 , 56 , 57 ]
Peptide‐templated acyl transfer relies on the presence of a cysteine residue. We successfully combined this with the SP derived from the ETBR, which is cleaved during biosynthesis. This SP was shown to be tolerant towards substitutions of the subsequent N‐terminal domains. [58] However, no such detailed characterization of the SP of the ETAR is available. Since a negative impact of the Cys‐P1/P3 modification on ETAR expression could not be excluded a priori, we characterized the ETAR‐derived constructs concerning membrane localization and downstream signaling. The Cys‐P1/P3‐tag were introduced into the ETAR between S20 and D21 and wt‐like Gq signaling profiles were determined and efficient staining of membrane‐embedded receptors was detected, indicating the presence of the SP. Some GPCR species display high sensitivity towards modification of their N‐termini due to interference with membrane insertion and intracellular transport. In the initial report on GPCR labeling by peptide‐templated acyl transfer, an increased intracellular retention was observed for the N‐terminally modified neuropeptide Y4 receptor and neuropeptide FF receptors 1 and 2. [27] We combined the SP of the ETBR with the Cys‐P1/P3‐tag sequences to improve on intracellular transport and membrane insertion mechanisms. The SPETBR was shown previously to be transferable to other GPCRs like the μ‐opioid receptor. [59] The fusion of SP‐P1‐ and SP‐P3‐tags to the AT1R and APJ allowed membrane insertion and extracellular exposure of the N‐terminus. The SPETBR of modified AT1R and APJ is cleaved, releasing the N‐terminal cysteine and allowing efficient labeling by peptide‐templated acyl transfer. The peptide tags did not interfere with activation by AngII and Ap13, respectively, as demonstrated by EC50/Emax values comparable to the wt receptors. We successfully applied different fluorescent dyes like TAMRA, Atto488 and Atto565 to visualize membrane‐embedded receptors. The application of diverse cargos demonstrates the adaptability and flexibility of the acyl transfer reaction to label different protein species in a one‐pot reaction. Contrary to other coiled‐coil methods like MiniVIPER, our approach covalently links the cargo to the protein targets, without intracellular separation of the reporter group from the POI. [60] The transferred dye is covalently bound to the POI, which is emphasized by the applied alkaline wash protocol. This hydrolyzes any thioester‐dye conjugates, which did not participate in an acyl transfer reaction at N‐terminal cysteine residues as demonstrated previously.[ 25 , 27 ]
Besides the labeling versatility, the efficiency of protein labeling is of crucial importance. Using the E3/K3 coiled‐coil system, we recently demonstrated efficient labeling of different GPCRs like the human neuropeptide Y2 receptor.[ 25 , 26 , 27 ] By transfer of a TAMRA dye onto the N‐terminus of this receptor, carrying a C‐terminal YFP, we employed the TAMRA/YFP ratio as a means to determine the labeling efficiency, which were comparable for different GPCR subpopulations, indicating a highly robust labeling platform. Here, we determined the TAMRA/GFP ratio for the P1/P2 and P3/P4 peptides. Although, this is only an estimation of labeling efficiency in live cell setups due to the consideration of the total receptor population, robust and unbiased protein modification was detected for both tag/probe pairs.
For the orthogonal peptide pairs P1/P2 and P3/P4, the transfer kinetics of peptide nucleic acids (PNA) from P1 to P2 were assessed in in vitro transfer reactions. [29] A product formation rate of 60 % was quickly achieved using a 11mer PNA as cargo. However, these experiments differ from the reaction conditions applied here. The determination of transfer rates was not carried out under cell culture conditions. The increased temperature we applied here (cell culture conditions) for the GPCR labeling in live cells, is likely to positively influence the reaction kinetics and increase the transfer rate. Additionally, the cargo size influences the reaction kinetics of the transfer reaction and smaller cargos enhance the reaction kinetics. [29] Since the PNA cargo displays a higher molecular weight (∼3 kDa) compared to the dyes (<1 kDa), which were exploited in our experiments, a fast transfer of the fluorophores is presumable.
No cross reactivity of the two different coiled‐coil pairs was observed in our labeling experiments, which is consistent with the in depth characterization the P1/P2 and P3/4 coiled‐coils by Gradišar and Jerala. [28] Using this system, orthogonal labeling has been recently described for live cell imaging. [61] We employed peptide‐templated labeling for proximity‐dependent FRET analyses of four different GPCRs for which we detected varying FRET signals. High FRET signals point towards a close proximity of the GPCRs. For example the AT1R, which displayed high FRETmax values in our experiments, was reported to form large complexes. [18] Both APJ and ETBR display lower observed FRETmax values, indicating a distant N‐terminal orientation. Interestingly, no difference between the observed and calculated FRETmax value was detected for the ETBR, indicating protein proximity even at low expression levels, which correlates well to the ETBR compartmentalization in caveolae. [62] For the hetero‐receptor co‐transfections, a saturation of the observed FRETmax was limited to the ETAR/ETBR setup. However, proximity of ETBR and AT1R was previously suggested, which we were not able to detect. [63] However, co‐trafficking of AT1R and ETBR subpopulations was observed in our studies due to a potential localization of both GPCRs to caveolae.[ 62 , 64 , 65 ]
To assess the impact of receptor activation in our experimental setup, ET‐1 was added to either ETAR, ETBR or ETAR/ETBR co‐expressing cells. A fast decline of the FRET signal was observed, which points towards N‐terminal sensitivity to receptor activation, which we also observed for AT1R and APJ. Generally, ligand binding to GPCRs involves N‐terminal movement. The N‐terminus of the ETBR and AT1R play a crucial role in ligand binding by adopting lid‐like structures.[ 66 , 67 ] This movement can change the environment of the respective dye, providing a basis for altered fluorescent signals in the absence of lateral protein movement.
A crucial determinant of protein interactions are the respective expression levels. Here, we focused on membrane‐embedded receptor subpopulations, and used fluorescent signals derived from only P1‐ and P3‐labeling (in co‐expression setups) to estimate the receptor population at the cell membrane. However, this approach depends on efficient transfer rates between the coiled‐coil peptides. Combining precise determination of POI expression levels with absolute labeling efficacies in the cellular environment might allow for a more detailed discrimination between the interaction patterns of different GPCR species in the future.
To validate the relevance of the potential ETAR/ETBR interaction, we characterized its impact on ETBR signaling. Taking advantage of our labeling platform to visualize only membrane‐embedded proteins, a constitutive internalization was observed for the ETBR, but not AT1R, APJ, and ETAR. The ligand‐independent internalization of the ETBR is significantly delayed by co‐expression of the ETAR. However, FRET between membrane‐embedded ETAR and ETBR can lead to an underestimated initial ETBR population at the cell membrane but does not impair internalized receptors. No delay is detectable by co‐transfecting AT1R or APJ. Ligand‐induced receptor internalization was not altered by hetero‐receptor expression and complete receptor internalization was observed for the ETBR. Further, the signaling data of hetero‐receptor expression imply that the interaction of the ETBR with the intracellular Gq protein is also impaired by co‐expression of the ETAR. The integrated cellular signal of both ETAR and ETBR by the dual‐agonist ET‐1 was comparable over all transfection ratios, but the Ca2+ release after selective ETBR activation was decreased, which is consistent with a previous study on failed ETBR signaling due to ETAR co‐expression. [68] We verified the impaired ETBR signaling in case of antagonist‐occupied ETAR and ETBR‐selective agonists. For activation of the ETBR the recently described monocyclic ET‐1 analog [4Ala1,3,11,15, Nle7]‐ET‐1 was used due to its missing ETAR activation properties. [69] As we demonstrated the reduction of ETBR signaling also for ETAR occupied by the competitive antagonist sitaxentan, ligand scavenging by ETAR can be excluded as reason for the reduced ETBR activation. Further studies demonstrated that ETAR expression does not alter the signaling profile of other Gq‐coupled GPCRs as demonstrated for the AT1R. Therefore, it is likely that ETAR selectively impairs ETBR signaling due to close proximity in the cell membrane e. g. by masking intracellular interaction interfaces and, thus, delaying the initial effector coupling, sequestering endothelin receptor signaling. Sequential activation of both GPCRs species in the cell membrane potentially allows long‐lasting constriction effects, which have been reported for vascular smooth muscle cells (VSMC).[ 70 , 71 , 72 ] VSMC of pulmonary arteries and resistance vessels express both endothelin receptor subtypes with ETAR being the dominant species.[ 73 , 74 , 75 , 76 , 77 ] The ETAR potentially prolongs the ETBR membrane residence time in tissues, expressing both receptors (e. g., lung tissue), emphasizing its function as scavenging receptor for circulating ET‐1 to prevent hypertension. [6]
Conclusion
We established a novel technique for the simultaneous labeling of two GPCR species in a rapid one‐pot reaction in live cells. The approach consists of two N‐terminal tags, carrying an N‐terminal cysteine residue, which do not interfere with membrane localization and activation of the ETAR, ETBR, AT1R, and APJ. Using the orthogonal coiled‐coil pairs P1/P2 and P3/P4 allows the analysis of membrane‐embedded proteins without intracellular background. Furthermore, we demonstrate the versatility of this system by applying it to study GPCR distribution. We show that all four homo‐receptors are closely associated in the plasma membrane. Interestingly, proximity between different GPCRs was restricted to ETAR and ETBR. We identified a prolonged membrane residence time and an impaired initial Gq signaling of the ETBR in the presence of co‐expressed ETAR. However, ETAR expression did not alter Gq signaling of other GPCRs, indicating an additional regulatory element in endothelin signaling in the cardiovascular system.
Experimental Section
Materials for peptide synthesis
N‐α‐Fmoc‐protected amino acids, ethyl 2‐cyano‐2‐(hydroxyimino)acetate (Oxyma), and N,N′‐diisopropylcarbodiimide (DIC) were purchased from Iris Biotech (Marktredwitz, Germany), and F‐Wang resin, NovaSyn® W‐TGA resin, O‐(7‐azabenzotriazolyl)‐tetramethyluronium hexafluorophosphate (HATU), and O‐(1H‐6‐chlorobenzotriazole‐1‐yl)‐1,1,3,3‐tetramethyluronium hexafluorophosphate (HCTU) were supplied from Novabiochem (Darmstadt, Germany). TentaGel R RAM resin was obtained from Rapp Polymere (Tübingen, Germany). 6‐Carboxytetramethylrhodamine (TAMRA) was purchased from ChemPep, Inc. (Wellington, Florida). Atto488 and Atto565 dyes were obtained from ATTO TEC (Siegen, Germany). Acetonitrile (ACN) was purchased from VWR (Darmstadt, Germany). Dimethylformamide (DMF) and dichloromethane (DCM) were obtained from Biosolve (Valkenswaard, The Netherlands). N,N‐Diisopropylethylamine (DIPEA), 1,2‐ethanedithiol (EDT), 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), hydrazine monohydrate, 4‐methoxytriphenylmethyl chloride (Mmt‐Cl), mercaptophenyl acetic acid (MPAA), 4‐methylmorpholine (NMM), piperidine, thioanisole (TA), trifluoroacetic acid (TFA), and triisopropylsilane (TIS) were purchased from Sigma‐Aldrich (Taufkirchen, Germany). Diethyl ether was from Merck (Darmstadt, Germany).
Solid phase peptide synthesis
Synthesis of peptide agonists: Peptide were synthesized by solid phase peptide synthesis (SPPS) based on the 9‐fluorenylmethyloxycarbonyl (Fmoc)/tert‐butyl (tBu) strategy, carried out at 15 μmol scale. An eightfold molar excess of N‐α‐Fmoc‐protected amino acid, Oxyma, and DIC in DMF was used for automated robot synthesis performed on a SYRO I peptide synthesizer (Witten, Germany). Couplings reactions were carried twice with a reaction time of 40 min each. For Fmoc cleavage, a solution of 40 % (v/v in DMF) piperidine was applied for 3 min and 20 % (v/v in DMF) piperidine for 10 min. Peptides were cleaved from the resin with TFA/EDT/TA (90 : 3 : 7, v/v/v) for 3 h at room temperature (RT) and precipitated from ice‐cold diethyl ether. Peptide purification was carried out on a RP‐HPLC system (Shimadzu) equipped with a Phenomenex Kinetex C18 100 Å column by applying a linear gradient of eluent A (0.1 % TFA in H20, v/v) and B (0.08 % TFA in ACN). Peptide purity was confirmed by analytical RP‐HPLC system, and peptide identity was verified by two mass spectrometry (MS) systems: MALDI‐ToF MS (UltraflexIII, Bruker Daltonics) and ESI‐ion trap MS (HCT, Bruker).
Synthesis of thioester‐linked dye peptide conjugates: SPPS of the peptides P2 and P4 was performed on a G‐TentaGel R RAM resin (1–2 μmol scale), based on the Fmoc/ t Bu strategy, as described previously. [25] The S‐Mmt‐protected MPAA and the N‐Mmt‐Gly handle were prepared as described..[ 78 , 79 ] The S‐Mmt‐MPAA‐OH and Mmt‐Gly‐OH (4.5 equiv.) were coupled twice for 45 min in DMF using HCTU (4 equiv.), and NMM (8 equiv.). Mmt deprotection was performed by incubation with DCM/TFA/TIS (96 : 2 : 2, v/v/v, 2×1 min). The dyes were coupled using 4 equiv. dye acid, 3.6 equiv. HCTU, and 8 equiv. NMM in DMF for 45 min at RT. Peptides were cleaved from the resin with TFA/H2O/TIS (96 : 2 : 2, v/v/v) and precipitated from ice‐cooled diethyl ether. Purification was carried out on a semi preparative HPLC (Agilent 1100 series; column: Varian Polaris C18 A 5 μ 250×100, pore size 220 Å) by applying a gradient of eluent A (98.9 % H2O, 1 % ACN, 0.1 % TFA) and B (98.9 % ACN, 1 % H2O, 0.1 % TFA). Peptide purity was determined by analytical HPLC (Merck‐Hitachi Elite LaChrom; column: Varian Polaris C18 A 5 μ 250×46, pore size 220 Å). Peptide identity was verified by MALDI‐ToF MS (Voyager‐DE Pro Biospectrometry Workstation) and ESI MS (Agilent 1100 series LC/MSD).
Materials for molecular biology
Lysogeny broth was obtained from Sigma‐Aldrich (Taufkirchen, Germany). The antibiotics (ampicillin, hygromycin, and kanamycin), Phusion polymerase, restriction enzymes, and T4 ligase were purchased from Thermo Fisher Scientific (Waltham, Massachusetts). Oligonucleotides used for cloning were obtained from biomers.net GmbH (Ulm, Germany). The pCMV3‐ETAR‐GFP and pCMV3‐ETBR‐GFP plasmids were purchased from Sino Biological Inc. (Beijing, China).
Plasmid construction
Primer sequences used for cloning are provided in Supporting Information Figure S1. The cDNA of the human angiotensin II receptor type 1 (AT1R; NM_009585.4) and apelin receptor (APJ; NM_005161.5) were C‐terminally fused to the enhanced yellow fluorescent protein (eYFP) and subcloned into the pVitro2‐hygro‐mcs vector. The Cys‐P1‐tag (primer: AT1R: 1 and 5; APJ: 3 and 5) and the Cys‐P3‐tag (primer for AT1R: 2 and 5; APJ: 4 and 6) were fused to the N‐terminus of the AT1R‐eYFP or APJ‐eYFP, respectively, by overlap extension (OE) PCR, and subcloned into the pVitro2‐hygro‐mcs vector. Removal of the eYFP‐tag was achieved by selective amplification of the Cys‐P1/P3‐GPCR cDNA (primer 8/9 and 10/11, respectively) and subcloning into the pVitro2 vector. Insertion of the signal peptide (SP), was achieved by restriction‐free ligation (RFL). [80] The RFL primer was amplified from the SP sequence from the pCMV3‐ETBR‐GFP plasmid (Cys‐P1: 12 and 13; Cys‐P3: 14 and 15). The cDNA of the Cys‐P1‐ and Cys‐P3‐tag were inserted behind the endogenous SPs of the ETAR and ETBR by RFL using the pVitro2‐SP‐Cys‐P1/P3‐AT1R‐eYFP as template (primer for Cys‐P1‐ETAR: 16 and 17; Cys‐P3‐ETAR: 18 and 19; Cys‐P3‐ETBR: 22 and 23). Generation of the Cys‐P1‐ETBR‐GFP plasmid has been described previously. [29] The C‐terminal fluorophore was removed by selective amplification of the SP‐Cys‐P1/P3‐ETAR/ETBR sequence and subcloning into the pVitro2 vector. Plasmids were verified by Sanger sequencing using an in‐house facility.
Materials for cell culture
Dulbecco's modified Eagle's medium (DMEM), Dulbecco's phosphate buffered saline (DPBS), Ham′s F12, Hank's balanced salt solution (HBSS), and trypsin/EDTA were purchased from Lonza (Basel, Switzerland). Fetal calf serum (FCS) was obtained from Biochrom (Berlin, Germany). Poly‐D‐lysine was obtained from Merck (Darmstadt, Germany). OptiMEM was purchased from Life Technologies (Carlsbad, California). Hoechst 33342, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), Pluronic® F‐127, probenecid, and lithium chloride were obtained from Sigma‐Aldrich (Taufkirchen, Germany). Bovine serum albumin (fraction V; BSA) was obtained from Carl Roth GmbH (Karlsruhe, Germany). Endothelin 1 (ET‐1) was ordered from Bachem (Bubendorf, Switzerland). 8‐Well ibiTreat μ‐slides were obtained from IBIDI (Martinsried, Germany). Lipofectamine® 2000 was supplied from Invitrogen (Carlsbad, California). Metafectene® Pro was obtained from Biontex Laboratories GmbH (Munich, Germany). Cell culture flasks were purchased from TPP (Trasadingen, Switzerland). CELLSTAR 6‐well and 384‐well plates, μCLEAR CELLSTAR 96‐well plates, black 96‐well plates were obtained from Greiner Bio‐One (Kremsmünster, Austria). Tris(2‐carboxyethyl) phosphine (TCEP) was obtained from Carl Roth GmbH & Co. KG (Karlsruhe, Germany). Fluo‐2AM was supplied from Abcam (Cambridge, United Kingdom). The IP‐one Gq kit was obtained from Cis‐Bio (Codolet, France).
Cell culture
All eukaryotic cell lines were cultivated at 37 °C, 95 % humidity, and 5 % CO2 (standard conditions). HEK293 cells were grown in DMEM/Ham′s F12 (1 : 1, v/v) supplemented with 15 % FCS (v/v). COS‐7 cells were grown in DMEM supplemented 10 % FCS (v/v). After reaching full confluency, cells were split into new culture flasks for further cultivation. All cell lines were tested negative for mycoplasma.
Peptide‐templated acyl transfer and live cell fluorescence microscopy
HEK293 cells were seeded in 8‐well ibiTreat μ‐slides (160,000 cells/well), coated with poly‐D‐lysine. After reaching 70–80 % confluency, cells were transiently transfected using Lipofectamine® 2000 (according to the manufacturer's protocol). For transfection a total amount of 1000 ng plasmid DNA was applied, balanced with empty pcDNA3.1 vector (mock). For co‐transfection, plasmids were applied in different ratios (1/1 or 1/3). For microscopy, the medium was replaced with 200 μL OptiMEM for 20 min. For peptide‐templated acyl transfer, cells were incubated with 200 μL 20 mM HEPES in HBSS (pH 7.2) for 10 min at standard conditions. Labeling probe was applied at 100 nM (in 20 mM HEPES in HBSS, supplemented with 0.1 mM TCEP) for 5 min at room temperature. For simultaneous labeling of two proteins, probes were applied simultaneously. Then, cells were treated with an alkaline wash (200 mM NaHCO3 in DPBS, pH 8.4) for 1.5 min, followed by two washing steps (20 mM HEPES in HBSS, pH 7.2). For nuclear staining, 1 μL of Hoechst 33342 (0.5 mg/mL) was added 30 min prior to image acquisition. A Zeiss Axio Observer.Z1 microscope (including ApoTome.2 Imaging System, AxioCamMRm camera, incubation chamber, and C‐Apochromat 63x/1.20 W objective) was used for image acquisition. A 46HE filter set (GFP: excitation 488–512 nm; emission 520–550 nm), a 31 filter set (TAMRA: excitation 550–580 nm; emission 590–650 nm), and a 49 filter set (nuclear stain: excitation 335–383 nm; emission 420–470 nm) were used for fluorescence detection. To quantify membrane fluorescence, the open‐access software ImageJ was applied to .TIFF files (8 Bit grayscale), using the segmented line function. Background fluorescence was subtracted for each picture individually. Data were normalized to no internalization (t=0 min post labeling; 100 %) and background fluorescence (0 %). Statistical analyses were performed using the software GraphPad PRISM 5.0 (San Diego, USA). Data represent mean±standard error of the mean (SEM) of at least two experiments and analyzing at least 10–15 different cells per time point and repetition.
Signal transduction assays
Calcium flux assay: COS‐7 or HEK293 cells were grown in 25 cm2 culture flasks until 70–80 % confluency was reached. Transient transfection was performed using Metafectene® Pro (according to the manufacturer's protocol) with a total of 4000 ng plasmid DNA per 25 cm2 flask (low ETBR expression: 1000 ng; intermediate ETBR expression: 2000 ng), balanced with empty pcDNA3.1 vector (mock). For co‐transfection, respective plasmids ratios were applied. 24 h post transfection, cells were seeded into black 96‐well plates (100,000 cells/well) and incubated overnight. For Ca2+ flux analysis cells were incubated with 1 μM Fluo2AM (in 20 mM HEPES and 2.5 mM probenecid in HBSS, pH 7.5) for 1 h at 37 C. The solution was replaced with assay buffer (20 mM HEPES and 2.5 mM probenecid in HBSS, pH 7.5). Peptides were applied in a concentration range (10−7–10−12 M), using the FlexStation (Molecular Devices) (excitation: 485 nm; emission: 525 nm). Concentration‐response curves were generated as x‐fold over basal and by normalization to the respective wild‐type receptor or mock control. Determination of EC50/pEC50, and Emax values was performed with the software GraphPad PRISM 5.0 (San Diego, USA). Curves represent mean±standard error of the mean (SEM) of at least three experiments, each performed in duplicates.
Peptide‐templated acyl transfer and GPCR proximity analyses: HEK293 cells were grown in 6‐well plates until 70–80 % confluency was reached. Transient transfection was performed by Metafectene® Pro (according to the manufacturer's protocol) with a total DNA amount of 4000 ng per well. For co‐transfection, plasmids were used in different ratios, balanced with empty pcDNA3.1 (mock). For titration experiments, 1000 ng or 2000 ng donor were co‐transfected with increasing amounts of acceptor. For kinetic FRET experiments, 1000 ng of donor and acceptor were applied. 24 h post transfection, cells were re‐seeded into black 96‐well plates (100,000 cells/well), coated with poly‐D‐lysine, and cultured overnight at standard conditions. For peptide‐templated labeling, the medium was replaced with 100 μL blocking buffer (1 % BSA in 20 mM HEPES in HBSS, pH 7.2) at 37 °C for 10 min. After aspiration of the blocking buffer, labeling was performed using 100 nM of each peptide probe dissolved in 20 mM HEPES in HBSS (pH 7.2), supplemented with 0.1 mM TCEP, and incubation for 5 min at standard conditions. Then, cells were treated with an alkaline wash (200 mM NaHCO3 in DPBS, pH 8.4) for 1 min, followed by two washing steps (20 mM HEPES in HBSS, pH 7.2). For saturation experiments, the acceptor/donor fluorescence (A/D) ratio was determined by labeling membrane‐embedded proteins on cells, co‐expressing P1‐ and P3‐tagged GPCRs, using only the Atto488‐P2 or Atto565‐P4 peptide and direct excitation of the respective fluorophore. Fluorescence detection was performed on a Tecan Spark plate reader (Tecan Group, Männedorf, Switzerland) (Atto488: excitation: 475–495 nm; emission: 515–525 nm; Atto565 excitation: 525–555 nm, emission: 580–600 nm):
For measuring FRET, both probes were simultaneously applied and the donor/acceptor fluorescence were measured after donor excitation:
For receptor titration experiments netFRET values were determined by subtraction of background fluorescence signals from cells expressing only the P1‐tagged donor receptor (no expression of the acceptor protein), treated with either Atto488‐P2 or Atto565‐P4, respectively, to account for fluorescent bleed‐through and probe retention after washing:
For kinetic measurements the baseline was recorded for 3 min prior to ligand addition and changes in FRET were recorded for 10 min at 37 °C. FRET ratios were determined as A/D ratio after donor excitation. Normalization to the buffer control was performed to account for donor dye photobleaching. FRET curves represent mean±standard error of the mean (SEM) of at least two experiments, each performed in quadruplicates.
Statistical analysis
Statistical analyses were performed with the software GraphPad PRISM 5.0 (San Diego, USA). Significances were calculated by one‐way ANOVA and Tukey's t‐test.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank K. Löbner, J. Schwesinger, R. Müller, R. Reppich‐Sacher and C. Dammann for their expert technical assistance. Funding from the “Fonds der chemischen Industrie” (FCI). Funding by the German Science Foundation (SPP1623, BE1264‐15, SE819‐17; and project number 421152132, SFB1423 TP B01), and the Graduate School “Leipzig School of Natural Sciences – Building with Molecules and Nanoobjects” (BuildMoNa) is gratefully acknowledged. Open Access funding enabled and organized by Projekt DEAL.
P. Wolf, A. Mohr, G. Gavins, V. Behr, K. Mörl, O. Seitz, A. G. Beck-Sickinger, ChemBioChem 2022, 23, e202100340.
References
- 1. Ritter S. L., Hall R. A., Nat. Rev. Mol. Cell Biol. 2009, 10, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mahoney J. P., Sunahara R. K., Curr. Opin. Struct. Biol. 2016, 41, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim K., Chung K. Y., Arch. Pharmacal Res. 2020, 43, 890–899. [DOI] [PubMed] [Google Scholar]
- 4. Davenport A. P., Hyndman K. A., Dhaun N., Southan C., Kohan D. E., Pollock J. S., Pollock D. M., Webb D. J., Maguire J. J., Pharmacol. Rev. 2016, 68, 357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maguire J. J., Davenport A. P., Br. J. Pharmacol. 1995, 115, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukuroda T., Fujikawa T., Ozaki S., Ishikawa K., Yano M., et al., Biochem. Biophys. Res. Commun. 1994, 199, 1461–1465. [DOI] [PubMed] [Google Scholar]
- 7. Plumpton C., Ferro C. J., Haynes W. G., Webb D. J., Davenport A. P., Br. J. Pharmacol. 1996, 119, 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verhaar M. C., Strachan F. E., Newby D. E., Cruden N. L., Koomans H. A., Rabelink T. J., Webb D. J., Circulation 1998, 97, 752–756. [DOI] [PubMed] [Google Scholar]
- 9. Kiowski W., Lüscher T. F., Linder L., Bühler F. R., Circulation 1991, 83, 469–475. [DOI] [PubMed] [Google Scholar]
- 10. Wright C. E., Fozard J. R., Eur. J. Pharmacol. 1988, 155, 201–203. [DOI] [PubMed] [Google Scholar]
- 11. Vukelic S., Griendling K. K., Circ. Res. 2014, 114, 754–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mughal A., O′Rourke S. T., Pharmacol. Ther. 2018, 190, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andersen C. U., Markvardsen L. H., Hilberg O., Simonsen U., Respir. Med. 2009, 103, 1663–1671. [DOI] [PubMed] [Google Scholar]
- 14. Schinzari F., Veneziani A., Mores N., Barini A., Di Daniele N., Cardillo C., Tesauro M., Hypertension 2017, 69, 942–949. [DOI] [PubMed] [Google Scholar]
- 15. Gregan B., Schaefer M., Rosenthal W., Oksche A., J. Cardiovasc. Pharmacol. 2004, 44 Suppl 1, 3. [DOI] [PubMed] [Google Scholar]
- 16. Evans N. J., Walker J. W., Biophys. J. 2008, 95, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansen J. L., Theilade J., Haunsø S., Sheikh S. P., J. Biol. Chem. 2004, 279, 24108–24115. [DOI] [PubMed] [Google Scholar]
- 18. Young B. M., Nguyen E., Chedrawe M. A. J., Rainey J. K., Dupré D. J., J. Biol. Chem. 2017, 292, 3341–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai X., Bai B., Zhang R., Wang C., Chen J., Sci. Rep. 2017, 7, 40335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Işbilir A., Serfling R., Möller J., Thomas R., de Faveri C., Zabel U., Scarselli M., Beck-Sickinger A. G., Bock A., Coin I., Lohse M. J., Annibale P., Nat. Protoc. 2021, 16, 1419–1451. [DOI] [PubMed] [Google Scholar]
- 21. Wolf P., Gavins G., Beck-Sickinger A., Seitz O., ChemBioChem 2021, 22, 1717–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milligan G., Ramsay D., Pascal G., Carrillo J. J., Life Sci. 2003, 74, 181–188. [DOI] [PubMed] [Google Scholar]
- 23. Cottet M., Faklaris O., Maurel D., Scholler P., Doumazane E., Trinquet E., Pin J.-P., Durroux T., Front. Endocrinol. 2012, 3, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo H., An S., Ward R., Yang Y., Liu Y., Guo X.-X., Hao Q., Xu T.-R., Biosci. Rep. 2017, 37, BSR20160547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reinhardt U., Lotze J., Zernia S., Mörl K., Beck-Sickinger A. G., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 10237–10241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10402–10406. [Google Scholar]
- 26. Lotze J., Wolf P., Reinhardt U., Seitz O., Mörl K., et al., ACS Chem. Biol. 2018, 13, 618–627. [DOI] [PubMed] [Google Scholar]
- 27. Reinhardt U., Lotze J., Mörl K., Beck-Sickinger A. G., Seitz O., Bioconjugate Chem. 2015, 26, 2106–2117. [DOI] [PubMed] [Google Scholar]
- 28. Gradišar H., Jerala R., J. Pept. Sci. 2011, 17, 100–106. [DOI] [PubMed] [Google Scholar]
- 29. Gavins G. C., Gröger K., Bartoschek M. D., Wolf P., Beck-Sickinger A. G., et al., Nat. Chem. 2021, 13, 15–23. [DOI] [PubMed] [Google Scholar]
- 30. Nielsen H., Engelbrecht J., Brunak S., von Heijne G., Protein Eng. 1997, 10, 1–6. [DOI] [PubMed] [Google Scholar]
- 31. Almagro Armenteros J. J., Tsirigos K. D., Sønderby C. K., Petersen T. N., Winther O., et al., Nat. Biotechnol. 2019, 37, 420–423. [DOI] [PubMed] [Google Scholar]
- 32. Saito Y., Mizuno T., Itakura M., Suzuki Y., Ito T., et al., J. Biol. Chem. 1991, 266, 23433–23437. [PubMed] [Google Scholar]
- 33. Abdel-Sayed S., Am. J. Hypertens. 2003, 16, 515–521. [DOI] [PubMed] [Google Scholar]
- 34. Schulz A., Jankowski J., Zidek W., Jankowski V., Clin. Proteomics 2014, 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Owen N. E., Nyimanu D., Kuc R. E., Upton P. D., Morrell N. W., et al., Peptides 2021, 136, 170440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maguire J. J., Kuc R. E., Davenport A. P., Life Sci. 2012, 91, 681–686. [DOI] [PubMed] [Google Scholar]
- 37. Jonas K. C., Huhtaniemi I., Hanyaloglu A. C., Methods Cell Biol. 2016, 132, 55–72. [DOI] [PubMed] [Google Scholar]
- 38. Scarselli M., Annibale P., Radenovic A., J. Biol. Chem. 2012, 287, 16768–16780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jha A., van Zanten T. S., Philippe J.-M., Mayor S., Lecuit T., Curr. Biol. 2018, 28, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weinberg Z. Y., Puthenveedu M. A., Traffic 2019, 20, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Savi P., Zachayus J.-L., Delesque-Touchard N., Labouret C., Hervé C., et al., Proc. Natl. Acad. Sci. USA 2006, 103, 11069–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Toneatti R., Shin J. M., Shah U. H., Mayer C. R., Saunders J. M., et al., Sci. Signaling 2020, 13, eaaw3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song Y., Xu C., Liu J., Li Y., Wang H., et al., Circ. Res. 2021, 128, 262–277. [DOI] [PubMed] [Google Scholar]
- 44. Wu B., Chien E. Y. T., Mol C. D., Fenalti G., Liu W., et al., Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., et al., Nature 2012, 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., et al., Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang J., Chen S., Zhang J. J., Huang X.-Y., Nat. Struct. Mol. Biol. 2013, 20, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. AbdAlla S., Lother H., Abdel-tawab A. M., Quitterer U., J. Biol. Chem. 2001, 276, 39721–39726. [DOI] [PubMed] [Google Scholar]
- 49. Santos E. L., Reis R. I., Silva R. G., Shimuta S. I., Pecher C., et al., Regul. Pept. 2007, 141, 159–167. [DOI] [PubMed] [Google Scholar]
- 50. Bai B., Liu L., Zhang N., Wang C., Jiang Y., et al., Cell. Signalling 2014, 26, 1549–1559. [DOI] [PubMed] [Google Scholar]
- 51. Bai B., Cai X., Jiang Y., Karteris E., Chen J., J. Cell. Mol. Med. 2014, 18, 2071–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoare B. L., Kocan M., Bruell S., Scott D. J., Bathgate R. A. D., Pharmacol. Res. Perspect. 2019, 7, e00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gronemeyer T., Chidley C., Juillerat A., Heinis C., Johnsson K., Protein Eng. Des. Sel. 2006, 19, 309–316. [DOI] [PubMed] [Google Scholar]
- 54. Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., et al., ACS Chem. Biol. 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- 55. Jain R. K., Joyce P. B., Molinete M., Halban P. A., Gorr S. U., Biochem. J. 2001, 360, 645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yanushevich Y. G., Staroverov D. B., Savitsky A. P., Fradkov A. F., Gurskaya N. G., et al., FEBS Lett. 2002, 511, 11–14. [DOI] [PubMed] [Google Scholar]
- 57. Serfling R., Seidel L., Bock A., Lohse M. J., Annibale P., et al., ACS Chem. Biol. 2019, 14, 1141–1149. [DOI] [PubMed] [Google Scholar]
- 58. Alken M., Schmidt A., Rutz C., Furkert J., Kleinau G., et al., Mol. Pharmacol. 2009, 75, 801–811. [DOI] [PubMed] [Google Scholar]
- 59. Westendorf C., Schmidt A., Coin I., Furkert J., Ridelis I., et al., J. Biol. Chem. 2011, 286, 35588–35600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Doh J. K., Tobin S. J., Beatty K. E., Biochemistry 2020, 59, 3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gavins G. C., Gröger K., Reimann M., Bartoschek M. D., Bultmann S., et al., RSC Chem. Biol. 2021, 2, 1291–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamaguchi T., Murata Y., Fujiyoshi Y., Doi T., Eur. J. Biochem. 2003, 270, 1816–1827. [DOI] [PubMed] [Google Scholar]
- 63. Zeng C., Hopfer U., Asico L. D., Eisner G. M., Felder R. A., et al., Hypertension 2005, 46, 926–931. [DOI] [PubMed] [Google Scholar]
- 64. Wyse B. D., Prior I. A., Qian H., Morrow I. C., Nixon S., et al., J. Biol. Chem. 2003, 278, 23738–23746. [DOI] [PubMed] [Google Scholar]
- 65. Czikora I., Feher A., Lucas R., Fulton D. J. R., Bagi Z., Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shihoya W., Nishizawa T., Okuta A., Tani K., Dohmae N., et al., Nature 2016, 537, 363–368. [DOI] [PubMed] [Google Scholar]
- 67. Singh K. D., Unal H., Desnoyer R., Karnik S. S., J. Chem. Inf. Model. 2019, 59, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Evans N. J., Walker J. W., Can. J. Physiol. Pharmacol. 2008, 86, 526–535. [DOI] [PubMed] [Google Scholar]
- 69. Wolf P., Beck-Sickinger A. G., J. Pept. Sci. 2021, 27, e3325. [DOI] [PubMed] [Google Scholar]
- 70. Batra V. K., McNeill J. R., Xu Y., Wilson T. W., Gopalakrishnan V., Am. J. Physiol. 1993, 264, 479–484. [DOI] [PubMed] [Google Scholar]
- 71. Fellner S. K., Arendshorst W. J., Kidney Int. 2004, 65, 1810–1817. [DOI] [PubMed] [Google Scholar]
- 72. Miller E., Czopek A., Duthie K. M., Kirkby N. S., van de Putte, Fransen E., et al., Hypertension 2017, 69, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maguire J. J., Kuc R. E., Pell V. R., Green A., Brown M., et al., Life Sci. 2012, 91, 544–549. [DOI] [PubMed] [Google Scholar]
- 74. Hay D. W., Luttmann M. A., Hubbard W. C., Undem B. J., Br. J. Pharmacol. 1993, 110, 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bax W. A., Bruinvels A. T., van Suylen R. J., Saxena P. R., Hoyer D., Naunyn-Schmiedeberg′s Arch. Pharmacol. 1993, 348, 403–410. [DOI] [PubMed] [Google Scholar]
- 76. Molenaar P., Kuc R. E., Davenport A. P., Br. J. Pharmacol. 1992, 107, 637–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Peter M. G., Davenport A. P., J. Cardiovasc. Pharmacol. 1995, 26 Suppl 3, 7. [PubMed] [Google Scholar]
- 78. Mourtas S., Gatos D., Kalaitzi V., Katakalou C., Barlos K., Tetrahedron Lett. 2001, 42, 6965–6967. [Google Scholar]
- 79. Canle M., Clegg L. W., Demirtas I., Elsegood M. R. J., Maskill H., J. Chem. Soc. Perkin Trans. 2 2000, 85–92. [Google Scholar]
- 80. van den Ent F., Löwe J., J. Biochem. Biophys. Methods 2006, 67, 67–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information