

Abstract
The incidence of metastatic breast cancer (MBC) is increasing and the therapeutic arsenal available to fight it is insufficient. Brain metastases, in particular, represent a major challenge for chemotherapy as the impermeable nature of the blood–brain barrier (BBB) prevents most drugs from targeting cells in the brain. For their ability to transpose biological membranes and transport a broad spectrum of bioactive cargoes, cell‐penetrating peptides (CPPs) have been hailed as ideal candidates to deliver drugs across biological barriers. A more ambitious approach is to have the CPP as a drug itself, capable of both killing cancer cells and interacting with the blood/brain interface, therefore blocking the onset of brain metastases. vCPP2319, a viral protein‐derived CPP, has both properties as it: (a) is selective toward human breast cancer cells (MDA‐MB‐231) and increases cell stiffness compared to breast epithelial cells (MCF 10A) hindering the progression of metastases; and (b) adsorbs at the surface of human brain endothelial cells potentially counteracting metastatic cells from reaching the brain. Overall, the results reveal the selective anticancer activity of the peptide vCPP2319, which is also able to reside at the blood–brain interface, therefore counteracting brain penetration by metastatic cancer cells.
Keywords: anticancer activity, biomechanics, blood–brain barrier, cell‐penetrating peptide, metastatic breast cancer
The viral protein‐derived cell‐penetrating peptide vCPP2319 can be used as a promising candidate for drug development for metastatic breast cancer treatment. Breast cancer cells treated with vCPP2319 become stiffer, which hinders metastases formation. vCPP2319 is retained at the surface of brain endothelial cells, where it can target incoming breast cancer cells, impeding metastatic cancer cells from reaching the brain.
Abbreviations
- ACP
anticancer peptide
- AFM
atomic force microscopy
- AMP
antimicrobial peptide
- CF
carboxyfluorescein
- CPP
cell‐penetrating peptide
- DDS
drug delivery system
- FS
force spectroscopy
- GAG
glycosaminoglycans
- HBMEC
human brain microvascular endothelial cells
- LDH
lactate dehydrogenase
- LUV
large unilamellar vesicle
- MBC
metastatic breast cancer
- MLVs
multilamellar vesicles
- PS
phosphatidylserine
- RP‐HPLC
reversed‐phase high‐performance liquid chromatography
- SA
sialic acid
- SPPS
solid‐phase peptide synthesis
- TNBC
triple‐negative breast cancer
Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of cancer‐related death among women, in more than 100 countries worldwide [1]. Extensive screening programs and improvements in the therapeutic management of the systemic disease contributed to the increase of the median survival of women diagnosed with metastatic breast cancer (MBC) [2, 3, 4]. However, MBC incidence is increasing and, as it remains incurable, more patients will keep succumbing to this disease [2, 5]. The most frequent metastatic sites in MBC are lungs, brain, bones and liver [3]. First‐line therapies for MBC rely on chemotherapy, which can be partially effective toward metastases development in bones, soft tissues and viscera [2]. Yet, most of these drugs show an insufficient selectivity–that leads to unspecific cell targeting and drastic side‐effects–and very poor penetration in the central nervous system (CNS) due to the impermeability of the blood–brain barrier (BBB) [2, 6, 7, 8]. Therefore, there is an urgent need to develop more effective strategies to control MBC progression.
Cell‐penetrating peptides (CPPs) are small molecules of 4–40 amino acid residues with the ability to enter cells using different membrane translocation mechanisms [9]. These peptides promote intracellular effects either by themselves or by delivering conjugated bioactive cargoes, such as small RNA or DNA oligomers, other peptides, proteins, plasmids, antibodies or nanoparticles [9, 10, 11]. CPPs are thus considered as prime candidates for the development of new drugs or drug‐delivery systems (DDS) [12, 13]. In fact, there are several examples of the application of CPPs in DDS development for cancer treatment [13], including the use of CPPs conjugated with doxorubicin to transpose the BBB or to penetrate breast cancer cells [14, 15, 16, 17, 18]. Considering the potential impact in cancer treatment, it is of paramount importance to characterize the pharmacokinetics and pharmacodynamics of CPPs as potential drugs or drug delivery applications [13], or a combination of both.
Viruses have emerged as important tools in the fields of nanoengineering and nanomedicine for their natural ability to transport cargoes into cells. Our group had previously used a viral protein database to search for new potential CPPs [19]. A group of CPP candidates was selected and tested experimentally, resulting in a set of six CPPs with high percentage of cell internalization (> 73%) [19]. In the present work, the hemolytic activity of the CPPs was studied in parallel with their anticancer activity toward several breast cancer cell lines, one epithelial breast cell line, and one fibroblast cell line. This screening showed that vCPP2319, a cationic peptide derived from the capsid protein of the Torque teno douroucouli virus [19, 20], was the best candidate for drug development strategies as it was the most efficient regarding toxicity toward cancer cells. In subsequent assays, the effect of this peptide in cells from tissues relevant to the development of drugs targeted to MBC–namely a breast cancer cell line, a breast epithelial cell line, and one cell line derived from primary cell cultures of the brain endothelium–was unraveled. vCPP2319 has selective anticancer activity and the potential to strengthen the protection of the blood–brain interface against the invasion of brain by metastatic cancer cells.
Results
Anticancer and hemolytic activity of CPPs
The in vitro activity of all peptides was initially studied toward two triple‐negative breast cancer (TNBC) cell lines (MDA‐MB‐231 and BT‐20), two HER2+ breast cancer cell lines (BT‐474 and SKBR3), one human healthy epithelial breast cell line (MCF 10A), and one fibroblast cell line (Hs68). Cell lines were exposed to increasing concentrations of peptides for 24 h and cell viability was assessed by quantifying the metabolic conversion of resazurin to resorufin through fluorescence intensity using the CellTiter‐Blue® assay (Table 1). Upon incubation with TNBC cell lines, the lowest IC50 values were obtained with vCPP0769 and vCPP2319; vCPP0769 showed an IC50 of 9.0 ± 0.1 µm and 3.5 ± 1.5 µm toward MDA‐MB‐231 and BT‐20, respectively; vCPP2319 showed an IC50 of 4.5 ± 0.1 µm and 4.2 ± 2.2 µm toward MDA‐MB‐231 and BT‐20, respectively. Concerning HER2+ cancer cell lines, vCPP2319 revealed the highest effectiveness, with an IC50 of 15.7 ± 1.5 µm and 14.1 ± 0.1 µm toward BT‐474 and SKBR3, respectively. The remaining CPPs demonstrated an IC50 > 20 µm for all the breast cancer cells lines used in this study. All the CPPs were also tested toward MCF 10A cells and Hs68 fibroblasts. The IC50 obtained for vCPP0769 and vCPP2319 with MCF 10A cells were 55.4 ± 1.7 and 16.7 ± 1.3 µm, respectively. The incubation of Hs68 with vCPP0769 or vCPP2319 revealed IC50 of 53.4 ± 0.7 and 10.8 ± 0.2 µm, respectively. For both healthy cell lines MCF 10A and Hs68, the remaining CPPs showed no toxicity up to 100 µm.
Table 1.
Cytotoxicity of peptides toward human cell lines. Cultures from different cell lines were incubated with various peptide concentrations ranging from 0.01 to 100 µm, for 24 h. Percentage of cell viability was detected using the CellTiter®‐Blue assay.
| IC50 (µm) | ||||||
|---|---|---|---|---|---|---|
| MDA‐MB‐231 | BT‐20 | BT‐474 | SKBR3 | MCF 10A | Hs68 | |
| vCPP0275 | > 100 | 44.8 ± 1.1 | > 100 | 34.0 ± 1.0 | > 100 | > 100 |
| vCPP0417 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| vCPP0667 | > 100 | 88.8 ± 1.0 | 92.8 ± 1.1 | 71.2 ± 1.1 | > 100 | > 100 |
| vCPP0769 | 9.0 ± 0.1 | 3.5 ± 1.5 | 37.5 ± 1.7 | 21.1 ± 2.0 | 55.4 ± 1.7 | 53.4 ± 0.7 |
| vCPP1779 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| vCPP2319 | 4.5 ± 0.1 | 4.2 ± 2.2 | 15.7 ± 1.5 | 14.1 ± 1.0 | 16.7 ± 1.3 | 10.8 ± 0.3 |
The hemolytic properties of peptides were tested in human red blood cells (RBCs). The peptides vCPP0275, vCPP0417, vCPP0667, and vCPP1779 were not hemolytic up to 100 µm (Fig. 1 and Table 2). The maximum hemolysis obtained at this concentration was observed with vCPP0275 (14.6 ± 0.9%). vCPP0769 and vCPP2319 demonstrated mild hemolytic activity with HC50 of 93.7 ± 1.1 µm and 44.8 ± 1.0 µm, respectively, which contrasts with the result obtained with the gold standard hemolytic peptide melittin that shows an HC50 of 5.7 ± 1.1 µm.
Fig. 1.
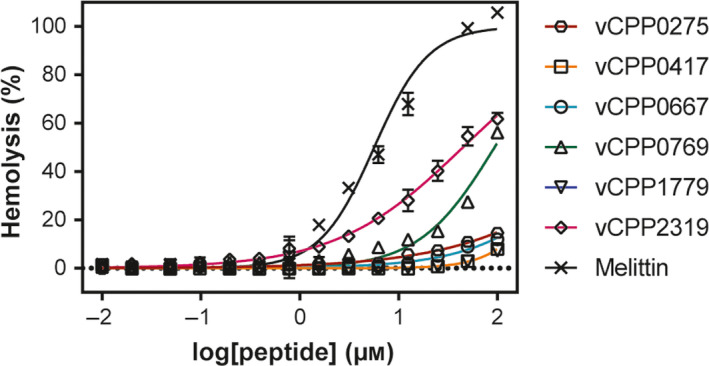
Toxicity of peptides toward RBCs. A suspension of RBCs (0.25%, v/v) was incubated with various concentrations of peptides (ranging from 0.01 to 100 μm). Percentage of hemolysis was detected by absorbance of hemoglobin released into the supernatant. Melittin was included as a positive control for hemolysis. Error bars refer to the standard deviation obtained from different experiments, performed in different days with independent blood samples (n = 3 for all vCPP peptides and n = 2 for melittin).
Table 2.
Hemolytic activity of the peptides. HC50 was defined as the concentration of peptide causing 50% hemolysis on erythrocytes. H max was defined as the percentage (%) hemolysis of erythrocytes at the highest concentration tested (100 µm).
| Peptide | HC50 (µm) | H max (%) |
|---|---|---|
| vCPP0275 | > 100 | 14.6 ± 0.9 |
| vCPP0417 | > 100 | 7.9 ± 0.8 |
| vCPP0667 | > 100 | 12.0 ± 0.4 |
| vCPP0769 | 93.7 ± 1.1 | 56.2 ± 1.5 |
| vCPP1779 | > 100 | 7.2 ± 0.4 |
| vCPP2319 | 44.8 ± 1.0 | 61.8 ± 2.0 |
| Melittin | 5.7 ± 1.1 | 105.8 ± 1.6 |
Circular dichroism (CD) was used to further investigate the hypothesis that differential hemolytic activity is caused by differences in secondary structure. Despite the differences in the hemolytic activity, the spectra of all peptides were characteristic of random‐coil conformation in aqueous solution, in the absence or in the presence of liposomes with different lipid compositions (Fig. S1).
Following the initial screening of the CPPs, vCPP2319 was selected to proceed with this study along with the cell lines MDA‐MB‐231 and MCF 10A. Considering the challenge posed by TNBC and the fact vCPP2319 has a more powerful activity against this cancer subtype, we decided to unveil the peptide’s mode of action in TNBC breast cancer cells. From the two TNBC cell lines used in the initial screening, MDA‐MB‐231 was used to proceed with this study, since it is considered a gold standard TNBC model used in preclinical research and its phenotype is highly metastatic and more aggressive than that of BT‐20 [21]. MCF 10A cells were used as an healthy counterpart [22, 23]. The cytotoxic activity was confirmed for vCPP2319 toward MDA‐MB‐231 and MCF 10A cells using a different assay and the refined IC50 values are in agreement with those in the initial screening, again showing that vCPP2319 has selective anticancer activity as its IC50 is significantly lower for MDA‐MB‐231 than MCF 10A cells. To increase data precision and robustness, the cytotoxic activity of vCPP2319 was once more investigated in vitro using the two selected breast cell lines, this time using more data points in a specific range of concentrations for MDA‐MB‐231 cells. Cell viability assays were performed by exposing the cells to a range of different peptide concentrations for 24 h and quantifying the metabolic conversion of 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide salt (MTT) to formazan through UV‐Vis electronic absorption. The dose–response curves obtained are presented in Fig. 2A,B. The IC50 determined for vCPP2319 was 5.2 ± 0.3 µm for MDA‐MB‐231 and 18.2 ± 0.8 µm for MCF 10A cells.
Fig. 2.
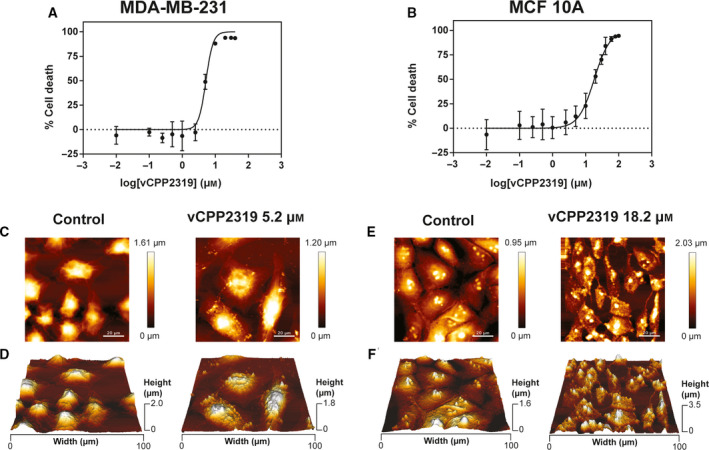
Cytotoxic activity of vCPP2319 and evaluation of the morphological/topographical effect on human breast cancer and healthy cells. The in vitro cytotoxic activity of vCPP2319 against MDA‐MB‐231 (A) and MCF 10A cells (B) was studied. AFM representative height images and 3D projections are shown for MDA‐MB‐231 (C and D, respectively) and for MCF 10A cells (E and F, respectively) in the absence and presence of vCPP2319, at the IC50 obtained for each cell line. Error bars in A and B refer to the standard deviation obtained from three different experiments (n = 3), performed in different days with independently grown cell cultures. Scale bar represents 20 μm.
vCPP2319 interaction with breast cell membranes and cell internalization
To evaluate the morphological and topographical effect of vCPP2319 in both MDA‐MB‐231 and MCF 10A cells and gain insights on the peptide mode of action, images of untreated and treated cells were acquired using atomic force microscopy (AFM), at the IC50 determined for each cell line. Representative height images (C and E) and respective 3D projections (D and F) are shown in Fig. 2. AFM images show intact MDA‐MB‐231 cells with filopodia and pseudopodia, which are associated with highly invasive behavior [24, 25]. These structures were not observed in MCF 10A cells. In the presence of vCPP2319, MDA‐MB‐231 showed nuclear collapse concomitant with the appearance of several vesicular bodies, cell shrinkage, and detectable cytoskeleton. In treated MCF 10A cells, there is a considerable level of cell shrinkage and loss of nuclear structure.
Cell height profiles were obtained from AFM images for both cell lines. Cell surface roughness (R ms) was determined for MDA‐MB‐231, in the absence and presence of the peptide. The same analysis was not possible for MCF 10A cells treated with vCPP2319 due to cell damage. Cell height profiles were in agreement with the morphological alterations observed in the AFM images (Fig. S2A,B) for both cell lines treated with the peptide. No significant changes were obtained for the values of cell height or R ms values in MDA‐MB‐231‐treated cells compared to untreated controls (Fig. S2C,D).
To investigate the hypothesis of cell membrane crossing by vCPP2319 and its activity on an intracellular target, we decided to study the peptide‐membrane interaction and evaluate membrane integrity. To unveil peptide interaction with breast cancer and healthy cell membranes, the zeta potential of the cells was determined by electrophoretic light scattering and changes in the membrane dipole potential were studied using 4‐(2‐[6‐(dioctylamino)‐2‐naphthalenyl]ethenyl)‐1‐(3‐sulfopropyl)pyridinium inner salt (di‐8‐ANEPPS) fluorescence spectroscopy (Fig. 3A–F). This fluorescent membrane probe inserts in the outer leaflet of cell membranes and becomes sensitive to variations in the dipole potential formed by the alignment of the lipid head groups and water molecules [26]. Cell membrane dipole potential variations caused by the peptide are reported by the spectral shifts of the membrane‐inserted di‐8‐ANEPPS [26]. No significant changes were detected in zeta potential values for MDA‐MB‐231 and MCF 10A cells after 30 min incubation with vCPP2319 (Fig. 3A,B), in agreement with the high ability of the peptide to internalize cells [19]. The differential spectra and normalized intensity ratios (R norm) of di‐8‐ANEPPS obtained after 15 min incubation with the peptide are shown in Fig. 3C–F. The presence of vCPP2319 significantly perturbs the membrane dipole potential of MCF 10A cells, causing a red shift in the excitation spectra of the probe and a significant decrease in the R norm. In contrast, vCPP2319 does not perturb the membrane dipole potential of MDA‐MB‐231 cells. Eventual vCPP2319‐caused permeation of cell membranes was evaluated by extracellular lactate dehydrogenase (LDH) activity assay. After 15‐min incubation of MDA‐MB‐231 and MCF 10A cells with vCPP2319, the LDH activity in cell culture medium was not significant, in contrast with the results obtained with cell culture medium samples collected from melittin or Triton X‐100‐treated cells (Fig. 3G,H). It is worth mentioning that 15 min is long enough for cell damage to occur, prior to peptide internalization, as interaction between peptides and cell membrane occurs very rapidly [13, 27].
Fig. 3.
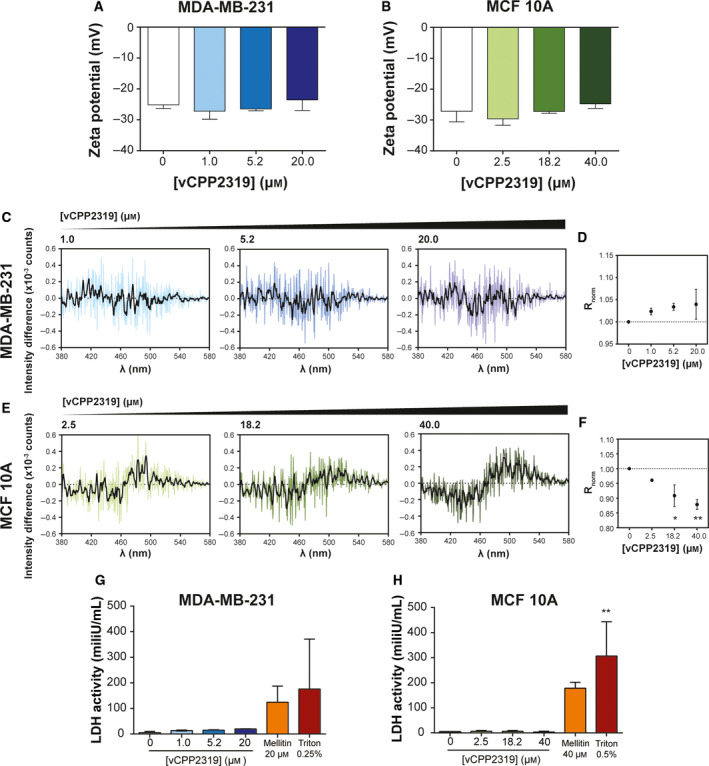
vCPP2319 interaction with human breast cell membranes. Zeta potential measurements of MDA‐MB‐231 (A) and MCF 10A (B) cells were performed in the absence (n = 13 and n = 8, respectively) and presence of vCPP2319, at increasing peptide concentrations (n = 2). The cell membrane dipole potential perturbation by vCPP2319 was studied by fluorescence spectroscopy using the fluorescent dye di‐8‐ANEPPS (C–F). The differential spectra were obtained by subtracting the normalized excitation spectra (to the integrated spectrum area) of the membrane‐inserted di‐8‐ANEPPS in the absence of vCPP2319 from the normalized excitation spectra of the membrane‐inserted di‐8‐ANEPPS in the presence of vCPP2319. A smoothened version of each differential spectrum is showed in each condition for clarity (C and E). The variation of the normalized excitation intensity ratios (R norm) with the increase in peptide concentration is also presented for MDA‐MB‐231 (D) and MCF 10A (F) cells (n = 2). This ratio enables for a better evaluation of the magnitude of membrane dipole potential perturbation. LDH activity was assessed in medium samples collected from untreated and treated MDA‐MB‐231 (G) and MCF 10A (H) cells (n = 2). Error bars refer to the standard deviation. Significance was assessed by ANOVA followed by Tukey’s (for zeta potential data) or Dunnett's post‐test. *P‐value ≤ 0.05; **P‐value ≤ 0.01.
As a final confirmation of cell membrane translocation by vCPP2319, we performed measurements of 5(6)‐carboxyfluorescein (CF)‐labeled peptide internalization in cells using flow cytometry combined with confocal microscopy imaging. Figure 4A,D–show flow cytometry results obtained with CF‐vCPP2319‐treated MDA‐MB‐231 and MCF 10A cells, respectively; fluorescence intensity variation over time is represented in Fig. 4C,F. Internalization occurs in 15 min for both cell lines. The results also reveal differences in the fluorescence intensity decay with time: for treated MDA‐MB‐231 the fluorescence intensity decreases slowly during 24 h while for treated MCF 10A the decay is complete in 6 h. Confocal imaging (Fig. 4B,E) are in line with the results observed in flow cytometry. In both cell lines, it is possible to observe the presence of CF‐vCPP2319 (yellow) inside the cells after 15 min. In treated MDA‐MB‐231, vCPP2319 accumulates inside the cells until 6 h of incubation; at 24 h the levels of internalized peptide decrease and are comparable to those obtained at 1 h. However, in treated MCF 10A, the peptide accumulates inside the cells until 1 h of incubation and the fluorescence intensity decreases thereafter. This difference between vCPP2319 accumulation inside MDA‐MB‐231 and MCF 10A cells suggests different modes of action which is in line with vCPP2319 selectivity for cancer cells.
Fig. 4.
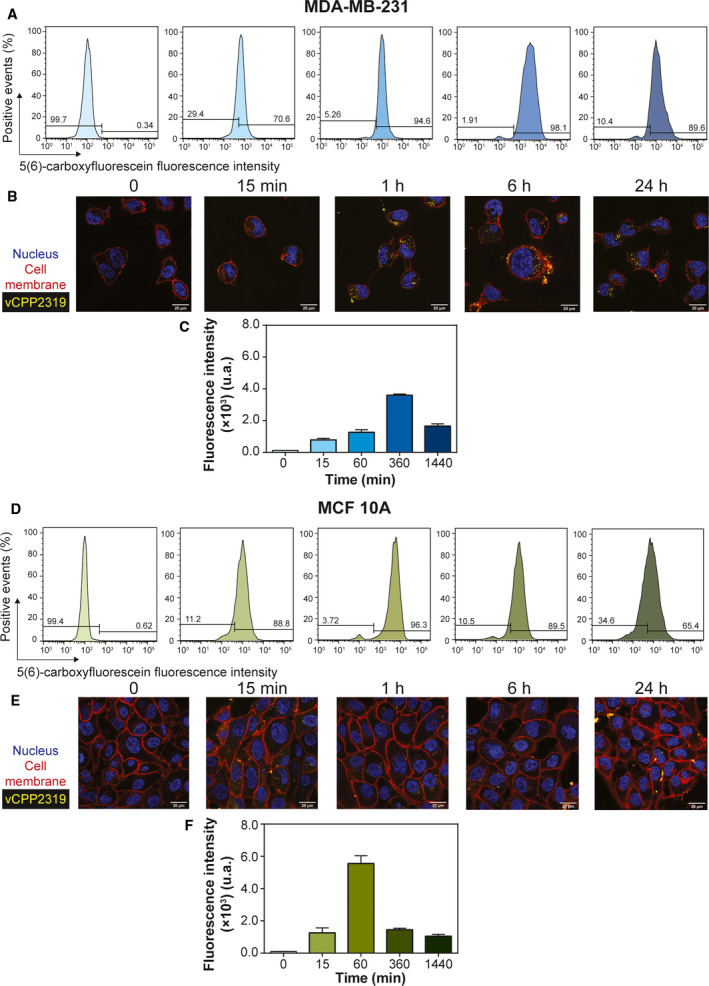
vCPP2319 internalization in human breast cells. Flow cytometry and confocal microscopy were used to evaluate the internalization of the 5(6)‐carboxyfluorescein‐labelled vCPP2319 (yellow) in MDA‐MB‐231 (A and B) and MCF 10A (D and E) cells. Cell nucleus (blue) was stained with Hoechst 33342, and cell membranes (red) were stained with CellMask™ Deep Red. The fluorescence intensity of the 5(6)‐carboxyfluorescein‐labeled vCPP2319 is also represented as function of time for treated MDA‐MB‐231 and MCF 10A cells (C and F, respectively). Error bars in C and F refer to the standard deviation obtained from three different experiments (n = 3), performed in different days with independently grown cell cultures. Scale bar represents 20 μm.
vCPP2319 effect on breast cells biomechanics
The force spectroscopy capability of the AFM equipment (AFM‐FS) was used to analyze live MDA‐MB‐231 and MCF 10A cells treated with vCPP2319 at 5.2 µm in terms of their biomechanical properties, by acquiring force curves, using the Quantitative Imaging (QI™) mode. The force‐displacement curves were converted into force‐distance (f‐d) curves and then corrected for offset and tilt. The Hertz/Sneddon model was applied to the extended segment of the f‐d curves to obtain the Young’s modulus. Figure 5A,B–show the distribution of the Young’s modulus values obtained from areas of 10 × 10 µm of the nuclear regions of MDA‐MB‐231 and MCF 10A cells. The distribution of the relative frequencies shows that, in both cases, the majority of the Young’s modulus values obtained range from 1 to 100 kPa (82.9% and 83.5% for MDA‐MB‐231 and MCF 10A, respectively). However, in the case of MDA‐MB‐231 cells, these values are homogeneously distributed in the interval while for MCF 10A cells most of the values are within 1–10 kPa. The treatment of both cell lines with vCPP2319 had a significant impact on the distribution of the Young’s modulus obtained. Treated MDA‐MB‐231 cells show an increase of Young’s modulus values in the range 10–100 kPa, suggesting that the treatment with the peptide resulted in stiffer cells. Conversely, MCF 10A cells treated with vCPP2319 exhibited a distribution of Young’s modulus that slightly shifted to smaller values. Yet, most of the Young’s modulus values of treated MCF 10A cells remain in the 1–10 kPa range.
Fig. 5.
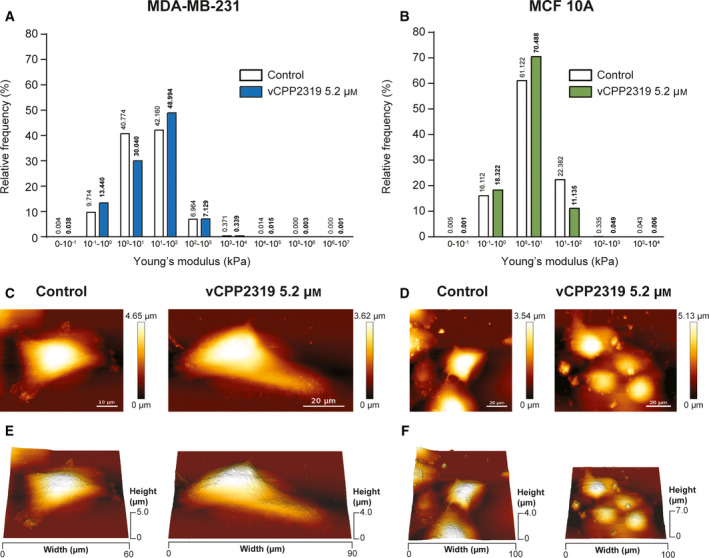
Effect of vCPP2319 on cell biomechanics assessed through AFM. Cell cultures from both cell lines were treated with 5.2 μM of vCPP2319, and live cells were analyzed in terms of their biomechanical properties using QITM mode to obtain force curves from 10 × 10 μm areas scanned over the nucleus. Young’s modulus values were obtained from force curves using the Hertz/Sneddon fit. The distribution of the Young’s modulus is displayed for MDA‐MB‐231 (A) and MCF 10A (B) cells untreated (n = 3 and n = 4, respectively) and treated with vCPP2319 (n = 3 and n = 2, respectively). The Mann–Whitney test was applied to the comparison between distributions obtained for treated and untreated samples from each cell line, and the level of significance was ****(P‐value < 0.0001) in each case. Live imaging of whole cells (100 × 100 μm) was performed also using QITM mode for untreated and treated MDA‐MB‐231 and MCF 10A cells. Representative height images (C and D) and 3D projections (E and F) for untreated and treated cells are exhibited for MDA‐MB‐231 and MCF 10A cells. Scale bar represents 10 μm in C–Control; and 20 μm in the C–vCPP2319 5.2 μM–and D.
Live imaging of whole cells was performed with the same cell cultures in order to qualitatively evaluate the effect of the peptide in cell morphology and topography, complementing the experiments performed with fixed cells (Fig. 2C–F). Live cell imaging has the advantage of eliminating artifacts associated with cell fixation [28, 29]. Representative AFM height images and 3D projections obtained with live MDA‐MB‐231 and MCF 10A cells are shown in Fig. 5C–F. In untreated live MDA‐MB‐231 cells it was possible to observe cytoskeletal structures mainly at the cell edges, while in treated MDA‐MB‐231 small round‐shaped irregularities were observed in thinner areas of the cells and no cytoskeletal structures were discernible. In untreated live MCF 10A cells it was not possible to observe the nucleoli as in fixed cells (Fig. 2E,F) and some cytoskeletal structures were observed. The magnitude of the cell damage observed in MCF 10A cells treated with 18.2 µm did not allow performing live cell imaging. Therefore, live cell images of treated MCF 10A cells were performed at 5.2 µm, the IC50 obtained for MDA‐MB‐231 cells. Despite the difference in image resolution obtained from fixed and live cell imaging, it was possible to observe a lesser extent of cell shrinkage in live MCF 10A cells treated with 5.2 µm than that observed in fixed MCF 10A cells treated with 18.2 µm.
Confocal microscopy images were acquired for both cell lines treated with increasing vCPP2319 concentrations to evaluate the peptide's effect in structural elements of the cytoskeleton, namely vimentin and actin. Representative images are shown Fig. 6A,C. From the acquired images, we calculated the corrected total cell fluorescence (CTCF) intensity for vimentin and actin and the results are shown in Fig. 6B,D. The results revealed higher levels of vimentin and actin in MDA‐MB‐231 cells in comparison with MCF 10A cells. This observation is in line with other studies that demonstrated that vimentin is overexpressed in several types of cancer and its overexpression is associated with increased invasion and migration of breast cancer cells [30, 31]. Nonetheless, in both cell lines a well‐organized cytoskeleton was observed, with an abundant presence of actin fibers in the cells' periphery and vimentin encircling the nucleus, but more widely distributed in MCF 10A cells. In treated MDA‐MB‐231 the CTCF intensity values for vimentin and actin decreased with the increase in peptide concentration (Fig. 6B). In contrast, in treated MCF 10A cells there is only a significant decrease in CTCF intensity values for vimentin and actin at the highest peptide concentration tested (18.2 µm) (Fig. 6D). Therefore, vCPP2319 impacts selectively on breast cancer cells cytoskeleton, as suggested by alteration in the Young's modulus values after peptide treatment and also by the AFM imaging of live and fixed cells in the presence of the peptide.
Fig. 6.
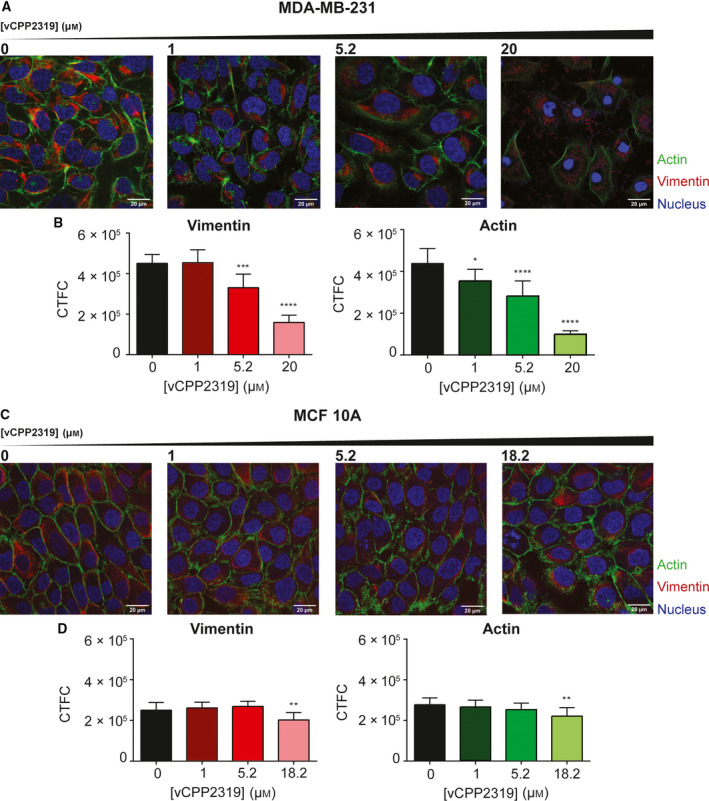
Impact of vCPP2319 on MDA‐MB‐231 and MCF 10A cytoskeleton studied by confocal microscopy. Vimentin filaments (red) and actin fibers (green) were stained with Vimentin Monoclonal Antibody (V9) eFluor 660 and CellMask™ Green Actin Tracking Stain, respectively. Cell nucleus (blue) were stained with Hoechst 33342. Images were obtained for both cells lines with increasing peptide concentrations (A and C), and corrected total cell fluorescence (CTCF) intensity was calculated for vimentin and actin in MDA‐MB‐231 (B) and MCF 10A (D) cells (n = 3). Error bars refer to the standard deviation. One‐way ANOVA followed by Tukey’s multiple comparison test was applied to infer on data significance. *P‐value ≤ 0.05; **P‐value ≤ 0.01; ***P‐value ≤ 0.001; ****P‐value ≤ 0.0001. Scale bar represents 20 μm.
vCPP2319 activity in primary cells derived from the blood–brain barrier
As breast cancer brain metastases are a major challenge in the clinics, the development of new therapeutic strategies to selectively target breast cancer brain metastases is urgent. Therefore, the cytotoxic activity of vCPP2319 was studied in primary human brain microvascular endothelial cells (HBMEC). MTT cell viability assays revealed an IC50 of 16.2 ± 1.4 µm for vCPP2319 toward HBMEC (Fig. 7A). To explore the biomedical potential of vCPP2319, further studies with HBMEC were carried at the IC50 obtained with MDA‐MB‐231 cells.
Fig. 7.
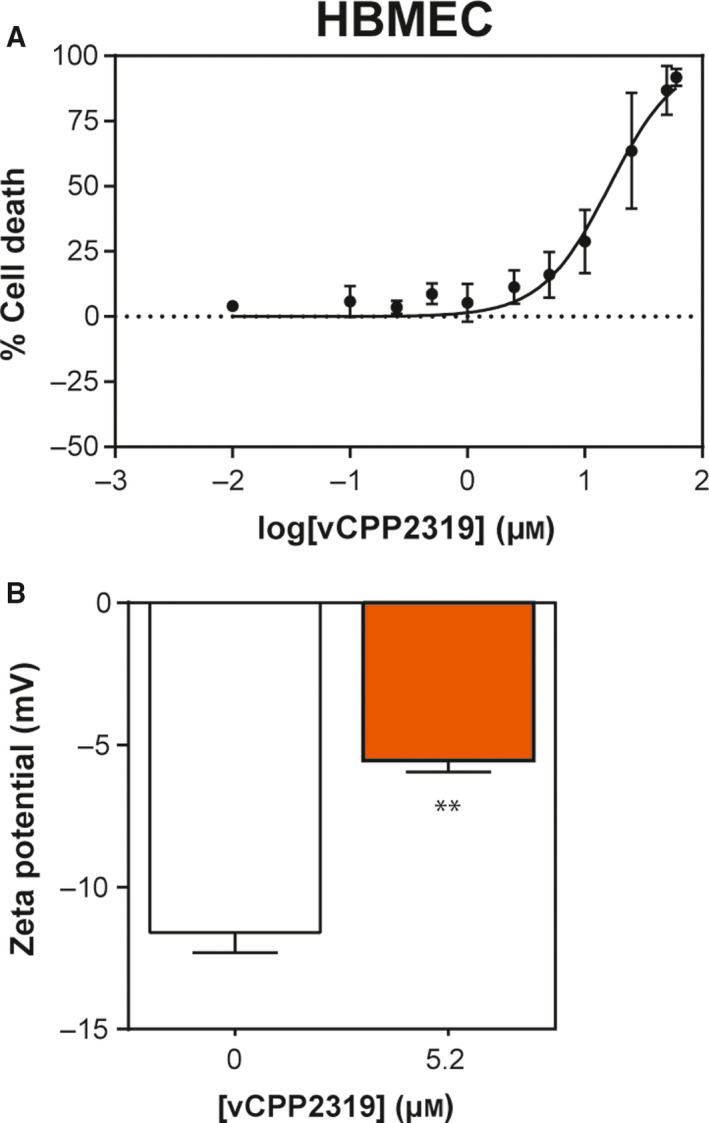
Cytotoxic activity and membrane effect of vCPP2319 in primary cell line from brain endothelium. The cytotoxic activity (A) of vCPP2319 toward HBMEC was evaluated (n = 4). Zeta potential measurements were performed in HBMEC treated with vCPP2319 at 5.2 μM (B) (n = 2). Error bars refer to the standard deviation obtained from different experiments, performed in different days with independently grown cell cultures. Significance was assessed by the unpaired t‐test. **P‐value ≤ 0.01.
In order to study the activity of the peptide in this cell line, measurements of zeta potential were performed (Fig. 7B) along with monitorization of cell membrane dipole potential variations through di‐8‐ANEPPS fluorescence in the absence and presence of vCPP2319 (Fig. S3). Zeta potential measurements of HBMEC treated with 5.2 µm of vCPP2319 showed a significant increase from −11.6 ± 0.7 to −5.6 ± 0.4 mV. Such an increase in zeta potential values suggests that vCPP2319 accumulates on the surface of the cells. Nevertheless, no significant shifts were obtained in the differential spectra obtained for the cells with incorporated di‐8‐ANEPPS dye in the presence of vCPP2319 (see Fig. S3). Therefore, despite the adsorption of the peptide in the surface of brain microvascular endothelial cells, vCPP2319 did not cause significant variations on the membrane dipole potential of these cells, meaning it did not perturb the membrane extensively.
Morphological and topographical alterations in HBMEC caused by vCPP2319
The effect of vCPP2319 in the morphology and topography of HBMEC was studied to evaluate the extent of the cellular damage at the IC50 determined with MDA‐MB‐231 cells. AFM imaging was performed with untreated and treated cells and representative height images, 3D projections and height profiles are shown in Fig. 8. Cell surface roughness was also studied. The values in the cytoplasm and the nucleus were normalized to the control and compared. The study of the maximum cell height was also performed, but no significant variations were observed between untreated and treated cells (Fig. S4).
Fig. 8.
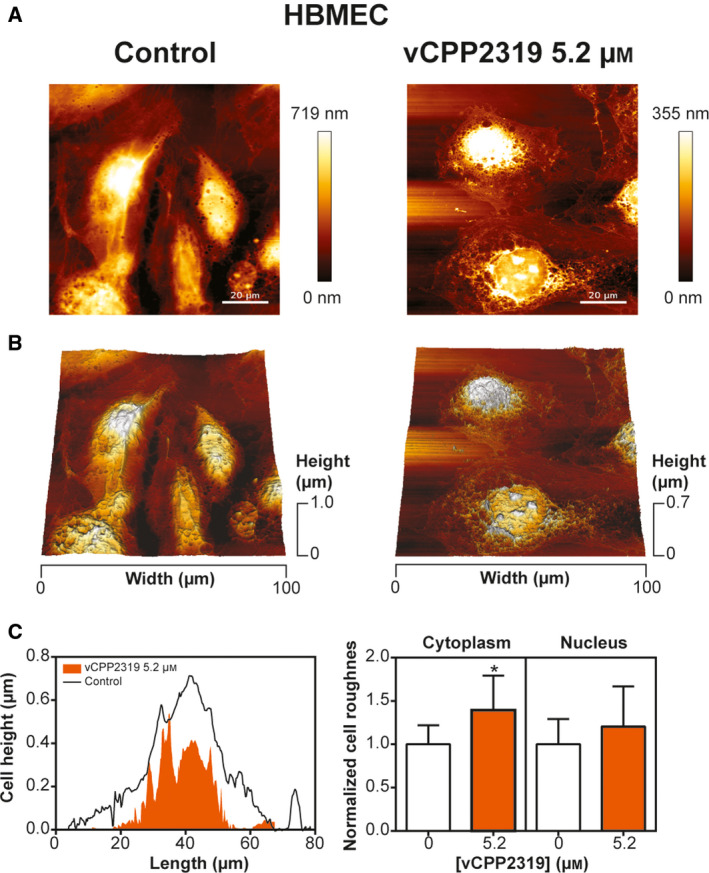
vCPP2319 effect on the morphology and topography of HBMEC. AFM was used to image untreated and treated HBMEC. Representative height images and 3D projections are shown (A and B, respectively). Representative height profiles and normalized cell roughness (n = 3 for control and n = 2 for cells treated with vCPP2319) are also represented (C). Error bars refer to the standard deviation. One‐way ANOVA followed by Tukey’s multiple comparison test was applied to infer on data significance. *P‐value ≤ 0.05. Scale bar represents 20 μm.
Qualitative image analysis (Fig. 8A,B) revealed a mild effect on treated cells' topography, while cell morphology did not seem to be critically affected. In treated HBMEC, it is possible to observe irregularities in the cytoplasm and nucleus. Quantitative analysis (Fig. 8C) for its part revealed that, at 5.2 µm and after 24 h incubation with vCPP2319, the HBMEC cytoplasm surface roughness is altered.
Discussion
Clinical decisions in breast cancer treatment depend on the presence/absence of three main different markers: endocrine receptors for estrogen (ER) and progesterone (PgR), and aberrant human epidermal growth factor receptor 2 (HER2) expression [32]. TNBC lacks all three markers, which are used as molecular targets in chemotherapeutic strategies. This breast cancer subtype has a very aggressive clinical profile, often associated with very poor prognosis [32, 33]. Local and distant relapse with the presence of visceral and/or brain metastases are frequent in TNBC patients [33]. Considering the challenge that TNBC poses in the clinic, two TNBC cell lines were used in this study–MDA‐MB‐231 and BT‐20. Nevertheless, since the risk of brain metastases is also higher in HER2+ patients when compared with breast cancer patients with a luminal phenotype [3], two HER2+ breast cancer cell lines were also included–BT‐474 and SKBR3.
Due to their characteristics, CPPs are considered prime candidates for DDS development and some applications of these peptides include BBB‐level activity and breast cancer cell penetration [13, 14, 15, 16, 17, 18]. As the combination of anticancer activity with BBB‐targeting is of paramount importance to tackle the challenges of MBC treatment, we were prompted to study the anticancer activity of a set of potent CPPs. A recent study performed in our lab with CPPs showed that they are likely to act as antimicrobial peptides (AMPs) [20], which raises the hypothesis of their activity also as anticancer peptides (ACPs) because the CPP, AMP, and ACP functionalities are interconnected [7, 11, 34, 35]. In particular, the ability of some CPPs to exhibit anticancer activity has been reported before [36, 37].
An initial screening of the CPPs toward the previously mentioned cell lines, in parallel with a healthy epithelial breast cell line–MCF 10A–and human fibroblasts–Hs68–revealed vCPP0769 and vCPP2319 with the highest anticancer activity and selectivity toward TNBC cell lines. Interestingly, these two CPPs are also the ones with the most prominent AMP activity, which further supports the AMP/CPP activity correlation [20]. The hemolysis assays revealed that vCPP0769 and vCPP2319 were hemolytic only at high concentrations, with HC50 above 40 μm. Despite the differences in the hemolytic activity, all CPPs displayed spectra characteristic from a random coil conformation, which showed no relationship between these two features. Since vCPP2319 was the only CPP that revealed a promising anticancer activity toward TNBC and HER2+ cell lines, and the lowest IC50 was obtained to TNBC, this CPP was selected to proceed with this study. Moreover, the HC50 (44.8 µm) is much higher than the IC50 (5.2 µm), which enables the use of a wide range of vCPP2319 concentrations that are not toxic to RBCs.
To gain insights on vCPP2319 mode of action in human breast cells, AFM imaging of both breast cancer and healthy cells was performed at the respective IC50. Due to its high resolution, AFM is a reference technique for the detection of changes in cell morphology and topography [38, 39, 40]. Overall, AFM images of treated breast cells showed intact cell membranes and structural alterations that suggest an intracellular target at variance with its mode of action as AMP [20]. Moreover, peptide‐induced cell shrinkage in both cell lines could be related to cell death by apoptosis [41] and the appearance of vesicular bodies in MDA‐MB‐231 treated with vCPP2319 could be related to the formation of apoptotic bodies. Despite the structural alterations observed in peptide‐treated cells, no significant changes were obtained for the cell surface roughness or maximum height values, which suggests that the cytoskeleton did not collapse and kept modulating cell morphology and topography [42].
Following the hypothesis of cell membrane crossing by vCPP2319 and activity in an intracellular target, the peptide‐membrane interaction and membrane integrity were investigated. Zeta potential measurements revealed a net anionic cell surface charge of MDA‐MB‐231, which is typical of cancer cells due to the accumulation of phosphatidylserine (PS) in the outer leaflet of the cell membrane, overexpression of O‐glycosylated mucins, glycosaminoglycans (GAGs) and the presence of high level of sialic acid (SA) residues [34, 43]. For MCF 10A cells, zeta potential was similar to that of MDA‐MB‐231 cells; in this case, sialomucins may contribute to the negative values [38, 44]. The anionic surface charge reported for both cell lines contributes to the electrostatic attraction of vCPP2319, which is cationic. When the accumulation of cationic peptides occurs at the cell membrane, the net anionic cell surface charge increases toward neutrality [38, 45], which was not the case. The results show that there is no accumulation of peptide in the surface of cells, retention in the interior of membranes or translocation to the cell cytoplasm being valid hypotheses. Nonetheless, di‐8‐ANEPPS differential fluorescence excitation spectra show that MDA‐MB‐231 has no perturbation of the membrane dipole potential, at variance with MCF 10A. Altogether, these observations reveal that cancer cells do not retain the peptide in the membrane whereas non‐cancer cells do. Interestingly, LDH activity assays revealed that neither MDA‐MB‐231 nor MCF 10A cells treated with vCPP2319 were leaking intracellular content and, therefore, their membrane integrity was not compromised [46, 47]. These results are deemed important because they show that the selective toxicity of vCPP2319 toward cancer cells reflects penetration and activity in the cytoplasm. To confirm cell membrane translocation by vCPP2319, flow cytometry combined with confocal microscopy was used to investigate CF‐labeled peptide internalization in both cell lines. The low peptide concentration used (1 µm) assures minimal cell damage. The results show that after 15 min vCPP2319 is already internalized in both cell lines and it resides longer inside breast cancer cells when compared to healthy epithelial cells, hence contributing to the peptides' selectivity. Overall, the results clearly show that vCPP2319 internalizes breast cells but has increased selectivity toward breast cancer cells as it resides for a longer period inside MDA‐MB‐231 than MCF 10A cells.
The key role of cell biomechanics in tumor progression has been highlighted over the past decades [48, 49]. ACPs, like AMPs, are expected to act by multitargeting different macromolecular structures rather than specific intracellular receptors [7]. Cell shape and mechanical deformation are thus markers of both intracellular activity and anticancer potential. Specifically, the cytoskeleton and its molecular components are of particular importance because they critically influence, not only cell mechanics, but also the interactions with the surrounding environment [49]. The ability of a molecule to modulate cell biomechanical properties may thus contribute to hinder metastatic cancer progression [50]. The effect of vCPP2319 in the biomechanics of both cell lines was evaluated using live cells and AFM‐FS. The results revealed significant alterations in the distributions of the Young's modulus obtained for treated MDA‐MB‐231 and MCF 10A cells. The treatment of MDA‐MB‐231 cells with vCPP2319 led to an increase in cell stiffness reported by the shift of the Young's modulus distribution toward higher values. The increase in cell stiffness could be associated with structural alterations in the cytoskeleton [51] and/or a result of apoptosis [49]. MCF 10A cells treated with vCPP2319 at 5.2 μm showed a mild decrease in the Young's modulus which translates in a slight decrease in cell stiffness. The differential action of vCPP2319 on cancer and non‐cancer cells is striking, in line with the differential action detected before on cell membrane interaction and cell penetration. To further ascertain potential structural alterations in the cytoskeleton suggested by the shifts in the Young's modulus values and AFM imaging of fixed and live cells, confocal microscopy was used to image MDA‐MB‐231 and MCF 10A cells with stained actin and vimentin, which are two components of the cytoskeleton with critical motile and structural functions [49]. Actin participates in a wide variety of cellular processes, such as cell motility–as it constitutes the lamellipodia, filopodia and invadopodia–adhesion, differentiation, division and membrane organization [50, 52]. Vimentin is associated with the acquisition of a migratory and invasive phenotype by tumor cells, and it is commonly used as an epithelial‐mesenchymal transition (EMT) marker [53, 54]. Interestingly, vimentin overexpression is found in several tumor types and it correlates with aggressiveness and poor prognosis [31, 54]. The treatment of MDA‐MB‐231 cells led to a notable decrease in the fluorescence intensity associated with actin and vimentin while treated MCF 10A cells only showed a significant decrease with the highest peptide concentration. The difference in the extent of vCPP2319 impact on the cytoskeleton supports the selective anticancer activity of the peptide. More importantly, when in presence of applied strain, vimentin disruption hinders cells' compressibility [55, 56]. This supports the correlation between the results obtained for the Young's modulus values and the confocal microscopy since MDA‐MB‐231 cell treated with vCPP2319 became stiffer and showed a decrease in the fluorescence intensity associated to vimentin. Overall, the effect of vCPP2319 in breast cancer cells cytoskeleton strongly suggests that the metastization process of these cells is compromised due to disruption of the actin cytoskeleton and vimentin intermediate filaments. Moreover, the loss of these cytoskeleton components could be a result of proteolysis induced by caspases during cell apoptosis, which is associated with cell retraction, as observed in AFM images (Fig. 2C–F) [54, 57]. Additionally, there are numerous round‐shaped structures surrounded by vimentin in MDA‐MB‐231 cells treated with 18.2 µm of vCPP2319. Vimentin staining at the site of apoptotic bodies formation was observed before [58]. Lastly, the impact of vCPP2319 is notorious in MDA‐MB‐231 at 5.2 µm, while at the same concentration and for MCF 10A, the peptide had no significant impact on the cells' cytoskeleton.
The BBB is the CNS protective barrier with a very predominant role in modulating the brain microenvironment [59]. The brain endothelium differs from the peripheral endothelium mainly due to the abundant presence of tight junctions [59, 60]. Given our interest in studying the antimetastatic breast cancer action of vCPP2319, we decided to address the interaction of this peptide with brain endothelial cells, as these cells are key to brain penetration by cancer cells [61]. The impact of vCPP2319 toward the BBB was evaluated using primary HBMEC. We decided to use a cell line derived from primary cell cultures for toxicity studies, since the morphology and physiology of these cells better resembles the ones found in vivo [62]. The IC50 of vCPP2319 in HBMEC revealed similar toxicity of the peptide for HBMEC and MCF 10A. The pronounced difference between the IC50 values obtained for this peptide toward breast cancer cells and the healthy cell lines used in this study shows selectivity toward cancer cell lines and reveals an overall potential safety profile for therapeutic applications. Zeta potential measurements of HBMEC revealed a net negative surface charge, consistent with previous findings [63]. However, zeta potential measurements of HBMEC treated with vCPP2319 at 5.2 μm resulted in a significant increase in the zeta potential of the cell surface toward neutrality, showing peptide accumulation at the cells' surface. Because it was not possible to detect significant membrane dipole potential alterations, it is possible to conclude that even though vCPP2319 accumulate on HBMEC surface, it does not insert and reside in the membrane of these cells. The qualitative and quantitative analysis of AFM images obtained from treated HBMEC revealed that the cytoplasm surface roughness is altered. Whether such alteration is due to peptide clustering on the membrane or cytoskeleton rearrangements cannot be conclusively established. However, alterations in the cytoskeleton would strongly impact cell viability, which is not the case. In general, vCPP2319 revealed a mild impact on primary endothelial cells from the BBB, being retained at the surface of HBMEC with few alterations in cells' morphology and topography.
In summary, vCPP2319, with demonstrated cell internalization ability [19], acts selectively as an ACP in breast cancer cells. It acts intracellularly without causing membrane structural damages nor membrane permeation. Further studies of this peptide activity revealed a mild effect on the topography of human brain microvascular endothelial cells, relevant for brain targeting by metastatic cancer cells. However, vCPP2319 adsorbed to the surface of these cells, which was not observed with breast cancer cells. vCPP2319 can also hinder breast cancer metastases formation by modulating cell biomechanics. Treatment of breast cancer cells with vCPP2319 led to an increase in cell stiffness, which hampers cell deformation and hence should prevent metastases development and/or progression. In conclusion, vCPP2319 is a unique drug that combines anticancer action with BBB targeting so that extra brain protection is conferred, and thus it is a promising candidate for drug development for metastatic breast cancer treatment.
Materials and methods
Reagents
Fmoc‐protected amino acids, Fmoc‐Rink amide (MBHA) resin, 2‐(1H‐benzotriazol‐1‐yl)‐1, 1, 3, 3–tetramethyluronium hexafluorophosphate (HBTU), and N‐hydroxybenzotriazole (HOBt) were from Iris Biotech (Marktredwitz, Germany). HPLC‐grade acetonitrile (ACN), peptide‐synthesis grade N,N‐dimethylformamide (DMF), dichloromethane, N,N‐diisopropylethylamine (DIEA), N,N‐diisopropylcarbodiimide, trifluoroacetic acid (TFA), and triisopropylsilane (TIS) were from Carlo Erba (Sabadell, Spain). 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC), cholesterol (ChoI), egg sphingomyelin (SM), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE), and 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐L‐serine (POPS) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Dulbecco's Modified Eagle Medium (DMEM), heat‐inactivated fetal bovine serum (FBS), penicillin and streptomycin solution, and trypsin (TrypLE Express enzyme) were obtained from Life Technologies (Carlsbad, CA, USA). Mammary Epithelial Basal Medium (MEBM) and SingleQuots hydrocortisone, bovine pituitary extract (BPE), recombined human epidermal growth factor (rhEGF), and recombinant human insulin were purchased as the clonetics MEGM™ BulletKit™ from Lonza, Ltd. (Basel, Switzerland). Endothelial Cell Medium was purchased as a kit from Innoprot (Bizkaia, Spain), along with Primary Cells Detach Kit and fibronectin. Eagle's Minimum Essential Medium (EMEM), McCoy's 5A medium, cholera toxin from Vibrio cholera, trypsin inhibitor from Glycine max (soybean), glutaraldehyde solution (50% in water), dimethyl sulfoxide (DMSO) for cell culture, di‐8‐ANEPPS probe and lactate dehydrogenase activity assay kit were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Uvasol® DMSO (spectrophotometric grade) was purchased from Merck (Darmstadt, Germany). MTT was purchased from Invitrogen™ (Carlsbad, CA, USA).
Peptide synthesis
vCPP0275 (KKRYKKKYKAYKPYKKKKKF‐amide), vCPP0417 (SPRRRTPSPRRRRSQSPRRR‐amide), vCPP0667 (RPRRRATTRRRITTGTRRRR‐amide), vCPP0769 (RRLTLRQLLGLGSRRRRRSR‐amide), vCPP1779 (GRRGPRRANQNGTRRRRRRT‐amide), vCPP2319 (WRRRYRRWRRRRRQRRRPRR‐amide), and CF‐vCPP2319 (CF‐ WRRRYRRWRRRRRQRRRPRR‐amide) were synthesized in a Prelude instrument (Gyros Protein Technologies, Tuczon, AZ, USA) running Fmoc (FastMoc) SPPS protocols at 0.1 mmol scale on a Fmoc‐Rink‐amide ChemMatrix resin (Table 3). Side chain functionalities were protected with tert‐butyl (Glu, Ser, Thr, Tyr), NG‐2,2,4,6,7‐ pentamethyldihydrobenzofuran‐5‐sulfonyl (Arg), and N α ‐tert‐butoxycarbonyl (Trp) groups. Eight‐fold excess of Fmoc‐L‐amino acids and HBTU, in the presence of double molar amount of DIEA, was used for the coupling steps, with DMF as solvent. After chain assembly, full deprotection and cleavage were carried out with a 94 : 2.5 : 2.5 : 1 (% v/v) mixture of TFA : H2O : DODT : TIS for 90 min, at room temperature. The CF‐vCPP2319 peptide was similarly synthesized, except that 5(6)‐carboxyfluorescein (CF) was coupled manually in a four‐fold excess in the presence of an equivalent of DIPCDI in DMF to the deprotected N‐terminus prior to full deprotection and cleavage. After synthesis completion, the peptide was fully deprotected and cleaved with a 94 : 2.5 : 2.5 : 1 (% v/v) mixture of TFA : H2O : DODT : TIS for 90 min, at room temperature. Peptides were isolated by precipitation with cold diethyl ether and centrifugation at 4000 g , 4 °C for 20 min, taken up in H2O and lyophilized. Analytical reversed‐phase high‐performance liquid chromatography (RP‐HPLC) was performed on a Luna C18 column (4.6 mm × 50 mm, 3 µm) (Phenomenex, Torrance, CA, USA). Linear gradients of solvent B (0.036% TFA in ACN) into solvent A (0.045% TFA in H2O) were used at a flow rate of 1 mL·min−1 and with UV detection at 220 nm. Preparative HPLC runs were performed on a Luna C18 column (21.2 mm × 250 mm, 10 µm) (Phenomenex) using linear gradients of solvent B (0.1% TFA in ACN) into solvent A (0.1% TFA in H2O) at a flow rate of 25 mL·min−1 and with UV absorption detection at 220 nm. Fractions of adequate HPLC homogeneity and with the expected mass were combined and lyophilized. LC‐MS was performed in a LC‐MS 2010EV instrument (Shimadzu, Kyoto, Japan) fitted with an XBridge C18 column (4.6 mm × 150 mm, 3.5 µm) (Waters, Cerdanyola del Valles, Spain), eluting with linear gradients of formic acid/ACN (0.08% v/v) into formic acid/H2O (0.1% v/v) over 15 min at 1 mL·min−1. Peptide stock solutions (0.5 or 1 mm) in filtered H2O or PBS were stored at −20 °C.
Table 3.
Peptides used in this study.
| Peptide | Amino acid sequence |
Mass (Da) Calculated; found |
HPLC t R (min) a | Purity (%) b |
|---|---|---|---|---|
| vCPP0275 | KKRYKKKYKAYKPYKKKKKF‐amide | 2679.3; 2679.5 | 5.5 | 99.2 |
| vCPP0417 | SPRRRTPSPRRRRSQSPRRR‐amide | 2545.8; 2544.4 | 5.8 | 98.9 |
| vCPP0667 | RPRRRATTRRRITTGTRRRR‐amide | 2578.4; 2579.1 | 6.2 | 99.4 |
| vCPP0769 | RRLTLRQLLGLGSRRRRRSR‐amide | 2505.9; 2506.1 | 5.6 | 99.2 |
| vCPP1779 | GRRGPRRANQNGTRRRRRRT‐amide | 2476.4; 2476.8 | 5.1 | 99.5 |
| vCPP2319 | WRRRYRRWRRRRRWRRRPRR‐amide | 3178.7; 3179.8 | 6.9 | 99.1 |
| CF‐vCPP2319 | CF‐WRRRYRRWRRRRRWRRRPRR‐amide | 3536.1; 3537.8 | 7.8 | 98.1 |
See experimental method for details
Peptide purity was estimated by peak integration of the analytical HPLC chromatograms.
Lipid vesicle preparation
Large unilamellar vesicles (LUVs) (100 nm diameter) were prepared in 10 mm sodium phosphate (75.4 mm Na2HPO4, 24.6 mm NaH2PO4, pH 7.4), as described previously [64]. Briefly, lipids were dissolved in spectroscopy‐grade chloroform and mixed as required to prepare defined lipid compositions. The solvent was removed under a nitrogen stream to yield a lipid film which was left to dry under vacuum, overnight. The film was hydrated with 10 mm sodium phosphate (75.4 mm Na2HPO4, 24.6 mm NaH2PO4, pH 7.4); upon agitation and eight freeze–thaw cycles, suspensions of multilamellar vesicles (MLVs) were obtained. The MLVs were extruded through polycarbonate filters (100 nm pore size) eight times to obtain LUVs. Different lipid compositions were used in this study: POPC, POPC : ChoI (8 : 2), and POPC : SM : POPE : POPS : ChoI (4.35 :4.35 : 1 : 0.3 : 1).
Circular dichroism
Circular dichroism (CD) spectra of the different peptides were acquired in a J‐815 spectropolarimeter (Jasco, Tokyo, Japan) at 25 °C in the 190–260 nm wavelength range, with a bandwidth of 1 nm and a scan speed of 50 nm·min−1, using a 0.1 cm quartz cell. Samples containing 50 µm peptides with or without liposomes with different compositions (2 mm final lipid concentration) were analyzed. The final spectra for each peptide were the average of three consecutive scans per sample after subtraction of buffer baselines. Results were expressed as mean residue ellipticity ([θ]MRW) (deg·cm−2·dmol−1), as follows:
where θObs is the observed ellipticity in degrees, MRW is the mean residue weight, d is the cell path length, and c is the peptide mass concentration.
Hemolytic activity
Human blood samples were obtained from healthy donors with written informed consent. RBCs isolation followed a protocol described elsewhere [65]. Briefly, human blood samples were centrifuged at 1000 g for 10 min at 4 °C, washed three times, and resuspended in phosphate‐buffered saline (PBS) (1×, pH 7.4) at 0.25% (v/v). Then, RBC suspensions were incubated with increasing peptide concentrations (0.01–100 µm) for 24 h at 37 °C with gentle agitation, in a 96‐well round‐bottomed plate (Corning, New York, NY, USA). After incubation, the plate was centrifuged at 1000 g for 5 min at 4 °C. The supernatants were transferred to a 96‐well flat‐bottomed plate (Corning) and the presence of hemoglobin was measured by absorbance at 415 nm using the Infinite® M200 Tecan microplate reader (Männedorf, Zurich, Switzerland). PBS without peptide and Triton X‐100 at 1% (v/v, in H2O) were used as negative and positive controls for hemolysis, respectively. The well‐known hemolytic peptide melittin was included as a positive control for peptide‐induced hemolysis. Hemolytic activity (%) was determined using the following equation:
HC50 values were determined using the graphpad prim 7.0 software (San Diego, CA, USA) through non‐linear regression of a log(inhibitor) vs. normalized response equation to the experimental data. Experiments were performed on different days using independent human blood samples.
Cell culture
Human breast cell lines MDA‐MB‐231 (ATCC® HTB‐26™), BT‐20 (ATCC® HTB‐19™), BT‐474 (ATCC® HTB‐20™), SKBR3 (ATCC® HTB‐30™), MCF 10A (ATCC® CRL‐10317™) and human fibroblasts Hs68 (ATCC® CRL‐1635™) were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). Primary human brain microvascular endothelial cells (HBMEC) were purchased from Innoprot. The TNBC cell lines MDA‐MB‐231 and BT‐20 were cultured as a monolayer in DMEM and EMEM media, respectively. The HER2+ cell lines BT‐474 and SKBR3 were cultured as a monolayer in EMEM and McCoy's 5A media, respectively. The human fibroblasts Hs68 were cultured as a monolayer in DMEM medium. All the above‐mentioned media were supplemented with 10% FBS, and 1% penicillin/streptomycin (Gibco/Thermo Fisher Scientific, Waltham, MA, USA). MCF 10A breast epithelial cells were cultured as a monolayer in MEBM supplemented with SingleQuots, cholera toxin (100 ng·mL−1), and 1% penicillin‐streptomycin, according to ATCC instructions. HBMEC were cultured in T‐flasks coated with fibronectin (2 µg·cm−2) using endothelial cell basal medium with 5% FBS, 1% penicillin–streptomycin and 1% endothelial cell growth supplement. All cell cultures were maintained at 37 °C and 5% CO2, in a humidified atmosphere.
Cell proliferation
CellTiter‐Blue® assay
Peptide cytotoxicity against a panel of human cell lines was determined using CellTiter‐Blue® cell viability assay (Promega, Madrid, Spain), according to the manufacturer’s instructions. Briefly, MDA‐MB‐231, BT‐20, BT‐474, SKBR3, MCF 10A, and Hs68 cells were allowed to grow until 80% confluence in a 75‐T‐flask under standard conditions. Cells were then carefully detached and seeded at 1.0 × 105 cell·mL−1, 1.5 × 105 cell·mL−1, 3.0 × 105 cell·mL−1, 1.5 × 105 cell·mL−1, 4.0 × 105 cell·mL−1, and 1.5 × 105 cell·mL−1, respectively, in 96‐well flat‐bottomed plates, of 100 µL per well volume (Corning). After 24 h, medium was removed, cells were washed twice with PBS and incubated with 100 µL of previous diluted peptides in complete serum‐free medium (range 0.01–100 µm) for an additional 24 h in a humidified atmosphere of 5% CO2 at 37 °C. Then, 20 µL of CellTiter‐Blue® Reagent was added to each well and incubated for 3 h under the same conditions. Fluorescence intensity was measured with λexc = 560 nm and λem = 590 nm in an Infinite® M200 Tecan microplate reader. Medium without peptides and 1% Triton X‐100‐containing medium were used as positive controls (100% cell viability) and negative controls (0% cell viability), respectively. Cell viability (%) was determined by the following expression:
IC50 values were determined using the graphpad prim 7.0 software through non‐linear regression of a log(inhibitor) vs. normalized response equation to the experimental data. Experiments were performed on different days using independent cell cultures.
MTT assay
An MTT cell viability assay was also used to evaluate the cytotoxic activity of vCPP2319 toward human breast cell lines MDA‐MB‐231 and MCF 10A, and primary human brain microvascular endothelial cells–HBMEC. Briefly, MDA‐MB‐231 were seeded at 3.0 × 104 cells·mL−1 and MCF 10A and HBMEC were seeded at 5.0 × 105 cell·mL−1 into 96‐well flat‐bottomed plates with a 100 µL per well volume. After 24 h incubation, cells were washed once with PBS and complete serum‐free media with different vCPP2319 concentrations was added in a volume of 100 µL per well. The tested concentrations ranged between 0.01–40 µm for MDA‐MB‐231 cells, 0.01–100 µm for MCF 10A cells, and 0.01–60 µm for HBMEC. Following a 24 h incubation period with the peptide, 10 µL of a 5 mg·mL−1 solution of MTT was added to each well and allowed to incubate for 2 h in plates with MDA‐MB‐231 and MCF 10A and for 4 h in plates with HBMEC. Finally, the media containing the peptide and MTT was removed from each well, and 150 µL of spectrophotometric grade DMSO was added to each well, to dissolve the resulting formazan crystals. Absorbance was measured at 540 nm [66] in a Tecan Infinite® F500 microplate reader.
Positive controls (100% cell viability) were performed by adding serum‐free medium to untreated cells. Negative controls (0% cell viability) were performed by adding serum‐free medium with 20% DMSO to MDA‐MB‐23 cells and HBMEC and 30% DMSO to MCF 10A cells. Cell viability (%) was determined as:
and cell death (%) as:
IC50 values were assessed using graphpad prim 6.0 software package through non‐linear regression of a log(inhibitor) vs. normalized response equation to the experimental data. The experiments were performed on different days with independently grown cell cultures.
Zeta potential measurements
Cells were harvested from confluent cell cultures by trypsinization, washed, resuspended in PBS buffer, and diluted to final a concentration of 1.0 × 105 cell·mL−1 for MDA‐MB‐231 and MCF 10A cells and 2.5 × 105 cell·mL−1 for HBMEC. Cell suspensions with vCPP2319 were prepared at the following concentrations: 1.0, 5.2, and 20.0 µm for MDA‐MB‐231 cells; 2.5, 18.2, and 40.0 µm for MCF 10A and 5.2 µm for HBMEC. Samples with and without vCPP2319 were loaded into disposable zeta cells with gold electrodes and allowed to equilibrate for 30 min at 37 °C. Each experiment consisted of a set of 15 measurements with 40 subruns performed on a Malvern Zetasizer Nano ZS (Malvern, UK), at a constant voltage of 40 V, with a 90 s pause between measurements. The complete experiment was carried out at least two times using independent cellular suspensions and a control (untreated cells) was performed each day.
Fluorescence spectroscopy with di‐8‐ANEPPS
Confluent cell cultures were trypsinized, cells were resuspended in PBS, diluted to 1.5 × 106 cell·mL−1, and incubated for 1 h with 30 µm of di‐8‐ANNEPS with constant rotation, at room temperature. The cells were washed to remove non‐incorporated di‐8‐ANEPPS and diluted to a final concentration of 5.0 × 105 cell·mL−1. Cell membrane dipole potential variations were examined through di‐8‐ANEPPS excitation spectrum shifts in the presence of vCPP2319 after 10–15 min incubation. Di‐8‐ANEPPS fluorescence excitation spectra were collected on an Edinburgh FLS920 spectrofluorimeter (Livingston, UK) in the absence and presence of vCPP2319. In order to prevent membrane fluidity artifacts, the emission wavelength (λem) was set to 670 nm [67] and individual spectra were retrieved between 380 and 580 nm for untreated and treated cells. Excitation spectra were corrected for background intensity. Each spectrum was normalized to the respective integrated spectrum area. The differential excitation spectra of the dye in each cell line in the presence of vCPP2319 were obtained by subtracting the normalized excitation spectrum of the probe in the cells without the peptide from the normalized excitation spectra of the probe in the presence of vCPP2319. [26] To quantify di‐8‐ANEPPS spectral shifts, excitation intensity ratios (R) were calculated using the relationship and normalized to the control R (untreated cells). Normalized (R norm) intensity ratios were used as a quantitative parameter of di‐8‐ANEPPS spectral shifts, which correlates with variations in cell membrane dipole potential. This experiment was repeated on two different days using independently grown cell cultures.
LDH activity assay
MDA‐MB‐231 and MCF 10A cells were seeded at 1.0 × 105 cell·mL−1 into a 96‐well plate, with a volume of 100 µL per well, and incubated for 24 h. After media removal, cells were washed once with PBS and then incubated with different concentrations of vCPP2319, in DMEM without phenol red, in a volume of 100 µL per well, for 15 min, at 37 °C. The plates were centrifuged at 271 g or 174 g for MDA‐MB‐231 or MCF 10A cells, respectively, at 4 °C, and the supernatants were collected and kept on ice. The collected samples were centrifuged at 10 000 g at 4 °C for 15 min. The supernatants were collected and loaded into non‐treated 96‐well plates, with a 50 µL per well volume. The LDH activity assay kit was used following the manufacturer’s instructions. Briefly, a master mix was prepared with LDH assay buffer and LDH substrate mix and added to the previously loaded samples in a 1 : 1 ratio. The plates were incubated for 2 min at 37 °C and sample absorbance was measured at 450 nm every 5 min for at least 1 h, at a constant temperature, to monitor the enzymatic conversion of NAD+ into NADH. LDH activity was determined using the following expression:
where Reaction Time corresponds to T final – T initial (min) and V is the volume of sample added (mL). NADH standard curves and LDH positive controls were included in every plate and performed in each experiment. Negative controls were performed with supernatants collected from non‐treated cells. Positive controls were performed with supernatants collected from cells treated with melittin at 20 or 40 µm and Triton X‐100 at 0.25% or 0.5%, for MDA‐MB‐231 or MCF 10A cells, respectively. Sample handling was performed in a cold chamber to assure the lowest possible enzyme activity before assay initiation. Experiments were repeated into two different days, using independently grown cell cultures.
Flow cytometry
MDA‐MB‐231 and MCF 10A were seeded 5.0 × 104/300 µL and 1.0 × 105/300 µL, respectively, in 24‐well flat‐bottomed plates (Corning) for 24 h. Then, the medium was removed and cells were washed twice with PBS and once with medium, and incubated for different time‐points (15, 60, 360, and 1440 min) with CF‐vCPP2319, at a final concentration of 1 µm in a humidified atmosphere of 5% CO2 at 37 °C. Finally, cells were collected and washed twice with PBS. The fluorescence intensity of 10 000 cells was analyzed with a BD LSRFortessa X‐20 flow cytometer. The mean fluorescence of samples was obtained by subtracting the autofluorescence of cells. All experiments were performed in duplicates in three independent days.
Confocal microscopy
Cell internalization
MDA‐MB‐231 and MCF 10A cells were seeded 5.0 × 104/200 µL and 1.0 × 105/200 µL, respectively, on an ibiTreat‐coated 8‐well µ‐slide (Ibidi, Bayern, Germany) for 24 h. Then, cells were washed carefully twice with 1× PBS and once with medium, and incubated for different time‐points (15, 60, 360, and 1440 min) with CF‐vCPP2319, at a final concentration of 1 µm in a humidified atmosphere of 5% CO2 at 37 °C. Nucleus and cell membrane were stained with Hoechst 33342 (Sigma‐Aldrich) and CellMask™ Deep Red (Thermo Fisher), respectively. After the incubation period, cells were washed twice with PBS, and both the nucleus and the membrane dye were added to cells at a final concentration of 5 µg·mL−1 for 15 min at 37 °C. Finally, cells were washed twice with PBS and imaged immediately.
The acquisition was made on a confocal point‐scanning Zeiss LSM 880 microscope (Carl Zeiss, Oberkochen, Germany) equipped with an alpha Plan‐Apochromat X 63 oil immersion objective (1.40 numerical aperture). Diode 405‐30 laser was used to excite Hoechst 33342, argon laser in the 488‐nm line to excite CF‐vCPP2319, and HeNee633 to excite CellMask™ Deep Red. In the normal acquisition mode, × 0.6 zoom images were recorded at 4096 × 4096 resolution. ZEN software was used for image acquisition and fiji software [68] was used for image processing. At least five total images were acquired in three independent replicates.
Vimentin and actin distribution
MDA‐MB‐231 and MCF 10A cells were seeded 5.0 × 104/200 µL on an ibiTreat‐coated 8‐well µ‐slide (Ibidi) for 24 h. Then, cells were washed carefully three times with PBS and incubated for 24 h with CF‐vCPP2319 in complete serum‐free media in a humidified atmosphere of 5% CO2 at 37 °C. The final CF‐vCPP2319 concentrations tested were as follows: 1, 5.2, and 20 µm for MDA‐MB‐231 and 1, 5.2, and 18.2 µm for MCF 10A cells. The cells were washed three times with PBS and actin filaments were live stained with CellMask™ Green Actin Tracking Stain (Thermo Fisher) (1×) in DMEM without phenol, for 30 min, in a humidified atmosphere of 5% CO2 at 37 °C. After washing the cells three times with PBS, paraformaldehyde 3.75% in PBS was added to the cells followed by 10 min room temperature incubation. The cells were again washed three times with PBS and then incubated for 1 h with PBS with 1% BSA and 0.2% Triton X‐100, at room temperature. After three washes with PBS, Vimentin Monoclonal Antibody (V9) eFluor660 (Thermo Fisher) was added to the cells at a final concentration of 5 µg·mL−1 in PBS with 1% BSA, and incubated for 1 h, at room temperature. The cells were again washed three times with PBS, and the cells' nucleus was stained with Hoechst 33342 (Sigma‐Aldrich) (1 : 2000), for 10 min at room temperature. Finally, cells were washed three times with PBS and imaged.
The acquisition was made on a confocal point‐scanning Zeiss LSM 880 microscope (Carl Zeiss) equipped with an alpha Plan Apochromat X 63 oil immersion objective (1.40 numerical aperture). Diode 405‐30 laser was used to excite Hoechst 33342, argon laser in the 488‐nm line to excite CellMask™ Green Actin Tracking Stain, and HeNee633 to excite Vimentin Monoclonal Antibody (V9) eFluor 660. In the normal acquisition mode, ×0.60 zoom images were recorded 4096 × 4096 resolution. ZEN software was used for image acquisition and fiji software was used for image processing. At least 12 images were acquired in three independent replicates.
To compare the fluorescence intensities of the different cytoskeleton probes within MDA‐MB‐231 and MCF 10A cells, we calculated the corrected total cell fluorescence (CTCF) intensity using the following equation:
Atomic force microscopy (AFM)
Cell imaging
Cells were seeded into 40‐mm tissue culture dishes (TPP, Trasadingen, Switzerland) in 2 mL at concentrations of 1.0 × 104 cell·mL−1 for MDA‐MB‐231 cells and HBMEC and 1.0 × 105 cell·mL−1 for MCF 10A cells and allowed to grow for 24 h. After washing the cells with PBS, complete serum‐free medium with vCPP2319 was added to a final volume of 1 mL. Control cells were prepared with serum‐free medium only. Following a 24 h incubation, the cells were washed three times with PBS, three times with sterile milli‐Q water, and incubated with 1% glutaraldehyde for 10 min, at room temperature. The glutaraldehyde solution was then removed, and the cells were washed with PBS, sterile milli‐Q water, and finally air‐dried. AFM images were performed using a JPK NanoWizard® II for MDA‐MB‐231 and MCF 10A cells and a JPK NanoWizard® 4 (Berlin, Germany) for HBMEC. The equipment was mounted on a Zeiss Axiovert 200 inverted microscope (Carl Zeiss). The AFM head was equipped with a 15 µm z‐range linearized piezoelectric scanner and an infrared laser. Uncoated silicon ACL cantilevers from Applied NanoStructures (Mountain View, CA, USA) with typical resonance frequencies of 145–230 or 160–225 kHz and an average spring constant of 45 or 36–90 N·m−1 were used for breast and brain cell lines, respectively. Images were obtained using intermittent contact mode and contact mode. Scan speeds ranged between 0.1 and 0.4 Hz and total scan areas of 100 × 100 µm were imaged with a 512 × 512 pixel resolution.
Height profiles of the cells were acquired by drawing a cross section using the JPK SPM Data Processing version 4.2.61 or 6.0.55. For MDA‐MB‐231 and MCF 10A cells, 10 treated cells and 10 untreated were analyzed. For HBMEC, 11 treated cells and 13 untreated cells were analyzed. The surface roughness was defined as the root‐mean‐square roughness (R ms) and it was obtained from AFM height images using Gwyddion software version 2.24 [69]. R ms values were calculated for the nucleus and cytoplasm areas, from square areas of 2.5 × 2.5 µm. Within each cell, one R ms value is calculated for each cellular component (nuclei and cytoplasm) as the result of the average of the R ms values obtained from five square areas. The final R ms value is the result of the average of a total of at least five cells (treated and untreated). Cells were observed and imaged through at least two different days using independent grown cultures.
Force spectroscopy (AFM‐FS)
Indentation experiments were performed on live MDA‐MB‐231 and MCF 10A cultures prepared as described above. The cell culture medium was removed after the 24 h incubation with vCPP2319, and the cells were kept in PBS buffer at room temperature, after washing. A JPK NanoWizard® 4 mounted on a Zeiss Axiovert 200 inverted microscope was used to acquire force‐distance curves using QI™ mode. The cantilevers used in the experiment were qp‐BioAC‐50 with resonance frequency ranging from 24 to 36 kHz and force constant ranging between 0.03 and 0.09 N·m−1 (CB3). The calibration was performed through contact‐free method. QI™ mode settings are described as follows: Z‐length was 2500 nm, Z‐range was 15 µm, speed was 300.0 µm·s−1, and setpoint was defined as 0.6 and 0.7 nN for MDA‐MB‐231 and MCF 10A cells, respectively. Areas of 10 × 10 (in nuclear area) or 100 × 100 µm were scanned with a 256 × 256 pixel resolution. Hertz/Sneddon model was fitted to the extended segment of the obtained force‐distance curves obtained from the 10 × 10 µm areas to calculate the Young’s modulus. The distribution of the Young’s modulus values obtained correspond to the distribution of values collected from 11 untreated and 13 treated cells for MDA‐MB‐231 and 13 untreated and 10 treated cells for MCF 10A. This experiment was repeated on different days using independently grown cell cultures. QI™ mode results in large datasets since one force curve is acquired per pixel (256 × 256 pixel resolution means 65 536 force curves per cell) and, if fitted successfully, each force curve will result in a Young’s modulus value. These large datasets required a trimming process to reduce the amount of data and then applying a statistical test to compare the two groups of Young’s moduli (untreated and treated cells). Datasets were trimmed by prune row analysis and reduced by a factor of 100, resulting in 100 × smaller datasets, which were then compared for each cell line. Scanned areas of 100 × 100 µm were qualitatively analyzed as live images of treated and untreated cell lines.
Statistical analysis
Excell 2002 (Microsoft, Redmond, WA, USA) and graphpad prim 6.0 and 7.0 software packages were used to process quantitative data. Pairwise significances were assessed using one‐way ANOVA followed by Tukey’s or Dunnett’s multiple comparison test, and Mann–Whitney or unpaired t‐tests.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
FDO, MC, VN, DG, and MARBC designed the experiments. FDO, MC and DG performed the experimental work and data analysis. Synthetic peptides were made at UPF (Barcelona) with JV assistance, under DA supervision. TNF contributed to the fluorescence spectroscopy analysis. FDO, MC, and MARBC wrote the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16247.
Supporting information
Fig. S1. Secondary structure of peptides.
Fig. S2. Quantitative analysis of AFM images obtained for MDA‐MB‐231 and MCF 10A cells.
Fig. S3. vCPP2319 perturbation of the membrane dipole potential of brain endothelium cells derived from primary cultures.
Fig. S4. Quantitative analysis of the maximum cell height obtained from AFM images of brain endothelium cells derived from primary cell cultures.
Acknowledgements
FDO, MC, and DG acknowledge FCT I.P. for fellowships PD/BD/135046/2017, PD/BD/128281/2017, and SFRH/ BPD/73500/2010. FCT I.P. is also acknowledged for funding (Projects PTDC/BIA‐BQM/5027/2020 and PTDC/BBB‐NAN/1578/2014). Marie Skłodowska‐Curie Research and Innovation Staff Exchange (RISE) is also acknowledged for funding: call H2020‐MSCA‐RISE‐2014, grant agreement 644167, 2015–2019. Work at UPF was supported by grant AGL2017‐84097‐C2‐2‐R and the “María de Maeztu” Program for Units of Excellence in R&D (MDM‐2014‐0370) from the Spanish Ministry of Economy and Competitiveness (MINECO). The authors thank Dr Marco Domingues (IMM, Portugal) for technical guidance in AFM data analysis.
Contributor Information
David Andreu, Email: david.andreu@upf.edu.
Diana Gaspar, Email: diana.gaspar@gmail.com.
Miguel A. R. B. Castanho, Email: macastanho@medicina.ulisboa.pt.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA & Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- 2. Komorowski AS, Warner E, MacKay HJ, Sahgal A, Pritchard KI & Jerzak KJ (2020) Incidence of brain metastases in nonmetastatic and metastatic breast cancer: is there a role for screening? Clin Breast Cancer 20, e54–e64. [DOI] [PubMed] [Google Scholar]
- 3. Witzel I, Oliveira‐Ferrer L, Pantel K, Müller V & Wikman H (2016) Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res 18, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng X & Hung M‐C (2007) Breast cancer brain metastases. Cancer Metastasis Rev 26, 635–643. [DOI] [PubMed] [Google Scholar]
- 5. Bartsch R, Wenzel C & Steger GG (2007) Trastuzumab in the management of early and advanced stage breast cancer. Biol Targets Ther 1, 19–31. [PMC free article] [PubMed] [Google Scholar]
- 6. Riedl S, Zweytick D & Lohner K (2011) Membrane‐active host defense peptides – challenges and perspectives for the development of novel anticancer drugs. Chem Phys Lipids 164, 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gaspar D, Veiga AS & Castanho MARB (2013) From antimicrobial to anticancer peptides. A review. Front Microbiol 4, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palmieri D, Chambers AF, Felding‐Habermann B, Huang S & Steeg PS (2007) The biology of metastasis to a sanctuary site. Clin Cancer Res 13, 1656–1662. [DOI] [PubMed] [Google Scholar]
- 9. Langel Ü (2015) Cell‐Penetrating Peptides, 2nd edn. Springer New York, New York, NY. [Google Scholar]
- 10. Freire JM, Veiga AS, Conceição TM, Kowalczyk W, Mohana‐Borges R, Andreu D, Santos NC, Da Poian AT & Castanho MARB (2013) Intracellular nucleic acid delivery by the supercharged dengue virus capsid protein. PLoS One 8, e81450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Milletti F (2012) Cell‐penetrating peptides: classes, origin, and current landscape. Drug Discov Today 17, 850–860. [DOI] [PubMed] [Google Scholar]
- 12. de Figueiredo IR, Freire JM, Flores L, Veiga AS & Castanho MARB (2014) Cell‐penetrating peptides: a tool for effective delivery in gene‐targeted therapies. IUBMB Life 66, 182–194. [DOI] [PubMed] [Google Scholar]
- 13. Langel Ü (2019) CPP, Cell‐Penetrating Peptides. Springer Singapore, Singapore. [Google Scholar]
- 14. Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM & Temsamani J (2000) New advances in the transport of doxorubicin through the blood‐brain barrier by a peptide vector‐mediated strategy. Mol Pharmacol 57, 679–686. [DOI] [PubMed] [Google Scholar]
- 15. Rousselle C, Smirnova M, Clair P, Lefauconnier JM, Chavanieu A, Calas B, Scherrmann JM & Temsamani J (2001) Enhanced delivery of doxorubicin into the brain via a peptide‐vector‐mediated strategy: saturation kinetics and specificity. J Pharmacol Exp Ther 296, 124–131. [PubMed] [Google Scholar]
- 16. Li Y, Zheng X, Cao Z, Xu W, Zhang J & Gong M (2012) Self‐assembled peptide (CADY‐1) improved the clinical application of doxorubicin. Int J Pharm 434, 209–214. [DOI] [PubMed] [Google Scholar]
- 17. Aroui S, Brahim S, Hamelin J, De Waard M, Bréard J & Kenani A (2009) Conjugation of doxorubicin to cell penetrating peptides sensitizes human breast MDA‐MB 231 cancer cells to endogenous TRAIL‐induced apoptosis. Apoptosis 14, 1352–1365. [DOI] [PubMed] [Google Scholar]
- 18. Aroui S, Brahim S, Waard MD & Kenani A (2010) Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell‐penetrating peptides in different cell lines: a comparative study. Biochem Biophys Res Commun 391, 419–425. [DOI] [PubMed] [Google Scholar]
- 19. Freire JM, Dias SA, Flores L, Veiga AS & Castanho MARB (2015) Mining viral proteins for antimicrobial and cell‐penetrating drug delivery peptides. Bioinformatics 31, 2252–2256. [DOI] [PubMed] [Google Scholar]
- 20. Dias SA, Freire JM, Pérez‐Peinado C, Domingues MM, Gaspar D, Vale N, Gomes P, Andreu D, Henriques ST, Castanho MARB et al. (2017) New potent membrane‐targeting antibacterial peptides from viral capsid proteins. Front Microbiol 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma F, Li H, Wang H, Shi X, Fan Y, Ding X, Lin C, Zhan Q, Qian H & Xu B (2014) Enriched CD44+/CD24‐ population drives the aggressive phenotypes presented in triple‐negative breast cancer (TNBC). Cancer Lett 353, 153–159. [DOI] [PubMed] [Google Scholar]
- 22. Agus DB, Alexander JF, Arap W, Ashili S, Aslan JE, Austin RH, Backman V, Bethel KJ, Bonneau R, Chen W‐C et al. (2013) A physical sciences network characterization of non‐tumorigenic and metastatic cells. Sci Rep 3, 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chavez KJ, Garimella SV & Lipkowitz S (2011) Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis 32, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shankar J, Messenberg A, Chan J, Underhill TM, Foster LJ & Nabi IR (2010) Pseudopodial actin dynamics control epithelial‐mesenchymal transition in metastatic cancer cells. Cancer Res 70, 3780–3790. [DOI] [PubMed] [Google Scholar]
- 25. Mattila PK & Lappalainen P (2008) Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9, 446–454. [DOI] [PubMed] [Google Scholar]
- 26. Matos PM, Franquelim HG, Castanho MARB & Santos NC (2010) Quantitative assessment of peptide–lipid interactions. Biochim Biophys Acta Biomembr 1798, 1999–2012. [DOI] [PubMed] [Google Scholar]
- 27. Freire JM, Veiga AS, Rego de Figueiredo I, de la Torre BG, Santos NC, Andreu D, Da Poian AT & Castanho MARB (2014) Nucleic acid delivery by cell penetrating peptides derived from dengue virus capsid protein: design and mechanism of action. FEBS J 281, 191–215. [DOI] [PubMed] [Google Scholar]
- 28. Sinniah K, Paauw J & Ubels J (2002) Investigating live and fixed epithelial and fibroblast cells by atomic force microscopy. Curr Eye Res 25, 61–68. [DOI] [PubMed] [Google Scholar]
- 29. Henderson E (1994) Imaging of living cells by atomic force microscopy. Prog Surf Sci 46, 39–60. [Google Scholar]
- 30. Korsching E, Packeisen J, Liedtke C, Hungermann D, Wülfing P, van Diest PJ, Brandt B, Boecker W & Buerger H (2005) The origin of vimentin expression in invasive breast cancer: epithelial–mesenchymal transition, myoepithelial histogenesis or histogenesis from progenitor cells with bilinear differentiation potential? J Pathol 206, 451–457. [DOI] [PubMed] [Google Scholar]
- 31. Satelli A & Li S (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 68, 3033–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bianchini G, Balko JM, Mayer IA, Sanders ME & Gianni L (2016) Triple‐negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 13, 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collignon J, Lousberg L, Schroeder H & Jerusalem G (2016) Triple‐negative breast cancer: treatment challenges and solutions. Breast Cancer Targets Ther 8, 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoskin DW & Ramamoorthy A (2008) Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta Biomembr 1778, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Henriques ST, Melo MN & Castanho MARB (2006) Cell‐penetrating peptides and antimicrobial peptides: how different are they? Biochem J 399, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang E, Boire A, Agarwal A, Nguyen N, O’Callaghan K, Tu P, Kuliopulos A & Covic L (2009) Blockade of PAR1 signaling with cell‐penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res 69, 6223–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gronewold A, Horn M, Ranđelović I, Tóvári J, Muñoz Vázquez S, Schomäcker K & Neundorf I (2017) Characterization of a cell‐penetrating peptide with potential anticancer activity. ChemMedChem 12, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Figueira TN, Oliveira FD, Almeida I, Mello ÉO, Gomes VM, Castanho MARB & Gaspar D (2017) Challenging metastatic breast cancer with the natural defensin PvD 1. Nanoscale 9, 16887–16899. [DOI] [PubMed] [Google Scholar]
- 39. D Antonio P, Lasalvia M, Perna G & Capozzi V (2012) Scale‐independent roughness value of cell membranes studied by means of AFM technique. Biochim Biophys Acta Biomembr 1818, 3141–3148. [DOI] [PubMed] [Google Scholar]
- 40. Canetta E, Riches A, Borger E, Herrington S, Dholakia K & Adya AK (2014) Discrimination of bladder cancer cells from normal urothelial cells with high specificity and sensitivity: combined application of atomic force microscopy and modulated Raman spectroscopy. Acta Biomater 10, 2043–2055. [DOI] [PubMed] [Google Scholar]
- 41. Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Girasole M, Pompeo G, Cricenti A, Longo G, Boumis G, Bellelli A & Amiconi S (2010) The how, when, and why of the aging signals appearing on the human erythrocyte membrane: an atomic force microscopy study of surface roughness. Nanomedicine 6, 760–768. [DOI] [PubMed] [Google Scholar]
- 43. Dobrzynska I, Szachowicz‐Petelska B, Sulkowski S & Figaszewski Z (2005) Changes in electric charge and phospholipids composition in human colorectal cancer cells. Mol Cell Biochem 276, 113–119. [DOI] [PubMed] [Google Scholar]
- 44. Tait L, Soule HD & Russo J (1990) Ultrastructural and immunocytochemical characterization of an immortalized human breast epithelial cell line, MCF‐10. Cancer Res 50, 6087–6094. [PubMed] [Google Scholar]
- 45. Gaspar D, Veiga AS, Sinthuvanich C, Schneider JP & Castanho MARB (2012) Anticancer peptide SVS‐1: efficacy precedes membrane neutralization. Biochemistry 51, 6263–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johansson HJ, El‐Andaloussi S, Holm T, Mäe M, Jänes J, Maimets T & Langel Ü (2008) Characterization of a novel cytotoxic cell‐penetrating peptide derived from p14ARF protein. Mol Ther 16, 115–123. [DOI] [PubMed] [Google Scholar]
- 47. Stoddart MJ (2011) Cell viability assays: introduction. In Methods in Molecular Biology (Stoddart M, ed.), pp. 1–6. Humana Press, New York City, NY. [DOI] [PubMed] [Google Scholar]
- 48. Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti‐Dallenbach R, Oertle P, Hyotyla JT, Aebi U, Bentires‐Alj M, Lim RYH et al. (2012) The nanomechanical signature of breast cancer. Nat Nanotechnol 7, 757–765. [DOI] [PubMed] [Google Scholar]
- 49. Suresh S (2007) Biomechanics and biophysics of cancer cells. Acta Mater 55, 3989–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fife CM, McCarroll JA & Kavallaris M (2014) Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharmacol 171, 5507–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heu C, Berquand A, Elie‐Caille C & Nicod L (2012) Glyphosate‐induced stiffening of HaCaT keratinocytes, a Peak Force Tapping study on living cells. J Struct Biol 178, 1–7. [DOI] [PubMed] [Google Scholar]
- 52. Jiang P, Enomoto A & Takahashi M (2009) Cell biology of the movement of breast cancer cells: intracellular signalling and the actin cytoskeleton. Cancer Lett 284, 122–130. [DOI] [PubMed] [Google Scholar]
- 53. Chung B‐M, Rotty JD & Coulombe PA (2013) Networking galore: intermediate filaments and cell migration. Curr Opin Cell Biol 25, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strouhalova K, Přechová M, Gandalovičová A, Brábek J, Gregor M & Rosel D (2020) Vimentin intermediate filaments as potential target for cancer treatment. Cancers (Basel) 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mendez MG, Restle D & Janmey PA (2014) Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys J 107, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ofek G, Wiltz DC & Athanasiou KA (2009) Contribution of the cytoskeleton to the compressive properties and recovery behavior of single cells. Biophys J 97, 1873–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taylor RC, Cullen SP & Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9, 231–241. [DOI] [PubMed] [Google Scholar]
- 58. Grzanka A, Grzanka D & Orlikowska M (2003) Cytoskeletal reorganization during process of apoptosis induced by cytostatic drugs in K‐562 and HL‐60 leukemia cell lines. Biochem Pharmacol 66, 1611–1617. [DOI] [PubMed] [Google Scholar]
- 59. Abbott NJ, Rönnbäck L & Hansson E (2006) Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci 7, 41–53. [DOI] [PubMed] [Google Scholar]
- 60. Tajes M, Ramos‐Fernández E, Weng‐Jiang X, Bosch‐Morató M, Guivernau B, Eraso‐Pichot A, Salvador B, Fernàndez‐Busquets X, Roquer J & Muñoz FJ (2014) The blood‐brain barrier: structure, function and therapeutic approaches to cross it. Mol Membr Biol 31, 152–167. [DOI] [PubMed] [Google Scholar]
- 61. Langley RR & Fidler IJ (2013) The biology of brain metastasis. Clin Chem 59, 180–189. [DOI] [PubMed] [Google Scholar]
- 62. Qureshi H, Hamid SS, Ali SS, Anwar J, Siddiqui AA & Khan NA (2015) Cytotoxic effects of aflatoxin B1 on human brain microvascular endothelial cells of the blood‐brain barrier. Med Mycol 53, 409–416. [DOI] [PubMed] [Google Scholar]
- 63. Ribeiro MMB, Domingues MM, Freire JM, Santos NC & Castanho MARB (2012) Translocating the blood‐brain barrier using electrostatics. Front Cell Neurosci 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Henriques ST, Pattenden LK, Aguilar MI & Castanho MARB (2009) The toxicity of prion protein fragment PrP(106–126) is not mediated by membrane permeabilization as shown by a M112W substitution. Biochemistry 48, 4198–4208. [DOI] [PubMed] [Google Scholar]
- 65. Cavaco MC, Garcia JV, da Silva RDM, Correia JDG, Castanho MARB, Andreu Martinez D & Neves VLS (2020) DPepH3, an improved peptide shuttle for receptor‐independent transport across the blood‐brain barrier. Curr Pharm Des 26, 1–12. [DOI] [PubMed] [Google Scholar]
- 66. Morgan DML (1998) Tetrazolium (MTT) assay for cellular viability and activity. In Polyamine Protocols (Morgan DML, ed), pp. 179–184. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 67. Clarke RJ & Kane DJ (1997) Optical detection of membrane dipole potential: avoidance of fluidity and dye‐induced effects. Biochim Biophys Acta Biomembr 1323, 223–239. [DOI] [PubMed] [Google Scholar]
- 68. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nečas D & Klapetek P (2012) Gwyddion: an open‐source software for SPM data analysis. Cent Eur J Phys 10, 181–188. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Secondary structure of peptides.
Fig. S2. Quantitative analysis of AFM images obtained for MDA‐MB‐231 and MCF 10A cells.
Fig. S3. vCPP2319 perturbation of the membrane dipole potential of brain endothelium cells derived from primary cultures.
Fig. S4. Quantitative analysis of the maximum cell height obtained from AFM images of brain endothelium cells derived from primary cell cultures.