

Abstract
Objective
Macrophages mediate inflammation, angiogenesis, and tissue destruction in giant cell arteritis (GCA). Serum levels of the macrophage‐associated protein YKL‐40 (chitinase 3–like protein 1), previously linked to angiogenesis and tissue remodeling, remain elevated in GCA despite glucocorticoid treatment. This study was undertaken to investigate the contribution of YKL‐40 to vasculopathy in GCA.
Methods
Immunohistochemistry was performed on GCA temporal artery biopsy specimens (n = 12) and aortas (n = 10) for detection of YKL‐40, its receptor interleukin‐13 receptor α2 (IL‐13Rα2), macrophage markers PU.1 and CD206, and the tissue‐destructive protein matrix metalloproteinase 9 (MMP‐9). Ten noninflamed temporal artery biopsy specimens served as controls. In vitro experiments with granulocyte–macrophage colony‐stimulating factor (GM‐CSF)– or macrophage colony‐stimulating factor (M‐CSF)–skewed monocyte‐derived macrophages were conducted to study the dynamics of YKL‐40 production. Next, small interfering RNA–mediated knockdown of YKL‐40 in GM‐CSF–skewed macrophages was performed to study its effect on MMP‐9 production. Finally, the angiogenic potential of YKL‐40 was investigated by tube formation experiments using human microvascular endothelial cells (HMVECs).
Results
YKL‐40 was abundantly expressed by a CD206+MMP‐9+ macrophage subset in inflamed temporal arteries and aortas. GM‐CSF–skewed macrophages from GCA patients, but not healthy controls, released significantly higher levels of YKL‐40 compared to M‐CSF–skewed macrophages (P = 0.039). In inflamed temporal arteries, IL‐13Rα2 was expressed by macrophages and endothelial cells. Functionally, knockdown of YKL‐40 led to a 10–50% reduction in MMP‐9 production by macrophages, whereas exposure of HMVECS to YKL‐40 led to significantly increased tube formation.
Conclusion
In GCA, a GM‐CSF–skewed, CD206+MMP‐9+ macrophage subset expresses high levels of YKL‐40 which may stimulate tissue destruction and angiogenesis through IL‐13Rα2 signaling. Targeting YKL‐40 or GM‐CSF may inhibit macrophages that are currently insufficiently suppressed by glucocorticoids.
INTRODUCTION
Giant cell arteritis (GCA) is an inflammatory disease that affects the medium‐ and large‐sized arteries and has potential serious acute complications, such as blindness and stroke (1). Chronic complications can also occur, since long‐term aortic inflammation is associated with the development of aneurysms and aorta dissections (2, 3). GCA is commonly treated with glucocorticoids (GCs). More recently, tocilizumab (interleukin‐6 [IL‐6] receptor blockade) has become available as GC‐sparing therapy in GCA (4). Both GCs and tocilizumab treatment suppress disease symptoms. It is less clear, however, if GCs and tocilizumab suppress smoldering vascular inflammation, which is likely associated with relapses and chronic complications of GCA (5, 6, 7, 8, 9).
The inflamed vessel wall in GCA patients is characterized by a granulomatous tissue reaction involving mainly T cells and macrophages (1). Besides promoting ongoing inflammation, macrophages release factors leading to myofibroblast proliferation (e.g., platelet‐derived growth factor), angiogenesis (e.g., vascular endothelial growth factor [VEGF]), and tissue destruction (e.g., matrix metalloproteinase 9 [MMP‐9]) (10, 11). These processes are responsible for the pathologic changes associated with the serious complications of GCA, such as vascular occlusion due to intima hyperplasia and aneurysms due to media degradation. Importantly, several studies have shown that even with treatment, macrophage activity persists in GCA patients, indicating that currently available treatments do not sufficiently suppress the local inflammatory response (6, 12). Therefore, new strategies and targets are needed to adequately halt inflammation and destruction of the vessel wall.
We recently described functionally heterogeneous macrophage subsets in GCA lesions, likely due to local signals involving granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (13). We demonstrated the presence of a specific macrophage subset in and around the media layer that lacked folate receptor β expression but showed high expression of the mannose receptor CD206. Importantly, these CD206+ macrophages exclusively expressed MMP‐9, indicating that these cells are likely to be important in media destruction. Moreover, others reported MMP‐9 to be an important mediator of endothelial cell migration and neoangiogenesis, thus facilitating T cell and macrophage recruitment to the vessel wall, processes crucial in the pathogenesis of GCA (11). Subsequent in vitro experiments demonstrated that the CD206+ macrophage phenotype can be induced by culturing macrophages with GM‐CSF, a growth factor abundantly expressed in GCA lesions (13). Our findings on functional macrophage heterogeneity in GCA lesions, along with additional recent studies on GM‐CSF signaling in GCA (12), prompted us to investigate the phenotype and functioning of the CD206+ macrophage subset in GCA in more detail. To this end, we took clues from the field of cancer immunology on tumor‐associated macrophages.
Tumor‐associated macrophages promote tumor growth and are associated with poor survival (14). Prior studies indicated an important role for YKL‐40 (also known as chitinase 3–like protein 1) produced by tumor‐associated macrophages in various inflammatory and tissue remodeling processes, including angiogenesis and tissue destruction. Furthermore, YKL‐40 has been implicated as an upstream signal for MMP‐9 production (15). Although early reports described elevated serum levels of YKL‐40 in autoinflammatory conditions, including GCA (16), less is known about the role of YKL‐40 in the immunopathology of GCA. YKL‐40 is a chitinase‐like protein, meaning that it is able to bind to chitin but does not cleave it, owing to the lack of enzymatic activity of YKL‐40 (14). YKL‐40 production by innate immune cells, including macrophages, is induced by various stimuli, including the cytokines IL‐6, IL‐1β, and interferon‐γ (IFNγ) (17).
Interestingly, we previously showed that serum levels of YKL‐40 are elevated in GCA patients at diagnosis, but do not normalize after GC treatment. In contrast, acute‐phase markers such as C‐reactive protein are strongly suppressed by treatment with GCs and tocilizumab, since their levels are highly dependent on IL‐6 in GCA (7, 18). Moreover, abundant expression of YKL‐40 has been documented in GCA temporal artery biopsy specimens (7, 16). However, it is yet unknown which specific cell type(s) produce YKL‐40 and what the role of YKL‐40 is in the immunopathogenesis of GCA.
IL‐13 receptor α2 (IL‐13Rα2), a high‐affinity receptor for IL‐13 that is distinct from IL‐13Rα1, was previously thought to be a decoy receptor for IL‐13 due to the lack of a signal transducing cytoplasmic tail (19, 20). However, recent studies demonstrated the activation of MAPK, Akt, ERK, and STAT3 pathways in intestinal epithelial cells, nasal epithelial cells, glioblastoma cells, dendritic cells, and macrophages upon binding of either IL‐13 or YKL‐40 to IL‐13Rα2 (21, 22, 23, 24, 25). The expression of this receptor in the context of GCA has so far not been reported.
In this study, we aimed to determine the cellular source of YKL‐40 and to investigate its contribution to vascular pathology in GCA. Using immunohistochemistry (IHC) and immunofluorescence, we identified the subset of CD206+ macrophages as the main cellular source of YKL‐40 in inflamed temporal arteries and in the aorta. In the same tissues, we next examined the expression of IL‐13Rα2 to establish whether YKL‐40 can signal at the site of inflammation. To assess whether YKL‐40 is an upstream modulator of the tissue‐destructive MMP‐9, we performed in vitro experiments with monocyte‐derived macrophages on the dynamics of YKL‐40 and MMP‐9 expression. Finally, we confirmed the angiogenic potential of YKL‐40 in vitro in a tube formation assay. Our data support an important role for the YKL‐40/IL‐13Rα2 axis in tissue destruction and neovascularization in GCA.
PATIENTS AND METHODS
For more details on the methods, see the Supplementary Methods, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract.
Patients
Twelve inflamed temporal artery tissue samples from treatment‐naive patients with histologically proven GCA were studied. The diagnosis of GCA was based on a pathologist’s assessment of the temporal artery biopsy sample as positive for panarteritis. Ten inflamed aorta tissue samples from patients with an untreated GCA‐related aneurysm were also included (13). Ten noninflamed temporal artery biopsy specimens were included as controls. These noninflamed temporal artery biopsy specimens were obtained from patients with positron emission tomography (PET)–proven GCA (skip lesions; n = 3), patients with isolated polymyalgia rheumatica (PMR; n = 5), and individuals who had neither GCA nor PMR (n = 2). The study was approved by the institutional review board of the University Medical Center Groningen (METc2010/222), and written informed consent was obtained. All procedures were conducted in compliance with the Declaration of Helsinki.
Serum and frozen peripheral blood mononuclear cells (PBMCs) from treatment‐naive GCA patients and age‐ and sex‐matched healthy controls were used for Luminex assay and in vitro studies. (Baseline characteristics of the patients and controls are shown in Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract.) GCA diagnoses were confirmed by temporal artery biopsy and/or PET. Healthy controls were screened by health assessment questionnaires, physical examination, and laboratory tests for past and current morbidities and were excluded if they were not healthy. Follow‐up serum samples were obtained from GCA patients 1 year after the start of treatment and at treatment‐free remission for the Luminex analysis. Patients were considered to be in treatment‐free remission if it had been ≥2 years since the start of treatment, ≥3 months since the last treatment, and they did not have a relapse for ≥6 months after the sample was obtained.
IHC and triple fluorescence multispectral imaging
Formalin‐fixed, paraffin‐embedded tissue sections (3 μm) were deparaffinized and rehydrated, followed by antigen retrieval. For single staining IHC, tissues were stained with antibodies targeting YKL‐40 and IL‐13Rα2. Double staining for YKL‐40 and the macrophage transcription factor PU.1 was performed to confirm the expression of YKL‐40 by macrophages. Temporal artery biopsy specimens from GCA patients were semiquantitatively scored as previously described (26). Briefly, samples were scored on a 5‐point semiquantitative scale of 0–4, where 0 = no positive cells, 1 = occasional positive cells (0–1% estimated positive), 2 = small numbers of positive cells (>1–20%), 3 = moderate numbers of positive cells (>20–50%), and 4 = large numbers of positive cells (>50%). Scores represent the average of the scores of 2 independent investigators (YvS and WFJ).
For colocalization study, triple fluorescence stainings of CD206, YKL‐40, and MMP‐9 were performed (n = 5). Colocalization of all 3 fluorophores was assigned the color cyan. For lists of antibodies used in IHC and immunofluorescence staining, see the Supplementary Methods and Supplementary Tables 2 and 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract.
Monocyte‐derived macrophages
Monocytes were isolated from thawed PBMCs from GCA patients and healthy controls using an EasySep monocyte enrichment kit without CD16 depletion (StemCell Technologies). Isolated monocytes were cultured in the presence of GM‐CSF or macrophage colony‐stimulating factor (M‐CSF) to generate GM‐CSF–skewed or M‐CSF–skewed macrophages. Supernatants were collected for Luminex assay or enzyme‐linked immunosorbent assay (ELISA) (see the Supplementary Methods for details on the Luminex assay and ELISA methods). For the assessment of YKL‐40 levels, macrophages were activated by the addition of lipopolysaccharide (LPS) on day 5, and the supernatants were collected on day 7. Assessment of IL‐6 was included as stimulation control. For YKL‐40 and MMP‐9 kinetics experiments, GM‐CSF–skewed macrophages were generated and the supernatants were collected every 48 hours, with the final collection on day 8 for ELISAs.
Small interfering RNA (siRNA) knockdown of YKL‐40
Monocyte‐derived macrophages were generated in the presence of GM‐CSF. On day 6, macrophages were harvested and transfected with YKL‐40 siRNA (assay ID s2999, Silencer Select; ThermoFisher) or nontargeting control siRNA (catalog no. 4390843, Silencer Select; ThermoFisher) using INTERFERin (Polyplus Transfection). After 24 hours, the medium was replaced with fresh complete medium containing GM‐CSF and incubated for 24 hours. The medium was then refreshed with complete medium containing GM‐CSF with or without LPS. After 24 hours, medium was collected for ELISA and cells were lysed for RNA extraction and quantitative polymerase chain reaction analysis.
Tube formation assay
Human microvascular endothelial cells (HMVECs; Lonza) were treated with medium only, 150 ng/ml YKL‐40 (Organon; MSD), 1,500 ng/ml YKL‐40, or 20 ng/ml VEGF (PeproTech) in triplicate. HMVECs were cultured for 16 hours and then scanned on a TissueFAXS system (TissueGnostics). Tube formation was assessed by counting the number of visible enclosed fields in a blinded manner.
Flow cytometric analysis
HMVECs were stained for expression of endothelial cell marker CD31, IL‐13Rα2, and VEGF receptor (VEGFR) (all from Miltenyi Biotec). Three technical replicates containing the 3 markers were compared to “fluorescence minus one” controls, in which either IL‐13α2 or VEGFR antibody was omitted.
Statistical analysis
To analyze the differences between YKL‐40 expression in different layers of the vessel wall and the results for GM‐CSF–skewed macrophages and M‐CSF–skewed macrophages from the same donor, the paired Wilcoxon signed rank test was used. Correlation analyses were performed with Spearman’s correlation. To analyze the difference between groups in the tube formation assay, one‐way analysis of variance with Tukey’s post hoc test was used.
RESULTS
Elevation of serum levels of YKL‐40 in GCA patients, without normalization of YKL‐40 levels after GC treatment
To unravel the dynamics of YKL‐40 in GCA patients over time, we first examined serum levels of YKL‐40. In this expansion of our previous work (7), serum YKL‐40 levels were confirmed to be elevated in treatment‐naive GCA patients when compared to age‐matched healthy controls (P < 0.0001) (Figure 1A). Treatment with GCs for 1 year did not significantly decrease YKL‐40 levels. Moreover, even GCA patients in treatment‐free remission had elevated YKL‐40 levels. In contrast, the high levels of IL‐6 observed at baseline decreased significantly after 1 year of treatment with GCs and in treatment‐free remission (Figure 1B).
Figure 1.
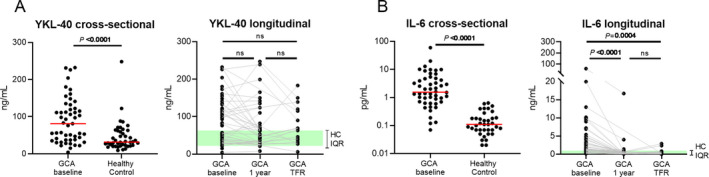
Serum levels of YKL‐40 and interleukin‐6 (IL‐6) in patients with giant cell arteritis (GCA) and healthy controls (HCs). A cross‐sectional analysis of serum levels of YKL‐40 (A) and IL‐6 (B) was conducted in treatment‐naive GCA patients (n = 51) and healthy controls (n = 42), and a longitudinal analysis of serum levels of YKL‐40 (A) and IL‐6 (B) was conducted in matched samples from GCA patients at baseline (n = 51), 1 year after the start of glucocorticoid treatment (n = 42), and at the time of treatment‐free remission (TFR; n = 17). Shading indicates the interquartile range (IQR) in healthy controls. Symbols represent individual subjects; horizontal lines show the median. In the cross‐sectional analysis, P values were determined by Mann‐Whitney U test. In the longitudinal analysis, P values were determined by Wilcoxon’s signed rank test. NS = not significant. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract.
Production of YKL‐40 by a distinct subset of CD206+MMP‐9+ macrophages in inflamed temporal arteries and aortas from GCA patients
IHC for the detection of YKL‐40 in inflamed temporal artery biopsy specimens from GCA patients (n = 12) revealed a distinct staining pattern characterized by abundant expression primarily at the intima‐media border (Figure 2A) (see Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract, for isotype control staining). Double staining for YKL‐40 and the transcription factor PU.1, which is highly expressed by macrophages, revealed that YKL‐40 was expressed predominantly by macrophages (Figure 2B). Interestingly, not all PU.1‐positive cells expressed YKL‐40, suggesting that YKL‐40 is produced by a specific subset of macrophages. Consecutive staining of YKL‐40 revealed a pattern similar to that of CD206 and MMP‐9 expression, suggesting that YKL‐40 is predominantly produced by tissue‐destructive CD206+MMP‐9+ macrophages (Figure 2A). Expression of YKL‐40 was not detected in noninflamed temporal arteries (Supplementary Figure 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract).
Figure 2.
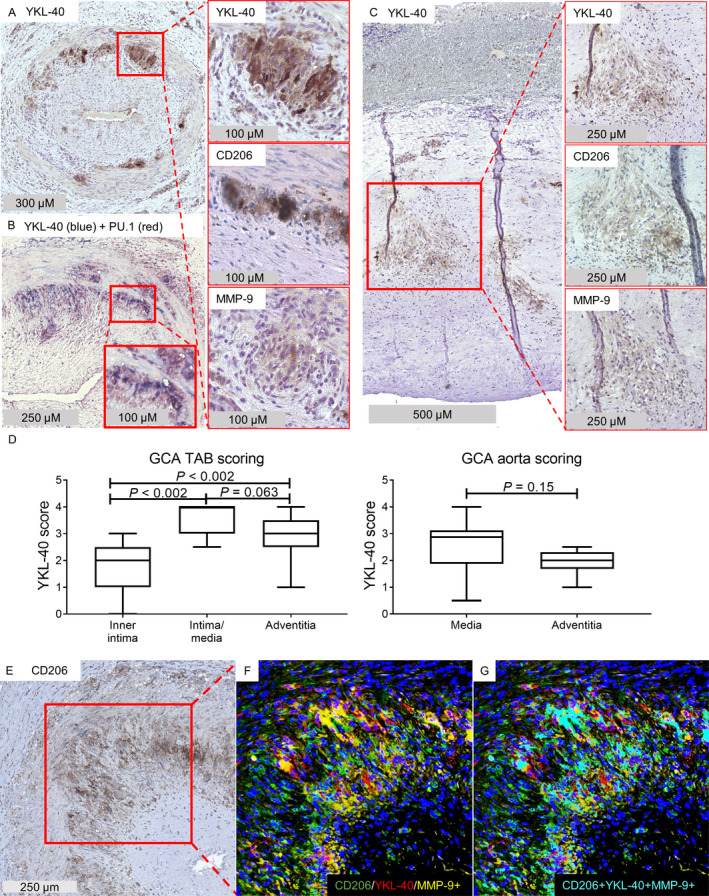
YKL‐40 expression in CD206+MMP‐9+ macrophage‐rich areas in inflamed medium‐ and large‐sized arteries from patients with giant cell arteritis (GCA). A, Temporal artery biopsy (TAB) specimens from treatment‐naive GCA patients were stained for detection of YKL‐40 by immunohistochemistry (IHC). Right panels are higher‐magnification views of the boxed area, showing expression of YKL‐40, CD206, and matrix metalloproteinase 9 (MMP‐9) within the same region of the temporal artery biopsy. B, Double staining for YKL‐40 and the transcription factor PU.1, which is predominantly expressed by macrophages, indicated that YKL‐40 was mainly expressed by macrophages. The bottom boxed area is a higher‐magnification view of the top boxed area, showing YKL‐40 staining in macrophages. C, Aortas from patients with GCA‐related aortic aneurysm were stained for detection of YKL‐40 by IHC. Right panels are higher‐magnification views of the boxed area, showing expression of YKL‐40, CD206, and MMP‐9. A pattern of YKL‐40 production similar to that shown in A was observed within the region of CD206‐expressing cells, located at the site of the granuloma in the media of inflamed aortas. D, YKL‐40 expression in GCA temporal artery biopsy specimens and aortas was scored semiquantitatively. Expression of YKL‐40 was higher in the intima‐media region of temporal artery biopsy specimens, but no significant differences were found between layers in the aortas. The intima was not scored in GCA aortas due to a lack of infiltrate. Data are shown as Tukey box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 75th percentile plus 1.5 times the interquartile range. P values were determined by Wilcoxon’s signed rank test. E–G, IHC staining of GCA temporal artery biopsy specimens was performed. Single staining IHC for CD206 (E), a merged image of triple immunofluorescence staining for CD206, YKL‐40, and MMP‐9 (F), and overlapping pixels (cyan) indicating colocalization of CD206, YKL‐40, and MMP‐9 (G) are shown. Results are representative of 12 samples in A and B, 10 samples in C, and 5 samples in E–G.
YKL‐40 expression was also observed in the aortas of patients with a GCA‐related aortic aneurysm (n = 10) (Figure 2C). In these aortas, YKL‐40 expression was mainly detected in areas of granulomatous inflammation in the media, a region containing CD206+MMP‐9+ macrophages (Figure 2C). Semiquantitative scoring indeed revealed an interesting spatial distribution of YKL‐40–expressing cells in inflamed temporal arteries, with the highest expression in the intima‐media region (Figure 2D). To formally prove expression of YKL‐40 by the CD206+MMP‐9+ macrophage subset, we performed triple fluorescence staining for these markers. These data confirmed expression of YKL‐40 by CD206+MMP‐9+ macrophages in the inflamed temporal artery biopsy specimens (Figures 2E–G; for single immunofluorescence staining of each marker, see Supplementary Figure 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract). Taken together, our results indicate that in GCA lesions, YKL‐40 is highly expressed primarily by a subset of CD206+MMP‐9+ tissue‐destructive macrophages located in the media and at its borders.
Production of higher levels of YKL‐40 by GM‐CSF–skewed macrophages than by M‐CSF–skewed macrophages from GCA patients
Previously, we demonstrated that GM‐CSF skews macrophages into a CD206+ phenotype (13). To determine whether GM‐CSF–skewed macrophages produce YKL‐40, monocytes from GCA patients (n = 8) were differentiated into macrophages in the presence of GM‐CSF or M‐CSF for 7 days. During the last 2 days of culture, cells were stimulated with LPS (Figure 3A). Higher levels of YKL‐40 were detected in the supernatants of GM‐CSF–skewed macrophages than in the supernatants of M‐CSF–skewed macrophages from GCA patients (P = 0.0391) (Figure 3B). This finding is consistent with the coexpression of CD206 and YKL‐40 observed in GCA temporal artery biopsy specimens. Interestingly, no significant difference in YKL‐40 levels was detected in the culture supernatant when comparing GM‐CSF–skewed macrophages and M‐CSF–skewed macrophages from healthy controls (n = 7).
Figure 3.
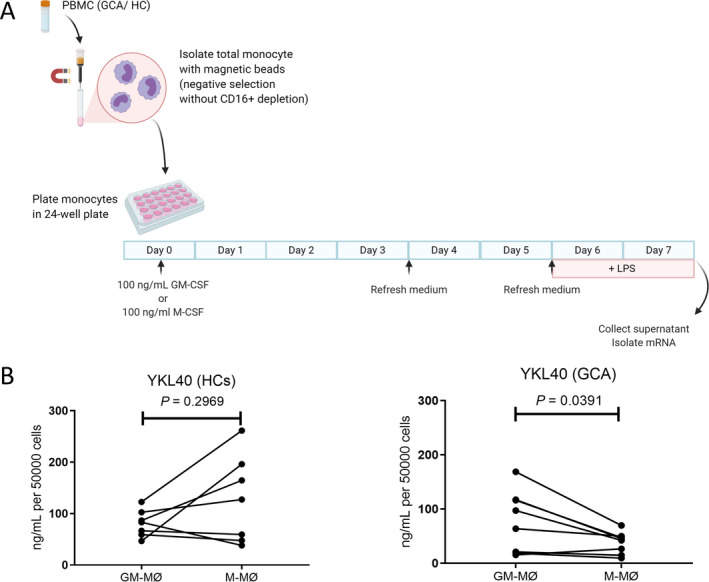
Elevated levels of YKL‐40 in granulocyte–macrophage colony‐stimulating factor (GM‐CSF)–skewed macrophages from patients with giant cell arteritis (GCA). A, Monocytes from peripheral blood mononuclear cells (PBMCs) from healthy controls (HCs; n = 7) and GCA patients (n = 8) were differentiated into macrophages and subsequently activated for 48 hours with lipopolysaccharide (LPS). B, YKL‐40 concentrations in the supernatants of GM‐CSF–skewed macrophages (GM‐MØ) and macrophage colony‐stimulating factor (M‐CSF)–skewed macrophages (M‐MØ) from healthy controls and GCA patients were determined by Luminex assay. YKL‐40 concentrations were significantly higher in GM‐CSF–skewed macrophages than in M‐CSF–skewed macrophages from GCA patients. P values were determined by Wilcoxon’s signed rank test. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract.
Assessment of IL‐6 levels as stimulation control revealed significant up‐regulation of IL‐6 production by GM‐CSF–skewed macrophages compared to M‐CSF–skewed macrophages (Supplementary Figure 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract), consistent with previous studies (27, 28), in both GCA patients and healthy controls. Our data suggest an altered GM‐CSF/M‐CSF signaling sensitivity in monocyte/macrophages from GCA patients compared to healthy controls.
Strong expression of IL‐13Rα2, the receptor for YKL‐40, in GCA lesions
Since YKL‐40 is highly expressed in GCA lesions, we next investigated the expression of IL‐13Rα2, a confirmed receptor for YKL‐40 (24, 25), in inflamed temporal artery biopsy specimens from GCA patients (n = 12). IHC detection revealed high expression of IL‐13Rα2 by endothelial cells, infiltrating leukocytes, vascular smooth muscle cells, and fibroblasts (Figure 4A), which is consistent with previous reports in the literature (19, 25, 29, 30, 31). Semiquantitative scoring of IL‐13Rα2 staining demonstrated high expression levels in all 3 layers of the vessel wall, most prominently in the adventitia and media‐intima borders (Figure 4B).
Figure 4.
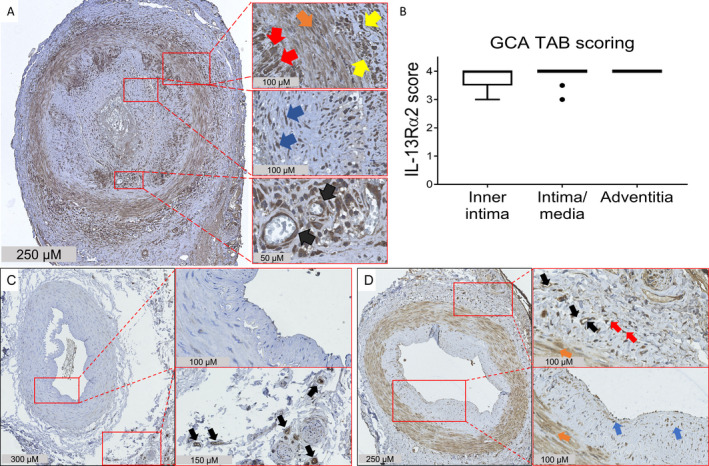
Elevated expression of interleukin‐13 receptor α2 (IL‐13Rα2) in the inflamed temporal arteries of patients with giant cell arteritis (GCA). A, Temporal artery biopsy (TAB) specimens from treatment‐naive GCA patients were stained for IL‐13Rα2. Expression of IL‐13Rα2 was detected in all 3 layers of the vessel wall. Right panels are higher‐magnification views of the boxed areas, showing expression of IL‐13Rα2 by macrophages (red arrows), vascular smooth muscle cells (orange arrow), infiltrating leukocytes (yellow arrows), fibroblasts (blue arrows), and endothelial cells (black arrows). B, IL‐13Rα2 expression in the vessel layers of GCA temporal artery biopsy specimens was scored semiquantitatively. The box represents the 25th to 75th percentiles. The line outside the box represents the 75th percentile plus 1.5 times the interquartile range (IQR). Horizontal lines represent the median. Circles indicate points outside the 25th or 75th percentile plus 1.5 times the IQR. C, Temporal artery biopsy specimens from individuals without GCA or polymyalgia rheumatica were stained for IL‐13Rα2. Right panels are higher‐magnification views of the boxed areas, showing minimal expression of IL‐13Rα2 in the medial vascular smooth muscle layer and the lumen, and stronger expression in the vasa vasorum endothelial cells (black arrows). D, Temporal artery biopsy specimens from patients with positron emission tomography–proven GCA were stained for IL‐13Rα2. Strong expression of IL‐13Rα2 was detected in all 3 layers of the vessel wall. Right panels are higher‐magnification views of the boxed areas, showing expression of IL‐13Rα2 by endothelial cells (black arrows), vascular smooth muscle cells (orange arrows), and presumably resident dendritic cells (red arrows) and fibroblasts (blue arrows). Results are representative of 12 samples in A, 2 samples in C, and 3 samples in D.
Interestingly, when analyzing the expression of IL‐13Rα2 in noninflamed temporal arteries with no or few infiltrating cells, a more restricted staining was seen. IL‐13Rα2 was only weakly expressed by endothelium and vascular smooth muscle layers in noninflamed temporal artery biopsy specimens from individuals without GCA or PMR (Figure 4C), while IL‐13Rα2 was strongly expressed by endothelial cells, vascular smooth muscle cells, and fibroblasts in noninflamed temporal artery biopsy specimens from patients with PET‐proven GCA (skip lesions) (Figure 4D) and patients with isolated PMR (Supplementary Figure 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract). Taken together, our data suggest a role for the YKL‐40/IL‐13Rα2 signaling axis in GCA lesions.
YKL‐40 as an upstream modulator of macrophage MMP‐9 production
To assess if YKL‐40 is an upstream modulator of the tissue‐destructive MMP‐9, we studied the kinetics of YKL‐40 and MMP‐9 production by differentiating monocytes from healthy controls (n = 8) into macrophages in the presence of 20 ng/ml or 100 ng/ml of GM‐CSF (Supplementary Figures 6A and B, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract). Both concentrations of GM‐CSF equally induced the up‐regulation of YKL‐40 and MMP‐9 as the monocytes differentiated into macrophages. Interestingly, the increase in YKL‐40 levels preceded the up‐regulation of MMP‐9 by 2–4 days. Moreover, the increase in YKL‐40 production from day 6 to day 8 strongly correlated with the increase in MMP‐9 production (Supplementary Figure 6C), suggesting that YKL‐40 could be an upstream modulator of MMP‐9 production.
To confirm the potential role of YKL‐40 as an upstream signal for MMP‐9 production in macrophages, we performed siRNA‐mediated knockdown of YKL‐40 using monocyte‐derived macrophages from healthy controls and GCA patients (n = 3 each), demonstrating a knockdown efficiency of 80–95% for YKL‐40 mRNA (Supplementary Figure 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41887/abstract). This knockdown in turn led to an 80–95% reduction in YKL‐40 protein levels compared to nonbinding siRNA control (Figure 5A). Interestingly, YKL‐40 knockdown reduced MMP‐9 protein levels by 10–50% when compared to siRNA control (Figure 5B), suggesting that (autocrine/paracrine) MMP‐9 secretion by macrophages is partially dependent on YKL‐40.
Figure 5.
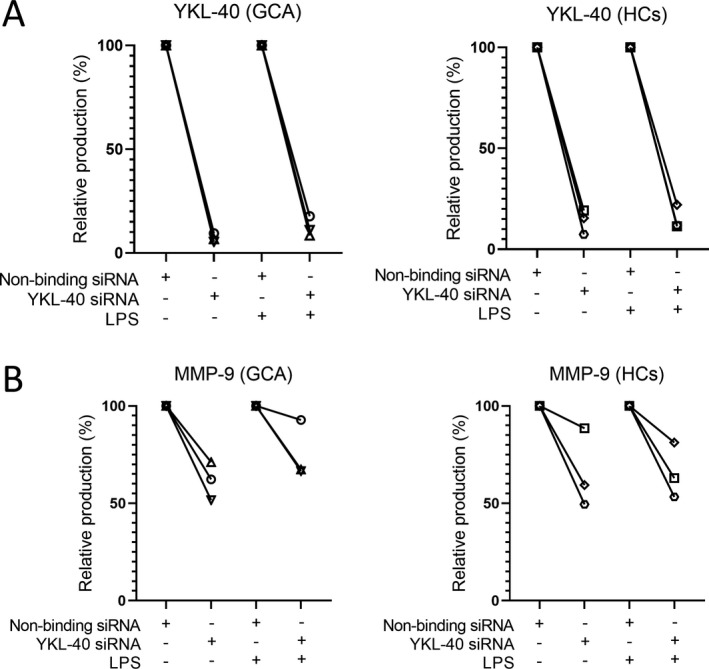
Reduction in macrophage YKL‐40 and matrix metalloproteinase 9 (MMP‐9) protein secretion upon small interfering RNA (siRNA)–mediated knockdown of YKL‐40 mRNA. A, YKL‐40 secretion in culture supernatants was reduced by 80–95% after siRNA‐mediated knockdown in macrophages from both patients with giant cell arteritis (GCA) and healthy controls (HCs), left unstimulated or stimulated with lipopolysaccharide (LPS; n = 3 per group). B, MMP‐9 secretion was reduced by 10–50% after siRNA‐mediated knockdown in macrophages from both GCA patients and healthy controls, left unstimulated or stimulated with LPS (n = 3 per group).
Promotion of endothelial tube formation by YKL‐40
YKL‐40 and VEGF likely stimulate neovascularization by interacting with their respective receptors, IL‐13Rα2 and VEGFR, both of which are abundantly expressed by HMVECs (Figure 6A). We aimed to confirm the proangiogenic potential of YKL‐40 by performing a tube formation assay, which is often used to measure the ability of endothelial cells to form capillary‐like structures. Previously, it has been demonstrated that YKL‐40 has high angiogenic potential, performing equally as well as VEGF in stimulating HMVEC tube formation (32). Indeed, we demonstrated potentiation of tube formation in the presence of YKL‐40 when compared to unstimulated HMVECs (Figures 6B and C). Moreover, the higher concentration of YKL‐40 had a stimulation potential equal to that of VEGF. Additionally, YKL‐40 and VEGF combined induced even higher tube formation. Taken together, these results further implicate YKL‐40 as playing a role in the inflammatory process in GCA by enhancing MMP‐9 production and promoting angiogenesis.
Figure 6.
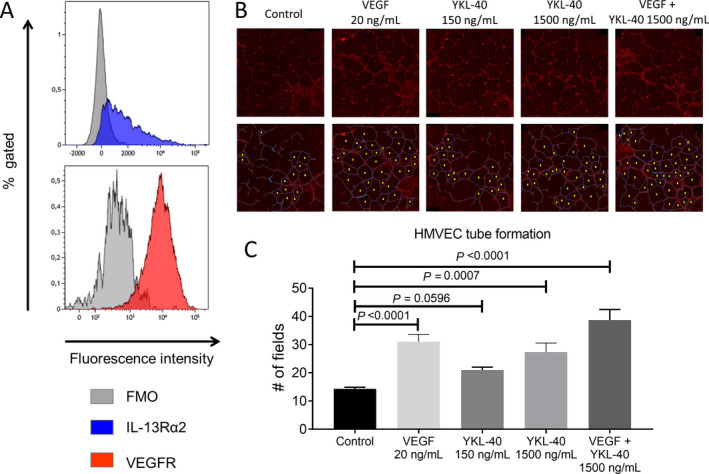
YKL‐40 promotion of human microvascular endothelial cell (HMVEC) tube formation. A, Flow cytometry staining of CD31+ HMVECs showed abundant expression of interleukin‐13 receptor α2 (IL‐13Rα2) and vascular endothelial growth factor receptor (VEGFR). Three technical replicates are shown, comparing IL‐13Rα2 and VEGFR stainings with “fluorescence minus one” (FMO) controls. B, HMVECs were treated with medium only (control) or the indicated concentrations of VEGF alone, YKL‐40 alone, or VEGF and YKL‐40. Representative images of tube formation are shown (top). Tube formation was assessed by counting the number of enclosed fields (bottom). The visible HMVEC membranes are indicated in blue, and the enclosed fields are marked with a yellow dot. C, Treatment of HMVECs with a higher concentration of YKL‐40 induced more tube formation. Bars show the mean ± SD number of enclosed fields formed by HMVEC tube formation (triplicate experiments). P values were determined by analysis of variance with Tukey’s post hoc test.
DISCUSSION
Whereas the presence of macrophages in GCA lesions is well established, their functional heterogeneity and associated role in GCA pathology is less well described. In this study, we identified a specific CD206+MMP‐9+ subset of macrophages that abundantly produces YKL‐40 in the GCA lesions. Moreover, in vitro studies revealed that YKL‐40 acts as an upstream signal that contributes to macrophage MMP‐9 production and exerts potent proangiogenic effects. Taken together, our results provide strong evidence that YKL‐40, secreted by CD206+MMP‐9+ macrophages and giant cells, mediates vascular pathology in GCA through its ability to stimulate MMP‐9 secretion and neoangiogenesis.
The presence of YKL‐40 in GCA lesions was first reported in 1999 (16), when expression of YKL‐40 by CD68+ macrophages located in the media borders was demonstrated. Our data confirm and extend those findings by revealing abundant expression of YKL‐40 predominantly by a specific CD206+MMP‐9+ subset of macrophages in the inflamed vessel walls. As recently described by our group, CD206+MMP‐9+ macrophages, likely induced by local GM‐CSF production, are mainly located in the media and its borders near sites of medial destruction (13). Hence, local GM‐CSF production is likely important in skewing this macrophage subset toward YKL‐40 production in GCA lesions as well.
Indeed, it has been reported previously that GM‐CSF–skewed macrophages derived from healthy donors produce higher levels of YKL‐40 than their M‐CSF–skewed counterparts (33). In this study, we also found higher YKL‐40 levels in GM‐CSF–skewed macrophages (compared to M‐CSF–skewed macrophages) derived from monocytes from GCA patients, but not in those from healthy controls. Although the exact reason for this apparent discrepancy is not known, it may relate to shifts in monocyte subset composition in conjunction with altered responsiveness to GM‐CSF stimulation. With aging, the proportion of classic monocytes decreases (34), whereas this subset is expanded in GCA patients (35). Since classic monocytes have the highest GM‐CSF receptor expression (13), these shifts in monocyte subset composition may underlie a heightened sensitivity to GM‐CSF skewing in GCA patients. Supporting this notion, monocyte‐derived macrophages from GCA patients also express higher levels of CD206, a marker of GM‐CSF skewing (13). Currently, the efficacy of mavrilimumab, a GM‐CSF receptor antagonist, is being evaluated for the treatment of GCA (ClinicalTrials.gov identifier: NCT03827018). The first encouraging results of this phase II trial have recently been reported, showing a 62% lower risk of flare by week 26 compared to placebo treatment. It is highly likely that mavrilimumab targets the YKL‐40–producing macrophage subset, a concept which warrants further investigation.
Our data further extend the notion that YKL‐40 is one of the upstream signals for MMP‐9 production by macrophages. Our experiments show that YKL‐40 and MMP‐9 are produced by the same subset of macrophages in GCA lesions and that knocking down YKL‐40 in in vitro differentiated macrophages substantially reduces their MMP‐9 production. Previously, YKL‐40 was found to enhance production of CCL2 and MMP‐9, while YKL‐40 gene silencing decreased the expression of these proteins in macrophages in mouse models bearing mammary tumors (15). In these mouse models, neutralization of YKL‐40 by administering chitin decreased serum YKL‐40 levels and decreased MMP‐9 production by isolated splenic T cells and macrophages. MMP‐9 is likely an important factor in the pathogenesis of GCA, not only in the context of medial destruction, but also as a mediator of T cell and monocyte invasion into the vessel wall (11). Taken together, our data implicate GM‐CSF–skewed CD206+ macrophages in GCA lesions in the production of high levels of YKL‐40 that could boost MMP‐9 expression in an autocrine or paracrine manner.
This is the first study to demonstrate that IL‐13Rα2, a confirmed receptor for YKL‐40 (24, 25), is highly expressed in GCA lesions. Previous studies have shown that the interaction of YKL‐40 with IL‐13Rα2 activates the Akt and ERK pathways, both required for MMP‐9 expression (36, 37, 38, 39). Although IL‐13Rα2 was also found to be expressed by endothelial cells, smooth muscle cells, and fibroblasts in the vessel walls of individuals without GCA, its expression was highly increased in active GCA lesions, predominantly by infiltrating leukocytes. Interestingly, we found that IL‐13Rα2 was only weakly expressed in temporal artery biopsy specimens from individuals without GCA or PMR (n = 2), indicating that up‐regulation of IL‐13Rα2 in vessel walls could be GCA/PMR specific. Further studies with more artery samples from individuals without GCA are warranted to confirm these findings.
Our data also suggest that YKL‐40 may be an important mediator of neovascularization in GCA, a process that fuels the inflammatory response. The inflamed arteries of GCA patients show an expanded vasa vasorum, extending into the media and intima layers (40). YKL‐40 may be one of the main instigators of this process, together with other proangiogenic molecules such as VEGF and angiopoietin 2 (10). Our tube formation assay confirms previous reports on the proangiogenic capacity of YKL‐40 alone and in combination with VEGF, which is also highly expressed in GCA lesions (10, 32). YKL‐40 is also produced by tumor‐associated macrophages, and likely plays a role in tumor angiogenesis and progression (14). Studies by Shao et al and Francescone et al showed increased CD31+ endothelial cell density in conjunction with increased expression of YKL‐40 in breast cancer and glioblastoma tumors, respectively (32, 41). Moreover, those studies also showed a significant reduction in CD31+ endothelial cell infiltration in tumors in mice treated with YKL‐40 siRNA and YKL‐40 neutralizing antibody. This tumor‐supportive effect is reflected by an association of YKL‐40 serum levels with a poor outcome in cancer patients. Consistent with these findings, we previously reported that high serum levels of YKL‐40 in baseline GCA patients predicted a longer time to discontinuation of GC treatment (7).
Our data suggest an involvement of YKL‐40 in tissue destruction and neoangiogenesis. YKL‐40 may signal via IL‐13Rα2, a known receptor for YKL‐40, which we found to be expressed by endothelial cells at the site of inflammation in GCA. Efforts to silence this receptor using siRNA in vitro led to reduced transcript levels, but unfortunately did not suppress cell surface protein expression, suggesting a long half‐life of this receptor (data not shown). Thus, we cannot exclude the possibility that the proangiogenic effects of YKL‐40 are mediated via another, not yet identified, receptor (32, 41). Further studies are required to definitely prove a role for the YKL‐40/IL‐13Rα2 axis in neovascularization in GCA.
Although YKL‐40 is becoming recognized as an important biomarker of inflammation, few mechanistic studies have been performed to investigate its role in autoinflammatory diseases such as GCA. In this study, we used a variety of approaches to identify the cellular source of YKL‐40 and its function. Given the substantial effects of GCs on inflammatory processes, it is important to emphasize that we used tissues and cells from treatment‐naive patients. Although our in vitro studies with cultured macrophages have elucidated YKL‐40–mediated effects on MMP‐9 production, the tissue microenvironment may interfere with these effects in more complex ways. Hence, further studies are required, in particular using in vivo or ex vivo models to improve our understanding of the role of YKL‐40 in GCA pathogenesis.
Given its implication in various pathologic pathways, YKL‐40 could be a promising target for treatment in macrophage‐driven diseases. A neutralizing antibody targeting YKL‐40 has shown potential in reducing angiogenesis and tumor progression (42). Blocking YKL‐40 or IL‐13Rα2 could also prove to be beneficial for the treatment of GCA. GCs, as well as new treatment options such as tocilizumab, may be able to temporarily repress symptoms in GCA patients (4). However, more emerging evidence has indicated that asymptomatic vessel wall inflammation persists, ultimately leading to relapses in a substantial subset of patients (6, 7, 8, 9). The persistently high YKL‐40 levels in spite of GC treatment, observed in our previous study (7) and confirmed in this study, suggest ongoing vascular inflammation and remodeling. This may be explained by the notion that YKL‐40 expression is driven by a multitude of cytokines, including IL‐6, GM‐CS, and IFNγ (17, 33). While IL‐6 signaling may be terminated specifically by treatment with tocilizumab and partially by GC treatment, it appears that GM‐CSF and IFNγ production are resistant to both drugs (12, 43, 44). Moreover, a study by Kunz et al revealed little effect of GC treatment on macrophage YKL‐40 expression (33). Taken together, these observations are consistent with the persistence of YKL‐40–mediated pathology in the vessel wall.
In conclusion, our findings show that a distinct subset of YKL‐40–producing CD206+ macrophages is a characteristic feature of the vasculopathy in GCA. These macrophages may fuel media destruction, vasa vasorum neovascularization, and leukocyte invasion into the vessel wall. This process, likely mediated by the YKL‐40/IL‐13Rα2 axis, initiates a positive forward loop of proinflammatory and tissue remodeling processes via MMP‐9 overexpression. Given its potential to promote several mechanisms involved in vessel wall injury, neutralizing YKL‐40 or its upstream pathways may prove to be an interesting treatment option for GCA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. van Sleen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
van Sleen, Jiemy, Pringle, Brouwer, Heeringa, Boots.
Acquisition of data
van Sleen, Jiemy, Pringle.
Analysis and interpretation of data
van Sleen, Jiemy, van der Geest, Abdulahad, Sandovici, Brouwer, Heeringa, Boots.
Supporting information
Supplementary Material
Supported by the Dutch Arthritis Foundation “ReumaNederland” (grant RF 14‐3‐401). Mr. Jiemy’s work was supported by a scholarship from the University Medical Center Groningen Graduate School of Medical Science and the Abel Tasman Talent Program.
Dr. van Sleen and Mr. Jiemy contributed equally to this work. Drs. Heeringa and Boots contributed equally to this work.
Dr. van der Geest has received consulting fees, speaking fees, and/or honoraria paid to his institution from Roche (less than $10,000). Dr. Brouwer has received consulting fees, speaking fees, and/or honoraria paid to her institution from Roche (less than $10,000). No other disclosures relevant to this article were reported.
REFERENCES
- 1. Samson M, Corbera‐Bellalta M, Audia S, Planas‐Rigol E, Martin L, Cid MC, et al. Recent advances in our understanding of giant cell arteritis pathogenesis [review]. Autoimmun Rev 2017;16:833–44. [DOI] [PubMed] [Google Scholar]
- 2. Gonzalez‐Gay MA, Garcia‐Porrua C, Piñeiro A, Pego‐Reigosa R, Llorca J, Hunder GG. Aortic aneurysm and dissection in patients with biopsy‐proven giant cell arteritis from northwestern Spain: a population‐based study. Medicine (Baltimore) 2004;83:335–41. [DOI] [PubMed] [Google Scholar]
- 3. Nuenninghoff DM, Hunder GG, Christianson TJ, McClelland RL, Matteson EL. Incidence and predictors of large‐artery complication (aortic aneurysm, aortic dissection, and/or large‐artery stenosis) in patients with giant cell arteritis: a population‐based study over 50 years. Arthritis Rheum 2003;48:3522–31. [DOI] [PubMed] [Google Scholar]
- 4. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant‐cell arteritis. N Engl J Med 2017;377:317–28. [DOI] [PubMed] [Google Scholar]
- 5. Schmidt WA. Ultrasound in the diagnosis and management of giant cell arteritis [review]. Rheumatology (Oxford) 2018;57 Suppl 2:ii22–31. [DOI] [PubMed] [Google Scholar]
- 6. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow‐up temporal artery biopsies in 40 treated patients. Mod Pathol 2017;30:788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Sleen Y, Sandovici M, Abdulahad W, Bijzet J, van der Geest K, Boots AM, et al. Markers of angiogenesis and macrophage products for predicting disease course and monitoring vascular inflammation in giant cell arteritis. Rheumatology (Oxford) 2019;58:1383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gloor AD, Yerly D, Adler S, Reichenbach S, Kuchen S, Seitz M, et al. Immuno‐monitoring reveals an extended subclinical disease activity in tocilizumab‐treated giant cell arteritis. Rheumatology (Oxford) 2018;57:1795–801. [DOI] [PubMed] [Google Scholar]
- 9. Reichenbach S, Adler S, Bonel H, Cullmann JL, Kuchen S, Bütikofer L, et al. Magnetic resonance angiography in giant cell arteritis: results of a randomized controlled trial of tocilizumab in giant cell arteritis. Rheumatology (Oxford) 2018;57:982–6. [DOI] [PubMed] [Google Scholar]
- 10. Kaiser M, Younge B, Björnsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis: production of angiogenic cytokines by multinucleated giant cells. Am J Pathol 1999;155:765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. MMP (matrix metalloprotease)‐9‐producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res 2018;31;123:700–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cid M, Muralidharan S, Corbera‐Bellalta M, Espigol‐Frigole G, Hernandez JM, Denuc A, et al. Fri0010 GM‐CSFR pathway is implicated in pathogenic inflammatory mechanisms in giant cell arteritis. Ann Rheum Dis 2020;79:576. [Google Scholar]
- 13. Jiemy WF, van Sleen Y, van der Geest KS, Ten Berge HA, Abdulahad WH, Sandovici M, et al. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony‐stimulating factor (GM‐CSF) and macrophage colony‐stimulating factor (M‐CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin Transl Immunology 2020;27;9:e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Libreros S, Iragavarapu‐Charyulu V. YKL‐40/CHI3L1 drives inflammation on the road of tumor progression [review]. J Leukoc Biol 2015;98:931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Libreros S, Garcia‐Areas R, Shibata Y, Carrio R, Torroella‐Kouri M, Iragavarapu‐Charyulu V. Induction of proinflammatory mediators by CHI3L1 is reduced by chitin treatment: decreased tumor metastasis in a breast cancer model. Int J Cancer 2012;15;131:377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johansen JS, Baslund B, Garbarsch C, Hansen M, Stoltenberg M, Lorenzen I, et al. YKL‐40 in giant cells and macrophages from patients with giant cell arteritis. Arthritis Rheum 1999;42:2624–30. [DOI] [PubMed] [Google Scholar]
- 17. Kzhyshkowska J, Mamidi S, Gratchev A, Kremmer E, Schmuttermaier C, Krusell L, et al. Novel stabilin‐1 interacting chitinase‐like protein (SI‐CLP) is up‐regulated in alternatively activated macrophages and secreted via lysosomal pathway. Blood 2006;15;107:3221–8. [DOI] [PubMed] [Google Scholar]
- 18. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Glucocorticoid dosages and acute‐phase reactant levels at giant cell arteritis flare in a randomized trial of tocilizumab. Arthritis Rheumatol 2019;71:1329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fichtner‐Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL‐13 signaling through the IL‐13α 2 receptor is involved in induction of TGF‐β 1 production and fibrosis. Nat Med 2006;12:99–106. [DOI] [PubMed] [Google Scholar]
- 20. Lupardus PJ, Birnbaum ME, Garcia KC. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL‐13 and IL‐13Rα2. Structure 2010;18:332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J, Li Y, Andiappan A, Yan Y, Tan K, Ong H, et al. Role of IL‐13Rα2 in modulating IL‐13‐induced MUC 5 AC and ciliary changes in healthy and CRS w NP mucosa. Allergy 2018;73:1673–85. [DOI] [PubMed] [Google Scholar]
- 22. Bhardwaj R, Suzuki A, Leland P, Joshi BH, Puri RK. Identification of a novel role of IL‐13Rα2 in human Glioblastoma multiforme: interleukin‐13 mediates signal transduction through AP‐1 pathway. J Transl Med 2018;20;16:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roy S, Liu H, Jaeson MI, Deimel LP, Ranasinghe C. Unique IL‐13Rα2/STAT3 mediated IL‐13 regulation detected in lung conventional dendritic cells, 24 h post viral vector vaccination. Sci Rep 2020;10:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He CH, Lee CG, Cruz CS, Lee C, Zhou Y, Ahangari F, et al. Chitinase 3‐like 1 regulates cellular and tissue responses via IL‐13 receptor α2. Cell Rep 2013;4:830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu N, Bo Q, Shao R, Liang J, Zhai Y, Yang S, et al. Chitinase‐3‐like‐1 promotes M2 macrophage differentiation and induces choroidal neovascularization in neovascular age‐related macular degeneration. Invest Ophthalmol Vis Sci 2019;60:4596–605. [DOI] [PubMed] [Google Scholar]
- 26. Van der Geest KS, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol 2014;66:1927–38. [DOI] [PubMed] [Google Scholar]
- 27. Samaniego R, Palacios BS, Domiguez‐Soto Á, Vidal C, Salas A, Matsuyama T, et al. Macrophage uptake and accumulation of folates are polarization‐dependent in vitro and in vivo and are regulated by activin A. J Leukoc Biol 2014;95:797–808. [DOI] [PubMed] [Google Scholar]
- 28. Lukic A, Larssen P, Fauland A, Samuelsson B, Wheelock CE, Gabrielsson S, et al. GM‐CSF–and M‐CSF–primed macrophages present similar resolving but distinct inflammatory lipid mediator signatures. FASEB J 2017;31:4370–81. [DOI] [PubMed] [Google Scholar]
- 29. Paquin‐Proulx D, Greenspun BC, Pasquet L, Strunz B, Aleman S, Falconer K, et al. IL13Rα2 expression identifies tissue‐resident IL‐22‐producing PLZF innate T cells in the human liver. Eur J Immunol 2018;48:1329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Badalyan V, Thompson R, Addo K, Borthwick LA, Fisher AJ, Ort T, et al. TNF‐α/IL‐17 synergy inhibits IL‐13 bioactivity via IL‐13Rα2 induction. J Allergy Clin Immunol 2014;134:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hecker M, Zasłona Z, Kwapiszewska G, Niess G, Zakrzewicz A, Hergenreider E, et al. Dysregulation of the IL‐13 receptor system: a novel pathomechanism in pulmonary arterial hypertension. Am J Respir Crit Care Med 2010;182:805–18. [DOI] [PubMed] [Google Scholar]
- 32. Shao R, Hamel K, Petersen L, Cao Q, Arenas RB, Bigelow C, et al. YKL‐40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene 2009;28:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunz LI, van't Wout EF, van Schadewijk A, Postma DS, Kerstjens HA, Sterk PJ, et al. Regulation of YKL‐40 expression by corticosteroids: effect on pro‐inflammatory macrophages in vitro and its modulation in COPD in vivo. Respir Res 2015;16:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F. Age‐dependent alterations of monocyte subsets and monocyte‐related chemokine pathways in healthy adults. BMC Immunol 2010;11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Sleen Y, Wang Q, van der Geest KS, Westra J, Abdulahad WH, Heeringa P, et al. Involvement of monocyte subsets in the immunopathology of giant cell arteritis. Sci Rep 2017;7:6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruhul Amin A, Senga T, Oo ML, Thant AA, Hamaguchi M. Secretion of matrix metalloproteinase‐9 by the proinflammatory cytokine, IL‐1β: a role for the dual signalling pathways, Akt and Erk. Genes Cells 2003;8:515–23. [DOI] [PubMed] [Google Scholar]
- 37. Lee SJ, Lee YS, Seo KW, Bae JU, Kim GH, Park SY, et al. Homocysteine enhances MMP‐9 production in murine macrophages via ERK and Akt signaling pathways. Toxicol Appl Pharmacol 2012;260:89–94. [DOI] [PubMed] [Google Scholar]
- 38. Shim Y, Kang B, Jeon H, Park I, Lee K, Lee I, et al. Clusterin induces matrix metalloproteinase‐9 expression via ERK1/2 and PI3K/Akt/NF‐κB pathways in monocytes/macrophages. J Leukoc Biol 2011;90:761–9. [DOI] [PubMed] [Google Scholar]
- 39. Lee Y, Tran HT, Van Ta Q. Regulation of expression of matrix metalloproteinase‐9 by JNK in Raw 264.7 cells: presence of inhibitory factor (s) suppressing MMP‐9 induction in serum and conditioned media. Exp Mol Med 2009;41:259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cid MC, Hernández‐Rodríguez J, Esteban M, Cebrián M, Gho YS, Font C, et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant‐cell arteritis. Circulation 2002;106:1664–71. [DOI] [PubMed] [Google Scholar]
- 41. Francescone RA, Scully S, Faibish M, Taylor SL, Oh D, Moral L, et al. Role of YKL‐40 in the angiogenesis, radioresistance, and progression of glioblastoma. J Biol Chem 2011;286:15332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Faibish M, Francescone R, Bentley B, Yan W, Shao R. A YKL‐40–neutralizing antibody blocks tumor angiogenesis and progression: a potential therapeutic agent in cancers. Mol Cancer Ther 2011;10:742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med 1994;121:484–91. [DOI] [PubMed] [Google Scholar]
- 44. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T‐cell responses in giant cell arteritis. Circulation 2010;23;121:906–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material