

Abstract
The advent of checkpoint immunotherapy, particularly with programmed death‐1 (PD‐1) and programmed death‐ligand 1 (PD‐L1) inhibitors, has provided ground‐breaking results in several advanced cancers. Substantial efforts are being made to extend these promising therapies to other refractory cancers such as gliomas, especially glioblastoma, which represents the most frequent and malignant glioma and carries an exceptionally grim prognosis. Thus, there is a need for new therapeutic strategies with related biomarkers. Gliomas have a profoundly immunosuppressive tumour micro‐environment and evade immunological destruction by several mechanisms, one being the expression of inhibitory immune checkpoint molecules such as PD‐L1. PD‐L1 is recognised as an important therapeutic target and its expression has been shown to hold prognostic value in different cancers. Several clinical trials have been launched and some already completed, but PD‐1/PD‐L1 inhibitors have yet to show convincing clinical efficacy in gliomas. Part of the explanation may reside in the vast molecular heterogeneity of gliomas and a complex interplay within the tumour micro‐environment. In parallel, critical knowledge about PD‐L1 expression is beginning to accumulate including knowledge on expression levels, testing methodology, co‐expression with other checkpoint molecules and prognostic and predictive value. This article reviews these aspects and points out areas where biomarker research is needed to develop more successful checkpoint‐related therapeutic strategies in gliomas.
Keywords: biomarker, checkpoint inhibition, glioma, glioblastoma multiforme, immunotherapy, PD‐L1, prognosis, programmed death‐ligand 1
PD‐L1 is a critical immune checkpoint in several malignancies and has demonstrated potential as a prognostic biomarker and a therapeutic target. In this review, we provide a comprehensive overview of PD‐L1 expression in gliomas with a particular focus on the tumour micro‐environment, testing methodology, co‐expression with other checkpoint molecules and the prognostic and predictive significance. Furthermore, we present clinical trial data on PD‐1/PD‐L1 inhibitors in glioma patients and discuss the future of PD‐L1 as a biomarker in checkpoint‐related therapeutic strategies.
Abbreviations
- BTLA
B and T lymphocyte attenuator
- CD
Cluster of differentiation
- CNS
Central nervous system
- CTLA‐4
Cytotoxic T‐lymphocyte associated antigen‐4
- dMMR
Mismatch repair deficiency
- DNA
Deoxyribonucleic acid
- HR
Hazard ratio
- IDH
Isocitrate dehydrogenase
- KPS
Karnofsky performance status
- LAG‐3
Lymphocyte activation gene 3 protein
- mAbs
Monoclonal antibodies
- MGMT
O6‐methylguanine‐DNA methyltransferase
- MSI
Microsatellite instability
- NK cells
Natural killer cells
- NSCLC
Non‐small cell lung cancer
- NY‐ESO‐1
New York oesophageal squamous cell carcinoma 1
- OR
Odds ratio
- ORR
Objective response rate
- OS
Overall survival
- PD‐1
Programmed death‐1
- PD‐L1
Programmed death‐ligand 1
- PD‐L2
Programmed death‐ligand 2
- PFS
Progression‐free survival
- RNA
Ribonucleic acid
- sPD‐L1
Soluble programmed death‐ligand 1
- TAMs
Tumour‐associated microglia/macrophages
- TCGA
The Cancer Genome Atlas
- TILs
Tumour‐infiltrating lymphocytes
- TIM‐3
T‐cell immunoglobulin and mucin‐domain containing protein 3
- TMAs
Tissue microarrays
- TMB
Tumour mutational burden
- TME
Tumour micro‐environment
- TRAEs
Treatment‐related adverse events
Key points
PD‐L1 is overexpressed in gliomas and seems to be associated with factors such as glioma grade and molecular subtype, but PD‐L1 expression has not demonstrated prognostic or predictive significance as a potential biomarker in gliomas.
Although clinical trials with PD‐1/PD‐L1 inhibitors have not yet shown significant clinical efficacy in glioma patients and there are no approved PD‐1/PD‐L1 inhibitors for therapeutic use in gliomas, trial data indicate potential efficacy in certain highly selected subgroups of patients.
A multi‐combinatorial approach with a particular focus on the lack in T‐cell infiltration and accumulation of immunosuppressive cells such as microglia/macrophages may be necessary to overcome the profoundly immunosuppressive tumour micro‐environment in gliomas.
Combined biomarker strategies with PD‐L1 in combination with other biomarkers reflecting either immune phenotype or tumour genotype are most likely needed to obtain successful PD‐1/PD‐L1 immune checkpoint‐related therapies.
INTRODUCTION
Gliomas are the most common primary brain tumours in adults, and glioblastoma multiforme constitutes the most malignant glioma, categorised as a World Health Organization grade IV tumour [1]. Standard of care for newly diagnosed glioblastoma is a multimodal regimen comprising maximal surgical resection, radiation therapy and chemotherapy [2, 3, 4, 5]. Unfortunately, the disease almost inevitably relapses due to its aggressive, diffusely infiltrative behaviour and inherent treatment resistance [5, 6]. In recurrent glioblastoma, treatment options are very limited and have minimal efficacy [4]. Despite advances made in the last decades, extensive research efforts have not translated into significant improvements in the prognosis, since the introduction of temozolomide in 2005 [2, 3, 4, 5]. Novel and more efficacious therapies for primary and recurrent disease are therefore urgently needed.
The interplay between cancer and the immune system is a hallmark of cancer biology [7]. A salient feature of gliomas is the ability to solicit profound local and systemic immunosuppression within the tumour micro‐environment (TME), thereby abolishing antitumoural immune defence [8, 9]. This has led to the advent of immunotherapy [9, 10, 11, 12]. While chemotherapy and targeted therapy seek to target tumour cells directly and ameliorate underlying signalling defects in tumour cells, immunotherapy fundamentally differs by modulating the native immune system, with the purpose of tilting immune balance from a pro‐tumourigenic towards an anti‐tumourigenic state, in order to mount an effective antitumour response [13]. In an era of personalised and precision medicine, attention is increasingly shifting from conventional cytotoxic and targeted therapy towards immunotherapy, which has emerged as a novel treatment paradigm [9, 14, 15, 16, 17]. In the realm of various immunotherapeutic strategies, the concept of immune checkpoint inhibition has attracted the most attention in recent years. In particular, therapeutic targeting of the immune checkpoints cytotoxic T‐lymphocyte associated antigen‐4 (CTLA‐4), programmed death‐1 (PD‐1, CD279) and programmed death‐ligand 1 (PD‐L1, B7‐H1) has been revolutionary [18, 19, 20, 21, 22].
Immune checkpoints are co‐inhibitory or co‐stimulatory molecular immune system modulators that normally ensure appropriate tolerance to self‐antigens and physiological immune homeostasis while minimising tissue damage and precluding autoimmunity [22, 23, 24, 25]. PD‐1 is a key immune inhibitory receptor predominantly located on the surface of activated CD3+/CD8+ T‐cells, B‐cells, natural killer (NK) cells and monocytes [23, 24, 25, 26]. PD‐1 primarily functions peripherally by negatively regulating T‐cell mediated signalling upon ligation to its transmembrane ligands, PD‐L1 and programmed death‐ligand 2 (PD‐L2, B7‐DC) [23, 24, 25]. (Figure 1). Under physiological, non‐inflammatory conditions, PD‐L1 is expressed in antigen‐presenting cells and some non‐lymphoid tissues [23, 24, 25, 26, 27]. Engagement of PD‐L1 to PD‐1 on T‐cells attenuates effector T‐cell activity by inducing exhaustion, anergy and apoptosis of activated CD8+ cytotoxic T‐cells, inhibiting proinflammatory cytokine production and recruiting CD4+CD25+FoxP3+ regulatory T‐cells, a potently immunosuppressive effector T‐cell subset [23, 24, 25, 26, 27, 28]. A multitude of cancers have the ability to harness and co‐opt the PD‐1/PD‐L1 pathway as a maladaptive shield by upregulating PD‐L1 expression to escape immune surveillance and eradication, favouring tumour growth [26, 27, 28, 29]. This constitutes one of multiple operational mechanisms of tumour immune evasion [29].
FIGURE 1.
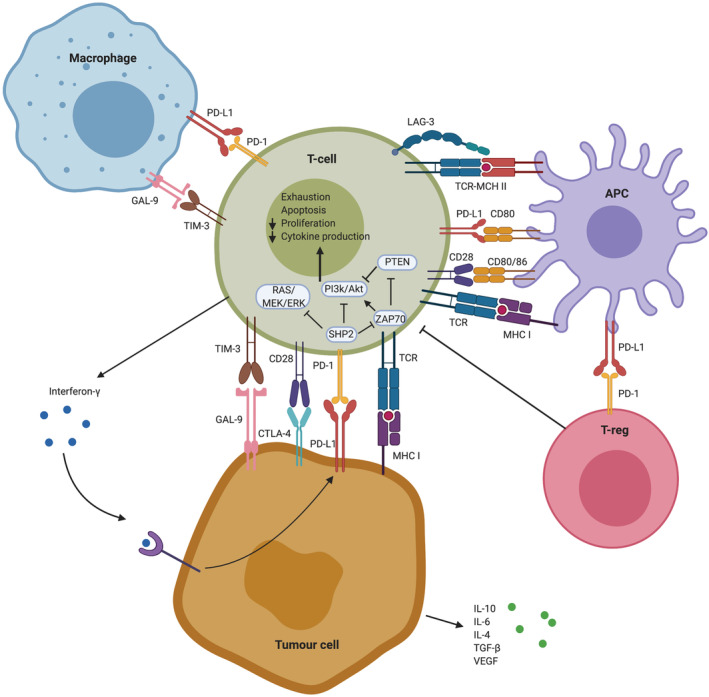
Mechanisms of immune evasion mediated by the PD‐1/PD‐L1 signalling pathway. PD‐1/PD‐L1 interaction in the immunosuppressive tumour micro‐environment of gliomas leads to decreased proliferation and exhaustion of effector T‐cells as well as decreased cytokine production, providing the tumour cell with survival benefits. Blocking the PD‐1/PD‐L1 pathway with anti‐PD‐1 or anti‐PD‐L1 monoclonal antibodies leads to differentiation of effector T‐cells by stimulating the PI3K/Akt or Ras/MAPK pathways and suppression of T‐reg differentiation, which ultimately enhances anti‐tumour immunity. Abbreviations: PD‐L1 Programmed death‐ligand 1, PD‐1 Programmed death‐1, GAL‐9 Galectin 9, TIM‐3 T‐cell immunoglobulin mucin‐3, LAG‐3 Lymphocyte‐activation gene 3, TCR T‐cell receptor, MHC Major histocompatibility complex, APC Antigen presenting cell, CD Cluster of differentiation, T‐reg Regulatory T‐cell, IL Interleukin, VEGF Vascular endothelial growth factor, TGF Transforming growth factor. Created with BioRender.com
PD‐1/PD‐L1 inhibitors are therapeutic monoclonal antibodies (mAbs) that antagonise PD‐1 or PD‐L1 to reverse immunosuppression and restore antitumour immunity [30]. PD‐1/PD‐L1 inhibitors in non‐small cell lung cancer (NSCLC) [31] and melanoma [32] have altered the prognostic landscape, demonstrating remarkable clinical efficacy with the prospect of durable remission alongside acceptable toxicity. This has led to clinical implementation of PD‐1/PD‐L1 inhibitors in various cancers assuming an integral, frontline position next to conventional therapies [18, 19, 20, 21, 22]. Great efforts are therefore being made to extend these therapies to refractory cancers including gliomas [10, 12]. However, to apply personalised immunotherapy successfully in clinical practice, accurate biomarkers are needed in therapeutic decision‐making and prognosis stratification, and PD‐L1 assessed by immunohistochemistry has shown compelling potential as a biomarker in some cancers [33, 34, 35, 36]. Understanding the role of PD‐L1 and similar co‐expressed markers in gliomas may lead to new successful therapeutic strategies.
Herein we summarise current evidence on PD‐L1 expression in gliomas, including expression levels, differential expression on various cell types, co‐expression with other immune checkpoint molecules, and technical caveats and challenges related to PD‐L1 immunohistochemistry. Furthermore, the prognostic and predictive potential of PD‐L1 as a biomarker is reviewed. Last, the future of biomarker‐based immune checkpoint inhibitor strategies in gliomas is discussed.
THE ROLE OF THE PD‐1/PD‐L1 PATHWAY IN THE BRAIN
Though there is still much to be learned, neurodegenerative and neuroinflammatory animal models have provided invaluable insights into the role of PD‐1/PD‐L1 in the normal brain. Thus, it is recognised that neurons, microglia/macrophages, astrocytes, oligodendrocytes and endothelial cells in the brain can express PD‐L1 under normal conditions [37, 38, 39]. During neuroinflammatory conditions, PD‐L1 is upregulated in activated resident glial cells due to cytokine release. Through the upregulation of PD‐L1, they govern infiltrating T‐cell activity and diminish T‐cell responses, thereby confining and limiting detrimental CNS damage [38, 39, 40]. PD‐L1 deficient mice exposed to coronavirus demonstrated an increased inflammatory response resulting in more rapid clearance of the virus, but with the costs of earlier symptom onset and increased morbidity [41]. Genetic ablation of PD‐L1 in T‐cells, in addition to increased incidence and aggravated disease, caused local endothelial dysfunction in a spontaneous mouse model of CNS autoimmunity [42]. These data point towards a critical immune modulatory checkpoint role for PD‐L1 in multiple aspects of brain function.
Knowledge of PD‐L1 expression in various CNS compartments is exceedingly sparse. Interestingly though, there have been reports on soluble PD‐L1 (sPD‐L1), which is believed to be released from PD‐L1 positive cells [43]. sPD‐L1 is reported to contribute to systemic immunosuppression and is proposed as a potential minimally invasive biomarker. In gliomas, elevated sPD‐L1 levels have been reported in peripheral blood and cerebrospinal fluid and associated with aggressive behaviour [43, 44].
PD‐L1 EXPRESSION IN GLIOMAS
Frequency and distribution of PD‐L1 expression
Expression of PD‐L1 in tumour cells has been consistently documented in cancers such as NSCLC, melanoma, renal cell carcinoma, urothelial carcinoma and colorectal cancer [18, 19, 20, 33, 36]. In gliomas, preclinical models have demonstrated PD‐L1 expression [45, 46, 47, 48, 49, 50]. Table 1 provides a comprehensive overview of studies evaluating PD‐L1 protein and/or gene expression in human gliomas [24, 44, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68]. These studies reported highly variable tumour cell PD‐L1 expression rates in gliomas ranging from 6.1%–88% (Table 1). In 2003, Wintterle et al. [24] provided the first evidence of PD‐L1 protein expression in malignant glioma specimens in 10 out of 10 patients (nine glioblastomas and one mixed glioma, which is not further specified in the article) using immunohistochemistry. However, neither staining patterns nor scoring cut‐offs were specified. A study by Berghoff et al. [55] reported diffuse/fibrillary PD‐L1 expression in 88% and membranous PD‐L1 expression in 37.6% of newly diagnosed glioblastoma patients using full histological slides in contrast to other studies in Table 1 using tumour microarrays (TMAs). Interestingly, the two abovementioned studies both utilised the 5H1 clone and reported some of the highest expression rates among all the studies in Table 1. 5H1 is a non‐commercial antibody clone and was one of the most frequently used clones among the studies. In comparison, two commercial antibody clones, SP142 and SP263, gave much lower expression frequencies. A microarray study using SP263 reported a rate of membranous PD‐L1 protein expression of 23.4% in diffuse gliomas with a 5% threshold [68]. Garber et al. [57] conducted a study comprising 347 patients with gliomas of all grades and found a 6.1% expression rate using the SP142 clone, but positivity was confined to glioblastomas only. Another study reported similarly low rates using SP142 [65].
TABLE 1.
Characterisation of studies evaluating PD‐L1 expression and/or the prognostic significance of PD‐L1 expression in gliomas
| Reference (year) | Number of patients/diagnosis (WHO grade) | Tissue preparation/sample size | Assay | PD‐L1 antibody clone | Evaluated tumoral PD‐L1 staining pattern | PD‐L1 positivity cut‐off | Tumoral PD‐L1 expression rate (%) | Prognostic impact of tumoral PD‐L1 expression | Other findings |
|---|---|---|---|---|---|---|---|---|---|
| Wintterle et al. (2003) [24] | 10/glioma (III–IV) | Frozen section/full slide | IHC | 5H1 | NR | NR, positivity semiquanti tatively 5%, 25%–50%, 50%–75%, >75% | NR | NR |
Constitutive mRNA PD‐L1 expression in glioma cell lines >50% of tumour cells expressed PD‐L1 in all specimens |
| Wilmotte et al. (2005) [51] | 54/glioma (II–IV) | Frozen section/ full slide | IHC | MIH1 | Membranous/cytoplasmic |
NR, positivity semiquantitatively assessed: >50%, 30%–50%, 10%–30%, 1%–10%, 0% |
NR | NR | Increased PD‐L1 expression in HGG compared to LGG |
| Yao et al.(2009) [52] | 48/astrocytic tumour (I–IV) | Frozen section/full slide | IHC | MIH1 | Membranous, cytoplasmic | NR | NR | NR |
Increased PD‐L1 expression in HGG compared to LGG. Increased PD‐L1 expression in tumour edge compared to tumour core PD‐L1 expression in TILs |
| Fresh tissue | Western blot | Anti‐B7‐H1 | Membranous, cytoplasmic | NR | NR | NR | |||
| Liu et al. (2013) [53] | 17/glioma (III–IV) | Frozen section/full slide |
Immune‐flourescence histochemistry |
Mouse anti‐human PD‐L1 | NR | NR | NR | Adverse OS |
TABT neuronal PD‐L1 expression associated with prolonged survival |
| Baral et al. (2014) [44] | 78/glioma | Frozen section/full slide | IHC | MIH1 | NR | NR | NR | NR |
Increased PD‐L1 expression in HGG compared to LGG |
| Berghoff et al. (2015) [55] | 117 (including 18 matched local recurrences)/ND GBM(IV) | FFPE/full slide |
IHC |
5H1 |
Membranous |
>5% |
ND: 37.6% RC: 16.7% |
No impact on OS No impact on OS |
Neuronal PD‐L1 expression not associated with survival Increased PD‐L1 expression in mesenchymal GBM compared to other subtypes |
|
Diffuse/ fibrillary |
NR, semiquantitatively assessed: none, <25%, 25%–50%, 50%–75%, >75% |
ND: 88% RC: 72.2% |
|||||||
| 446/GBM (IV) | TCGA/gene | Agilent microarray | — | — | NR | NR | |||
| Ndoum et al. (2016) [56] | 94 GBM (IV) | FFPE/TMA |
IHC, flow cytometry |
ERP1161 (2) |
Membranous (digital quantification) |
≥1% ≥5% ≥25% ≥50% |
60.6% 38.3% 17% 5.32% |
Adverse OS (5% cut‐off) |
PD‐L1 expression in TILs in 26.8% of patients |
| 149/GBM (IV) | TCGA/gene | Illumina RNAseq | — | — |
0.37 |
NR |
Adverse OS (HR = 1.52, 95% CI 1.03–2.25, 0.0343) |
‐ |
|
| Garber et al. (2016) [57] | 345/ glioma (I–IV) | FFPE/full slide | IHC | SP142 | Membranous (moderate‐strong staining intensity) | >5% |
6.1% (grade IV gliomas only) |
NR |
PD‐L1 expression correlated with grade IV gliomas |
| Zeng et al. (2016) [58] | 229/ glioma (I–IV) | FFPE/TMA | IHC | Rabbit poly‐clonal anti‐PD‐L1 | Membranous, cytoplasmic | >5% |
51.1% |
No impact on OS |
PD‐L1 expression did not correlate with malignancy grade |
| Wang et al. (2016) [59] | 301/glioma (II–IV) | CGGA/gene | Agilent microarray | — | — | NR | NR | Adverse OS |
Increased PD‐L1 expression in GBM compared to grade II and III Increased PD‐L1 expression in IDH‐ wt compared to IDH‐mutant GBM Increased PD‐L1 expression in mesenchymal GBM compared to other subtypes |
| 675/glioma (II–IV) | TCGA/gene |
Illumina RNAseq |
— | — |
NR |
NR |
Adverse OS |
||
| Miyazaki et al. (2017) [60] | 16/GBM (IV) | FFPE/full slide |
IHC |
28‐8 |
Membranous |
NR, semiquantitatively assessed: Absent staining, <25%, 25–50% and >50% |
NR |
No impact on OS |
No difference in PD‐L1 expression between initially and secondary resected tumours |
| Heiland et al. (2017) [61] | 48/GBM (IV) | FFPE/full slide | IHC | E1LRN |
NR (digital quantification) |
NR | NR |
No impact on OS (HR = 0.94, P>0.05) |
Increased PD‐L1 expression in GBM compared to LGG. Increased PD‐L1 expression in mesenchymal GBM compared to other subtypes. Increased PD‐L1 expression in IDH1/2 wt glioma |
| NR/glioma (I–IV) | TCGA/gene | Agilent array, Illumina RNeq | — | — | NR | NR |
No impact on OS (HR = 0.98, P>0.05) |
||
| Xue et al. (2017) [62] | 64/glioma (I–IV) | FFPE/full slide | IHC | Ab 58810 | Membranous, cytoplasmic (digital quantification) | >5% | 78.12% | NR | Increased PD‐L1 expression in HGG compared to LGG |
| Heynckes et al. (2017) [63] | 64 (38 matched samples with de‐novo and recurrent disease)/GBM (IV) | FFPE/full slide | IHC immunofluorescence | E1LRN | Membranous, diffuse/fibrillary, cytoplasmic | NR | NR | NR | Reduced PD‐L1 expression in first recurrent GBM compared to de‐novo GBM |
| 874/GBM (IV) | 6 publicly available databases/gene | NR | — | — | NR | NR | NR | ||
| Berghoff et al. (2017) [64] | 174/glioma (II–IV) | FFPE/full slide | IHC | 5H1 |
Membranous (strong intensity) Diffuse/fibrillary |
≥1% >25% |
IDH‐wt: 56.2% IDH‐mut: 5.7% IDH‐wt: 56.2% IDH‐mut: 5.7% |
NR | IDH‐wt gliomas have more prominent TIL infiltration |
| 677/glioma (II‐–IV) | TCGA/gene | RNAseq | — | — | NR | NR | NR | ||
|
Hodges et al. (2017) [65] |
310/glioma (I–IV) | FFPE/full slide | IHC | SP142 | Membranous (moderate‐strong staining intensity) | >5% | 7.7% | NR |
PD‐L1 expression does not correlate with TML or PD‐1/PD‐L1 expression |
| Han et al. (2017) [66] | 54/GBM (IV) | FFPE/TMA | IHC | Anti‐PD‐L1 antibody | Membranous, cytoplasmic (any staining intensity) | ≥5% | 31.5% | Adverse OS (HR 4.958, 95% CI, 1.557–15.79 P = 0.007) |
PD‐L1+/PD‐1+ TIMC low group had poorer OS compared to the three other groups PD‐L1 expression in TILs in 0.04% of patients |
| Lee et al. (2018) [67] | 115/ND GBM (IV) | FFPE/TMA | IHC | E1L3N | Membranous, fibrillary (any staining intensity) | >5% | 32.3% |
No impact on OS (HR 1.204, P = 0.615) |
PD‐L1 expression in immune cells in 5.2% of patients |
| Pratt et al. (2018) [69] | 183/glioma (II–IV) | FFPE/TMA | IHC | SP263 | Membranous (digital quantification) | ≥5% | 23.4% | Adverse OS in GBM, which remained significant in subgroup analysis of recurrent glioblastoma, IDH‐wild type | PD‐L1 expression in TILs |
| 444/GBM (IV) | TCGA/gene | Agilent array | — | — | Median | NR | Adverse OS in non‐C‐GIMP glioblastoma | ||
| Knudsen et al. (2021) [54] | 163/GBM | FFPE/Full slide | IHC | EPR19759 | Membranous (digital quantification) | NR | NR, but mean membrane fraction 0.049% (0.002–0.14) | No impact on OS (HR 1.05, 95%CI, 0.8–1.5, P = 0.8) |
All tumours expressed PD‐L1 and Galectin‐3 with a positive correlation No prognostic value of PD‐L1 and Galectin‐3 separately or combined |
Abbreviations: IHC Immunohistochemistry, NR Not reported, HGG High‐grade glioma, LGG Low‐grade glioma, OS Overall survival, TABT Tumour‐associated brain tissue, ND Newly diagnosed, GBM Glioblastoma multiforme, FFPE Formalin fixed paraffin embedded, TCGA The Cancer Genome Atlas, CGGA Chinese Glioma Genome Atlas, RC Recurrent, TMA Tissue microarray, HR Hazard ratio, wt Wildtype, mut Mutated, TIMC Tumour infiltrating mononuclear cells, IDH Isocitrate dehydrogenase, TML Tumour mutational load, RISH RNA in situ hybridisation, TILs Tumour infiltrating lymphocytes.
PD‐L1 expression has primarily been studied in newly diagnosed gliomas. However, some studies have compared PD‐L1 expression in primary and recurrent disease and have reached differing conclusions. Heynckes et al. [63] reported significant reductions in PD‐L1 gene and protein expression in recurrent glioblastoma compared to de novo glioblastoma. In a sub‐cohort of 18 patients with recurrent glioblastoma, Berghoff et al. [55] found membranous PD‐L1 protein expression to be more frequent in newly diagnosed glioblastoma than matched recurrent glioblastoma specimens, while no such difference was detected for diffuse/fibrillary PD‐L1 expression. Meanwhile, in a small cohort (n = 16), Miyazaki et al. [60] did not observe reductions in the frequency of PD‐L1 expression in recurrent glioblastoma compared to the initial tumour. However, some patients had received immunotherapy prior to recurrence, potentially introducing biases.
It is noticeable that while some of the studies in Table 1 report high PD‐L1 protein expression in gliomas, PD‐L1 expression at the mRNA level in gliomas in The Cancer Genome Atlas (TCGA) is one of the lowest median levels across various cancers [69]. Chen et al. [70] examined PD‐L1 protein expression in different cancers and found glioblastoma to have a moderate frequency of PD‐L1 protein expression when positivity was defined as ≥ 1% of tumours cells being positive. It is well‐known that correlation between mRNA and protein expression can be low. The discrepant expression levels could be explained by differential post‐transcriptional modifications in different cancers combined with the limited samples sizes in the protein studies, compared to the large TCGA datasets.
Yao et al. [52] made an interesting discovery of significantly upregulated expression of PD‐L1 at the tumour edge compared to the tumour core. Such findings prompt concerns over heterogeneity of PD‐L1 expression. Spatial intratumoural expression heterogeneity is a well‐known phenomenon in PD‐L1 immunohistochemistry and represents a sampling bias in small tissue materials [71, 72]. Therefore, biopsies compared to resection specimens, and TMAs compared to full histological slides, may be more prone to sampling bias and ultimately erroneous PD‐L1 classification [71, 72, 73].
A threshold for PD‐L1 positivity on immunohistochemistry has not been established yet in gliomas [68]. In the studies listed in Table 1, the cut‐offs applied to categorise a sample as positive ranged from ≥1% to ≥25% of tumour cells positive for PD‐L1, albeit several studies did not sufficiently describe their cut‐off thresholds [24, 44, 51, 52, 53, 54, 59, 60, 61, 63].
The most commonly evaluated staining pattern was membranous staining compared to diffuse/fibrillary and cytoplasmic staining. In general, there is a tendency towards higher PD‐L1 expression rates in studies evaluating diffuse/fibrillary staining compared to those using membranous staining. A prevailing theory is that diffuse/fibrillary staining reflects PD‐L1 expression in the pathognomic neurofibrillary matrix of gliomas, a histomorphological characteristic not seen in other cancers [55, 73]. The biological significance of the differing staining patterns remains unresolved, though some state that only membranous PD‐L1 expression has biological pertinence [34, 73].
Correlation of PD‐L1 expression to grade and molecular subtype
Several studies have demonstrated a significant correlation between tumoural PD‐L1 expression and glioma grade [44, 51, 52, 57, 59, 61, 62]. A large study comprising a total of 976 samples reported higher PD‐L1 gene expression levels at the level of mRNA using mRNA microarray and RNAseq data in glioblastomas compared to low‐grade gliomas [59], which is corroborated by several other studies evaluating PD‐L1 protein expression [44, 51, 57, 61, 62]. Using western blot analysis, Yao et al. [52] demonstrated increased PD‐L1 protein expression in high‐grade gliomas compared to low‐grade gliomas.
In three studies [55, 59, 61], PD‐L1 was significantly upregulated in the mesenchymal subtype compared to the other transcriptional glioblastoma subtypes, namely classic, neural and proneural [74]. Doucette et al. [74] reported a concordant preferential enrichment of immunosuppressive and immune effector genes within mesenchymal glioblastoma. Taken together, these findings imply differential immunogenicity in glioblastoma subtypes, with mesenchymal glioblastoma being particularly immunologically reactive. This may be a potential confounding variable in evaluation of therapeutic response [61, 62, 74]. In other cancers, similar associations with specific molecular subgroups have been revealed. For example, triple negative breast cancer and microsatellite instability (MSI)‐high colorectal cancer have been found to display higher levels of tumour cell PD‐L1 expression than other subtypes [75].
The role of the tumour micro‐environment
The historical and somewhat archaic dogma of central nervous system (CNS) immune privilege has evolved over time and been replaced by an understanding of a dynamic crosstalk between the CNS and the immune system [16].
The TME of gliomas is a critical determinant and regulator of tumourigenesis, immunosuppression and disease progression, and harbours a plethora of non‐neoplastic cells including tumour infiltrating immune cells [76, 77].
Tumour‐associated microglia/macrophages (TAMs) are the dominant immune cell subset in glioblastoma, estimated to constitute up to 30% of glioblastoma tissue cells [77, 78, 79, 80]. Infiltration by TAMs, particularly those exhibiting M2‐phenotype, has been found to hold unfavourable prognostic impact in gliomas [78, 79, 80].
Tumour‐infiltrating lymphocytes (TILs) are another essential component of the immune infiltrate in gliomas, although present in substantially lower numbers than in other malignancies [55, 56]. CD8+ TILs have been reported to be favourably prognostic in gliomas [81, 82]. Regulatory T‐cells, an immunosuppressive T‐cell subtype, constitute a substantial proportion of TILs and an elevated CD8+ T‐cells/regulatory T‐cells ratio has been correlated with improved prognosis [12, 55].
While the primary focus has been on PD‐L1 expression by tumour cells, there is an increasing awareness of PD‐L1 expression in non‐neoplastic cells of the TME [22]. PD‐L1 expression in tumour infiltrating immune cells is described in several cancers including glioblastoma [20, 53, 78, 83, 84, 85, 86]. Bloch et al. [80] reported that glioma cells upregulate PD‐L1 expression in tumour‐infiltrating macrophages and circulating monocytes through interleukin‐10 signalling, thereby acquiring an immunosuppressive phenotype. Microglial PD‐L1 expression has immune inhibitory capabilities and can down‐regulate T‐cell activation [86]. Liu et al. [53] found neuronal PD‐L1 expression in brain tissue adjacent to glioblastoma to be associated with a favourable prognosis, whereas a lack in neuronal PD‐L1 expression was associated with high tumoural PD‐L1 expression and poor prognosis. The prognostic impact of neuronal PD‐L1 expression is however disputed by Berghoff et al. [55].
The clinical impact of immune cell PD‐L1 expression is inconsistently reported. However, a meta‐analysis across various cancers found PD‐L1 positive tumour infiltrating immune cells to predict improved survival (HR = 0.784, 95% CI: 0.616–0.997, P = 0.047). Immune cell PD‐L1 expression was even associated with enhanced response to PD‐1/PD‐L1 inhibitors [87]. Despite the essential role of the TME, only few of the glioma studies in Table 1 have evaluated PD‐L1 expression in non‐neoplastic cells, and these studies have generally reported limited expression [44, 51, 64, 65, 66].
The significance of immune cell PD‐L1 expression in defining PD‐L1 positivity is reflected in the differing PD‐L1 scoring algorithms. While some cancers (NSCLC, melanoma) use the tumour proportion score in which only membranous staining of viable tumour cells is evaluated, others (urothelial carcinoma) use the combined positivity score or inflammatory cell scoring, which include or are restricted to PD‐L1 expression in specific immune cells, respectively [88].
The concept of microenvironmental heterogeneity has arisen from intriguing research demonstrating a close association between immunologic phenotype/TME composition and molecular tumour status [76, 77]. In accordance with other studies [59, 64, 89], Pratt et al. [68] found isocitrate dehydrogenase (IDH)‐wildtype gliomas to express PD‐L1 more frequently than IDH‐mutant tumours. Berghoff et al. [64] additionally reported IDH‐wildtype gliomas to have significantly more CD3+PD‐1+ TIL infiltration. The clinical implications of such a PD‐L1/IDH‐wildtype association could be that IDH‐mutated gliomas are deemed unsuitable for PD‐1/PD‐L1 inhibitor therapy.
PD‐L1 regulation
The regulatory mechanisms of PD‐L1 expression are poorly understood but are mediated through two fundamental mechanisms—innate immune resistance, which mediates constitutive expression, and adaptive immune resistance mediating inducible expression [13].
PD‐L1 expression is determined by exogenous and endogenous regulatory factors and regulation is performed at several levels including the level of transcription, post‐transcription and post‐translation [90, 91, 92, 93]. Inducible PD‐L1 expression is mediated by proinflammatory exogenous factors derived from tumour cells and immune cells of the TME. Interferon‐γ (IFN‐γ) is the most potent inducer of PD‐L1 expression, and is derived mainly from T‐cells and possibly NK cells [90, 91, 92, 93, 94]. Upon binding of IFN‐γ to its receptor, the IFN‐γ receptor, the upregulation of PD‐L1 is mediated by downstream activation of the JAK‐STAT pathway in a negative feedback mechanism [90, 91]. In gliomas, IFN‐γ has been found to play an important role in PD‐L1 expression in tumour cells, as well as the TME, including infiltrating microglia/macrophages and myeloid cells. PD‐L1 serves as a negative feedback mechanism following, rather than preceding, T‐cell activation and IFN‐γ secretion [95].
STAT3, another regulator, is a transcription factor acting downstream of cytokines such as interleukin‐6, interleukin‐10 and growth factors and can also upregulate PD‐L1 [90, 91, 92]. In addition to inflammatory stimuli, intrinsic signals such as aberrant activation of oncogenic signalling pathways [90, 91, 92, 93, 94], including the MAPK and PTEN/PI3K/AKT pathways, are regulators of constitutive PD‐L1 expression. Although not consistently verified in other studies [55, 57, 61], Parsa et al. [96] found loss of phosphatase and tensin homolog (PTEN), a negative regulator of the phosphoinositide‐3‐kinase (PI3K) pathway, which regulates cellular proliferation, to confer oncogenic overactivation of the PI3K/PTEN/AKT/mTOR pathway and induce PD‐L1 expression. Lastly, epigenetic regulation of PD‐L1 includes DNA methylation and microRNA regulation [91, 92, 93].
Quantification of immunohistochemical PD‐L1 expression
The studies in Table 1 evaluating PD‐L1 expression predominantly employed a pathologist‐based manual scoring applied to PD‐L1 immunohistochemistry. Five studies have performed automated analyses [54, 56, 61, 62, 68]. Knudsen et al. [54] performed digital quantification of membranous PD‐L1 staining yielding a PD‐L1 mean membrane fraction of 0.049%. Manual scoring by visual quantification of immunohistochemistry is limited by subjectivity and prone to great inter‐ and intraobserver variability, thus representing a potential pitfall in the interpretation of PD‐L1 expression [97]. Automated digital image analysis is increasingly being implemented for immunohistochemical quantification of tissue biomarkers to obtain accurate, reliable and reproducible estimates, provide accelerated workflow of an otherwise tedious process and prevent erroneous classification [97, 98]. Automated analysis also has the potential of recapitulating the full scope of PD‐L1 expression and better account for spatial heterogeneity. Ultimately, it is expected to facilitate more accurate allocation of precision medicine to eligible patients. Although validation is needed, digital image analysis of PD‐L1 expression in a melanoma study displayed excellent concordance with manual scoring [99].
PD‐L1 EXPRESSION AND PATIENT OUTCOME
Prognostic value of PD‐L1 expression
In general, results are highly inconsistent as to whether tumoural PD‐L1 expression correlates with prognosis in different cancers. However, a meta‐analysis comprising studies across multiple solid tumours has documented a significant association of tumoural PD‐L1 expression with poor prognosis [33]. Among others, PD‐L1 protein expression confers unfavourable prognosis in NSCLC, renal cell carcinoma and ovarian cancer [33, 36, 100]. The differing prognostic significance of PD‐L1 expression across different tumours may be inextricably linked to regulatory mechanisms. Some tumours are classified as “immunologically hot” and present with abundant effector T‐cell infiltration, high IFN‐γ levels and thus have high cytokine‐driven PD‐L1 expression in tumour cells and the TME. These tumours typically respond better to PD‐1/PD‐L1 inhibitors and could be predicted to have better prognoses [101]. Other tumours are classified as “immunologically cold” with sparse T‐cell infiltration and low tumoural PD‐L1 expression, primarily induced by oncogenic activation and thus potentially a worse prognosis [101]. It would be very interesting for future studies examining regulation of PD‐L1 to incorporate analysis of TME to gain more insights into the effect of IFN‐γ on the TME.
In Table 1, there are 11 studies that have examined the prognostic value of PD‐L1 expression in gliomas [53, 54, 55, 56, 58, 59, 60, 61, 66, 67, 68]. Approximately half of these reported elevated PD‐L1 expression levels to be associated with poor survival [53, 56, 59, 66, 68].
Applying a 5% cut‐off, Ndoum et al. [56] found high PD‐L1 protein expression to be significantly associated with poorer overall survival (OS) in glioblastoma. O6‐methylguanine‐DNA methyltransferase (MGMT) promoter methylation status was excluded as a covariate due to incomplete patient data. Pratt et al. [68] applied a 5% cut‐off for membranous PD‐L1 expression in recurrent IDH‐wildtype glioblastoma and reported PD‐L1 expression to be adversely associated with OS in multivariate analysis (HR = 1.957, 95% CI 1.11–3.45, P = 0.0208). In 54 glioblastoma cases, Han et al. [66] also found high PD‐L1 protein expression to be associated with poor OS (HR = 4.958, 95% CI 1.557–15.79, P = 0.007). Additional patient stratification according to PD‐L1 status and PD‐1 positive tumour infiltrating mononuclear cell density revealed that the group with high PD‐L1 expression and low PD‐1 positive tumour infiltrating mononuclear cell density had significantly worse OS compared with other groups. This led the authors to suggest that the combination of tumoural PD‐L1 expression and PD‐1 positive tumour infiltrating mononuclear cell density could provide greater and more accurate prognostic value than PD‐L1 expression alone. Contrary to the abovementioned studies, Knudsen et al. [54] and Berghoff et al. [55] did not find a correlation between PD‐L1 protein expression and OS in glioblastoma patients. Similar to Ndoum et al. [56], Berghoff et al. [55] excluded MGMT methylation as a covariate.
Several studies have also examined the prognostic significance of PD‐L1 gene expression by using gene expression profiling data from TCGA, but reaching contradictory results. Ndoum et al. [56] reported a worse prognosis in glioblastomas exhibiting high PD‐L1 gene expression in multivariate analysis (HR = 1.52, 95 CI% 1.03–2.25, P = 0.0343), and Wang et al. [59] also found PD‐L1 to predict worse survival in gliomas. Pratt et al. [68] demonstrated an association between high PD‐L1 expression and adverse OS in a small group of recurrent non‐glioma CpG island methylator phenotype glioblastomas (IDH‐wildtype) supporting the findings at the protein level. Conversely, Berghoff et al. [55] did not find high PD‐L1 gene expression to predict poorer OS in gliomas. However, the results are not entirely comparable because of the use of different parts of the TCGA dataset generated by various platforms. For example, Ndoum et al. [56] used Illumina RNAseq data. However, when analysing Agilent microarray data, the same data used by Berghoff et al. [55], Ndoum et al. [56] reached the same conclusion of no survival difference between PD‐L1 low and high expressers. Poor correlation has previously been reported between Agilent microarray and RNAseq data [73].
A meta‐analysis of six glioma studies reported a significant association between high PD‐L1 expression (protein or gene) and worse OS (HR = 1.30, 95% CI: 1.02–1.65, P = 0.032), which remained significant in subgroup analysis of glioblastomas (HR = 1.40, 95% CI: 1.03–1.90, P = 0.030) [102]. Nevertheless, the results should be interpreted cautiously due to limited sample size, the lack of analysis for publication bias and insufficient stratification.
The discrepant findings on prognostic significance could be explained by several factors of which retrospective study design and immense methodological heterogeneity are among the most important and are discussed in the “Pitfalls and challenges” section. Another issue is that it is unclear whether any of the studies were adequately powered. A pivotal caveat is that none of the studies fully took account of or controlled for established prognostic factors such as Karnofsky performance status (KPS), steroid use, MGMT‐ and IDH‐status, potentially confounding the results. However, Berghoff et al. [55] presented particularly solid results. They were able to control for several relevant factors (KPS, age, extent of resection, presence of PD‐L1 positive neurons, PD‐L1 staining type, PD‐1 density) using multivariate analysis in a cohort with 117 patients. Another robust study by Knudsen et al. [54], the most recent study, also performed multivariate analysis in a cohort of 163 patients utilising digital PD‐L1 quantification. Both of these studies came to the same conclusion that there is no prognostic value of tumoural PD‐L1 in glioblastoma.
PD‐1/PD‐L1 inhibitor clinical trials in gliomas
The prominent role of PD‐1/PD‐L1 in cancer biology has prompted the development of therapeutic high‐affinity mAbs targeting PD‐1 or PD‐L1 [30]. Figure 2 illustrates the fast pace at which approvals by the Food and Drug Administration have been granted to PD‐1/PD‐L1 inhibitors over time. There are currently no approved immune checkpoint inhibitors for use in gliomas [68], but proof‐of‐concept for the efficacy of PD‐1/PD‐L1 inhibitors has been provided in preclinical glioma models in which PD‐1/PD‐L1 inhibition has shown to restore anti‐tumour T‐cell activity, generate tumour cell regression and improve survival, thus paving the way for clinical trials [45, 46, 47, 48, 49, 50].
FIGURE 2.
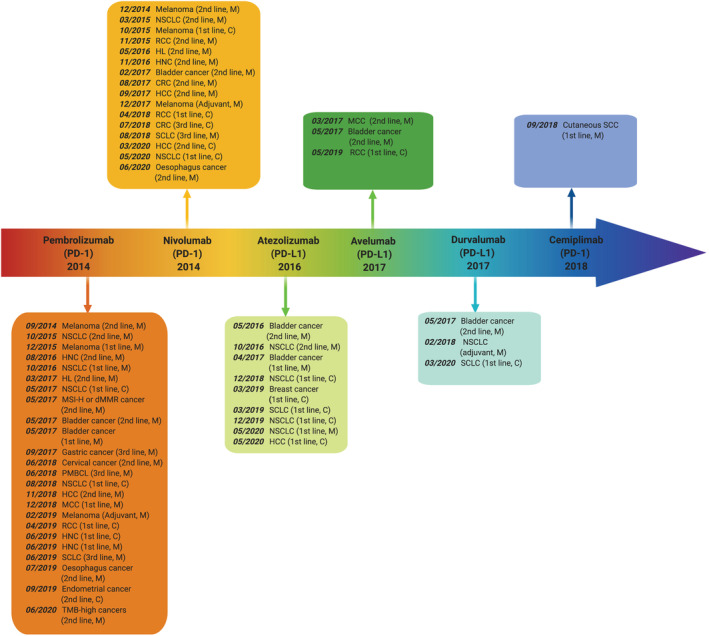
Timeline illustrating the rapidly progressive development in FDA approvals of PD‐1 and PD‐L1 inhibitor‐based therapies in different cancers since pembrolizumab was approved as the first PD‐1/PD‐L1 inhibitor in 2014. Data was retrieved from https://www.cancerresearch.org. Abbreviations: M Monotherapy, NSCLC Non‐small cell lung cancer, HNC Head and neck cancer, HL Hodgkin lymphoma, C Combination therapy, MSI‐H Microsatellite instability high, dMMR Mismatch repair deficiency, PMBCL Primary mediastinal large B‐cell lymphoma, HCC Hepatocellular carcinoma, MCC Merkel cell carcinoma, SCC squamous cell carcinoma, SLCL Small‐cell lung cancer, TMB Tumour mutational burden, RCC Renal cell carcinoma, CRC Colorectal cancer. Created with BioRender.com
Kim et al. [50], using a syngeneic orthotopic murine glioma model, found that triple therapy with an anti‐T‐cell immunoglobulin and mucin‐domain containing protein 3 (TIM‐3) mAb, anti‐PD‐1 mAb and stereotactic radiosurgery resulted in 100% OS (P < 0.05) compared with dual therapy with anti‐TIM‐3 mAb plus radiation (63.2%) or a PD‐1 inhibitor (57.9%), or monotherapy with anti‐TIM‐3 mAb (0% OS). Zeng et al. [49] demonstrated that combined stereotactic radiosurgery and PD‐1 inhibition generated a significant and synergistic survival improvement conferring a median OS of 52 days compared to 27 and 30 days with radiation or PD‐1 inhibition as single arm modalities, respectively. Furthermore, 10%–40% of the mice became long‐time survivors (>90 days after implantation) in the combination group. In a study with orthotopically GL261‐injected mice, simultaneous blockade of PD‐L1 and CTLA‐4 led to a remarkable long‐term survival rate of 90% [45]. Interestingly, survival rate increased to 100% by targeting indoleamine 2,3‐dioxygenase simultaneously with CTLA‐4 and PD‐L1 with a significant decrease in regulatory T‐cells compared to dual therapy.
These preclinical studies have paved the way for the currently active clinical trials with PD‐1/PD‐L1 inhibitors in gliomas summarised in Table 2 [103]. In Table 3, preliminary and final data from PD‐1/PD‐L1 inhibitor clinical trials in gliomas are listed [103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120] and some of the most significant studies are discussed below.
TABLE 2.
Active clinical trials evaluating PD‐1/PD‐L1 inhibitors in gliomas
| Trial identifier | Disease | PD‐1/PD‐L1 target | PD‐1/PD‐L1 inhibitor | Additional treatment | Study size | Trial status | Phase | Year start/end |
|---|---|---|---|---|---|---|---|---|
| NCT02017717 (CheckMate 143) | Recurrent glioblastoma | PD‐1 | Nivolumab | Bevacizumab, ipilimumab | N = 626 | Active, not recruiting | III | 2014/2020 |
| NCT02287428 | Newly diagnosed MGMT‐unmethylated glioblastoma | PD‐1 | Pembrolizumab | Personalised neoantigen vaccine, radiation therapy temozolomide | N = 56 | Recruiting | I | 2014/2022 |
| NCT02311920 | Newly diagnosed glioblastoma or gliosarcoma | PD‐1 | Nivolumab | Ipilimumab, temozolomide | N = 32 | Active, not recruiting | I | 2015/2017 |
| NCT02336165 | Glioblastoma | PD‐L1 | Durvalumab | Radiation therapy, bevacizumab | N = 159 | Active, not recruiting | II | 2015/2021 |
| NCT02337686 | Recurrent glioblastoma or gliosarcoma | PD‐1 | Pembrolizumab | Conventional surgery | N = 20 | Active, not recruiting | II | 2015/2020 |
| NCT02530502 | Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | Temozolomide, radiation therapy | N = 4 | Active, not recruiting | I | 2015/2020 |
| NCT02617589 (CheckMate 498) | Newly diagnosed MGMT‐unmethylated glioblastoma | PD‐1 | Nivolumab | Temozolomide, radiation therapy | N = 550 | Active, not recruiting | III | 2016/2020 |
| NCT02628067 (KEYNOTE‐158) | Different tumours including brain tumours (histological subtype not specified) | PD‐1 | Pembrolizumab | — | N = 13 (in total N = 233) | Recruiting | II | 2015/2026 |
| NCT02658981 | Recurrent glioblastoma | PD‐1 | Nivolumab | Anti‐LAG‐3 antibody BMS‐986016, urelumab | N = 63 | Active, not recruiting | I | 2016/2022 |
| NCT02667587 (Checkmate548) | Newly diagnosed MGMT‐methylated glioblastoma | PD‐1 | Nivolumab | Temozolomide, radiation therapy, nivolumab placebo | N = 693 | Active, not recruiting | III | 2016/2023 |
| NCT02794883 | Recurrent malignant glioma | PD‐L1 | Durvalumab | Tremelimumab, surgery | N = 36 | Active, not recruiting | II | 2016/2020 |
| NCT02798406 | Recurrent glioblastoma or gliosarcoma | PD‐1 | Pembrolizumab | DNX‐2401 | N = 49 | Active, not recruiting | II | 2016/2021 |
| NCT02866747 | Recurrent glioblastoma | PD‐L1 | Durvalumab | Hypofractionated stereotactic radiation therapy | N = 112 | Recruiting | I/II | 2017/2024 |
| NCT03018288 | Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | HSPPC‐96, temozolomide, placebo | N = 108 | Recruiting | II | 2017/2024 |
| NCT03047473 | Newly diagnosed glioblastoma | PD‐L1 | Avelumab | ‐ | N = 30 | Active, not recruiting | II | 2017/2022 |
| NCT03174197 | Newly diagnosed glioblastoma, gliosarcoma | PD‐L1 | Atezolizumab | Temozolomide, radiation therapy | N = 80 | Active, not recruiting | I/II | 2017/2021 |
| NCT03197506 | Glioblastoma, gliosarcoma | PD‐1 | Pembrolizumab | Temozolomide, radiation therapy, conventional surgery | N = 90 | Recruiting | II | 2017/2022 |
| NCT03233152 | Recurrent glioblastoma | PD‐1 | Nivolumab | Ipilimumab | N = 6 | Recruiting | I | 2016/2019 |
| NCT03277638 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | Laser interstitial thermotherapy | N = 34 | Recruiting | I/II | 2017/2021 |
| NCT03341806 | Recurrent glioblastoma | PD‐L1 | Avelumab | Laser interstitial thermal therapy | N = 30 | Recruiting | I | 2018/2021 |
| NCT03347617 | Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | Ferumoxytol MRI | N = 45 | Recruiting | II | 2017/2022 |
| NCT03367715 | Newly diagnosed MGMT‐unmethylated glioblastoma | PD‐1 | Nivolumab | Ipilimumab, radiation therapy | N = 24 | Recruiting | II | 2018/2020 |
| NCT03405792 | Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | Temozolomide, tumour treating fields | N = 29 | Recruiting | II | 2018/2023 |
| NCT03422094 | Newly diagnosed unmethylated glioblastoma | PD‐1 | Nivolumab | NeoVax, ipilimumab | N = 3 | Active, not recruiting | I | 2018/2021 |
| NCT03425292 | Newly diagnosed high grade glioma | PD‐1 | Nivolumab | Ipilimumab, bevacizumab, temozolomide, metronomic temozolomide, conformal brain radiation therapy | N = 90 | Recruiting | I | 2018/2022 |
| NCT03426891 | Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | Temozolomide, vorinostat, radiation therapy | N = 32 | Recruiting | I | 2018/2022 |
| NCT03430791 | Recurrent glioblastoma | PD‐1 | Nivolumab | Ipilimumab tumour treating fields | N = 60 | Recruiting | II | 2018/2021 |
| NCT03452579 | Recurrent glioblastoma | PD‐1 | Nivolumab | Bevacizumab | N = 90 | Active, not recruiting | II | 2018/2022 |
| NCT03491683 | Newly diagnosed glioblastoma | PD‐1 | Cemiplimab | INO‐5401, INO‐9012 temozolomide, radiation therapy | N = 52 | Active, not recruiting | I/II | 2018/2021 |
| NCT03493932 | Glioblastoma | PD‐1 | Nivolumab | BMS‐986016 | N = 25 | Recruiting | I | 2018/2021 |
| NCT03576612 | Newly diagnosed high‐grade glioma | PD‐1 | Nivolumab | Aglatimagene besadenovec, valacyclovir, temozolomide, radiation therapy | N = 36 | Recruiting | I | 2018/2021 |
| NCT03636477 | Recurrent or progressive glioblastoma | PD‐1 | Nivolumab | Veledimex,Ad‐RTS‐hIL‐12 | N = 21 | Active, not recruiting | I | 2018/2021 |
| NCT03661723 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | Bevacizumab, re‐irradiation | N = 60 | Recruiting | II | 2018/2021 |
| NCT03665545 | Relapsing glioblastoma | PD‐1 | Pembrolizumab | IMA950/Poly‐ICLC | N = 24 | Recruiting | I/II | 2018/2023 |
| NCT03673787 | Glioblastoma, metastatic prostate cancer, advanced solid tumours | PD‐L1 | Atezolizumab | Ipatasertib | N = 51 | Active, not recruiting | I/II | 2018/2020 |
| NCT03684811 | IDH1 mutated glioma and advanced solid tumours | PD‐1 | Nivolumab | FT‐2102, azacitidine, gemcitabine, cisplatin | N = 200 | Active, not recruiting | I/II | 2018/2022 |
| NCT03718767 | IDH‐mutant glioma | PD‐1 | Nivolumab | — | N = 95 | Recruiting | II | 2019/2025 |
| NCT03722342 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | TTAC‐0001 | N = 9 | Active, not recruiting | I | 2019/2020 |
| NCT03726515 | Newly diagnosed, MGMT‐unmethylated glioblastoma | PD‐1 | Pembrolizumab | CART‐EGFRvIII T cells | N = 7 | Active, not recruiting | I | 2019/2034 |
| NCT03743662 | Recurrent MGMT methylated glioblastoma | PD‐1 | Nivolumab | Bevacizumab, re‐irradiation, re‐resection | N = 94 | Recruiting | II | 2018/2021 |
| NCT03750071 | Progressive glioblastoma | PD‐L1 | Avelumab | VXM01 | N = 30 | Recruiting | I/II | 2018/2020 |
| NCT03797326 | Triple negative breast cancer, ovarian cancer, gastric cancer, colorectal cancer, glioblastoma, biliary tract cancers | PD‐1 | Pembrolizumab | Lenvatinib | N = 600 | Recruiting | II | 2019/2024 |
| NCT03890952 | Recurrent glioblastoma | PD‐1 | Nivolumab | Bevacizumab | N = 40 | Recruiting | II | 2018/2022 |
|
(PERGOLA) |
Newly diagnosed glioblastoma | PD‐1 | Pembrolizumab | — | N = 56 | Not yet recruiting | II | 2019/2023 |
| NCT04003649 | Recurrent/refractory glioblastoma | PD‐1 | Nivolumab | Ipilimumab, IL13Ralpha2‐specific Hinge‐optimised 4‐1BB‐co‐stimulatory CAR/Truncated CD19‐expressing Autologous TN/MEM Cells | N = 60 | Recruiting | I | 2019/2022 |
| NCT04006119 | Recurrent/progressive glioblastoma | PD‐1 | Cemiplimab | Veledimex, Ad‐RTS‐hIL‐12 | N = 36 | Active, not recruiting | II | 2019/2022 |
| NCT04013672 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | SurVaxM, sargramostim, montanide ISA 51 | N = 51 | Recruiting | II | 2020/2021 |
| NCT04047706 | Newly diagnosed glioblastoma | PD‐1 | Nivolumab | Temozolomide, radiation therapy, BMS‐986205 | N = 30 | Recruiting | I | 2019/2023 |
| NCT04145115 | Recurrent glioblastoma | PD‐1 | Nivolumab | Ipilimumab | N = 37 | Not yet recruiting | II | 2020/2023 |
| NCT04160494 | Recurrent grade IV malignant glioma | PD‐L1 | Atezolizumab | D2C7‐IT | N = 18 | Recruiting | I | 2020/2025 |
| NCT04195139 | Newly diagnosed glioblastoma | PD‐1 | Nivolumab | Temozolomide | N = 102 | Recruiting | II | 2018/2022 |
| NCT04201873 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | Dendritic cell tumour cell lysate vaccine, placebo, Poly ICLC | N = 40 | Recruiting | I | 2020/2025 |
| NCT04323046 | Recurrent or progressive high‐grade glioma | PD‐1 | Nivolumab | Ipilimumab, placebo | N = 45 | Not yet recruiting | I | 2020/2025 |
| NCT04396860 | Newly diagnosed MGMT unmethylated glioblastoma | PD‐1 | Nivolumab | Ipilimumab, NovoTTF‐100A Device, radiation therapy, temozolomide | N = 485 | Recruiting | II/III | 2020/2024 |
| NCT04429542 | Advanced solid tumours including glioblastoma | PD‐1 | Pembrolizumab | BCA101 | N = 292 | Recruiting | I | 2020/2023 |
| NCT04479241 | Recurrent glioblastoma | PD‐1 | Pembrolizumab | PVSRIPO | N = 10 | Not yet recruiting | I | 2020/2023 |
| NCT04583020 | Newly diagnosed glioblastoma | PD‐1 | Camrelizumab | Temozolomide, radiation | N = 42 | Recruiting | II | 2020/2023 |
| NCT04606316 | Recurrent glioblastoma | PD‐1 | Nivolumab | Ipilimumab, surgery | N = 60 | Recruiting | I | 2021/2023 |
| NCT04729959 | Recurrent glioblastoma | PD‐L1 | Atezolizumab | Surgery, fractionated stereotactic radiation, tocilizumab | N = 53 | Not yet recruiting | II |
Note: Data retrieved from ClinicalTrials.gov. 103 A search was last conducted on 1 February 2021.
Abbreviations: PD‐1 Programmed death‐1, PD‐L1 programmed death‐ligand 1, N Number, MGMT O6‐methylguanine‐DNA methyltransferase, MRI Magnetic Resonance Imaging, EGFR Epidermal Growth Factor Receptor, CD Cluster of differentiation.
TABLE 3.
Preliminary and final data from clinical trials evaluating PD‐1/PD‐L1 inhibitors in gliomas
| Trial identifier | Design/phase | Patients/Diagnosis | Intervention | ORR | mPFS (95% CI) | mOS (95% CI) | RR/HR (95% CI) | Status | Comments | |
|---|---|---|---|---|---|---|---|---|---|---|
| Newly diagnosed glioblastoma | ||||||||||
| NCT02617589 (Checkmate 498) [103, 104] | Randomized, open‐label/III | 560/MGMT‐unmethylated GBM | 280 | Arm A: Nivolumab + radiation therapy | NR | 6.01 months (5.65–6.21) | 13.40 months (12.62–14.29) |
HR OS:1.31 (1.09 –1.58) P = 0.0037 HR PFS: 1.38 (1.15–1.65) (P = 0.0005) |
Active, not recruiting |
Did not meet primary endpoint of OS. Complete evaluation is ongoing TMB not evaluated due to limited tissue availability |
| 280 | Arm B: Temozolomide + radiation therapy | NR |
6.21 months (5.91–6.74) |
14.88 months (13.27–16.13) |
||||||
| NCT02667587 (Checkmate 548) [103, 105] | Randomized, single blind/III | 693/MGMT‐methylated GBM | NR | Arm A: Temozolomide + radiation therapy + nivolumab | NR | NR | NR | NR | Active, not recruiting | Did not meet primary endpoint of PFS. Complete evaluation is ongoing |
| NR | Arm B: Temozolomide + radiation therapy + placebo | NR | NR | NR | NR | |||||
| NCT03174197 [103, 106] | Non‐randomized, open‐label/I/II | 60/GBM | NR | Atezolizumab + temozolomide + radiation therapy | NR |
9.7 months (7.6–15) MGMT methylated: 16.7 months (7.85—not reached) MGMT unmethylated: 7.9 months (6.70–12.4) |
17.1 months (13.9—not reached) |
NR | Active, not recruiting |
Concurrent use was tolerated and yielded modest efficacy Tumour immunocorrelative studies on PD‐L1 are pending |
|
Recurrent glioblastoma | ||||||||||
| Lombardi et al. [107] | Monocentric, observational pilot study | 13/HGG with partial or complete MMR protein expression loss | NR | Pembrolizumab | NR | 2.2 months (1.6–2.8) | 5.6 months (0.1–11.9) | NR | Completed |
No effect of pembrolizumab CD8+ T‐cells, CD68+ macrophages and TMB was not associated with pembrolizumab activity |
| NCT03291314 (GliAvAx) [108] | Non‐randomized, open‐label/II | 52/GBM | 27 | Cohort 1 (low baseline corticosteroids): Axitinib + avelumab | 33.3 % | 12.0 weeks (8.2–15.8) | 26.6 weeks (20.8–32.4) | NR | Completed | The combination did not reach prespecified activity threshold justifying further investigation of the treatment |
| 27 | Cohort 2 (high baseline corticosteroids): Axitinib (+ avelumab after 6 weeks) | 22.2 % | 10.7 weeks (5.3–16.1) | 18 weeks (12.5–23.5) | ||||||
| NCT02017717 (CheckMate 143) [109] | Randomized, open‐label/III | 369/GBM | 184 | Arm A: Nivolumab | 7.8% (1.4–13.3) | 1.5 months (1.5–1.6) | 9.8 months (8.2–11.8) |
HR OS: 1.04 (0.83–1.30) P = 0.76 HR PFS: 1.97 (1.57–2.48) P < 0.01 |
Active, not recruiting |
Did not meet primary endpoint of OS. Patients with MGMT‐methylation and no baseline corticosteroid use may be more likely to derive benefit Similar OS between patients with PD‐L1 <1% and ≥1% |
| 185 | Arm B: Bevacizumab | 23.1 % (16.7–30.5) | 3.5 months (2.9–4.6) | 10.0 months (9.0–11.8) | ||||||
| NCT02529072 (AVERT) [103] | Randomized open‐label/I | 6/HGG | 3 | Arm 1: Presurgical nivolumab and postsurgical nivolumab + DC vaccine | NR | 4.3 months (2.1–5.3) | 8.0 months (5.7–8.3) | NR | Completed |
Safety of nivolumab + DC vaccine is similar to nivolumab monotherapy Study terminated early due to results from Checkmate 143 showing no benefit from nivolumab |
| 3 | Arm 2: Presurgical nivolumab + DC vaccine and postsurgical nivolumab + DC vaccine | NR | 6.3 months (4.7–10.7) | 15.3 months (4.73 to NA a ) | ||||||
| NCT01375842 [110] | Randomized, open‐label/ I | 16/GBM | 16 | Atezolizumab | 6% | 1.2 months (0.7–10.7) | 4.2 months (1.2–18.8+) | NR | Completed |
Atezolizumab was well‐tolerated No changes in PD‐L1 expression (TC and IC) in archival and post‐progression samples |
| NCT02337491 [111] | Randomized open‐label/II | 80/bevacizumab naïve GBM | 50 | Cohort A: Pembrolizumab + bevacizumab | 20% | 4.1 months (2.8–5.5) | 8.8 months (7.7–14.2) | NR | Completed |
Pembrolizumab monotherapy and combination therapy with bevacizumab has limited activity PD‐L1 (positivity: membranous staining in ≥ 1% of TC), TILs, GEP were evaluated but did not predict outcome |
| 30 | Cohort B: Pembrolizumab | 0% | 1.43 months (1.4–2.7) | 10.3 months (8.5–12.5) | ||||||
| NCT02054806 (KEYNOTE‐028) [112] | Single group assignment, open label/Ib | 477/20 different advanced tumours including GBM | 26 PD‐L1 positive GBM | Pembrolizumab | 8% (1–26) | 2.8 months (1.9–8.1) | 13.1 months (8.0–26.6) | NR | Completed |
Manageable safety profile and durable response in a subset of patients PD‐L1 positivity: membranous staining in ≥1% of TC and associated IC or positive staining in stroma |
| NCT02313272 [113] | Single group assignment, open‐label/I | 32/HGG |
24 8 |
Bevacizumab‐naïve: HFSRT + bevacizumab + pembrolizumab Bevacizumab‐resistant: HFSRT + bevacizumab + pembrolizumab |
83% (63–95) 62.5% (24.5–91.5) |
7.92 months (6.31–12.45) 6.54 months (5.95–18.86) |
13.45 months (9.46–18.46) 9.3 months (8.97–18.86) |
NR | Completed |
Well tolerated and safe combination Majority had tumour/TME PD‐L1 expression <1% |
| NCT02337686 [114] | Single group assignment, open‐label/II | 15/GBM | 15 | Pembrolizumab before and after surgery | NR | 4.5 months (2.27–6.83) |
20.3 months (8.64–28.45) |
NR | Active, not recruiting |
Pembrolizumab alone cannot induce effector immunologic response Poor T‐cell infiltration and marked enrichment of CD68+ macrophages |
| Cloughsey et al. b [115] | Randomized, open‐label pilot study | 35/GBM | 16 | Neoadjuvant pembrolizumab followed by surgery and adjuvant pembrolizumab | NR | 3.3 months | 13.7 months |
HR OS: 0.39 (0.17–0.94) P = 0.04 HR PFS: 0.43 (0.20–0.90) 0.03 |
Completed | Neoadjuvant therapy improved OS and was associated with changes in the TME including enhanced PD‐L1 expression in the TME |
| 19 | Adjuvant post‐surgical pembrolizumab | 2.4 months | 7.5 months | |||||||
| Newly diagnosed and recurrent glioblastoma | ||||||||||
| NCT02550249 b (Neo‐Nivo) [116] | Single group assignment, open‐label/II | 29/GBM | 29 | Neoadjuvant nivolumab followed by surgery and adjuvant nivolumab | NR | 4.1 months | 7.3 months | NR | Completed |
Neoadjuvant nivolumab is safe and induces changes in TME No changes in PD‐L1 levels with nivolumab |
| NCT02336165 [117, 118, 119] |
Non‐randomized, open‐label/ II |
159/ GBM (5 cohorts) A: Newly diagnosed and MGMT unmethylated B+B2+B3: Bevacizumab‐ naïve recurrent C: Bevacizumab‐refractory recurrent |
A: 40 | Durvalumab + radiation therapy | NR | A: NR | A: 15.1 months (12–18.4) | NR |
Active, not recruiting B: NR B2: NR B3: NR C: NR |
A: Immunocorrelative studies with PD‐L1 pending B: Well tolerated and durable response C: Well tolerated with preliminary efficacy |
| B:31 | Durvalumab | B: 6‐months PFS: 20% | B: 12‐months OS: 44.4% | |||||||
| B2:34 | Durvalumab + bevacizumab |
B2: NR B3: NR |
B2: NR B3:NR |
|||||||
| B3:34 | Durvalumab + bevacizumab | |||||||||
| C: 22 | Durvalumab + continuing bevacizumab |
C: PFS range 0.9–24.4 weeks |
C: OS range 0.9–51.6 weeks |
|||||||
| NCT02628067(KEYNOTE 158) [120] | Non‐randomized, open‐label/II | 233/H‐MSI/dMMR cancers | 13 brain tumour patients | Pembrolizumab | 0% (0–24.7) | 1.1 months (0.7–2.1) | 5.6 months (1.5–16.2) | NR | Recruiting | Histological subtype of brain tumours not specified |
Abbreviations: ORR Objective response rate, mPFS Median progression‐free survival, mOS Median overall survival, RR Relative risk, HR Hazard ratio, CI Confidence interval, GBM Glioblastoma, NR Not reported, HGG High‐grade glioma, DC Dendritic cell, TC Tumour cells, IC Immune cells, dMMR Mismatch repair deficiency, TMB Tumour mutational burden, CR Complete response, PR Partial response, TILs Tumour‐infiltrating lymphocytes, GEP Gene expression profile, HFSRT Hypofractionated stereotactic radiation therapy, TME Tumour micro‐environment, H‐MSI Microsatellite instability high.
Upper limit not available
Neoadjuvant PD‐1/PD‐L1 therapy
Nivolumab
Nivolumab is a fully human IgG4 PD‐1 immune checkpoint inhibitor [109]. The open‐label phase III trial, Checkmate 143 (NCT02017717) was the first large‐scale randomised clinical trial to examine PD‐1/PD‐L1 inhibitors in gliomas [109]. Patients with recurrent glioblastoma were randomised to receive either nivolumab or bevacizumab until disease progression, unacceptable toxicity or death. At completion, the study did not meet its primary endpoint of OS with no significant improvements in survival in patients receiving nivolumab (mOS 9.8 months, 95% CI, 8.2–11.8) compared to bevacizumab (mOS 10.0 months, 95% CI, 9.0–11.8) with HR = 1.04 (95% CI, 0.83–1.3; P = 0.76.). Secondary outcomes of PFS (1.5 months vs. 3.5 months) and objective response rate (ORR) were significantly higher (7.8% vs. 23.1%) with bevacizumab. Interestingly though, patients receiving nivolumab displayed more durable responses providing hopes for a small subgroup of long‐term survivors. Exploratory, multivariate subgroup analyses revealed that MGMT‐methylation and lack of baseline corticosteroid use were associated with improved OS in the nivolumab group. In the bevacizumab group, MGMT‐methylation was also associated with improved survival. Subsequently, combined analyses of MGMT‐methylation and lack of corticosteroid use yielded a trend towards longer mOS in patients receiving nivolumab compared to bevacizumab (17 months vs. 10 months, HR = 0.58, 95% CI, 0.30–1.11). These results could potentially identify a subgroup of responders. Particularly, the results on lack of baseline corticosteroid use are intriguing. While it could be that patients with baseline corticosteroid use have more progressive disease, a potential effect of steroids on the immune system and activation of T‐cells is also plausible.
Patients were classified according to baseline tumoural PD‐L1 expression <1% or ≥1% with positivity defined as membranous staining in ≥1% of tumour cells and survival was reported to be similar between the two subgroups. Subgroup analyses should be interpreted cautiously though, because of the limited number of patients. A classification of the patients based on expression of PD‐L1 in tumour cells and/or microglial cells and macrophages would have been interesting as well as evaluation of tumour mutational burden (TMB) and mismatch repair (MMR).
Pembrolizumab
Pembrolizumab is a humanised anti–PD‐1 antibody. The phase II trial by Nayak et al. [111] (NCT02337491) did not find any significant difference in PFS and OS when pembrolizumab was administered alone or combined with bevacizumab compared to a historical cohort. The study performed immunocorrelative studies with potential tumour microenvironmental biomarkers such as PD‐L1 expression and TIL density. PD‐L1 was evaluated by immunohistochemistry using the 28‐8 clone with the same threshold as in Checkmate 143 [109]. None of the biomarkers predicted outcome but analyses were based on relatively small sample sizes and, in part, archival samples. Analyses of MGMT‐status and baseline corticosteroid use corroborated the results from Checkmate 143 [109].
The multi‐cohort phase 1 trial, Keynote‐028, evaluated pembrolizumab monotherapy across various tumour types including recurrent glioblastoma (n = 26) with some of the key eligibility criteria being bevacizumab‐naïve glioblastoma and PD‐L1 positive [112]. Though utilising the same threshold of ≥1%, the definition of PD‐L1 positivity differed somewhat from the two aforementioned studies [109, 111]. Keynote‐028 included membranous staining in stromal cells and staining of the stroma in addition to tumour cells, which is a less explored approach in gliomas. Thus, this PD‐L1 assessment might be less robust. Overall, only modest efficacy in a small subgroup of patients was found, similar to Checkmate 143 [109, 112]. Correlative analysis of PD‐L1 to outcome is pending, but the exclusion of PD‐L1 negative glioblastomas from the trial precludes definitive conclusions on the predictive value of PD‐L1 expression.
Neoadjuvant PD‐1/PD‐L1 inhibition
Interestingly, recently published results of neoadjuvant PD‐1/PD‐L1 inhibition have reinvigorated optimism [115, 116] and shed light on the importance of timing of checkpoint immunotherapy. In a randomised, multi‐institutional study by Cloughsey et al. [115] comprising 35 patients with recurrent, surgically resectable glioblastoma, patients receiving neoadjuvant pembrolizumab followed by adjuvant pembrolizumab displayed significantly prolonged median OS (417 days vs. 228.5 days) and PFS (99.5 days vs. 77.5 days) compared to patients receiving adjuvant therapy only. By leveraging T‐cell receptor sequencing, gene expression profiling, mass cytometry and quantitative multiplex immunofluorescence, several biological alterations of the tumoural immune landscape were documented including upregulated T‐cell and IFN‐γ related gene expression, focally upregulated PD‐L1 expression in the TME and enhanced T‐cell clonal expansion, suggestive of enhanced anti‐tumour response in neoadjuvant settings of PD‐1 blockade. Although not documenting similar clinical efficacy, a single‐arm phase II clinical trial of neoadjuvant and adjuvant nivolumab also reported alterations within the TME including enhanced T‐cell receptor clonal diversity in tumour‐infiltrating T‐cells and increased immune cell infiltration [116].
Taken together, the preliminary clinical results in gliomas have been disparate and disappointing. Definitive conclusions should, however, not be made because of the paucity and, in part, immaturity of the results. More data from completed, ongoing and pending trials are eagerly anticipated to provide more clarification.
Predictive value of PD‐L1 expression
In cancers highly amenable to PD‐1/PD‐L1 inhibition, the ORR has generally proven to be low to moderate [19, 20, 21, 35, 121]. Accordingly, PD‐1/PD‐L1 inhibition in gliomas is expected only to be applicable in a highly selected subset of patients, which further underscores the urgent need for predictive biomarkers in refining the selection of eligible patients to derive benefits from checkpoint inhibitor therapy [57, 65].
Emerging data supports the idea that tumoural PD‐L1 expression enriches for response to PD‐1/PD‐L1 inhibition in some cancers [21, 34, 35, 121]. A study by Topalian et al. [19] reported PD‐L1–positive tumours to have a 36% ORR (P = 0.006) using a 5% positivity cut‐off for membranous PD‐L1 expression, while no PD‐L1 negative patients displayed objective responses. In NSCLC, a pooled analysis found PD‐L1 positivity (tumour cell staining ≥1%) to be associated with higher overall ORR to PD‐1 inhibitors (OR = 2.44, 95% CI: 1.61–3.68) compared to PD‐L1 negative tumours. However, determining the predictive value of PD‐L1 is still a burgeoning field. If PD‐L1 expression in gliomas is a predictor of responsiveness to PD‐1/PD‐L1 blockade, the association is likely to be non‐linear. Interestingly, treatment response to PD‐1/PD‐L1 inhibitors has also been documented in up to 17% of PD‐L1 negative cancer patients, some even displaying advantageous long‐term outcomes [121]. While an underlying explanation could be intratumoural heterogeneity of expression, it could also point towards PD‐L1 not being a dichotomous, exclusionary biomarker that only may provide adequate predictive significance in conjunction with other biomarkers [34]. PD‐1+ TILs, TMB, and neoantigen load have been proposed as other candidate biomarkers [34].
Tumour mutational burden and mismatch repair deficiency as biomarkers
In several cancers, TMB and/or MMR deficiency (dMMR) have been identified as potential markers of response to immune checkpoint blockade, presumably due to enhanced neoantigen formation, heightened immunogenicity and immune infiltration [64, 65, 123, 124, 125, 126]. TMB‐high tumours such as lung cancer (9.9 mutations/megabase) and melanoma (12.9 mutations/megabase) have documented high response rates [64, 65, 123, 124, 125]. Gliomas, particularly glioblastomas (2.2 mutations/megabase), are considered poorly immunogenic harbouring comparatively lower TMB than other malignancies [64, 65, 124]. Nevertheless, in the setting of dMMR syndromes, patients with hypermutated gliomas have benefitted from immune checkpoint blockade [127, 128]. Tumours with dMMR are TMB‐high tumours. Collective data suggest that dMMR renders greater responsiveness to immune checkpoint inhibitors compared to MMR proficient tumours [65, 123], which has led to a tissue agnostic FDA approval of pembrolizumab in dMMR tumours [107]. An association between dMMR and hypermutation has been reported in gliomas, although the exact underlying mechanisms are unclear [129].
In a recent comprehensive study with more than 10,000 gliomas, Touat et al. [129] delineated two pathways by which gliomas acquire a hypermutated state—through de novo hypermutation and a more common post‐therapy pathway due to acquired resistance by a temozolomide driven expansion of MMR‐deficient cells and accumulation of temozolomide induced mutations in recurrent chemo‐sensitive glioblastoma. Retrospectively, the study did not find any efficacy of PD‐1 inhibitor therapy in a subgroup of hypermutated gliomas.
Samstein et al. [125] documented high TMB to be associated with improved survival in patients receiving checkpoint inhibitors across various malignancies except for gliomas in a retrospective study.
The recent phase 2 study KEYNOTE‐158 (NCT02628067) evaluated pembrolizumab in various non‐colorectal cancers (n = 233) [120]. Tumours were classified as dMMR/MSI‐high based on immunohistochemical loss of at least one MMR protein or MSI at polymerase chain reaction. Among various cancers, only patients with brain tumours (histological subtype was not specified) failed to show radiological responses. Lombardi et al. [107] obtained similar results and concluded that immunohistochemical MMR loss may be uncorrelated with dMMR and cannot be used as a predictive biomarker in high‐grade gliomas for checkpoint inhibitor therapy.
We may speculate as to why hypermutated gliomas differ in response to immunotherapy compared to other hypermutated cancers. As stated by Touat et al. [129], the fact that the events leading to dMMR are late events in post‐treatment gliomas compared to other cancers could be an explanation. Thus, it might be expected that patients with constitutional mismatch repair deficiency (CMMRD), a rare autosomal recessive hereditary cancer predisposition, would respond and benefit to checkpoint inhibition. In fact, there have been case‐reports describing favourable effect of checkpoint inhibition in CMMRD patients [127, 130]. Such beneficial effects could indeed be due the early onset and accumulation of genetic alterations, ultimately leading to sufficient immune responses within this group of patients [130].
Other reasons for the unresponsiveness to immunotherapy in hypermutated gliomas compared to other hypermutated cancers could be that hypermutated gliomas might harbour poor neoantigen quality (missense mutations versus frameshift‐producing insertions‐deletions) making quantity alone insufficient as a predictor in gliomas [129]. It could also be because of an extensive lack in CD8+ T‐cell infiltration, which has been repeatedly reported [65, 107, 114, 129]. Thus, even though tumour genotype can shape the TME, hypermutation does not necessarily equal increased immune cell infiltration, and TMB high tumours do not always harbour an immunologically hot phenotype [114, 129]. The effect of TMB/dMMR on the TME requires in‐depth characterisation in future studies. Lastly, the threshold for defining TMB‐high could be too low compared to other cancers, as noted by Samstein et al. [124] and thus only ultra‐mutated glioma may benefit from checkpoint immunotherapy as seen in MMR deficiency syndromes [127, 128].
IMMUNE CHECKPOINT CO‐EXPRESSION
Several other inhibitory immune checkpoint receptors involved in regulating T‐cell functions such as CTLA‐4, TIM‐3, lymphocyte activation gene 3 protein (LAG‐3), 2B4 (CD244) and B and T lymphocyte attenuator (BTLA) have been identified and studied individually, but their co‐expression and relative contribution to T‐cell exhaustion is poorly elucidated and data is only beginning to emerge [131].
Matsuzaki et al. [132] isolated TILs from ovarian cancer patients and found TILs expressing New York oesophageal squamous cell carcinoma 1 (NY‐ESO‐1) to co‐express PD‐1 and LAG‐3. By inhibiting these two checkpoints during T‐cell priming, an augmented in vitro proliferation and cytokine production in NY‐ESO‐1 specific CD8+ TILs were generated. Similar findings have been reported in melanoma with TIM‐3 and PD‐1 in T‐cells [133].
Early data from Baitch et al. [134] described extended co‐expression of 4 or more of the inhibitory immune checkpoints BTLA, TIM‐3, LAG‐3, KRLG‐1, 2B4, CD160, PD‐1 and CTLA‐4 in tumour‐antigen specific CD8+ effector T‐cells contrary to naïve T‐cells in metastatic lesions of melanoma patients. They also found many of the corresponding ligands to be expressed in tumour cells and/or cells of the TME. Upregulation of alternative immune checkpoints has also been described as an adaptive mechanism in response to PD‐1 inhibition [135]. Knudsen et al. [55] reported PD‐L1 and Galectin‐3 co‐expression in tumours cells and microglia/macrophages in glioblastoma patients, though without prognostic value.
These data suggest that intervention at multiple levels of the immune response will be needed to effectively ameliorate immunosuppression and elicit T‐cell immunity, which most likely cannot be achieved through single‐agent therapy [94, 135, 136, 137, 138].
THERAPEUTIC STRATEGIES WITH PD‐1/PD‐L1 INHIBITORS
Preclinical studies, as previously discussed, and clinical data suggest that inhibition of PD‐1/PD‐L1 concurrently or sequentially with other immune checkpoints or other modalities may augment antitumour activity [48, 49, 50].
In the CheckMate 067 melanoma trial, the combination of nivolumab and ipilimumab, compared to monotherapy with either, provided higher antitumour activity and longer OS [32]. Similar trials of multiple immune checkpoint inhibition are currently being conducted in gliomas, as illustrated in Table 2.
The possibility of combining PD‐1/PD‐L1 inhibitors with other immunotherapies, such as tumour vaccines, adoptive T‐cell therapies and oncolytic virotherapies are also being investigated [8, 9, 10, 11, 12].
Combination therapy could also implicate other modalities, as preclinically documented by Zeng et al. [49]. In a phase I clinical trial (NCT02313272) evaluating the combination of pembrolizumab, hypofractionated stereotactic radiation and bevacizumab in recurrent high‐grade gliomas, ORR was 83% (95% CI: 63–95) and 62.5% (95% CI: 24.5–91.5) and mOS 13.45 months (95% CI: 9.46–18.46), and 9.3 months (95% CI: 8.97–18.86) in bevacizumab‐naïve and bevacizumab‐resistant patients, respectively [113]. Radiation therapy is known to counteract tumour‐induced immunosuppression by increasing expression of the major histocompatibility complex class I and proinflammatory cytokines, and by enhanced tumour antigen‐presentation [62].
Combinatorial strategies with PD‐1/PD‐L1 inhibitors and molecularly targeted therapies such as bevacizumab are also being investigated. However, combination therapy with bevacizumab has not yielded successful results, as reported by Nayak et al. [111].
One of the most recent avenues being explored is the effect of the microbiome on response to checkpoint inhibition [139].
Arguably, the search for immunologic antitumour potency should be counterbalanced by vigilance for additive immune‐related toxicities, which could be a critical limiting factor of combination therapy [4, 10]. Exemplified by Larkin et al. [32], grade 3 and 4 treatment‐related adverse events (TRAEs) in melanoma patients increased from 16.3% and 27.3% with nivolumab and ipilimumab monotherapy, respectively, to 55% with combination therapy.
PITFALLS AND CHALLENGES
PD‐1/PD‐L1 is recognised as a crucial targetable pathway in several cancers [17, 18, 19, 20, 21]. Nevertheless, the significance in gliomas is uncertain. A growing body of evidence documents upregulated PD‐L1 expression in gliomas, albeit yielding highly variable expression, as well as inconsistencies in prognostic value across different studies. This may reflect variability in the immunohistochemical methodology, such as differences in antibody clones with distinct target epitopes and affinities, assays, platforms, staining protocols, evaluated staining patterns, staining intensity, cut‐off definitions, evaluated cell types and scoring algorithms, as well as differences in study characteristics such as retrospective study design, heterogeneous cohorts, antecedent therapies, grading and primary versus recurrent disease [35]. Besides spatial heterogeneity, temporal heterogeneity is another concern [140]; PD‐L1 expression at a given time point is a dynamic process believed to evolve over time and be influenced by therapies. Therefore, considerations of tissue source, preparation, and timing of sample collection are also pertinent [24].
An important barrier is the singularity in which each therapeutic PD‐1/PD‐L1 inhibitor has been co‐developed with a specific PD‐L1 immunohistochemistry assay using a specific PD‐L1 antibody clone, platform, staining protocol and scoring criteria [141, 142]. Such one‐drug‐one‐assay paradigm entails the risk of divergent patient PD‐L1 classification depending on the assay used [142]. Harmonisation and interchangeability of the different assays are a prerequisite in order to obtain comparable data and a common basis for therapeutic decision making. In NSCLC, comparative studies of diagnostic PD‐L1 assays have demonstrated high concordance in membranous tumour cell PD‐L1 expression evaluation between the 22C3, 28‐8 and SP263 assays, strongly indicative of interchangeability, while SP142 was as a clear outlier detecting lower PD‐L1 levels [142, 143]. On the contrary, immune cell PD‐L1 assessment displayed poor interobserver concordance. This could be due to a lack of pre‐specified assessment criteria, and because immune cell PD‐L1 assessment is a more unfamiliar and untrained skill among pathologists [142, 143].
Comprehensive preclinical data support the feasibility and potential efficacy of PD‐1/PD‐L1 inhibition in gliomas, but enthusiasm has been tempered by inconsistent preliminary clinical findings. The lack of success may be ascribed to multiple factors. It could in part pertain to deficient preclinical animal models unable to sufficiently replicate the immunosuppressive TME [36, 144]. Presumably, the most important impediment is the complex immunosuppressive milieu. While PD‐1/PD‐L1 inhibitors remove “the brakes” placed on the immune system, the effects of PD‐1/PD‐L1 inhibitors are mediated by a severely defective host cellular immunity, due to compromised T‐cell functions and a scarcity of effector cells in the TME, which instead is dominated by immunosuppressive myeloid cells and regulatory T‐cells [114, 137, 138, 144].
Gliomas comprise a group of tumours that exhibit enormous inter‐ and intratumoural heterogeneity [5]. In future studies, consistent stratification according to molecular subclasses and prognostic factors (e.g., MGMT, IDH, KPS, steroid use, prior therapy) could in part eliminate confounders and thereby disclose true clinical responders to checkpoint immunotherapy and elucidate the prognostic value of PD‐L1 [5].
The selective permeability of the blood‐brain‐barrier and other constraints of intracranial localisation are potential major hindrances to the benefits of checkpoint inhibitors in glioma. The integrity of the blood‐brain‐barrier is compromised and disrupted in cancer, thus increasing its permeability and allowing for trafficking into the brain [144, 145]. This is supported by a phase II trial in which pembrolizumab displayed intracranial effects on untreated or progressive cerebral metastases in metastatic melanoma and NSCLC patients without significant toxicity [146]. Though encouraging and indicative of intracranial accessibility, methods to bypass the blood‐brain‐barrier are being investigated, such as intracranial catheter applied therapies [10, 144, 145].
The widespread use of supportive steroids in glioma patients has several undesirable immunological consequences that compound pre‐existing immunosuppression, potentially abrogating the effects of checkpoint immunotherapy. Dexamethasone is known to deplete T‐ and B‐cells, decrease permeability of the blood‐brain‐barrier and dampen inflammatory cytokines [145]. Steroid use has even been associated with compromised survival and treatment inefficiency in gliomas [147, 148], as underscored by CheckMate 143 [109].
A further complicating aspect is the additional interactions within the PD‐1/PD‐L1 axis. In addition to PD‐1, PD‐L1 also binds CD80 (B7‐1) on activated T‐cells [13, 22]. PD‐L2 is another PD‐1 ligand and inhibits T‐cell activation by binding with higher affinity than PD‐L1 [149]. These findings suggest different functional effects of PD‐1 and PD‐L1 inhibitors, which underpin the need to understand the functional significance of such interactions.
CONCLUSION
Checkpoint immunotherapy has ushered in a new era in precision medicine, but preliminary clinical data in gliomas have been modest and disappointing. Nonetheless, the astounding efficacy seen in other cancers justifies continued research of PD‐1/PD‐L1 as a therapeutic target and biomarker in gliomas, especially glioblastomas. In particular, there is hope to be found in the efficacy demonstrated in certain highly selected subgroups of patients. Given the wide‐ranging heterogeneity of gliomas and the multi‐tiered immunosuppressive mechanisms employed, the future success of PD‐1/PD‐L1 inhibitors in gliomas undoubtedly relies on rational combinatorial therapies. The key to unleashing anti‐tumour immunity lies within the micro‐environment and future studies are warranted to more comprehensively evaluate expression of PD‐L1 and other biomarkers in the TME. Research indicates a lack of T‐cell infiltration in the TME and an enrichment of immunosuppressive microglia/macrophages in gliomas. Thus, approaches aimed at increasing cytotoxic T‐cell infiltration and targeting TAMs and other myeloid‐derived cells expressing potential biomarkers such as CD204, which has been associated with poor prognosis in glioblastoma, should be explored in combination with PD‐1/PD‐L1 inhibitors as potential immunotherapeutic targets [78].
To identify potential responders to checkpoint immunotherapy, a single parameter will most likely not suffice either. Instead, a multimodal approach with combined biomarkers, particularly combinations reflecting both immune/TME phenotype and tumour genotype should be explored. An IFN‐γ signature has been proposed as a new predictive biomarker, which would reflect immune/TME phenotype [95]. In addition to PD‐L1, the genomic biomarkers TMB and MMR are some of the most successful predictive biomarkers in cancer checkpoint immunotherapy. Similar to PD‐L1, TMB and MMR have not yet provided convincing results in glioma, but they have primarily been evaluated in single biomarker settings.
An impending transition and redefinition of the biomarker arena is underway based upon technological advances, which are expected to generate more elaborate, integrated and multi‐dimensional immune signatures and genetic profiles characterising the tumoural and microenvironmental landscape of gliomas. The hope is that such merging of various biomarker strategies will designate a new generation of combined biomarker signatures with high clinical value in glioma patients [11, 150]. Further large‐scale biomarker‐driven clinical trials are warranted to elucidate the biomarker potential of PD‐L1 in gliomas.
AUTHOR CONTRIBUTION
Gayaththri Vimalathas conceptualised, drafted and edited the manuscript. Bjarne Winther Kristensen conceptualised and edited the manuscript. All authors have read and approved the final manuscript.
CONFLICT OF INTERESTS
The authors declare that they have no conflicts of interest.
ACKNOWLEDGEMENTS
This study received no specific grant from any funding agency.
All figures have been created with BioRender.com.
Vimalathas G, Kristensen BW. Expression, prognostic significance and therapeutic implications of PD‐L1 in gliomas. Neuropathol Appl Neurobiol. 2022;48(1):e12767. doi: 10.1111/nan.12767
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO Classification of Tumors of the Central Nervous System. Lyon, France: IARC Press; 2007. [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987‐996. [DOI] [PubMed] [Google Scholar]
- 3. Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15:e395‐e403. [DOI] [PubMed] [Google Scholar]
- 4. Theeler BJ, Gilbert MR. Advances in the treatment of newly diagnosed glioblastoma. BMC Med. 2015;13:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro‐oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aum DJ, Kim DH, Beaumont TL, Leuthardt EC, Dunn GP, Kim AH. Molecular and cellular heterogeneity: the hallmark of glioblastoma. Neurosurg Focus. 2014;37:E11. [DOI] [PubMed] [Google Scholar]
- 7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 8. Reardon DA, Freeman G, Wu C, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 2014;16:1441‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res. 2014;20:3651‐3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. 2014;20:5620‐5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prados MD, Byron SA, Tran NL, et al. Toward precision medicine in glioblastoma: the promise and the challenges. Neuro Oncol. 2015;17:1051‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coleman N, Ameratunga M, Lopez J. Development of molecularly targeted agents and immunotherapies in glioblastoma: a personalized approach. Clin Med Insights Oncol. 2018;12:179554918759079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Momtaz P, Postow MA. Immunologic checkpoints in cancer therapy: focus on the programmed death‐1 (PD‐1) receptor pathway. Pharmgenomics Pers Med. 2014;7:357‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dahlrot RH, Dowsett J, Fosmark S, et al. Prognostic value of O‐6‐methylguanine‐DNA methyltransferase (MGMT) protein expression in glioblastoma excluding nontumour cells from the analysis. Neuropathol Appl Neurobiol. 2018;44:172‐184. [DOI] [PubMed] [Google Scholar]
- 15. Music D, Dahlrot RH, Hermansen SK, et al. Expression and prognostic value of the WEE1 kinase in gliomas. J Neurooncol. 2016;127:381‐389. [DOI] [PubMed] [Google Scholar]
- 16. Thomas AA, Ernstoff MS, Fadul CE. Immunotherapy for the treatment of glioblastoma. Cancer J. 2012;18:59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polivka J Jr, Polivka J, Holubec L, et al. Advances in experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res. 2017;37:21‐33. [DOI] [PubMed] [Google Scholar]
- 18. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti–PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366:2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taube JM, Klein A, Brahmer JR, et al. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti‐PD‐1 therapy. Clin Cancer Res. 2014;20:5064‐5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDermott DF, Atkins MB. PD‐1 as a potential target in cancer therapy. Cancer Med. 2013;2:662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wintterle S, Schreiner B, Mitsdoerffer M, et al. Expression of the B7‐related molecule B7‐H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462‐7467. [PubMed] [Google Scholar]
- 25. Dong H, Zhu G, Tamada K, Chen L. B7‐H1, a third member of the B7 family, co‐stimulates T‐cell proliferation and interleukin‐10 secretion. Nat Med. 1999;5:1365‐1369. [DOI] [PubMed] [Google Scholar]
- 26. Dong H, Strome SE, Salomao DR, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793‐800. [DOI] [PubMed] [Google Scholar]
- 27. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293‐11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blank C, Gajewski TF, Mackensen A. Interaction of PD‐L1 on tumor cells with PD‐1 on tumor‐specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51‐81. [DOI] [PubMed] [Google Scholar]
- 30. Sznol M, Chen L. Antagonist antibodies to PD‐1 and B7‐H1 (PD‐L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013;19:1021‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus docetaxel in previously treated patients with advanced non‐small‐cell lung cancer: two‐year outcomes from two randomized, open‐label, phase iii trials (checkmate 017 and checkmate 057). J Clin Oncol. 2017;35:3924‐3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu P, Wu D, Li L, Chai Y, Huang J. PD‐L1 and Survival in Solid Tumors: A Meta‐Analysis. PLoS One. 2015;10:e0131403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847‐856. [DOI] [PubMed] [Google Scholar]
- 35. Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD‐1/PD‐L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Teng F, Kong L, Yu J. PD‐L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016;9:5023‐5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chauhan P, Lokensgard JR. Glial cell expression of PD‐L1. Int J Mol Sci. 2019;20:1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schachtele SJ, Hu S, Sheng WS, Mutnal MB, Lokensgard JR. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD‐L1. Glia. 2014;62:1582‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ortler S, Leder C, Mittelbronn M, et al. B7‐H1 restricts neuroantigen‐specific T cell responses and confines inflammatory CNS damage: implications for the lesion pathogenesis of multiple sclerosis. Eur J Immunol. 2008;38:1734‐1744. [DOI] [PubMed] [Google Scholar]
- 40. Latchman YE, Liang SC, Wu Y, et al. PD‐L1‐deficient mice show that PD‐L1 on T cells, antigen‐presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691‐10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Phares TW, Ramakrishna C, Parra GI, et al. Target‐dependent B7‐H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J Immunol. 2009;182:5430‐5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klotz L, Kuzmanov I, Hucke S, et al. B7‐H1 shapes T‐cell‐mediated brain endothelial cell dysfunction and regional encephalitogenicity in spontaneous CNS autoimmunity. Proc Natl Acad Sci U S A. 2016;113:E6182‐E6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu S, Zhu Y, Zhang C, et al. The clinical significance of soluble programmed cell death‐ligand 1 (sPD‐L1) in patients with gliomas. Front Oncol. 2020;10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baral A, Ye HX, Jiang PC, Yao Y, Mao Y. B7‐H3 and B7‐H1 expression in cerebral spinal fluid and tumor tissue correlates with the malignancy grade of glioma patients. Oncol Lett. 2014;8:1195‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4:124‐135. [DOI] [PubMed] [Google Scholar]
- 46. Huang BY, Zhan YP, Zong WJ, et al. The PD‐1/B7‐H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One. 2015;10:e0134715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA‐4, and PD‐L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290‐5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ladomersky E, Zhai L, Lenzen A, et al. IDO1 inhibition synergizes with radiation and PD‐1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res. 2018;24:2559‐2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zeng J, See AP, Phallen J, et al. Anti‐PD‐1 blockade and stereotactic radiation produce long‐term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti‐PD‐1, anti‐TIM‐3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23:124‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilmotte R, Burkhardt K, Kindler V, et al. B7‐homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005;16:1081‐1085. [DOI] [PubMed] [Google Scholar]
- 52. Yao Y, Tao R, Wang X, Wang Y, Mao Y, Zhou LF. B7‐H1 is correlated with malignancy‐grade gliomas but is not expressed exclusively on tumor stem‐like cells. Neuro Oncol. 2009;11:757‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu Y, Carlsson R, Ambjørn M, et al. PD‐L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J Neurosci. 2013;33:14231‐14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Knudsen AM, Rudkjøbing SR, Sørensen MD, Dahlrot RH, Kristensen BW. Expression and prognostic value of the immune checkpoints Galectin‐9 and PD‐L1 in glioblastomas. J Neuropathol Exp Neurol. 2021;nlab041. [DOI] [PubMed] [Google Scholar]
- 55. Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor‐infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17:1064‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nduom EK, Wei J, Yaghi NK, et al. PD‐L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18:195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garber ST, Hashimoto Y, Weathers SP, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 2016;18:1357‐1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeng J, Zhang XK, Chen HD, Zhong ZH, Wu QL, Lin SX. Expression of programmed cell death‐ligand 1 and its correlation with clinical outcomes in gliomas. Oncotarget. 2016;7:8944‐8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Z, Zhang C, Liu X, et al. Molecular and clinical characterization of PD‐L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. 2016;5:e1196310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miyazaki T, Ishikawa E, Matsuda M, et al. Assessment of PD‐1 positive cells on initial and secondary resected tumor specimens of newly diagnosed glioblastoma and its implications on patient outcome. J Neurooncol. 2017;133:277‐285. [DOI] [PubMed] [Google Scholar]
- 61. Heiland DH, Haaker G, Delev D, et al. Comprehensive analysis of PD‐L1 expression in glioblastoma multiforme. Oncotarget. 2017;8:42214‐42225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xue S, Hu M, Li P, et al. Relationship between expression of PD‐L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget. 2017;8:49702‐49712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Heynckes S, Gaebelein A, Haaker G, et al. Expression differences of programmed death ligand 1 in de‐novo and recurrent glioblastoma multiforme. Oncotarget. 2017;8:74170‐74177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berghoff AS, Kiesel B, Widhalm G, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017;19:1460‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19:1047‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Han J, Hong Y, Lee YS. PD‐L1 expression and combined status of PD‐L1/PD‐1‐positive tumor infiltrating mononuclear cell density predict prognosis in glioblastoma patients. J Pathol Transl Med. 2017;51:40‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee KS, Lee K, Yun S, et al. Prognostic relevance of programmed cell death ligand 1 expression in glioblastoma. J Neurooncol. 2018;136:453‐461. [DOI] [PubMed] [Google Scholar]
- 68. Pratt D, Dominah G, Lobel G, et al. Programmed death ligand 1 is a negative prognostic marker in recurrent isocitrate dehydrogenase‐wildtype glioblastoma. Neurosurgery. 2019;85:280‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. The Cancer Genome Atlas Program [Internet] . National Cancer Institute. 2021. [cited 21 May 2021]. Available from: http://cancergenome.nih.gov/
- 70. Chen Y, Wang Y, Luo H, et al. The frequency and inter‐relationship of PD‐L1 expression and tumour mutational burden across multiple types of advanced solid tumours in China. Exp Hematol Oncol. 2020;9:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Madore J, Vilain RE, Menzies AM, et al. PD‐L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti‐PD‐1/PD‐L1 clinical trials. Pigment Cell Melanoma Res. 2015;28:245‐253. [DOI] [PubMed] [Google Scholar]
- 72. Ilie M, Long‐Mira E, Bence C, et al. Comparative study of the PD‐L1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances: a potential issue for anti‐PD‐L1 therapeutic strategies. Ann Oncol. 2016;27:147‐153. [DOI] [PubMed] [Google Scholar]
- 73. Preusser M, Berghoff AS, Wick W, Weller M. Clinical Neuropathology mini‐review 6‐2015: PD‐L1: emerging biomarker in glioblastoma? Clin Neuropathol. 2015;34:313‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Doucette T, Rao G, Rao A, et al. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res. 2013;1:112‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gatalica Z, Snyder C, Maney T, et al. Programmed cell death 1 (PD‐1) and its ligand (PD‐L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014;23:2965‐2970. [DOI] [PubMed] [Google Scholar]
- 76. Perus LJM, Walsh LA. Microenvironmental heterogeneity in brain malignancies. Front Immunol. 2019;10:2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen Z, Hambardzumyan D. Immune Microenvironment in Glioblastoma Subtypes. Front Immunol. 2018;9:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sørensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour‐associated microglia/macrophages predict poor prognosis in high‐grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44:185‐206. [DOI] [PubMed] [Google Scholar]
- 79. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7‐H1 expression in tumor‐associated macrophages. Clin Cancer Res. 2013;19:3165‐3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mostafa H, Pala A, Högel J, et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J Hematol Oncol. 2016;9:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kmiecik J, Poli A, Brons NH, et al. Elevated CD3+ and CD8+ tumor‐infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71‐83. [DOI] [PubMed] [Google Scholar]
- 83. Ren M, Dai B, Kong YY, Lv JJ, Cai X. PD‐L1 expression in tumour‐infiltrating lymphocytes is a poor prognostic factor for primary acral melanoma patients. Histopathology. 2018;73:386‐396. [DOI] [PubMed] [Google Scholar]
- 84. Kim KH, Choi KU, Kim A, et al. PD‐L1 expression on stromal tumor‐infiltrating lymphocytes is a favorable prognostic factor in ovarian serous carcinoma. J Ovarian Res. 2019;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bellmunt J, Mullane SA, Werner L, et al. Association of PD‐L1 expression on tumor‐infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol. 2015;26:812‐817. [DOI] [PubMed] [Google Scholar]
- 86. Magnus T, Schreiner B, Korn T, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci. 2005;25:2537‐2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhao T, Li C, Wu Y, Li B, Zhang B. Prognostic value of PD‐L1 expression in tumor infiltrating immune cells in cancers: A meta‐analysis. PLoS One. 2017;12:e0176822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guo H, Ding Q, Gong Y, et al. Comparison of three scoring methods using the FDA‐approved 22C3 immunohistochemistry assay to evaluate PD‐L1 expression in breast cancer and their association with clinicopathologic factors. Breast Cancer Res. 2020;22:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang L, Sorensen MD, Kristensen BW, Reifenberger G, McIntyre TM, Lin F. D‐2‐hydroxyglutarate is an intercellular mediator in IDH‐mutant gliomas inhibiting complement and T cells. Clin Cancer Res. 2018;24:5381‐5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD‐L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD‐L1 checkpoint. Immunity. 2018;48:434‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD‐L1: a novel role of pro‐survival signalling in cancer. Ann Oncol. 2016;27:409‐416. [DOI] [PubMed] [Google Scholar]
- 93. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD‐L1/PD‐1‐mediated tumor immune escape. Mol Cancer. 2019;18:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Qian J, Wang C, Wang B, et al. The IFN‐γ/PD‐L1 axis between T cells and tumor microenvironment: hints for glioma anti‐PD‐1/PD‐L1 therapy. J Neuroinflammation. 2018;15:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7‐H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84‐88. [DOI] [PubMed] [Google Scholar]
- 97. Bankhead P, Fernández JA, McArt DG, et al. Integrated tumor identification and automated scoring minimizes pathologist involvement and provides new insights to key biomarkers in breast cancer. Lab Invest. 2018;98:15‐26. [DOI] [PubMed] [Google Scholar]
- 98. Qaiser T, Mukherjee A, Reddy Pb C, et al. HER2 challenge contest: a detailed assessment of automated HER2 scoring algorithms in whole slide images of breast cancer tissues. Histopathology. 2018;72:227‐238. [DOI] [PubMed] [Google Scholar]
- 99. Koelzer VH, Gisler A, Hanhart JC, et al. Digital image analysis improves precision of PD‐L1 scoring in cutaneous melanoma. Histopathology. 2018;73:397‐406. [DOI] [PubMed] [Google Scholar]
- 100. Thompson RH, Kuntz SM, Leibovich BC, et al. Tumor B7‐H1 is associated with poor prognosis in renal cell carcinoma patients with long‐term follow‐up. Cancer Res. 2006;66:3381‐3385. [DOI] [PubMed] [Google Scholar]
- 101. Shergold AL, Millar R, Nibbs RJB. Understanding and overcoming the resistance of cancer to PD‐1/PD‐L1 blockade. Pharmacol Res. 2019;145:104258. [DOI] [PubMed] [Google Scholar]
- 102. Xue S, Song G, Yu J. The prognostic significance of PD‐L1 expression in patients with glioma: a meta‐analysis. Sci Rep. 2017;7:4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Home—ClinicalTrials.gov [Internet] . Clinicaltrials.gov. 2020. [cited 1 May 2021]. Available from: https://clinicaltrials.gov/
- 104. Bristol‐Myers Squibb Announces Phase 3 CheckMate‐498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo (nivolumab) Plus Radiation in Patients with Newly Diagnosed MGMT‐Unmethylated Glioblastoma Multiforme [press release]. Princeton, NJ: Bristol‐Myers Squibb Company; May 9; 2019. https://bit.ly/2V9ceag Accessed May 1, 2021. [Google Scholar]
- 105. Bristol Myers Squibb Announces Update on Phase 3 CheckMate ‐548 Trial Evaluating Patients with Newly Diagnosed MGMT‐Methylated Glioblastoma Multiforme [press release]. Princeton, NJ: Bristol‐Myers Squibb Company; May 9; 2019: https://news.bms.com/news/details/2020/Bristol-Myers-Squibb-Announces-Update-on-Phase-3-CheckMate--548-Trial-Evaluating-Patients-with-Newly-Diagnosed-MGMT-Methylated-Glioblastoma-Multiforme/default.aspx Accessed 1 May 2021. [Google Scholar]
- 106. Weathers SPS, Kamiya‐Matsuoka C, Harrison RA, et al. Phase I/II study to evaluate the safety and clinical efficacy of atezolizumab (atezo; aPDL1) in combination with temozolomide (TMZ) and radiation in patients with newly diagnosed glioblastoma (GBM). J Clin Oncol. 2020;38:2511‐2511. [Google Scholar]
- 107. Lombardi G, Barresi V, Indraccolo S, et al. Pembrolizumab activity in recurrent high‐grade gliomas with partial or complete loss of mismatch repair protein expression: a monocentric, observational and prospective pilot study. Cancers (Basel). 2020;12:2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Awada G, Ben Salama L, De Cremer J, et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: a stratified, open‐label, single‐center phase 2 clinical trial (GliAvAx). J Immunother Cancer. 2020;8:e001146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lukas RV, Rodon J, Becker K, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol. 2018;140:317‐328. [DOI] [PubMed] [Google Scholar]
- 111. Nayak L, Molinaro AM, Peters K, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27:1048‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Reardon DA, Kim TM, Frenel JS, et al. Treatment with pembrolizumab in programmed death ligand 1‐positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE‐028 trial. Cancer. 2021;127:1620‐1629. [DOI] [PubMed] [Google Scholar]
- 113. Sahebjam S, Forsyth PA, Tran ND, et al. Hypofractionated stereotactic re‐irradiation with pembrolizumab and bevacizumab in patients with recurrent high‐grade gliomas: results from a phase I study. Neuro Oncol. 2021;23:677‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. de Groot J, Penas‐Prado M, Alfaro‐Munoz K, et al. Window‐of‐opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune‐suppressive macrophages. Neuro Oncol. 2020;22:539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti‐PD‐1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schalper KA, Rodriguez‐Ruiz ME, Diez‐Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470‐476. [DOI] [PubMed] [Google Scholar]
- 117. Reardon DA, Kaley TJ, Dietrich J, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J Clin Oncol. 2019;37:2032‐2032. [Google Scholar]
- 118. Reardon DA, Kaley TJ, Dietrich J, et al. Phase 2 study to evaluate safety and efficacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: An update. J Clin Oncol. 2017;35:2042‐2042. [Google Scholar]
- 119. Reardon DA, Kaley T, Dietrich J, et al. ATIM‐12. phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (Durvalumab [DUR]) In Patients with bevacizumab (BEV)‐Refractory Recurrent Glioblastoma (GBM). J Clin Onco. 2017;19:vi28. [Google Scholar]
- 120. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair‐deficient cancer: results from the phase II KEYNOTE‐158 study. J Clin Oncol. 2020;38:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Shen X, Zhao B. Efficacy of PD‐1 or PD‐L1 inhibitors and PD‐L1 expression status in cancer: meta‐analysis. BMJ. 2018;362:k3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Passiglia F, Bronte G, Bazan V, et al. PD‐L1 expression as predictive biomarker in patients with NSCLC: a pooled analysis. Oncotarget. 2016;7:19738‐19747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Colli LM, Machiela MJ, Myers TA, Jessop L, Yu K, Chanock S. Burden of nonsynonymous mutations among TCGA cancers and candidate immune checkpoint inhibitor responses. Cancer Res. 2016;76:3767‐3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206‐2211. [DOI] [PubMed] [Google Scholar]
- 128. Johanns TM, Miller CA, Dorward IG, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6:1230‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Touat M, Li YY, Boynton AN, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580(7804):517‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rittberg R, Harlos C, Rothenmund H, et al. Immune checkpoint inhibition as primary adjuvant therapy for an IDH1‐mutant anaplastic astrocytoma in a patient with CMMRD: a case report‐usage of immune checkpoint inhibition in CMMRD. Curr Oncol. 2021;28:757‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013;19:4917‐4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Matsuzaki J, Gnjatic S, Mhawech‐Fauceglia P, et al. Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107:7875‐7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Baitsch L, Legat A, Barba L, et al. Extended co‐expression of inhibitory receptors by human CD8 T‐cells depending on differentiation, antigen‐specificity and anatomical localization. PLoS One. 2012;7:e30852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fecci PE, Sampson JH. The current state of immunotherapy for gliomas: an eye toward the future. J Neurosurg. 2019;131:657‐666. [DOI] [PubMed] [Google Scholar]
- 137. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T‐cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24:4175‐4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T‐cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res. 2018;24:3792‐3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sears CL, Pardoll DM. The intestinal microbiome influences checkpoint blockade. Nat Med. 2018;24:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mansfield AS, Aubry MC, Moser JC, et al. Temporal and spatial discordance of programmed cell death‐ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann Oncol. 2016;27:1953‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ionescu DN, Downes MR, Christofides A, Tsao MS. Harmonization of PD‐L1 testing in oncology: a Canadian pathology perspective. Curr Oncol. 2018;25:e209‐e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Büttner R, Gosney JR, Skov BG, et al. Programmed death‐ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in non‐small‐cell lung cancer. J Clin Oncol. 2017;35:3867‐3876. [DOI] [PubMed] [Google Scholar]
- 143. Hirsch FR, McElhinny A, Stanforth D, et al. PD‐L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD‐L1 IHC assay comparison project. J Thorac Oncol. 2017;12:208‐222. [DOI] [PubMed] [Google Scholar]
- 144. Sanders S, Debinski W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int J Mol Sci. 2020;21:2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Adhikaree J, Moreno‐Vicente J, Kaur AP, Jackson AM, Patel PM. Resistance mechanisms and barriers to successful immunotherapy for treating glioblastoma. Cells. 2020;9:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Goldberg SB, Gettinger SN, Mahajan A, et al. Pembrolizumab for patients with melanoma or non‐small‐cell lung cancer and untreated brain metastases: early analysis of a non‐randomised, open‐label, phase 2 trial. Lancet Oncol. 2016;17:976‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pitter KL, Tamagno I, Alikhanyan K, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139:1458‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer. 2015;113:232‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Latchman Y, Wood CR, Chernova T, et al. PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol. 2001;2:261‐268. [DOI] [PubMed] [Google Scholar]
- 150. Sims JS, Ung TH, Neira JA, Canoll P, Bruce JN. Biomarkers for glioma immunotherapy: the next generation. J Neurooncol. 2015;123:359‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.