

Abstract
Introduction/Aims
ASP0367, or bocidelpar sulfate, is an orally administered small molecule that potently and selectively modulates peroxisome proliferator–activated receptor δ (PPARδ) to address mitochondrial dysfunction occurring in diseases including primary mitochondrial myopathy and Duchenne muscular dystrophy. The objectives of this first‐in‐human trial were to evaluate the safety/tolerability, pharmacokinetics, and pharmacodynamics of ASP0367 in healthy participants.
Methods
In this double‐blind phase 1 study, adult participants were randomized to single or multiple ascending oral doses of ASP0367 or placebo. The study duration was 1 and 14 days, respectively. Pharmacokinetic parameters under fed conditions were also evaluated.
Results
A total of 64 (single‐dose cohort) and 37 (multiple‐dose cohort) participants were included in the study. After single doses of 1 to 120 mg, ASP0367 was rapidly absorbed, with median time to maximum plasma concentration (tmax) of 1.50 to 2.24 hours under fasting conditions; ASP0367 concentrations declined in a multiphasic manner after reaching maximum plasma concentration. Under fed conditions, tmax was delayed 1.7 hours. After multiple once‐daily doses, mean half‐life of ASP0367 10 to 75 mg ranged from 14.1 to 17.5 hours; steady state was reached after 4 days. Negligible accumulation was observed after repeated dosing. No participants receiving ASP0367 discontinued treatment, and all treatment‐emergent adverse events were mild to moderate in severity; none were considered drug‐related. No clinically significant changes were observed on laboratory or electrocardiographic evaluation. Treatment‐ and dose‐dependent upregulation of six PPARδ target genes was observed with single and multiple doses of ASP0367.
Discussion
ASP0367, or bocidelpar sulfate, was well tolerated; rapid absorption, roughly dose‐proportional bioavailability, and effects on PPARδ target genes were demonstrated in healthy adult participants.
Keywords: Duchenne muscular dystrophy, mitochondrial disease, mitochondrial myopathy, peroxisome proliferator–activated receptors, PPARδ agonist
Abbreviations
- AE
adverse event
- AUC
area under the concentration‐time curve
- AUC24
area under the concentration‐time curve over 24 hours
- AUCinf
area under the concentration‐time curve from time zero to infinity
- AUCƮ
area under the concentration‐time curve from time of dosing to start of next dosing interval
- BMI
body mass index
- CK
creatine kinase
- Cmax
maximum concentration
- DMD
Duchenne muscular dystrophy
- ECG
electrocardiogram
- FAO
fatty acid oxidation
- FDA
US Food and Drug Administration
- ICH
International Council for Harmonisation
- PD
pharmacodynamic
- PK
pharmacokinetic
- PMM
primary mitochondrial myopathy
- PPAR
peroxisome proliferator–activated receptor
- QTcF
QT interval using the Fridericia correction
- t1/2
terminal elimination half‐life
- TEAE
treatment‐emergent adverse event
- tmax
maximum plasma concentration
- ULN
upper limit of normal
1. INTRODUCTION
Mitochondrial dysfunction and the associated impairments in energy production are involved in both inherited and acquired diseases. 1 , 2 Unfortunately, for some diseases, including primary mitochondrial myopathy (PMM) and Duchenne muscular dystrophy (DMD), in which mitochondrial dysfunction has been implicated, all or most existing therapies are aimed at managing certain aspects of the symptomatology associated with the disease or optimizing quality of life. Agents targeting the mitochondrial pathway in various muscular dystrophies were ineffective in phase 3 trials (ie, idebenone) or are undergoing investigation, including (−)‐epicatechin. 3 , 4 , 5 Therapies approved by the United States Food and Drug Administration (FDA) for DMD treatment primarily address dystrophin protein restoration and/or symptom management; they are not curative. 6 , 7 , 8 , 9 , 10 To date, no treatments exist that target the potential mitochondrial dysfunction hypothesized to play a key role in the underlying disease etiologies. 11 , 12 , 13 , 14 , 15 Although mitochondrial myopathies are caused predominantly by impaired oxidative phosphorylation in the mitochondria that leads to a deficit in energy production, especially in skeletal muscle, 11 DMD involves plasma membrane instability due to loss of dystrophin. It has been hypothesized that this triggers mitochondrial stress secondary to excessive calcium ion influx. 16 , 17 In addition, a significant reduction or deregulation of mitochondrial gene expression has been identified in muscles of patients with DMD and PMM. 18 , 19 , 20 Muscle mass is lost to different degrees in genetic abnormalities commonly seen in some myopathies and muscular dystrophies. 21 In vitro repair of injured muscle myofiber is inhibited when mitochondrial oxidative phosphorylation is impaired, indicating a role for targeting mitochondrial dysfunction to address skeletal muscle deterioration. 22
Peroxisome proliferator–activated receptors (PPARs) are nuclear hormone receptors that act as transcription factors, thereby affecting biological processes by modifying gene expression. 23 , 24 Of the three distinct types of PPARs (α, γ, and δ), PPARδ is expressed in skeletal muscle up to 50‐fold higher than PPARα and PPARγ, specifically enhancing fatty acid oxidation (FAO), activating energy uncoupling proteins (to provide energy for oxidative phosphorylation), and mitochondrial biogenesis. 23 , 25 In addition, synthetic PPARδ agonists increase expression of skeletal muscle genes, including those involved in mitochondrial respiration and oxidative metabolism. 23 Given the involvement of PPARδ in mitochondrial bioenergetics, PPARδ modulation could provide intriguing and promising potential for treatment of primary as well as secondary mitochondrial diseases, including, but not limited to, PMM and DMD, in which patients exhibit metabolic impairment in mitochondria that manifests as exercise intolerance, fatigue, and muscle wasting. 1 , 11 , 16 Moreover, in a murine model of DMD (mdx mice), synthetic PPARδ modulators that increased gene expression of putative PPARδ target genes upregulated mitochondrial regulatory pathways and increased running endurance. 25 , 26 Mitochondrial dysfunction leading to decreased cellular bioenergetics was evident in mdx myoblasts 25 and, importantly, this impairment was partially reversed with PPARδ modulation, 26 supporting the concept of PPARδ as a therapeutic target in DMD.
ASP0367, also known as bocidelpar sulfate and formerly known as MA‐0211, is a small molecule that potently and selectively modulates human PPARδ and, based on preclinical findings, is expected to address mitochondrial dysfunction, improve muscle abnormalities, and amplify function in patients with DMD. Clinical effects of ASP0367 are also of interest in mitochondrial disorders, such as PMM, which similarly have no curative treatments and can also affect skeletal muscle and cause muscle weakness. 27 , 28 In preclinical studies, ASP0367 increased mRNA expression and protein levels of PPARδ target genes in mdx mice and human DMD myotubes (data on file). In three separate studies, once‐daily ASP0367 treatment for up to 35 days increased the exercise endurance of mdx mice relative to the performance seen with mdx mice given vehicle (data on file). ASP0367‐mediated activation of PPARδ in mouse cells as well as human healthy and DMD muscle cells increased FAO (data on file), indicating preliminary amelioration of one key aspect of mitochondrial dysfunction. The aims of this first‐in‐human study were to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of single and multiple ascending oral doses of ASP0367 in healthy adults.
2. METHODS
2.1 Study design
This was a single‐center, two‐part, randomized, double‐blind study composed of separate single ascending oral dose (part 1) and multiple ascending oral dose (part 2) escalations (NCT03682484). This study was conducted in the United States (Parexel International in Baltimore, Maryland). Study participants were recruited via outdoor events, radio and internet advertisements, and email blasts.
Part 1 consisted of eight double‐blind dose cohorts and one open‐label food effect cohort with eight healthy participants per cohort. The study duration was 1 day. Participants in the double‐blind cohorts were randomized to receive a single oral dose of ASP0367 (n = 6) or matching placebo (n = 2) under fasting conditions (Table S1). The first cohort (cohort 1.1) received the maximum recommended starting dose of 1 mg ASP0367 or matching placebo based on ASP0367 preclinical pharmacology data. Sentinel dosing was used for double‐blind cohorts wherein two participants (one each on ASP0367 or placebo) were dosed 5 minutes apart and monitored for safety; the remaining participants in the cohort were dosed at least 24 hours later. Planned doses thereafter were not fixed; dose levels were flexible and could be adapted depending on emergent and cumulative safety and PK data. Dose escalation was halted if any of the planned next dose levels were expected to exceed the mean exposure levels (maximum concentration [Cmax] and area under the concentration‐time curve [AUC] over 24 hours [AUC24]) agreed upon with the regulatory authorities. All participants in the food effect cohort (cohort 1.8) received ASP0367 under fed conditions (high‐fat breakfast of approximately 1000 kcal with 50%‐60% from fat). In the food effect cohort, the dose given corresponded to a dose tested in one of the previous single ascending dose cohorts and was confirmed by the dose‐escalation committee. Decisions to advance or modify dose levels and initiate part 2 were guided by blinded evaluation by a dose‐escalation committee with representation from the sponsor and principal investigator or delegate.
Part 2 consisted of five double‐blind dose cohorts with 12 healthy participants per cohort randomized to multiple ascending oral doses of ASP0367 (n = 9) or matching placebo (n = 3), given once daily for 14 days under fasting conditions. Dose selection in part 2 was dependent on safety and PK data (including terminal elimination half‐life [t1/2], Cmax, and area under the concentration‐time curve from time zero to infinity [AUCinf]) from part 1, including data from participants taking extemporaneously prepared capsules (described in what follows).
This study was approved by the institutional review board and conducted in accordance with Good Clinical Practice, International Council for Harmonisation (ICH) guidelines, and relevant regulations and guidelines on study conduct and ethical principles with origins in the Declaration of Helsinki. Written informed consent was obtained from all participants.
2.2. Investigational product preparation
Two ASP0367 formulations, pre‐prepared capsules (1, 3, 10, and 30 mg) and extemporaneously prepared capsules (30, 60, and 120 mg), were administered during part 1 of the study because the pre‐prepared study drug capsules could not be administered at doses greater than 100 mg due to exceeding daily maximum impurity levels set forth by the ICH of Technical Requirements for Pharmaceuticals for Human Use Harmonised Tripartite Guideline, Impurities in New Drug Substances, Q3A and FDA Guidance for Industry, Q3B Impurities in New Drug Products. Therefore, extemporaneously prepared formulations were manufactured at the clinical unit for administration to cohorts 1.5 to 1.8 in part 1. Only extemporaneously prepared capsules (10, 40, and 75 mg) were used in part 2.
2.3. Study participants
Healthy male and female participants, aged 18 to 55 years, were enrolled in the study. Participants were required to have a body mass index (BMI) ranging from 18.5 to 32.0 kg/m2 and weigh at least 50 kg at screening. Females of non‐childbearing potential, childbearing potential who were not pregnant if using contraception, and who were lactating but not breastfeeding, were eligible. Sexually active males were eligible if using contraception. Participants with clinically significant allergic conditions (including drug allergies, asthma, eczema, or anaphylactic reactions, but excluding untreated, asymptomatic, seasonal allergies), family history of long QT syndrome, alcohol/substance abuse (including recent use of drugs of abuse), recent infection, and use of PPAR ligands in the previous 4 weeks were not eligible. In addition, participants using metabolism inducers in the previous 3 months or any medications (prescribed or nonprescribed medicinal product) in the previous 2 weeks were excluded.
2.4. Objectives and assessments
The primary objective of both parts 1 and 2 was to evaluate the safety and tolerability of single (part 1) or multiple (part 2) ascending oral doses of ASP0367. All observed and participant‐reported adverse events (AEs), including abnormal physical exam and test results, were recorded. Safety assessments included vital signs, laboratory tests (blood samples for hematology/biochemistry, urine samples for urinalysis), routine and continuous 12‐lead electrocardiograms, and real‐time cardiac monitoring. All evaluations were prespecified (Tables S2 and S3).
Secondary objectives of parts 1 and 2 also included evaluating PK and effect on QT interval using the Fridericia correction (QTcF). In part 1, blood samples to determine plasma concentrations of ASP0367 were obtained up to 72 hours postdose. In part 2, blood samples were drawn predose on day 1, then up to 16 hours postdose; predose on days 2, 4, 6, 8, 10, and 12; and predose on day 14, then up to 72 hours postdose. Samples were analyzed via liquid chromatography–tandem mass spectrometry in positive ion mode (SCIEX Triple Quad 5500 TurboIonSpray; CMIC, Inc, Hoffman Estates, IL). Noncompartmental analysis was used to calculate plasma PK parameters using Phoenix version 6.3 or higher (Certara L.P., Princeton, NJ). Additional bioanalytical methodology is presented in the Supporting Information online.
In part 1, two additional secondary objectives included: (1) evaluation of PD effect assessed by differential expression of 12 PPARδ target genes (ABCA1, ACAA2, ACADVL, CAT, CPT1a, HIST2H2BE, PDK4, PSMB10, RAB11B, SEMA7A, SLC25A20, and ZCCHC11, normalized against three control housekeeping genes); and (2) determination of the effect of food on PK parameters. Blood samples for determination of gene expression in parts 1 and 2 and serum myostatin, serum follistatin, and plasma acylcarnitine levels in part 2 were obtained according to the schedule of outcomes assessments displayed in the Supporting Information (Tables S2 and S3). For PPARδ target gene expression, RNA was isolated from blood samples collected in PAXgene tubes and subsequently evaluated on a custom chip (NanoString TE_A0367; NeoGenomics Laboratories, Houston, TX). RNA sample concentration was assessed using a spectrophotometer (NanoDrop; Thermo Fisher Scientific, Waltham, MA) and for fragmentation using a bioanalyzer (Model 2100; Agilent, Santa Clara, CA). Results were analyzed with nSolver software version 3.0 (NanoString Technologies, Seattle, WA) using manufacturer‐recommended settings. Analyses of myostatin and follistatin were conducted using enzyme‐linked immunosorbent assay and analysis of acyl‐carnitines was performed using flow‐injection analysis tandem mass spectrometry.
2.5. Statistical methods
Safety assessments were evaluated using descriptive statistics and QT prolongation was evaluated using linear mixed‐effect models. Descriptive statistics were also used to characterize PK parameters, plasma concentrations, and PD effects. Steady state in part 2 was evaluated using visual inspection of trough concentrations of individual participants by day overlaid with a mean profile on a spaghetti plot, and dose proportionality was estimated graphically using natural log‐transformed scatterplots (part 1) and linear regression of natural log‐transformed parameter and dose (power model; part 2). Dose proportionality was concluded in the specified dose range if the 90% confidence interval (CI) of the slope was entirely within the prespecified limits of 0.656 to 1.34. Analysis of variance with the food condition (ie, fed or fasted) as a fixed effect was fitted on natural log‐transformed PK parameters and least‐squares mean differences between conditions were estimated. Normalized gene expression for the 12 target genes was evaluated and summarized using log‐transformed data and change from baseline by time‐point. Graphical representations were produced to show the anti‐log of the difference, which was used to compare differences between postbaseline and baseline ratio, also known as the fold change.
The safety analysis population included all participants who took at least one dose of ASP0367. PK analyses were conducted in all participants from the safety analysis population assigned to ASP0367 with data available to evaluate at least one primary PK parameter; PD analyses were conducted in participants with sufficient PD measurements. All data processing, summarization, and analyses were performed using SAS version 9.2 or higher (SAS Institute, Inc, Cary, NC).
3. RESULTS
3.2. Participant disposition and study population
Overall, part 1 included eight cohorts (Table 1) and part 2 included three cohorts (Table 2). A total of 64 (part 1) and 37 (part 2) participants were assessed for safety, PK, and PD. Of note, most participants in parts 1 and 2 were black or African American (88% and 73%, respectively).
TABLE 1.
Part 1 study population
| Parameter | PBO (n = 14) | 1 mg PP (n = 6) | 3 mg PP (n = 6) | 10 mg PP (n = 6) | 30 mg PP (n = 6) | 30 mg EP (n = 6) | 30 mg EP FE (n = 8) | 60 mg EP (n = 6) | 120 mg EP (n = 6) | Total (N = 64) |
|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | ||||||||||
| Median | 35 | 30 | 32 | 34 | 30 | 35 | 27 | 29 | 46 | 32 |
| Min, max | 23, 44 | 20, 47 | 24, 41 | 30, 45 | 21, 40 | 19, 55 | 20, 43 | 26, 34 | 32, 53 | 19, 55 |
| Male sex a | 7 (50) | 3 (50) | 1 (17) | 2 (33) | 1 (17) | 4 (67) | 6 (75) | 6 (100) | 5 (83) | 35 (55) |
| Hispanic or Latino a | 3 (21) | 1 (17) | 1 (17) | 2 (33) | 0 | 0 | 1 (13) | 0 | 2 (33) | 10 (16) |
| Race | ||||||||||
| White a | 0 | 1 (17) | 0 | 1 (17) | 1 (17) | 1 (17) | 2 (25) | 0 | 1 (17) | 7 (11) |
| Black or AA a | 13 (93) | 5 (83) | 6 (100) | 5 (83) | 5 (83) | 5 (83) | 6 (75) | 6 (100) | 5 (83) | 56 (88) |
| Asian a | 1 (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2) |
| Other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BMI (kg/m2) | ||||||||||
| Median | 28 | 24 | 29 | 25 | 27 | 26 | 27 | 26 | 27 | 27 |
| Min, max | 19, 31 | 22, 28 | 22, 31 | 21, 29 | 24, 30 | 22, 31 | 19, 31 | 22, 30 | 24, 31 | 18, 31 |
Abbreviations: AA, African American; BMI, body mass index; EP, extemporaneously prepared; FE, food effect; PBO, placebo; PP, pre‐prepared.
Data expressed as number (%).
TABLE 2.
Part 2 study population
| Parameter | PBO (n = 10) | 10 mg EP (n = 9) | 40 mg EP (n = 9) | 75 mg EP (n = 9) | Total (N = 37) |
|---|---|---|---|---|---|
| Age (years) | |||||
| Median | 41 | 36 | 35 | 33 | 36 |
| Min, max | 23, 55 | 26, 51 | 25, 48 | 24, 51 | 23, 55 |
| Male sex a | 4 (40) | 7 (78) | 8 (89) | 7 (78) | 26 (70) |
| Hispanic or Latino a | 2 (20) | 3 (33) | 2 (22) | 1 (11) | 8 (22) |
| Race a | |||||
| White | 2 (20) | 3 (33) | 2 (22) | 2 (22) | 9 (24) |
| Black or AA | 7 (70) | 6 (67) | 7 (78) | 7 (78) | 27 (73) |
| Asian | 0 | 0 | 0 | 0 | 0 |
| Other | 1 (10) | 0 | 0 | 0 | 1 (3) |
| BMI (kg/m2) | |||||
| Median | 25 | 27 | 26 | 27 | 27 |
| Min, max | 21, 29 | 22, 31 | 20, 31 | 22, 31 | 20, 31 |
Abbreviations: AA, African American; BMI, body mass index; EP, extemporaneously prepared; PBO, placebo; PP, pre‐prepared.
Data expressed as number (%).
In part 2, although stopping criteria were not met at the ASP0367 75‐mg dose, day 14 exposure approached 80% of the mean exposure limit with only 20% dose increments permitted, thus further dose escalation in additional cohorts was not conducted.
3.3. Pharmacokinetics for ASP0367 single doses (part 1)
After single oral doses of 1 to 120 mg, ASP0367 was rapidly absorbed across the dose range (Figure 1A) with a median time to maximum plasma concentration (tmax) of 1.50 to 2.24 hours under fasting conditions (Table 3). Most ASP0367 plasma concentrations at 15 minutes postdose (across all doses) in the absorption phase were below the limit of quantification (0.3 ng/mL). After reaching Cmax, ASP0367 plasma concentrations appeared to decline in a multiphasic manner. In the elimination phase, plasma concentrations were measurable for 1‐ and 3‐mg doses through 12 to 16 hours postdose. This increased to 72 hours postdose for doses higher than 10 mg. At 120 mg, all samples had measurable concentrations at 72 hours postdose. At ASP0367 doses of 10 mg or higher, estimated mean t1/2 was similar and ranged from 12.0 to 16.1 hours. The AUCinf and Cmax appeared consistent and comparable between pre‐prepared and extemporaneously prepared formulations at the 30‐mg dose.
FIGURE 1.
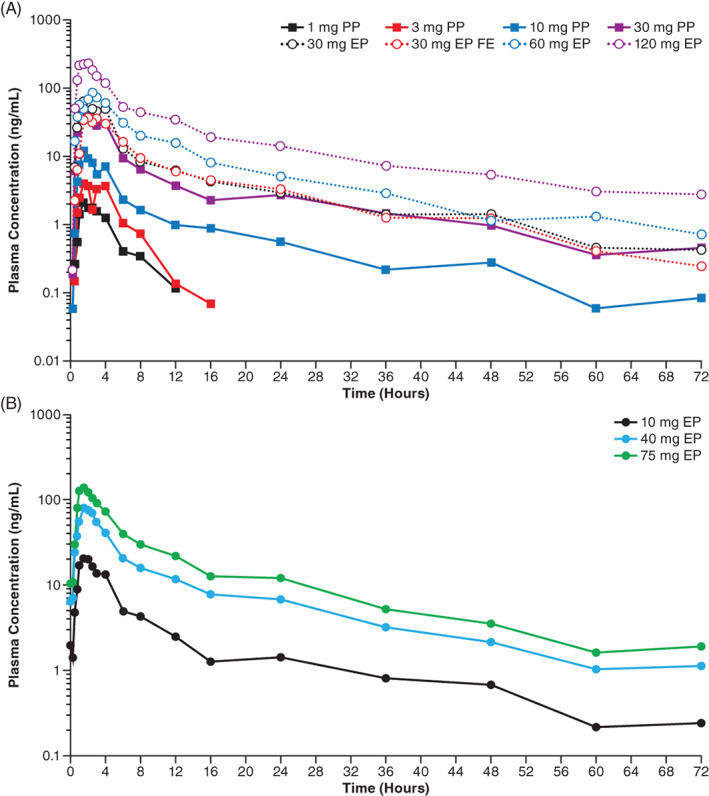
Mean plasma concentration‐time profiles of ASP0367. A, Mean plasma concentration‐time profiles of single oral doses of ASP0367. B, Mean plasma concentration‐time profiles of multiple, oral, once‐daily doses of ASP0367 on day 14. Abbreviations; EP, extemporaneously prepared; FE, food effect; PP, pre‐prepared
TABLE 3.
Pharmacokinetic parameters of single ASP0367 oral doses a
| Parameter | 1 mg PP (n = 6) | 3 mg PP (n = 6) | 10 mg PP (n = 6) | 30 mg PP (n = 6) | 30 mg EP (n = 6) | 30 mg EP FE (n = 8) | 60 mg EP (n = 6) | 120 mg EP (n = 6) |
|---|---|---|---|---|---|---|---|---|
| AUC24 (h∙ng/mL) | 8.77 (5.61) | 19.9 (9.26) | 53.8 (28.3) | 229 (118) | 320 (166) | 258 (56.3) | 533 (272) | 1250 (480) |
| AUCinf (h∙ng/mL) | 10.1 (NA) | 23.0 (9.00) | 71.7 (37.9) | 308 (160) | 342 (227) | 299 (52.5) | 634 (315) | 1810 (534) |
| AUClast (h∙ng/mL) | 8.05 (5.44) | 18.9 (9.17) | 62.2 (35.0) | 279 (145) | 369 (213) | 307 (56.4) | 632 (286) | 1540 (533) |
| CL/F (L/h) | 121 (NA) | 147 (58.9) | 182 (99.1) | 230 (333) | 111 (49.2) | 103 (16.9) | 114 (48.1) | 69.9 (17.2) |
| Cmax (ng/mL) | 2.50 (1.54) | 4.76 (2.20) | 12.7 (8.97) | 66.8 (40.0) | 77.4 (40.4) | 62.7 (36.6) | 93.5 (54.3) | 257 (86.0) |
| t1/2 (h) | 3.19 (NA) | 2.47 (0.615) | 15.0 (7.20) | 14.7 (2.94) | 13.8 (5.78) | 14.1 (1.92) | 12.0 (4.51) | 16.1 (1.98) |
| tmax (h), median (min, max) | 1.50 (0.750, 3.00) | 2.00 (1.50, 3.98) | 1.75 (1.02, 4.02) | 1.50 (1.00, 2.50) | 1.76 (0.983, 4.02) | 3.49 (1.50, 15.9) | 2.24 (1.53, 4.00) | 1.52 (0.983, 2.03) |
| Vz/F (L) | 335 (NA) | 498 (150) | 3270 (858) | 3850 (4440) | 1920 (442) | 2080 (439) | 2070 (1330) | 1660 (529) |
Note: Data for pharmacokinetic analysis set; some dose and parameter values are based on lower n (not shown).
Abbreviations: AUC24, area under the concentration‐time curve over 24 hours; AUCinf, area under the concentration‐time curve from time zero to infinity; AUClast, area under the concentration‐time curve from time 0 to the time of last quantifiable. concentration; CL/F, apparent oral clearance; Cmax, maximum concentration; EP, extemporaneously prepared; FE, food effect; NA, not applicable; PP, pre‐prepared; SD, standard deviation; t1/2, terminal elimination half‐life; tmax, time to maximum concentration; Vz/F, apparent volume of distribution.
Data expressed as mean (SD), unless noted otherwise.
When 30 mg of ASP0367 was administered with a high‐fat meal, tmax was delayed 1.7 hours, resulting in a median tmax of 3.49 hours, with individual values ranging from 1.50 to 15.9 hours. Administration of ASP0367 30 mg with a high‐fat meal slightly reduced Cmax, possibly due to delayed absorption; however, AUCinf was not affected by food consumption (Table 4).
TABLE 4.
Food effects on pharmacokinetic parameters
| Parameter | 30‐mg ASP0367 EP fed | 30‐mg ASP0367 EP fasted | Mean ratio (%) | 90% CI (%) | ||
|---|---|---|---|---|---|---|
| n | Geometric LS mean | n | Geometric LS mean | |||
| AUCinf (h∙ng/mL) | 5 | 295.60 | 5 | 298.84 | 98.92 | 61.38‐159.42 |
| AUClast (h∙ng/mL) | 8 | 302.95 | 6 | 323.31 | 98.71 | 64.62‐135.89 |
| Cmax (ng/mL) | 8 | 50.43 | 6 | 68.87 | 73.22 | 37.65‐142.44 |
Note: Pharmacokinetic analysis set; assessment is based on an analysis of variance performed on natural logarithmic‐transformed parameters with food condition as a fixed effect.
Abbreviations: AUCinf, area under the concentration‐time curve from time zero to infinity; AUClast, area under the concentration‐time curve from time 0 to the time of last quantifiable concentration; CI, confidence interval; Cmax, maximum concentration; EP, extemporaneously prepared; LS, least squares.
3.4. Pharmacokinetics of multiple ASP0367 doses (part 2)
After multiple oral doses of 10 to 75 mg, ASP0367 was absorbed across the dose range within 2 hours on day 14 (Figure 1B and Table 5). On day 14, mean t1/2 values were similar across the dose range and ranged from 14.1 to 17.5 hours; mean values for oral clearance and volume of distribution also appeared consistent with repeat dosing. Negligible accumulation was observed with repeat dosing based on the AUC accumulation ratio ranging from 1.25 to 1.36. Mean peak‐trough ratio values calculated over the dosing period ranged between 15.0 and 18.7. With negligible accumulation, steady‐state ASP0367 concentrations after daily dosing were achieved soon after the initial dose. Steady state was reached after 4 days of ASP0367 administration, irrespective of dose level.
TABLE 5.
Pharmacokinetic parameters of multiple ASP0367 oral doses
| Parameter, mean (SD) a | Day 1 | Day 14 | ||||
|---|---|---|---|---|---|---|
| 10 mg EP (n = 9) | 40 mg EP (n = 9) | 75 mg EP (n = 9) | 10 mg EP (n = 9) b | 40 mg EP (n = 9) | 75 mg EP (n = 9) | |
| AUC24 (h∙ng/mL) | 97.1 (55.0) | 349 (98.0) | 666 (266) | NA | NA | NA |
| AUCƮ (h∙ng/mL) | NA | NA | NA | 114 (52.3) | 469 (178) | 822 (351) |
| Cmax (ng/mL) | 23.6 (13.1) | 72.9 (33.0) | 152 (99.3) | 23.8 (13.7) | 92.8 (34.8) | 149 (74.6) |
| tmax (h), median (min, max) | 1.52 (1.00, 3.00) | 2.00 (1.00, 3.97) | 1.48 (0.983, 2.50) | 1.50 (1.00, 4.00) | 1.50 (0.750, 4.07) | 1.50 (0.750, 3.00) |
| Rac (AUC) | NA | NA | NA | 1.25 (0.337) | 1.36 (0.478) | 1.26 (0.318) |
| CLss/F (L/h) | NA | NA | NA | 111 (65.3) | 97.1 (35.8) | 104 (36.2) |
| PTR | NA | NA | NA | 18.7 (14.1) | 16.3 (6.56) | 15.0 (7.79) |
| t1/2 (h) | NA | NA | NA | 17.5 (6.29) | 14.1 (7.93) | 14.5 (1.93) |
| Vz/F (L) | NA | NA | NA | 2780 (1510) | 1770 (706) | 2150 (700) |
Abbreviations: AUCƮ, area under the concentration‐time curve from the last time of dosing to the start of the next dosing interval; AUC24, area under the concentration‐time curve over 24 hours; CL/F, apparent oral clearance; Cmax, maximum concentration; EP, extemporaneously prepared; NA, not applicable; PP, pre‐prepared; PTR, peak‐trough ratio; Rac, accumulation ratio; SD, standard deviation; ss, steady state; t1/2, terminal elimination half‐life; tmax, time to maximum concentration; Vz/F, apparent volume of distribution.
All parameters expressed as mean (SD), unless noted otherwise.
n = 8 for t1/2 and Vz/F.
Both AUC24 and Cmax increased in a dose‐proportional manner with parameters for days 1 and 14 showing the 90% CI of the slope to be within the prespecified limits (Table S4). Dose escalation was stopped at ASP0367 75 mg due to mean AUC from the time of dosing to the start of the next dosing interval (AUCƮ) and Cmax of 822 h·ng/mL and 149 ng/mL, respectively, reaching prespecified exposure limits.
3.5. Safety and tolerability
No participants discontinued treatment during part 1. In part 2, two participants randomized to placebo discontinued treatment and withdrew from the study; one of the participants was replaced. In both parts 1 and 2, all treatment‐emergent AEs (TEAEs) were considered mild to moderate in severity. No clinically relevant dose‐ or treatment‐related trends in AEs were observed and no dose‐limiting safety findings were reported. No TEAE was determined to be study drug‐related. No deaths, serious AEs, or TEAEs leading to withdrawal of treatment occurred in this study.
In part 1, of the participants who received ASP0367, three reported medical device site reactions (dermatitis due to electrocardiogram [ECG] leads) and one participant each reported vessel puncture site pain, constipation, an arthropod bite, pain in the extremity, and headache. No TEAEs were reported with the ASP0367 60‐ and 120‐mg doses. All AEs in the placebo group were also medical device site reactions arising from dermatitis due to ECG leads. Similarly, most part 2 participants had medical device site reactions (dermatitis due to ECG leads); other AEs included a laceration (n = 1) in the ASP0367 10‐mg dose group; headache (n = 2), dry skin (n = 2), and nausea (n = 1) with the ASP0367 40‐mg dose; and rhinorrhea (n = 1) with the ASP0367 75‐mg dose.
No clinically relevant results or changes from baseline were observed in clinical laboratory analyses. A similar distribution of participants with amylase or lipase elevations above the upper limit of normal (ULN) were reported in both parts 1 and 2 (ASP0367, n = 11; placebo, n = 4). In part 1, four participants had lipase elevations (ASP0367, n = 3; placebo, n = 1) that peaked at ≥2× ULN that subsequently returned to normal levels within 1 day or by last follow‐up assessment. In part 2, four participants who received ASP0367 (and one participant receiving placebo) had lipase elevations ≥2× ULN that also subsequently returned to normal or near normal during at least one time‐point; minor elevations in amylase were observed intermittently. Participants in both parts were asymptomatic and these elevations resolved without treatment. None of the amylase or lipase elevations were reported as TEAEs. One participant in part 1 at ASP0367 60 mg also had transient elevations for troponin I (creatine kinase–myoglobin binding and troponin T were within normal limits throughout the study) above the ULN on day 9 and on day 21 poststudy, but returned to normal by day 30. One participant in part 2 at ASP0367 40 mg had creatine kinase (CK) elevations (laboratory reference range of >308 U/L) from a baseline level of 267 U/L to a maximum value of 723 U/L on day 7 (predose); the participant was walking extensively in the clinical unit and when instructed not to exercise strenuously per study restrictions, the CK trended toward baseline and was within the reference range by day 12. No participants had clinically significant elevations of liver enzymes (alanine aminotransferase or aspartate aminotransferase) during the study.
From routine 12‐lead ECG readings, no clinically significant TEAEs were attributable to study treatments. For participants in both parts, neither QTcF values over 450 milliseconds, nor QTcF changes from baseline over 30 milliseconds, were observed across time‐points and treatment groups. Although changes in PR interval over 200 milliseconds and QRS duration of at least 110 milliseconds were observed, no clinically relevant dose‐ or treatment‐related trends were observed and no TEAEs were reported. Based on results of the analysis for plasma ASP0367 concentration and QTc relationship using a linear mixed‐effect model, ASP0367 did not significantly prolong the QTcF interval within the concentration range up to 420 ng/mL (Table S5). The magnitude of the placebo‐corrected baseline‐adjusted QTcF prolongation at the predefined exposure limit (175 ng/mL) and observed Cmax (420 ng/mL) did not reach the threshold of 10 milliseconds required for regulatory significance (Figure S1).
3.6. Pharmacodynamic effect of ASP0367 on PPARδ target gene expression
Expression of 12 PPARδ target genes was assessed in blood samples collected from participants in cohorts 1.3 through 1.7 in part 1 and in all three cohorts in part 2. Fold change from baseline was assessed at four time‐points (2, 4, 8, and 24 hours) postdosing. Treatment with a single dose of ASP0367 at 10 mg or higher showed consistent treatment‐ and dose‐dependent upregulation of six PPARδ target genes (ABCA1, ACAA2, ACADVL, CPT1a, PDK4, and SLC25A20), including those involved in FAO (Figure S2). Upregulation appeared rapid and persisted over the 24‐hour sampling period. Although the time to peak expression varied between individuals, the kinetics of gene expression upregulation suggested a saturated dose response as there was no significant increase in participants who received 60 mg ASP0367 compared with those who received 120 mg ASP0367.
The pattern of ASP0367‐mediated targeting of PPARδ target gene expression was also similar in participants who received daily dosing of ASP0367. In part 2, dose‐related effects on the same six PPARδ target genes as in part 1 samples demonstrated upregulation of gene expression in blood samples from both days 1 and 14 (Figure 2). The gene upregulation was also rapid and persisted over the 24‐hour sampling period on both days. Last, multiple doses of ASP0367 up to 75 mg did not appear to have a consistent treatment‐ or dose‐related effect on plasma acyl‐carnitines, serum follistatin, or serum myostatin.
FIGURE 2.
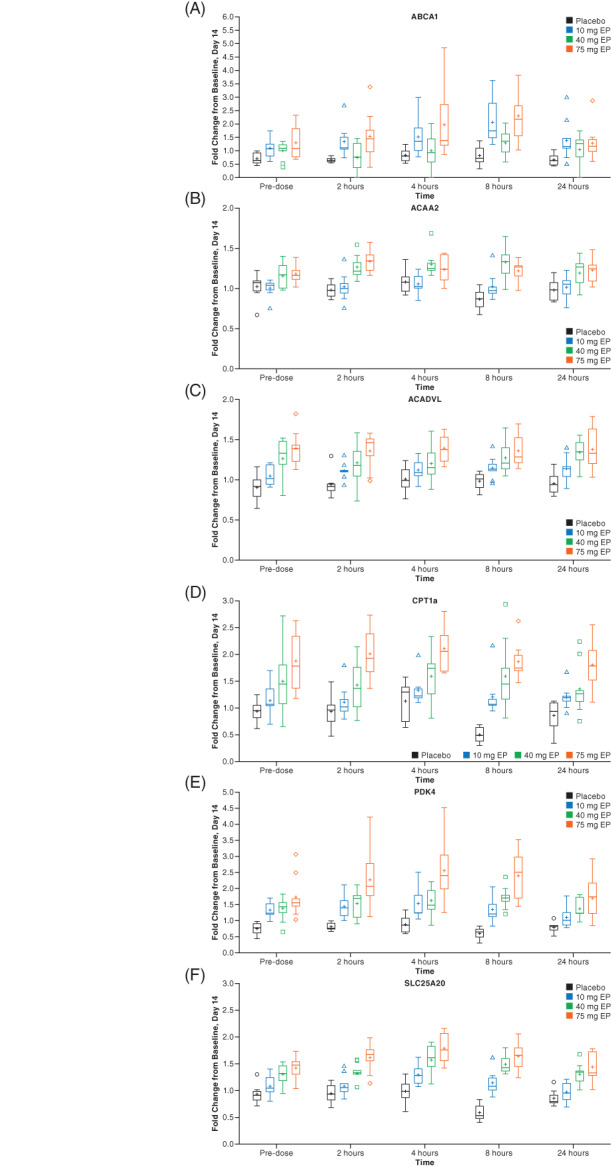
Gene expression graphs for six PPARδ target genes with multiple ascending once‐daily doses of ASP0367 during part 2, day 14. The boxplots for fold change from baseline to day 14 are shown for ABCA1 (A), ACAA2 (B), ACADVL (C), CPT1a (D), PDK4 (E), and SLC25A20 (F). There were eight participants in the placebo group and nine participants in each ASP0367 group at each of the doses administered 10, 40, and 75 mg. Abbreviation: EP, extemporaneously prepared
4. DISCUSSION
In this study of ASP0367, single ascending doses from 1 to 120 mg and multiple doses from 10 to 75 mg/day were considered safe and well tolerated in healthy adult participants given that no AEs were considered related to study treatments and no AEs led to discontinuation of study treatments. Although elevations in lipase and amylase were observed, participants remained asymptomatic and the elevated values returned to baseline values within 1 day or by the last follow‐up assessment without treatment. The CK elevations reported in one participant were of clinical interest until it was determined that the participant had increased physical activity while at the study site. In addition, this study population was predominantly (>80%) black/African American; higher baseline and subsequent CK values have been identified in African Americans compared with other racial groups. 29 Of note, immunosuppressive effects observed with deflazacort, a corticosteroid used in DMD management, 30 were not observed with ASP0367, as judged by lack of substantial changes to neutrophil and monocyte counts. Overall, there was no evidence for clinically relevant changes on laboratory parameters and QT interval from ASP0367.
ASP0367 displayed rapid absorption, minimal accumulation, and exposure in a dose‐proportional manner, with a t1/2 ranging from 12.0 to 16.1 hours. When administered with a high‐fat meal, the Cmax of ASP0367 was delayed by about 1.7 hours, which could have contributed to the slight effects on the Cmax.
Treatment‐ and dose‐dependent effects on PPARδ target genes were observed. These effects were similar to those identified in nonclinical studies. In participants, no effect on serum follistatin, serum myostatin, or plasma acyl‐carnitine was observed. Of all the PPAR subtypes, PPARδ is most abundant in skeletal muscle and its activation increases skeletal muscle lipid oxidation in addition to regulating genes involved in lipid metabolism. 24 As expected, ASP0367‐mediated PPARδ modulation impacted the expression of genes associated with mitochondrial FAO. 25 The elevated expression in six PPARδ‐responsive genes included FAO‐related genes, which were consistent with preclinical in vivo and human ex vivo findings. As mitochondrial impairment is a key physiological parameter in PMM and DMD, the PD effect of ASP0367 on PPARδ target genes suggests that addressing metabolic insufficiency may play a role in improving patient outcomes and quality of life for individuals with these conditions. Although increased expression was observed for only some of the PPARδ target genes associated with mitochondrial function, the role of the upregulation of these selected pathways in overcoming mitochondrial defects in individuals with PMM and DMD is currently unclear and remains to be tested.
Limitations to this study include that it was single center and conducted in healthy adult participants, with most participants being black or African American. Subsequently, generalizability to patients with mitochondrial dysfunction, including those with DMD and PMM, may be limited. However, in October 2020, the US FDA granted a “fast track” designation for development of ASP0367 as a potential treatment option for primary mitochondrial myopathies. 31 At the time of submission of this manuscript, a phase 2/3 study in patients with PMM (NCT04641962) and a phase 1b study in pediatric male patients with DMD (NCT04184882) are ongoing to further evaluate the safety, tolerability, and efficacy of ASP0367. Future ASP0367 evaluation in dynamic mitochondrial myopathies are of interest, considering the proposed effects of PPARδ modulation in mitochondrial bioenergetics.
In conclusion, single and repeated doses of ASP0367 demonstrated an acceptable safety profile and were well tolerated in healthy participants. Based on the positive gene expression data in this study, continued clinical development and evaluation of ASP0367 in the target populations of patients with diseases involving primary or secondary mitochondrial dysfunction are warranted.
CONFLICT OF INTEREST
M.I., S.T.‐W., R.A.S., T.W., A.Y., A.K., T.U., and G.J.M. are employees of Astellas Pharma Global Development, Inc. R.D.G. is an employee of Parexel International, which received funding from Astellas Pharma, Inc.
AUTHOR CONTRIBUTIONS
M.I., S.T.‐W., R.A.S., T.W., A.Y., A.K., and R.D.G. contributed to the study design and acquisition of study data. T.W., A.Y., and A.K. contributed to the analysis of study data. All authors interpreted the study data, critically reviewed the manuscript, and provided final approval.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
FIGURE S1 Scatterplot of ASP0367 concentrations against baseline‐adjusted QTcF (dQTcF) intervals grouped by dose. Analysis was performed on the safety analysis population excluding all participants from the food effect cohort in part 1 and two placebo participants from part 2. Points (circle, diamond, square, star) indicate the plasma concentration at each dose/preparation (per participant) and the corresponding dQTcF. Asterisks indicate placebo. Participants who received placebo were included in the analysis with concentration set to zero. Plasma concentrations that were below quantification limit (BQL) were treated as zero in the analysis and were included in the analysis. Black and gray bands are the 90% confidence interval around the predicted values in part 1 (CI_P1) and part 2 (CI_P2), respectively. Solid black and gray lines are the predicted value in part 1 and part 2 based on the final linear mixed‐effect model. Black medium dashed (L1) and black medium‐short‐medium t dashed lines (L2) form the locally estimated scatterplot smoothing regression line for parts 1 and 2, respectively. Abbreviations: dQTcF, baseline‐adjusted QTcF; EP, extemporaneously prepared; PP, pre‐prepared; QTcF, QT corrected using the Fridericia formula.
FIGURE S2 Gene expression graphs for six PPARδ target genes with single oral doses of ASP0367 during part 1. The boxplots for fold change from baseline are shown for ABCA1 (A), ACAA2 (B), ACADVL (C), CPT1a (D), PDK4 (E), and SLC25A20 (F). The number of patients in the placebo group and each ASP0367 dose group were 10 and 6, respectively, except for ABCA1: placebo, n = 8; ASP0367 10 mg PP, n = 4; ASP0367 30 mg PP, n = 5; ASP0367 120 mg PP, n = 4 and for PDK4: ASP0367 10 mg PP, n = 5. Abbreviations: EP, extemporaneously prepared; PP, pre‐prepared.
TABLE S1 Final dose scheme
TABLE S2 Schedule of outcomes assessments for part 1
TABLE S3 Schedule of outcomes assessments for part 2
TABLE S4 Statistical assessment of dose proportionality for ASP0367 on day 1 and after multiple dosing day 14 for part 2
TABLE S5 Effect of ASP0367 on ddQTcF at the predefined exposure limit concentration and maximum observed concentration
ACKNOWLEDGMENTS
Medical writing/editorial support was provided by Stephanie Phan, PharmD, Amit Lugade, PhD, and Elizabeth Hermans, PhD (Peloton Advantage, LLC, an OPEN Health Co, Parsippany, NJ), and funded by the study sponsor.
Ito M, Tauscher‐Wisniewski S, Smulders RA, et al. Single‐ and multiple‐dose safety, tolerability, pharmacokinetic, and pharmacodynamic profiles of ASP0367, or bocidelpar sulfate, a novel modulator of peroxisome proliferator‐activated receptor delta in healthy adults: Results from a phase 1 study. Muscle & Nerve. 2022;65(1):110-120. doi: 10.1002/mus.27436
Funding information Astellas Pharma, Inc.
DATA AVAILABILITY STATEMENT
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
REFERENCES
- 1. Cohen BH. Neuromuscular and systemic presentations in adults: diagnoses beyond MERRF and MELAS. Neurotherapeutics. 2013;10:227‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Komen JC, Thorburn DR. Turn up the power–pharmacological activation of mitochondrial biogenesis in mouse models. Br J Pharmacol. 2014;171:1818‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buyse GM, Voit T, Schara U, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double‐blind randomised placebo‐controlled phase 3 trial. Lancet. 2015;385:1748‐1757. [DOI] [PubMed] [Google Scholar]
- 4.Santhera to discontinue phase 3 SIDEROS study and development of Puldysa® in Duchenne muscular dystrophy (DMD) and focus on vamorolone. https://www.santhera.com/assets/files/press-releases/2020-10-06_siderospuldysa_e_final.pdf. Updated October 6, 2020. Accessed July 15, 2021.
- 5. McDonald CM, Ramirez‐Sanchez I, Oskarsson B, et al. (‐)‐Epicatechin induces mitochondrial biogenesis and markers of muscle regeneration in adults with Becker muscular dystrophy. Muscle Nerve. 2021;63:239‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amondys 45 (casimersen) injection prescribing information. https://www.amondys45.com/amondys45_(casimersen)_prescribing_information.pdf. Updated February 2021. Accessed July 15, 2021.
- 7.Exondys 51 (eteplirsen) injection prescribing information. https://www.exondys51.com/modules/exondys/files/exondys51pi.pdf. Updated July 2020. Accessed July 15, 2021.
- 8.Vyondys 53 (golodirsen) injection prescribing information. https://www.vyondys53.com/static/patient/assets/vyondys53_(golodirsen)_prescribing_Information.pdf. Updated February 2021. Accessed July 15, 2021.
- 9.Emflaza® (deflazacort) tablets prescribing information. https://emflaza.com/wp-content/uploads/2020/10/prescribing-information.pdf. Updated June 2021. Accessed July 15, 2021.
- 10.Viltepso (viltolarsen) injection prescribing information. https://www.viltepso.com/prescribing-information. Updated March 2021. Accessed July 15, 2021.
- 11. Ahmed ST, Craven L, Russell OM, Turnbull DM, Vincent AE. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics. 2018;15:943‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vila MC, Rayavarapu S, Hogarth MW, et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017;24:330‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang PB, Morrison L, Iannaccone ST, et al; Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic MedicineEvidence‐based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;84:1369‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes (Basel). 2020;11:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Timpani CA, Hayes A, Rybalka E. Revisiting the dystrophin‐ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses. 2015;85:1021‐1033. [DOI] [PubMed] [Google Scholar]
- 17. Kornegay JN, Spurney CF, Nghiem PP, Brinkmeyer‐Langford CL, Hoffman EP, Nagaraju K. Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials. ILAR J. 2014;55:119‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baron D, Magot A, Ramstein G, et al. Immune response and mitochondrial metabolism are commonly deregulated in DMD and aging skeletal muscle. PLoS One. 2011;6:e26952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen YW, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151:1321‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Barcelos IP, Emmanuele V, Hirano M. Advances in primary mitochondrial myopathies. Curr Opin Neurol. 2019;32:715‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manickam R, Duszka K, Wahli W. PPARs and microbiota in skeletal muscle health and wasting. Int J Mol Sci. 2020;21:8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma N, Medikayala S, Defour A, et al. Use of quantitative membrane proteomics identifies a novel role of mitochondria in healing injured muscles. J Biol Chem. 2012;287:30455‐30467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barish GD, Narkar VA, Evans RM. PPARδ: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116:590‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng YZ, Nikolic N, Bakke SS, et al. PPARδ activation in human myotubes increases mitochondrial fatty acid oxidative capacity and reduces glucose utilization by a switch in substrate preference. Arch Physiol Biochem. 2014;120:12‐21. [DOI] [PubMed] [Google Scholar]
- 25. Bell EL, Shine RW, Dwyer P, et al. PPARδ modulation rescues mitochondrial fatty acid oxidation defects in the mdx model of muscular dystrophy. Mitochondrion. 2019;46:51‐58. [DOI] [PubMed] [Google Scholar]
- 26. Narkar VA, Downes M, Yu RT, et al. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008;134:405‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karaa A, Haas R, Goldstein A, Vockley J, Weaver WD, Cohen BH. Randomized dose‐escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology. 2018;90:e1212‐e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kenney K, Landau ME, Gonzalez RS, Hundertmark J, O'Brien K, Campbell WW. Serum creatine kinase after exercise: drawing the line between physiological response and exertional rhabdomyolysis. Muscle Nerve. 2012;45:356‐362. [DOI] [PubMed] [Google Scholar]
- 30. Hahn BH, Pletscher LS, Muniain M. Immunosuppressive effects of deflazacort–a new glucocorticoid with bone‐sparing and carbohydrate‐sparing properties: comparison with prednisone. J Rheumatol. 1981;8:783‐790. [PubMed] [Google Scholar]
- 31.U.S. FDA grants fast track designation for ASP0367/MA‐0211, a selective PPARδ modulator being developed for the treatment of primary mitochondrial myopathies. https://www.astellas.com/en/news/16136. Updated October 20, 2020. Accessed July 15, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Scatterplot of ASP0367 concentrations against baseline‐adjusted QTcF (dQTcF) intervals grouped by dose. Analysis was performed on the safety analysis population excluding all participants from the food effect cohort in part 1 and two placebo participants from part 2. Points (circle, diamond, square, star) indicate the plasma concentration at each dose/preparation (per participant) and the corresponding dQTcF. Asterisks indicate placebo. Participants who received placebo were included in the analysis with concentration set to zero. Plasma concentrations that were below quantification limit (BQL) were treated as zero in the analysis and were included in the analysis. Black and gray bands are the 90% confidence interval around the predicted values in part 1 (CI_P1) and part 2 (CI_P2), respectively. Solid black and gray lines are the predicted value in part 1 and part 2 based on the final linear mixed‐effect model. Black medium dashed (L1) and black medium‐short‐medium t dashed lines (L2) form the locally estimated scatterplot smoothing regression line for parts 1 and 2, respectively. Abbreviations: dQTcF, baseline‐adjusted QTcF; EP, extemporaneously prepared; PP, pre‐prepared; QTcF, QT corrected using the Fridericia formula.
FIGURE S2 Gene expression graphs for six PPARδ target genes with single oral doses of ASP0367 during part 1. The boxplots for fold change from baseline are shown for ABCA1 (A), ACAA2 (B), ACADVL (C), CPT1a (D), PDK4 (E), and SLC25A20 (F). The number of patients in the placebo group and each ASP0367 dose group were 10 and 6, respectively, except for ABCA1: placebo, n = 8; ASP0367 10 mg PP, n = 4; ASP0367 30 mg PP, n = 5; ASP0367 120 mg PP, n = 4 and for PDK4: ASP0367 10 mg PP, n = 5. Abbreviations: EP, extemporaneously prepared; PP, pre‐prepared.
TABLE S1 Final dose scheme
TABLE S2 Schedule of outcomes assessments for part 1
TABLE S3 Schedule of outcomes assessments for part 2
TABLE S4 Statistical assessment of dose proportionality for ASP0367 on day 1 and after multiple dosing day 14 for part 2
TABLE S5 Effect of ASP0367 on ddQTcF at the predefined exposure limit concentration and maximum observed concentration
Data Availability Statement
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.