

ABSTRACT
Alzheimer disease (AD) is the most common neurodegenerative disease. Unfortunately, current effective therapeutics for AD are limited and thus the discovery of novel anti-AD agents is urgently needed. A key pathological hallmark of AD is the accumulation of phosphorylated MAPT/tau (microtubule associated protein tau) aggregates to form neurofibrillary tangles. Autophagy is a conserved catabolic process that degrades protein aggregates or organelles via lysosomes. TFEB (transcription factor EB), a master regulator of autophagy, transcriptionally regulates multiple autophagy, and lysosomal-related genes. A compromised autophagy-lysosomal pathway (ALP) has been implicated in AD progression, and enhancing TFEB-mediated ALP to degrade MAPT/tau aggregates is a promising anti-AD strategy. In a recent study, we showed that celastrol, a natural small molecule with an anti-obesity effect, is a novel TFEB activator, which enhances autophagy and lysosomal biogenesis both in vitro and in animal brains. Consequently, celastrol promotes the degradation of phosphorylated MAPT/tau aggregates both in cells and in the brain of P301S MAPT/tau and 3XTg mice, two commonly used AD animal models. Interestingly, celastrol also alleviates memory deficits in these mice. Altogether, celastrol enhances TFEB-mediated autophagy and lysosomal biogenesis to ameliorate MAPT/tau pathology, suggesting that celastrol represents a novel anti-AD and other tauopathies drug candidate.
Abbreviations: AD: Alzheimer disease; ALP: autophagy-lysosomal pathway; MAPT/tau: microtubule-associated protein tau; MTORC1: mechanistic target of rapamycin kinase complex 1; TFEB: transcription factor EB
KEYWORDS: Alzheimer disease, autophagy, celastrol, lysosome, MTOR, tau, TFEB
Alzheimer disease (AD), the most common neurodegenerative disease, induces more than half of the cases of dementia in the elderly. It is estimated that approximately 10% of people aged above 65-year-old are living with AD. It is perhaps the most terrible disease because the exact etiologies are not fully elucidated, and there are extremely limited effective therapeutic agents for AD. A key pathology hallmark of AD is the accumulation of neurofibrillary tangles that are composed of phosphorylated MAPT/tau (microtubule associated protein tau) protein aggregates. Macroautophagy/autophagy is a major lysosomal-mediated pathway for the degradation of protein aggregates. Recently, a compromised autophagy-lysosomal pathway (ALP) has been implicated in AD pathogenesis and progression, and enhancing ALP to degrade protein aggregates holds great promise for developing disease-modified anti-AD agents. TFEB (transcription factor EB) is a key regulator of ALP by regulating the expression of multiple critical genes for autophagosome formation and lysosomal functions. Thus, there has been an increased interest in identifying novel small molecule autophagy enhancers by targeting TFEB for developing novel therapeutics for neurodegenerative diseases including AD.
In our recent study, we discovered that the natural small molecule celastrol activates TFEB-mediated autophagy and lysosomal biogenesis to attenuate MAPT/tau pathology (Figure 1) [1]. Specifically, to identify novel ALP activators, we first monitored TFEB subcellular distribution by using HeLa cells stably expressing 3XFlag-TFEB as a model; we discovered that celastrol promotes TFEB translocation from the cytoplasm into the nucleus. Subsequently, celastrol promotes autophagy flux (increased microtubule-associated protein 1 light chain 3B-II in the presence of the lysosome inhibitor chloroquine, and elevated autolysosomes) and enhances lysosome biogenesis (as reflected by LysoTracker Red staining and increased expression of lysosomal markers). TFEB deficiency attenuates celastrol-induced autophagy flux, suggesting that celastrol-induced autophagy depends on TFEB.
Figure 1.
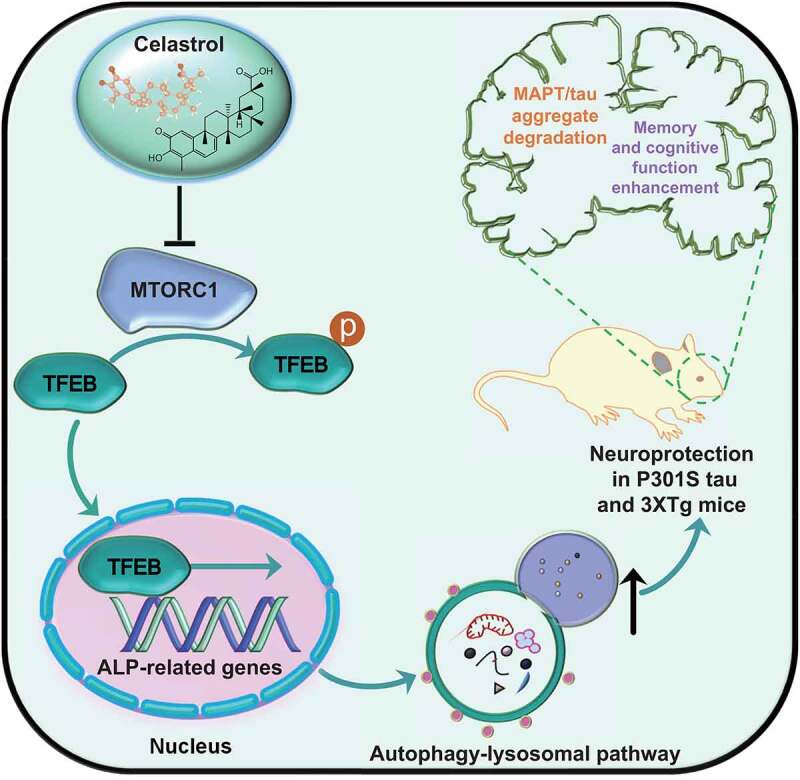
A schematic model of the neuroprotective effects of celastrol in experimental AD. Celastrol promotes the nuclear translocation of TFEB via MTORC1 inactivation. In the nucleus, dephosphorylated TFEB transcriptionally upregulates multiple autophagy-related genes to activate the autophagy-lysosomal pathway. Subsequently, celastrol promotes the degradation of phosphorylated MAPT/tau aggregates to improve memory and cognitive dysfunctions in AD animal models.
Because dephosphorylation of TFEB at Ser211 or Ser142 is critical for its nuclear accumulation, MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) phosphorylates TFEB, calcium-dependent PPP3/calcineurin and PPP2/PP2A (protein phosphatase 2) dephosphorylates TFEB, and they are major regulators of TFEB, we next determined whether celastrol promotes the accumulation of TFEB in the nucleus by regulating its phosphorylation status-mediated by this kinase and these phosphatases. We revealed that celastrol induces TFEB dephosphorylation at both Ser211 and Ser142 sites, and phosphomimetic TFEB mutants attenuate celastrol-induced nuclear accumulation of TFEB, indicating that celastrol-induced nuclear accumulation of TFEB depends on its dephosphorylation at these two serine residues. In addition, we found that celastrol induces MTORC1 inhibition; and blocking calcium signaling by calcium chelating agent, pharmacological inhibition of PPP3/calcineurin, or PPP2/PP2A does not attenuate celastrol-induced nuclear accumulation of TFEB, indicating that celastrol may activate TFEB via MTORC1 inactivation. Continued exploration of how celastrol inhibits MTOR and if there are additional MTOR-independent mechanisms involved in TFEB activation would provide novel information regarding the therapeutic potential of celastrol. Investigating whether and how celastrol activates TFE3 (transcription factor E3), another member of the MiTF/TFE family, which also plays a critical role in regulating ALP, would expand our understanding of the role of celastrol-induced ALP.
We further revealed that celastrol passes through the blood-brain barrier, and enhances TFEB-mediated autophagy and lysosomal biogenesis in mouse brains. These findings prompted us to explore the anti-AD effects of celastrol. Because the neurofibrillary tangles formed by phosphorylated MAPT/tau aggregates are tightly linked to memory deficits in AD, we mainly focused on determining the roles of celastrol in mitigating MAPT/tau pathology in AD animal models. Then, we evaluated the neuroprotective effects of celastrol in P301S MAPT/tau mice (expressing four-repeat human MAPT/tau protein with the P301S mutation in nerve cells), and 3XTg mice (harboring three human mutant genes encoding: APP with the Swedish mutation, PSEN1 [presenilin 1]-KI, and MAPT/tauP301L). Celastrol alleviates memory impairment and cognitive deficits in both P301S MAPT/tau and 3XTg mice as revealed by a series of behaviors tests such as the contextual fear conditioning test, open-field test, and Morris water maze test. Interestingly, celastrol further increases autophagy and reduces phosphorylated MAPT/tau aggregates, and reduces sarcosyl-insoluble phosphorylated MAPT/tau and autophagy substrate SQSTM1/p62 as shown by western blotting analysis and as shown by immunostaining assays. These results revealed that celastrol improves memory deficiency and specifically reduces insoluble, phosphorylated MAPT/tau aggregates in AD mice. Mechanistically, celastrol promotes phosphorylated MAPT/tau aggregate degradation in a TFEB- and autophagy-dependent manner in the mouse neuroblastoma cell line N2a cells. Future studies using brain-specific TFEB deficient mice to further elucidate the anti-AD effects of celastrol are warranted. Overall, celastrol enhances the degradation of phosphorylated MAPT/tau aggregates to alleviate MAPT/tau pathology via activating TFEB-mediated autophagy.
The most important finding of our work is the identification of a previously unexplored role of celastrol in promoting TFEB-mediated autophagy and lysosome biogenesis, and subsequently degradation of phosphorylated MAPT/tau aggregates, which consequently improves memory dysfunction in AD mice. Our results offer novel insight for further developing celastrol as a potential drug candidate for preventing and/or treating AD and other tauopathies. Nevertheless, many open questions remain regarding the pharmacological activities of celastrol in AD. First, whether celastrol alleviates amyloid-beta pathology in AD animal models is still unclear because enhancing autophagy also reduces amyloid-beta production. Second, whether celastrol activates TFEB in glia cells, and, if so, whether this event also contributes to the anti-AD effects of celastrol. Third, whether the reported anti-inflammatory effects of celastrol also contribute to its anti-AD effect needs to be further clarified. Finally, celastrol is well-known for its anti-obesity effects. Exploring the intrinsic links of anti-obesity, anti-AD, and the activation of autophagy effects of celastrol is an interesting topic. Addressing these outstanding questions and understanding the mechanistic and therapeutic aspects of celastrol-induced autophagy-lysosome induction will provide key evidence for developing celastrol as an anti-AD agent.
Acknowledgments
We thank Dr. Daniel J. Klionsky for editing this paper.
Funding Statement
This work was supported by the National Natural Science Foundation of China (82003721), Shenzhen Science and Technology Innovation Commission (JCYJ20210324114014039, JCYJ20210324115800001), China Postdoctoral Science Foundation (2020M683182), and Guangdong Basic and Applied Basic Research Foundation (2020A1515110549), National Key Research and Development Program of China (2020YFA0908000), the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No: ZYYCXTD-C-202002).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Yang C, Su C, Iyaswamy A, et al. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: implications for Alzheimer’s disease therapy. Acta Pharm Sin B. 2022. DOI: 10.1016/j.apsb.2022.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]