

Abstract
Cyclopropanes are an important class of building blocks in organic synthesis. Herein, a ring‐opening/arylcarboxylation/acylation cascade reaction for the 1,3‐difunctionalization of aryl cyclopropanes enabled by cooperative NHC and organophotoredox catalysis is reported. The cascade works on monosubstituted cyclopropanes that are in contrast to the heavily investigated donor–acceptor cyclopropanes more challenging to be difunctionalized. The key step is a radical/radical cross coupling of a benzylic radical generated in the photoredox catalysis cycle with a ketyl radical from the NHC catalysis cycle. The transformation features metal‐free reaction conditions and tolerates a diverse range of functionalities.
Keywords: cascade reactions, cyclopropanes, N-heterocyclic carbenes, photoredox catalysis, reaction mechanisms
Ring‐opening/arylcarboxylation/acylation of mono, di and trisubstituted cyclopropanes enabled by cooperative NHC and organophotoredox catalysis for the preparation of γ‐aroyloxy ketones is reported. Key step is the radical/radical cross‐ coupling of a benzylic radical generated in the photoredox catalysis cycle and a ketyl radical formed in the NHC catalysis cycle.

Cyclopropanes are characterized by a strained ring system and have been identified as versatile and powerful C3 building blocks in synthesis. [1] The inefficient orbital overlap of the C−C δ‐bonds renders the cyclopropane moiety reactive towards ring‐opening. If opening occurs with concomitant 1,3‐difunctionalization, the cyclopropane expresses formal C−C double bond character. [2] Along these lines, cyclopropane ring‐opening and further transformations such as cycloaddition, 1,3‐difunctionalization and rearrangement have been achieved. [3] For example, Lewis‐acid catalyzed 1,3‐difunctionalization of donor–acceptor (D–A) cyclopropanes has been intensively investigated (Scheme 1 a). [4] The “push–pull” substitution pattern in vicinal D–A cyclopropanes polarizes the C−C bond, strongly facilitating the ring‐opening. Hence, most methods work only on doubly‐activated D–A cyclopropanes and ring‐opening/1,3‐difunctionalization of monosubstituted cyclopropanes has not been well investigated. [5] Few pioneering works demonstrated ring opening/1,3‐difunctionalization of mono‐acceptor‐substituted cyclopropanes, which can be activated with Lewis acids or transition‐metals (Rh, Ni) (Scheme 1 b).[ 5e , 5h , 5j ] In addition, it was found that mono‐donor‐substituted cyclopropanes can be activated and difunctionalized with transition‐metals (Rh, Pd).[ 5f , 5g ] However, it is still desirable to develop efficient and versatile catalytic routes to perform the ring opening/1,3‐difunctionalization of arylcyclopropanes under mild conditions.
Scheme 1.

Ionic, transition‐metal‐catalyzed and radical 1,3‐difunctionalization of cyclopropanes.
In recent years, visible‐light photoredox catalysis has emerged as a powerful tool in synthesis. [6] In 2019, the König and Feng groups demonstrated visible‐light promoted ring‐opening/1,3‐difunctionalization of arylcyclopropanes (Scheme 1 d).[ 7a , 7b ] The cyclopropane radical cation, generated through single‐electron‐transfer (SET) oxidation, could be attacked by the Cl‐anion or pyrazole to induce ring‐opening and the thus generated benzylic radical is oxidized by dioxygen to eventually afford β‐chloro or β‐pyrazyl ketones.
Recently, we reported a three‐component coupling of aroyl fluorides, styrenes and the Langlois reagent (CF3SO2Na) to give various β‐trifluoromethylated alkyl aryl ketones by cooperative photoredox/NHC catalysis. [8] These cascades proceed via radical/radical cross coupling of ketyl radicals with benzylic C‐radicals. Inspired by this work, the König/Feng investigations[ 7a , 7b ] and recent studies on radical NHC catalysis, [9] we wondered whether SET‐oxidative cyclopropane ring‐opening can be used for the preparation γ‐functionalized ketones via nucleophile‐mediated ring‐opening of cyclopropyl radical cations and subsequent radical/radical cross coupling with aroylfluoride/NHC‐derived ketyl radicals (Scheme 1 e).
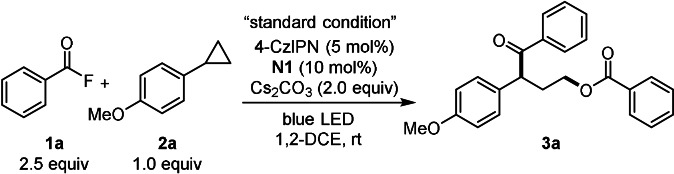
The study was initiated by employing benzoyl fluoride (1 a) and 1‐cyclopropyl‐4‐methoxybenzene (2 a) as model substrates. Careful optimization revealed that the ring‐opening product 3 a can be obtained in 81 % isolated yield by using 1,2,3,5‐tetrakis(carbazol‐9‐yl)‐4,6‐dicyanobenzene (4CzIPN) (5 mol %) as photocatalyst, triazolium salt N1 (10 mol %) as NHC‐precursor and Cs2CO3 (2 equiv) in 1,2‐DCE under blue LED irradiation at room temperature (Table 1, entry 1). Importantly, γ‐benzoyloxy ketones of type 3 occur in bioactive compounds. [10] Other types of triazoliums, imidazoliums and thiazolium salts showed either inferior results or no activity (entries 2–4). Notably, the base plays a curial role and Na2CO3 or K2CO3 were found to be ineffective (entries 5,6). Benzoyl chloride in place of the fluoride was also not suitable (entry 7). In addition, some metal‐based photocatalysts were investigated and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which has a similar oxidation potential (E 1/2=+1.21 V vs. SCE) [11b] as 4CzIPN (E 1/2=+1.35 V vs. SCE), [11a] also worked well. Ketone 3 a was obtained in 84 % yield (entry 8). With Ru(bpy)3(PF6)2 (E 1/2=+0.77 V vs. SCE) [11b] or Ir(dF(CF3)ppy2)(5,5′‐dCF3bpy)PF6 (E 1/2=+1.68 V vs. SCE) [11b] the desired product was not formed (entries 9–10). Control experiments revealed the necessity of both catalysts and blue LED irradiation (entries 11–13).
Table 1.
Reaction optimization.[a]
|
Entry |
Variation from “standard conditions” |
Yield [%][b] |
|---|---|---|
|
1 |
None |
81 |
|
2 |
N2 instead of N1 |
55 |
|
3 |
N3 instead of N1 |
<5 |
|
4 |
N4 instead of N1 |
<5 |
|
5 |
Na2CO3 instead of Cs2CO3 |
<5 |
|
6 |
K2CO3 instead of Cs2CO3 |
<5 |
|
7 |
benzoyl chloride instead of benzoyl fluoride |
<5 |
|
8 |
[Ir(dF(CF3)ppy)2(dtbbpy)]PF6 instead of 4CzIPN |
84 |
|
9 |
Ru(bpy)3(PF6)2 instead of 4CzIPN |
<5 |
|
10 |
Ir(dF(CF3)ppy2)(5,5′‐dCF3bpy)PF6 instead of 4CzIPN |
<5 |
|
11 |
Without 4CzIPN |
<5 |
|
12 |
Without N1 |
<5 |
|
13 |
In dark |
<5 |
|
| ||
[a] Reaction conditions: 1 a (0.25 mmol), 2 a (0.10 mmol), N1 (10 mol %), 4CzIPN (5 mol %), Cs2CO3 (0.20 mmol) in anhydrous 1,2‐DCE (1.0 mL), irradiation with blue LED at room temperature for 12 h. [b] isolated yield.
With the optimal conditions established, the scope with respect to the acyl fluoride was explored first. Acyl fluorides bearing electron‐donating as well as electron withdrawing groups engaged in the reaction to afford the γ‐aroyloxy ketones 3 b–o in moderate to good yield (45–81 %, Scheme 2). Various functional groups such as chloro (3 c), bromo (3 d, 3 f, 3 h), iodo (3 i) and cyano (3 l) are tolerated. Moreover, substrates containing the medicinally relevant CF3‐group (3 j, 3 k) and heterocyclic moieties are also compatible (see 3 n and 3 o).
Scheme 2.

Variation of the acyl fluoride component.
The cyclopropane component was varied next (Scheme 3). For aryl cyclopropanes bearing alkyl groups at the 2‐position such as gem‐dimethyl and gem‐diethyl, ring‐opening occurred with complete regiocontrol to give the ketones 3 p and 3 q (57–85 % yield). Para‐substituents at the aryl moiety such as benzyloxy (3 r), cyclopropyl (3 s) and phenyl (3 t) are tolerated and also disubstituted congeners with methoxy (3 u, v) and chloro (3 y) groups engaged in the cascade (59–72 %). It should be noted that the substrate bearing two cyclopropyl groups at positions 1,4 of the benzene ring selectively delivered the mono‐ring opening product 3 s (64 %). Hence, the starting 1,4‐biscyclopropyl benzene is SET‐oxidized whereas 3 s is reluctant towards oxidation. The structure of 3 s was assigned by X‐ray analysis. [12] An ortho‐methoxy‐substituent at the aryl cyclopropane component is also tolerated (3 v, 72 %). However, cyclopropanes which lack an electron‐donating substituent at the aryl group could not be converted to the targeted products, likely due to their higher oxidation potentials (see 3 w and 3 x).
Scheme 3.

Variation of the cyclopropane component.
We also addressed the problem of regio‐ and diastereoselectivity by studying unsymmetric cyclopropanes. Pleasingly, the reaction with 2‐alkyl‐1‐arylcyclopropanes occurred with complete regiocontrol, albeit low diastereoselectivity (3 z–3 ab). For 1‐methoxy‐2‐phenylcycylopropane, highly regioselective ring‐opening provided the acetal 3 ac in 53 % yield. Control of the regioselectivity is more challenging for 1,2‐diaryl cyclopropanes. For 1‐para‐methoxyphenyl‐2‐phenylcyclopropane, benzoate attack occurred with a 3:1 regioselectivity at the more electron‐rich benzylic position to give 3 ae in good yield but poor diastereoselectivity (1.2:1). Regioselectivity was higher (7:1) for the p‐CF3C6H4/p‐CH3OC6H4 couple, but diastereoselectivity remained low (see 3 af). For 1‐para‐methoxyphenyl‐2‐naphthylcyclopropane, ring opening was achieved with a 4:1 regioselectivity (3 ag). As compared to 3 ae, regioselectivity was better, likely due to steric effects. The regioselectivities were confirmed by further derivatization (for details, see SI). Late‐stage functionalization of biorelevant molecules derived from estrone and isoxepac provided 3 ah and 3 ai (52 and 61 %).
To demonstrate robustness, a larger scale experiment was conducted and a comparable yield was obtained with lower catalyst loading (Scheme 4 a). Furthermore, follow‐up chemistry on 3 a was preformed (Scheme 4 b). Saponification of the ester 3 a afforded the γ‐hydroxy ketone 4 a (76 %). Ketone 3 a could be reduced to the corresponding 1,4‐diol 4 b in near quantitative yield and excellent diastereoselectivity. [13] Under acidic conditions, 4 b cyclized to the tetrahydrofuran derivative 4 c in excellent yield and complete trans‐selectivity. [14]
Scheme 4.

Large scale experiment and follow‐up transformations. (I) NaOH, THF/H2O, 80 °C; (II) LiAlH4 (5 equiv), THF, 0 °C–rt; (III) TfOH (10 mol %), 1,2‐DCE, 40 °C.
To gain insights into the mechanism, control experiments were conducted (Scheme 5). When adding 2.0 equivalents of the radical scavenger TEMPO (2,2,6,6‐tetramethyl‐1‐piperidinyloxy), product formation was fully suppressed, while the TEMPO‐trapping product (benzoyl‐TEMPO) was detected by HMRS analysis (Scheme 5 a). This result suggests that the reaction may proceed through a radical‐based mechanism. Moreover, reaction of acyl azolium ion 5, sodium benzoate 6 and aryl cyclopropane 2 a afforded 3 a (52 %), which indicates that acyl azoliums and the benzoate anion are competent intermediates (Scheme 5 b). A mechanism where the ring‐opened benzylic radical gets reduced by the photocatalyst to the corresponding anion that is then acylated in an ionic process is not likely, since no reaction with the acyl fluoride in absence of NHC catalyst was observed (see Table 1, entry 12). In addition, we subjected enantiomerically enriched arylcyclopropane (1S,2S)‐2 s (90 % ee) to the standard conditions and obtained 3 ab with 1.2:1 diastereoselectivity in 83 % and 82 % ee, respectively. This experiment indicated that the transformation proceeds with high stereospecificity through an SN2‐type nucleophilic ring‐opening pathway (Scheme 5 c, top).[ 7b , 15 ] However, the enantiomerically enriched diarylcyclopropane (1R,2R)‐2 l (78 % ee) reacted with poor stereospecifity (14 % ee for both diastereoisomers) (Scheme 5 c, bottom), presumably due to the rapid ring opening/closing of the intermediate radical cation. [16] Finally, Stern–Volmer quenching studies revealed that SET‐oxidation of 2 a (E 1/2=+1.35 V vs. SCE) [7a] by the excited 4CzIPN* (E *red=+1.35 V vs. SCE) is preferred over the SET‐reduction of the acyl azolium 5 (E 1/2=−1.29 V vs. SCE)[ 9h , 17 ] with 4CzIPN* (E *ox=−1.04 V vs. SCE) [11a] (for details, see SI).
Scheme 5.

Mechanistic studies and proposed mechanism.
Based on these results and pertinent literatures,[ 7 , 18 ] a mechanism is proposed (Scheme 5 d). LED irradiation leads to photoexcited 4CzIPN*, [19] which is reductively quenched by 2, generating the aryl cyclopropane radical cation IV and 4‐CzIPN⋅− (E 1/2 red=−1.21 V vs. SCE). [11a] The fluoride 1 reacts with Cs2CO3 to generate the bisacyl carbonate intermediate I [20] that then reacts with the NHC to the acyl azolium ion II along with the benzoate anion and CO2. The benzoate serves as nucleophile for ring‐opening of IV to give the benzylic radical V. At this juncture, the reduced photocatalyst reduces the azolium ion II (E 1/2=−1.29 V vs. SCE) to generate the persistent ketyl radical III and 4‐CzIPN. Radical/radical cross coupling [21] of the persistent III and the transient V leads to the NHC‐bound intermediate VI. Finally, NHC‐fragmentation affords 3 closing the NHC catalysis cycle.
In light of the possible mechanism, we used anhydrides 8 as bifunctional reagents and the desired γ‐aroyloxy ketones 3 a, 3 e, 3 aj, 3 ak were obtained in 36–61 % yield with an Ir‐catalyst under otherwise identical conditions (Scheme 6). For unsymmetrical anhydrides, regioselectivity can be high (see 3 aj). In this case, the NHC attacks the anhydride at the less sterically shielded carbonyl group. Electronic effects are not pronounced in controlling the regioselectivity as shown for 3 ak. In general, the anhydrides 8 are less reactive than the carbonates I, as unreacted anhydride could be identified in two cases.
Scheme 6.

Ring‐opening of aryl cyclopropanes with anhydrides.
In summary, we developed a novel approach for the 1,3‐difunctionalization of aryl cyclopropanes by applying NHC/photoredox cooperative catalysis. This method enables sequential C−O and C−C bond formation leading to various γ‐aroyloxy ketones in good to excellent yield and good functional group tolerance. The ease of post‐synthetic modifications further increases the value of the introduced method. Mechanistic studies revealed that the cascade proceeds through nucleophilic ring‐opening of a cyclopropyl radical cation with subsequent radical/radical cross coupling as key steps.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the European Research Council (Advanced Grant Agreement 692640) and the Deutsche Forschungsgemeinschaft (DFG) for supporting this work. We also thank Dr. Qingyuan Meng (WWU Münster) for generously providing some starting materials. Open Access funding enabled and organized by Projekt DEAL.
Z. Zuo, C. G. Daniliuc, A. Studer, Angew. Chem. Int. Ed. 2021, 60, 25252.
References
- 1.For a book: see:
- 1a. Kulinkovich O. G., Cyclopropanes in Organic Synthesis, Wiley-VCH, Weinheim, 2015; For selected reviews, see: [Google Scholar]
- 1b. de Meijere A., Angew. Chem. Int. Ed. Engl. 1979, 18, 809; [Google Scholar]; Angew. Chem. 1979, 91, 867; [Google Scholar]
- 1c. Wong H. N. C., Hon M. Y., Tse C. W., Yip Y. C., Tanko J., Hudlicky T., Chem. Rev. 1989, 89, 165; [Google Scholar]
- 1d. Rubin M., Rubina M., Gevorgyan V., Chem. Rev. 2007, 107, 3117; [DOI] [PubMed] [Google Scholar]
- 1e. Zhu Z., Wei Y., Shi M., Chem. Soc. Rev. 2011, 40, 5534; [DOI] [PubMed] [Google Scholar]
- 1f. Chen D. Y.-K., Pouwer R. H., Richard J.-A., Chem. Soc. Rev. 2012, 41, 4631; [DOI] [PubMed] [Google Scholar]
- 1g. Ebner C., Carreira E. M., Chem. Rev. 2017, 117, 11651; [DOI] [PubMed] [Google Scholar]
- 1h. Dian L., Marek I., Chem. Rev. 2018, 118, 8415. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Gordon M. S., J. Am. Chem. Soc. 1980, 102, 7419; [Google Scholar]
- 2b. Bach R. D., Dmitrenko O., J. Am. Chem. Soc. 2004, 126, 4444; [DOI] [PubMed] [Google Scholar]
- 2c. Galano A., Alvarez J. R., Vivier-Bunge A., Theor. Chem. Acc. 2007, 118, 597. [Google Scholar]
- 3.
- 3a. Fumagalli G., Stanton S., Bower J. F., Chem. Rev. 2017, 117, 9404; [DOI] [PubMed] [Google Scholar]
- 3b. Xuan J., He X., Xiao W., Chem. Soc. Rev. 2020, 49, 2546; [DOI] [PubMed] [Google Scholar]
- 3c. Sokolova O. O., Bower J. F., Chem. Rev. 2021, 121, 80; [DOI] [PubMed] [Google Scholar]
- 3d. Wang J., Blaszczyk S. A., Li X., Tang W., Chem. Rev. 2021, 121, 110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Cohen Y., Cohen A., Marek I., Chem. Rev. 2021, 121, 140; [DOI] [PubMed] [Google Scholar]
- 3f. Pirenne V., Muriel B., Waser J., Chem. Rev. 2021, 121, 227. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Reissig H. U., Zimmer R., Chem. Rev. 2003, 103, 1151; [DOI] [PubMed] [Google Scholar]
- 4b. Schneider T. F., Kaschel J., Werz D. B., Angew. Chem. Int. Ed. 2014, 53, 5504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5608; [Google Scholar]
- 4c. Cavitt M. A., Phun L. H., France S., Chem. Soc. Rev. 2014, 43, 804; [DOI] [PubMed] [Google Scholar]
- 4d. Augustin A. U., Werz D. B., Acc. Chem. Res. 2021, 54, 1528; [DOI] [PubMed] [Google Scholar]
- 4e. Grover H. K., Emmett M. R., Kerr M. A., Org. Biomol. Chem. 2015, 13, 655; [DOI] [PubMed] [Google Scholar]
- 4f. Xia Y., Liu X., Feng X., Angew. Chem. Int. Ed. 2021, 60, 9192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9276. [Google Scholar]
- 5.
- 5a. Hixson S. S., Garrett D. W., J. Am. Chem. Soc. 1974, 96, 4872; [Google Scholar]
- 5b. Rao V. R., Hixson S. S., J. Am. Chem. Soc. 1979, 101, 6458; [Google Scholar]
- 5c. Dinnocenzo J. P., Zuilhof H., Lieberman D. R., Simpson T. R., Mckechney M. W., J. Am. Chem. Soc. 1997, 119, 994; [Google Scholar]
- 5d. Maeda H., Matsukawa N., Shirai K., Mizuno K., Tetrahedron Lett. 2005, 46, 3057; [Google Scholar]
- 5e. Liu L., Montgomery J., J. Am. Chem. Soc. 2006, 128, 5348; [DOI] [PubMed] [Google Scholar]
- 5f. Jiao L., Ye S., Yu Z., J. Am. Chem. Soc. 2008, 130, 7178; [DOI] [PubMed] [Google Scholar]
- 5g. Bhargava G., Trillo B., Araya M., López F., Castedo L., Mascareñas J., Chem. Commun. 2010, 46, 270; [DOI] [PubMed] [Google Scholar]
- 5h. Shaw M. H., Melikhova E. Y., Kloer D. P., Whittingham W. G., Bower J. F., J. Am. Chem. Soc. 2013, 135, 4992; [DOI] [PubMed] [Google Scholar]
- 5i. Pitts C. R., Ling B., Synder J. A., Bragg A. E., Lectka T., J. Am. Chem. Soc. 2016, 138, 6598; [DOI] [PubMed] [Google Scholar]
- 5j. Banik S. M., Mennie K. M., Jacobsen E. N., J. Am. Chem. Soc. 2017, 139, 9152; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5k. Ilchenko N. O., Hedberg M., Szabó K. J., Chem. Sci. 2017, 8, 1056; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5l. Gieuw M. H., Ke Z., Yeung Y. Y., Angew. Chem. Int. Ed. 2018, 57, 3782; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3844; [Google Scholar]
- 5m. Wang D., Xue X., Houk K. N., Shi Z., Angew. Chem. Int. Ed. 2018, 57, 16861; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17103; [Google Scholar]
- 5n. Yang S., Wang L., Zhang H., Liu C., Zhang L., Wang X., Zhang G., Li Y., Zhang Q., ACS Catal. 2019, 9, 716. [Google Scholar]
- 6.
- 6a. Narayanam J. M. R., Stephenson C. R. J., Chem. Soc. Rev. 2011, 40, 102; [DOI] [PubMed] [Google Scholar]
- 6b. Xuan J., Xiao W., Angew. Chem. Int. Ed. 2012, 51, 6828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6934; [Google Scholar]
- 6c. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Gentry E. C., Knowles R. R., Acc. Chem. Res. 2016, 49, 1546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075; [DOI] [PubMed] [Google Scholar]
- 6g. Koike T., Akita M., Acc. Chem. Res. 2016, 49, 1937; [DOI] [PubMed] [Google Scholar]
- 6h. Xie J., Jin H., Hashmi A. S. K., Chem. Soc. Rev. 2017, 46, 5193; [DOI] [PubMed] [Google Scholar]
- 6i. Wang C., Dixneuf P. H., Soulé J. F., Chem. Rev. 2018, 118, 7532; [DOI] [PubMed] [Google Scholar]
- 6j. Silvi M., Melchiorre P., Nature 2018, 554, 41; [DOI] [PubMed] [Google Scholar]
- 6k. Marzo L., Pagire S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188; [Google Scholar]
- 6l. Cannalire R., Pelliccia S., Sancineto L., Novellino E., Tron G. C., Giustiniano M., Chem. Soc. Rev. 2021, 50, 766. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Petzold D., Singh P., Almqvist F., König B., Angew. Chem. Int. Ed. 2019, 58, 8577; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8665; [Google Scholar]
- 7b. Ge L., Wang D., Xing R., Ma D., Walsh P. J., Feng C., Nat. Commun. 2020, 10, 4367. For electrochemical oxidation of D–A cyclopropanes, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Ahlburg N. L., Jones P. G., Werz D. B., Org. Lett. 2020, 22, 6404; [DOI] [PubMed] [Google Scholar]
- 7d. Kolb S., Petzold M., Brandt F., Jones P. G., Jacob C. R., Werz D. B., Angew. Chem. Int. Ed. 2021, 60, 15928; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16064. [Google Scholar]
- 8. Meng Q., Döben N., Studer A., Angew. Chem. Int. Ed. 2020, 59, 19956; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 20129. [Google Scholar]
- 9.For review, see:
- 9a. Sumida Y., Ohmiya H., Chem. Soc. Rev. 2021, 50, 6320. For recent examples, see: [DOI] [PubMed] [Google Scholar]
- 9b. Yang W., Hu W., Dong X., Li X., Sun J., Angew. Chem. Int. Ed. 2016, 55, 15783; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16015; [Google Scholar]
- 9c. Ishii T., Kakeno Y., Nagao K., Ohmiya H., J. Am. Chem. Soc. 2019, 141, 3854; [DOI] [PubMed] [Google Scholar]
- 9d. Dai L., Xia Z., Gao Y., Gao Z., Ye S., Angew. Chem. Int. Ed. 2019, 58, 18124; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18292; [Google Scholar]
- 9e. Ishii T., Ota K., Nagao K., Ohmiya H., J. Am. Chem. Soc. 2019, 141, 14073; [DOI] [PubMed] [Google Scholar]
- 9f. Li J., Liu Y., Zou W., Zeng R., Zhang X., Liu Y., Han B., Angew. Chem. Int. Ed. 2020, 59, 1863; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1879; [Google Scholar]
- 9g. Mavroskoufis A., Rajes K., Golz P., Agrawal A., Ruß V., Götze J. P., Hopkinson M. N., Angew. Chem. Int. Ed. 2020, 59, 3190; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3216; [Google Scholar]
- 9h. Davies A. V., Fitzpatrick K. P., Betori R. C., Scheidt K. A., Angew. Chem. Int. Ed. 2020, 59, 9143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9228; [Google Scholar]
- 9i. Liu K., Studer A., J. Am. Chem. Soc. 2021, 143, 4903; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9j. Meng Q., Lezius L., Studer A., Nat. Commun. 2021, 12, 2068; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9k. Ren S., Lv W., Yang X., Yan J., Xu J., Wang F., Hao L., Chai H., Jin Z., Chi Y. R., ACS Catal. 2021, 11, 2925. [Google Scholar]
- 10.
- 10a. Palazzino G., Galeffi C., Federici E., Monache F. D., Cometa M. F., Palmery M., Phytochemistry 2000, 55, 411; [DOI] [PubMed] [Google Scholar]
- 10b. Chang C., Zhang L., Chen R., Wu C., Huang H., Roy M. C., Huang J., Wu Y., Kuo Y., Bioorg. Med. Chem. 2010, 18, 518; [DOI] [PubMed] [Google Scholar]
- 10c. Yang X., Deng X., Liu X., Wu C., Li X., Wu B., Luo H., Li Y., Xu H., Zhao Q., Xu G., Chem. Commun. 2012, 48, 5998. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Shang T., Lu L., Cao Z., Liu Y., He W., Yu B., Chem. Commun. 2019, 55, 5408; [DOI] [PubMed] [Google Scholar]
- 11b. Clarke A. K., Parkin A., Taylor R. J. K., Unsworth W. P., Rossi-Ashton J. A., ACS Catal. 2020, 10, 5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deposition Number 2094400 (for 3s) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 13. Chung J. Y. L., Mancheno D., Dormer P. G., Variankaval N., Bal R. G., Tsou N. N., Org. Lett. 2008, 10, 3037. [DOI] [PubMed] [Google Scholar]
- 14. Fries P., Müller M. K., Hartung J., Org. Biomol. Chem. 2013, 11, 2630. [DOI] [PubMed] [Google Scholar]
- 15. Augustin A. U., Sensse M., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2017, 56, 14293; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14481. [Google Scholar]
- 16.The experimental details see SI.
- 17. Bayly A. A., McDonald B. R., Mrksich M., Scheidt K. A., Proc. Natl. Acad. Sci. USA 2020, 117, 13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bay A. V., Fitzpatrick K. P., González-Montiel G. A., Farah A. O., Cheong P. H. Y., Scheidt K. A., Angew. Chem. Int. Ed. 2021, 60, 17925; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 18069. [Google Scholar]
- 19. Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C., Nature 2012, 492, 234. [DOI] [PubMed] [Google Scholar]
- 20. Ren L., Wang L., Lv Y., Li G., Gao S., Org. Lett. 2015, 17, 5172. [DOI] [PubMed] [Google Scholar]
- 21. Leifert D., Studer A., Angew. Chem. Int. Ed. 2020, 59, 74; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information