

ABSTRACT
Mice deficient in GHR (growth hormone receptor; ghr KO) have a dramatic lifespan extension and elevated levels of hepatic chaperone-mediated autophagy (CMA). Using quantitative proteomics to identify protein changes in purified liver lysosomes and whole liver lysates, we provide evidence that elevated CMA in ghr KO mice downregulates proteins involved in ribosomal structure, translation initiation and elongation, and nucleocytosolic acetyl-coA production. Following up on these initial proteomics findings, we used a cell culture approach to show that CMA is necessary and sufficient to regulate the abundance of ACLY and ACSS2, the two enzymes that produce nucleocytosolic (but not mitochondrial) acetyl-coA. Inhibition of CMA in NIH3T3 cells has been shown to lead to aberrant accumulation of lipid droplets. We show that this lipid droplet phenotype is rescued by knocking down ACLY or ACSS2, suggesting that CMA regulates lipid droplet formation by controlling ACLY and ACSS2. This evidence leads to a model of how constitutive activation of CMA can shape specific metabolic pathways in long-lived endocrine mutant mice.
Abbreviations: CMA: chaperone-mediated autophagy; DIA: data-independent acquisition; ghr KO: growth hormone receptor knockout; GO: gene ontology; I-WAT: inguinal white adipose tissue; KFERQ: a consensus sequence resembling Lys-Phe-Glu-Arg-Gln; LAMP2A: lysosomal-associated membrane protein 2A; LC3-I: non-lipidated MAP1LC3; LC3-II: lipidated MAP1LC3; PBS: phosphate-buffered saline; PI3K: phosphoinositide 3-kinase
KEYWORDS: Aging, autophagy, growth hormone, metabolism, proteomics
Introduction
Autophagy, once thought to be a nonselective bulk-recycling mechanism, is now understood to be a selective and targeted process. Selective lysosomal degradation of intracellular cargo is required for cellular adaptations to stress, remodeling the cytoplasm during differentiation, and maintaining proteomic and metabolic homeostasis [1–3]. The three primary forms of autophagy in mammalian cells, i.e., macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), are all capable of sophisticated selection of targets for degradation [4,5]. However, the most selective form of autophagy is CMA, which targets individual proteins for lysosomal degradation [6,7].
CMA target proteins contain a consensus motif resembling Lys-Phe-Glu-Arg-Gln (KFERQ), which is recognized by the cytosolic chaperone HSPA8/Hsc70. HSPA8 is the primary receptor for CMA [8–10]. HSPA8, along with other cytosolic chaperones, participates in the trafficking of the KFERQ-bearing target to the lysosomal surface, where it is unfolded and translocated across the lysosomal membrane, independent of vesicle fusion, in a manner dependent upon LAMP2A, one of the three splice variants of lamp2 [8,11,12].
The KFERQ motif is not recognized as an exact sequence, but rather by the relative charges of amino acid side chains. The first or fifth amino acid of the pentapeptide motif is glutamine. The remaining four amino acids must contain one or two positive, one negative, and one or two hydrophobic side chains, in any order [10,13]. The KFERQ motif can be activated or deactivated through the addition or removal of post-translational modifications, such as phosphorylation or acetylation, which alter side-chain charges or hydrophobicity [14]. In silico analysis of the mouse proteome suggests that approximately 47% of mouse proteins contain at least one “canonical” KFERQ motif that does not require a post-translational modification to be active [14]. When including motifs hypothetically activated by post-translational modification, approximately 78% of mouse proteins contain at least one KFERQ motif [14]. However, because the sequestration of hydrophobic amino acids away from water in the cytosol is a key step in protein folding [15], it remains unclear how many KFERQ motifs (which must contain at least one amino acid with a hydrophobic side chain) will be exposed on the protein surface to interact with cytosolic HSPA8. Thus, empirical analysis of CMA targetomes is necessary to determine which proteins contain CMA motifs that are genuinely functional.
CMA regulates the abundance of several proteins implicated in metabolism, disease, and the rate of aging, with novel CMA substrates discovered every year [16–18]. However, the extent to which CMA regulates the overall content and relative abundance of proteins within the proteome remains unclear. In an effort to understand how CMA shapes the proteome, recent proteomics studies have employed a variety of strategies, which broadly fall into two categories: (1) lysosomal targetomics and (2) analysis of whole-cell proteomic changes in response to CMA manipulation.
Lysosomal targetomics involves the isolation of lysosomal fractions from cells or tissues in which CMA has been induced and where intra-lysosomal proteolysis has been blocked pharmacologically, allowing for the identification of CMA targets that have accumulated in the lysosomal lumen [16,19]. Both major studies using this technique used lysosomes from cells or tissues genetically depleted of LAMP2A as negative controls [16,19], allowing them to distinguish CMA targets from the targets of microautophagy or macroautophagy. This style of assay robustly detects CMA target proteins. However, CMA is not sufficient to control the total abundance of many CMA target proteins. Thus, lysosomal targetomics must be supplemented by additional assays to address whether or not CMA is necessary and/or sufficient to regulate the total abundance of target proteins in the cells or tissues being analyzed.
Lysosomal targetomics studies have found that endogenous CMA substrates of mouse liver lysosomes cluster into distinct pathways, many of which modulate metabolism of carbohydrates and lipids [16]. Mice with a liver-specific Lamp2a deficiency progressively develop hepatic steatosis and a global metabolic dysregulation, suggesting that CMA is necessary to maintain homeostatic lipid and carbohydrate metabolism [16]. Lysosomal targetomics of SUM159 human breast cancer cells identified a surprising role for CMA in regulating the abundance of translation initiation factors (but did not find a role for CMA in regulating translation elongation factors). Moreover, pharmacological activation of CMA decreases the whole-cell abundance of these CMA-targeted translation initiation factors [19].
Studies relying on the second proteomics strategy, analysis of whole-cell proteomic changes in response to CMA manipulation, have identified putative CMA targets, many of which fit into the functional pathways identified by lysosomal targetomics. ES2 human ovarian cancer cells treated with inhibitors of FLT3 to activate CMA show significant decreases in the abundance of proteins involved in metabolism, cell cycle control, and translation initiation [20], suggesting that CMA is sufficient to regulate the abundance of these proteins. Proteomic analysis of cultured primary rat cortical neurons treated with shRNA for Lamp2a found that LAMP2A depletion causes significant increases in the abundance of several proteins associated with neurodegeneration [21], consistent with several previous studies suggesting that CMA is necessary to regulate the abundance of disease-related proteins in neuronal tissue [22,23]. Whereas the analysis of whole-cell proteomic changes after manipulation of CMA produces interesting results, several validation experiments must be performed to ensure that the observed changes are direct results of increased or decreased CMA, and not cellular adaptations to other CMA-related proteomic changes.
We have recently reported that two long-lived mouse stocks with reduced signaling through the GH (growth hormone)-IGF1 (insulin-like growth factor 1) signaling axis (ghr KO and pou1f1 mutant/Snell dwarf mice) have constitutively enhanced hepatic CMA in young adulthood, with an mRNA-independent reduction of the CMA target CIP2A in multiple tissues [24]. This suggests the hypothesis that enhanced CMA, in early life, might regulate the abundance of proteins in a way that contributes to the metabolic and lifespan phenotypes of these mutant mice.
To identify proteins regulated by selective lysosomal proteolysis in long-lived ghr KO mice, we performed lysosomal targetomics (on liver lysosomes) and parallel proteomic evaluation of whole liver lysates. We identified a small subset of the proteome that showed both increased lysosomal enrichment in response to leupeptin by ghr KO liver lysosomes and decreased total abundance in ghr KO liver lysates. These proteins, which are candidate CMA targets, cluster into distinct functional groups, including ribosomal proteins, translation initiation factors, translation elongation factors, and enzymes essential for the production of nuclear and cytosolic (nucleocytosolic) acetyl-coA, but not mitochondrial acetyl-coA. Using cell-based assays, we then demonstrated that CMA is both necessary and sufficient to coordinately control the abundance of four enzymes involved in nucleocytosolic acetyl-coA production: IDH1 (isocitrate dehydrogenase 1), ACO1 (aconitase 1), ACLY (ATP citrate lyase) and ACSS2 (acyl-coA synthetase short-chain family member 2) as well as two enzymes involved in the consumption of cytosolic acetyl-coA for fatty acid synthesis ACACA (acetyl-CoA carboxylase alpha) and FASN (fatty acid synthase). Inhibition of CMA causes accumulation of intracellular lipid droplets [25], and we show that knockdown of Acly or Acss2 reduces lipid droplet accumulation in CMA-deficient cells. This suggests that CMA negatively regulates lipid accumulation by controlling ACLY and ACSS2 protein levels. This work defines a novel role for CMA in regulating enzymes for nucleocytosolic acetyl-coA production and provides a foundation for future studies evaluating the role of CMA in regulating the abundance of ribosomal proteins and translation elongation factors.
Results
Ghr KO and control liver lysosomes have distinct compositions and targetomes
To identify differences in the lysosomal targetome between ghr KO mice and their normal littermate controls, we treated three male and three female mice of each genotype with leupeptin or sterile phosphate-buffered saline (PBS; solvent control) by intraperitoneal injection (Figure 1A). Leupeptin inhibits lysosomal proteolysis without preventing lysosomal substrate uptake, resulting in the lumenal accumulation of endogenous autophagy target proteins. Two hours after leupeptin or PBS injection, the mice were humanely euthanized, and the liver lysosomes were isolated by density centrifugation, using our previous methods [24,26].
Figure 1.
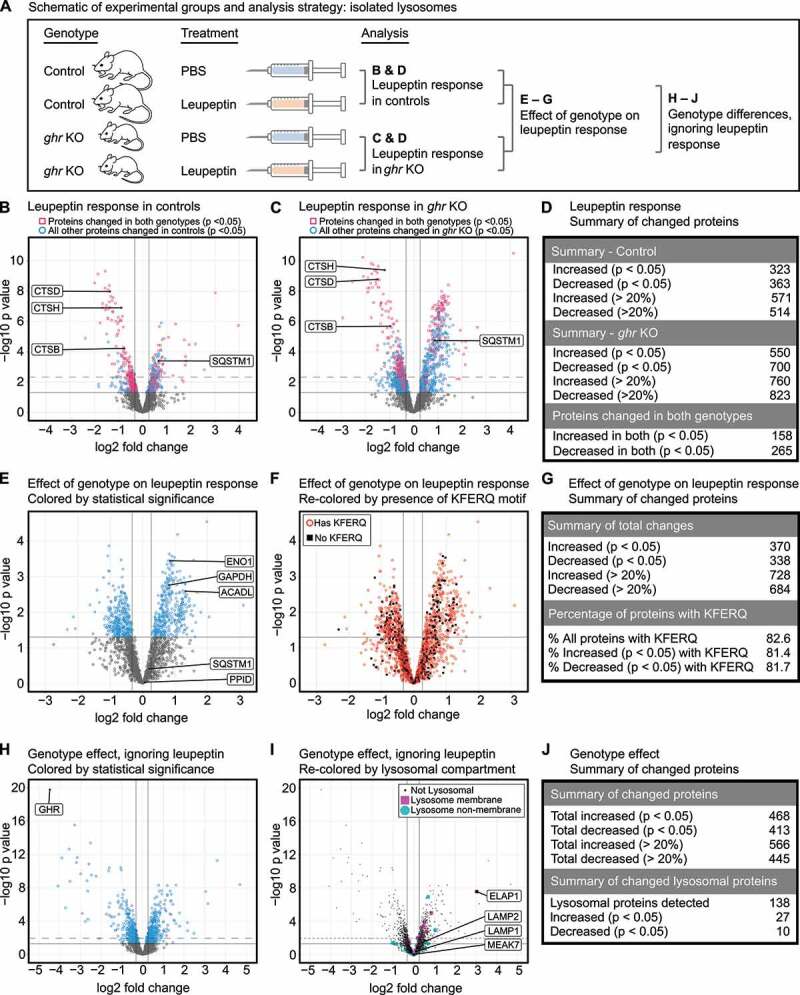
ghr KO and control lysosomes have distinct compositions and targetomes. (A) Schematic of the experimental groups and analysis. (B) Volcano plot showing the response to leupeptin in lysosomes from control mice. Proteins that are also significantly changed in ghr KO with identical directionality are shown as red squares. All other significantly changed proteins in controls are shown as blue circles. (C) Volcano plot showing the response to leupeptin in lysosomes from ghr KO mice. Proteins that are also significantly changed in controls with identical directionality are shown as red squares. All other significantly changed proteins in ghr KO are shown as blue circles. (D) Summary table for numbers of proteins changed in response to leupeptin in control and ghr KO lysosomes. (E) Volcano plot for a 2-factor analysis of the effects of the ghr KO genotype on the lysosomal leupeptin response. Proteins more enriched in ghr KO than in controls, in response to leupeptin, will have a positive log2 fold change. Proteins that are equally changed in both genotypes in response to leupeptin will have a near-zero value. (F) The volcano plot shown in (E), re-colored and relabeled to indicate proteins with at least one KFERQ motif (canonical, phosphorylation activated, or acetylation activated). (G) Summary table for numbers of proteins changed as determined by the 2-factor analysis. (H) Volcano plot showing the contribution of genotype to differences in lysosomal proteins, ignoring leupeptin as a variable. (I) The volcano plot shown in (H), re-colored to indicate proteins that have a lysosomal function. (J) Summary table for numbers of proteins changed between the ghr and control genotypes. For all volcano plots, dark vertical lines indicate a fold-change of change of 20%. Dark, solid horizontal lines indicate a threshold of p = 0.05. Where applicable, dashed horizontal lines indicate a false-discovery adjusted threshold of p = 0.05. Each of the four experimental groups shown in (A) has lysosomal samples from a total of 6 mice (3 of each sex).
Lysosomal fractions were evaluated for enrichment and purity, showing >50-fold enrichment for LAMP2A and LAMP1, without enrichment of mitochondrial, cytosolic, or nuclear markers, as expected (Fig. S1A). Isolated lysosomes were submitted to label-free quantitative proteomics, as a single batch, which identified 3345 proteins. Proteins that were identified based on the detection of a single peptide were excluded from the analysis. The remaining data were subjected to quantile normalization and the missing values, which were highly left-censored (i.e., almost entirely found in low-abundance proteins), were imputed for all proteins with <50% missing values. Proteins with >50% missing values were excluded, leaving 2430 proteins.
We first assessed the leupeptin response in each genotype separately (Figure 1B-D). Consistent with previous findings, leupeptin treatment decreased the lysosomal abundance of cathepsins and increased the macroautophagy substrate SQSTM1 in both genotypes [16,24]. The number of proteins changed (unadjusted p < 0.05) by leupeptin in ghr KO lysosomes was 182% of the number of proteins changed by leupeptin in control lysosomes, consistent with the increased CMA activity of the ghr KO lysosomes (Figure 1D).
To identify differences in the lysosomal targetomes between ghr KO and control, we performed a two-factor analysis, evaluating how the ghr KO genotype modified the lysosomal response to leupeptin (Figure 1A, E-G). We have previously reported that liver lysosomes of ghr KO mice, when compared to lysosomes from controls, have increased uptake of three well-characterized endogenous CMA substrates, ENO1, GAPDH, and ACADL [24], and these findings were replicated by the proteomics analysis (Figure 1E). Our previous work reported that the macroautophagy marker SQSTM1 and the endosomal microautophagy marker PPID were equally enriched in ghr KO and control lysosomes in response to leupeptin [24]. These findings were also confirmed by the proteomics analysis, which showed no preferential enrichment of PPID or SQSTM1 (Figure 1E). Despite the increased enrichment of known CMA substrates into ghr KO lysosomes in response to leupeptin, neither the presence of a KFERQ motif nor the number of KFERQ motifs was predictive of preferential increases or decreases in ghr KO lysosomes in response to leupeptin (Figure 1F-G, S1B).
While there is strong evidence to support the hypothesis that the increase in CMA in ghr KO mice results from decreased insulin-IGF1 signaling [24,26], the mechanism of the enhanced CMA in these mice is not completely understood. We hypothesized that ghr KO mice might have changes in the abundance of some signaling proteins associated with the cytosolic face of the lysosomal membrane. We assessed the effect of genotype, ignoring leupeptin (i.e., pooling leupeptin-treated and PBS-treated groups, and assessing for genotype differences), on the composition of the lysosomal fraction (Figure 1H-J). The most significantly changed protein in response to the ghr KO genotype was the GHR protein, which showed a > 20 fold decrease (Figure 1H). Of the 2430 proteins detected in the lysosomal fraction, 138 were known residents of the lysosomal compartment, according to Uniprot annotations. Known lysosomal proteins such as LAMP1, LAMP2, and MEAK7 showed no change with genotype. However, 37 of the 138 known lysosomal proteins identified in the data set significantly changed with genotype (Figure 1I-J, S1C). LAMTOR1 and LAMTOR5 were both significantly increased in the lysosomal fraction of ghr KO mice. LAMTOR1 and LAMTOR5 are components of the RAGULATOR complex, which serves context-specific roles in both the activation and inhibition of MTORC1 on the lysosomal membrane [27,28]. The most significantly changed of the known lysosomal proteins was ELAPOR1/ELAP1/EIG121, the product of an estrogen-induced gene that promotes autophagy and protects against apoptosis [29] which was increased more than 8-fold in ghr KO lysosomes (Fig. S1D). Determining whether or not these proteins play a role in the increased CMA observed in ghr KO mice will require significant follow-up work. However, these findings suggest that lysosomes from ghr KO and control mice have at least some important differences in composition.
Changes in ghr KO liver proteomics suggest increased mitochondrial respiration and decreased protein translation
To identify changes in the protein composition of the liver of ghr KO mice, liver tissue was isolated from six male and six female mice of both ghr KO and control genotypes. The mice were 5–6 months in age, fed ad libitum, and were not subjected to leupeptin treatment, to avoid any possible effects of leupeptin on tissue-wide protein abundance (Figure 2A). Liver samples were subjected to quantitative proteomics, and subjected to the same analysis pipeline described above.
Figure 2.
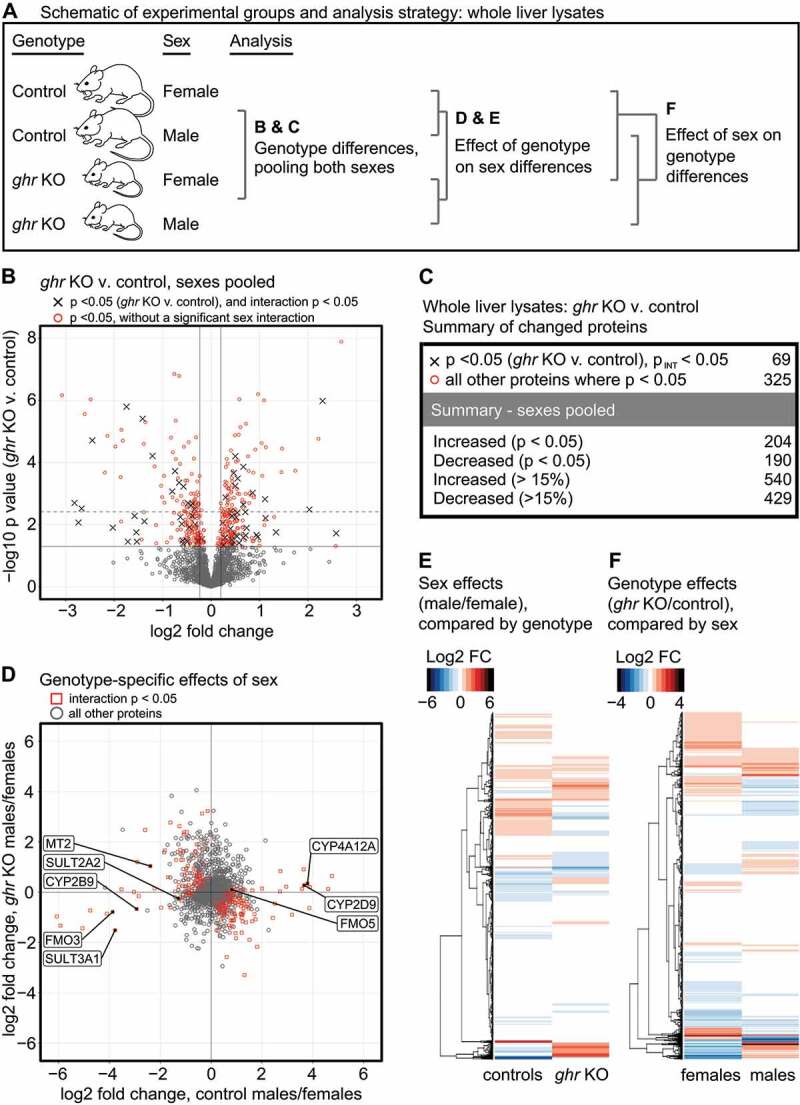
Proteomic changes in ghr KO Liver. (A) Schematic of the experimental groups and analysis. (B) Volcano plot showing proteins changed by the ghr KO genotype, with both sexes pooled. In parallel to the analysis shown in (B), a two-factor analysis was performed to identify proteins whose abundance was significantly affected by the interaction between sex and genotype. Proteins above the y-axis p < 0.05 threshold that are significantly affected by the sex v. genotype interaction are shown as “x” symbols. All other proteins above the y-axis p < 0.05 threshold are shown as red circles. Dark vertical lines indicate a fold-change of change of 15%. The dark solid horizontal line indicates a threshold of p = 0.05. The dashed horizontal line indicates a false-discovery adjusted threshold of p = 0.05. (C) Summary table for numbers of proteins changed by the ghr KO genotype. (D) Scatter plot showing the log2 fold-change in protein abundance with sex in ghr KO, plotted against the log2 fold-change in protein abundance with sex in controls. Proteins with a significant p value (p < 0.05) for the interaction between sex and genotype are shown as red squares. All other proteins are shown as gray circles. Proteins involved in xenobiotic metabolism that have been previously shown to have gene expression changes significantly affected by the interaction between sex and genotype are labeled. (E) Same data as shown in (D), presented as a heat map color-coded by log2 fold change, with a side-by-side comparison of sex differences, separated by genotype. (F) Heat map color-coded by log2 fold change, with a side-by-side comparison of genotype differences, separated by sex. Each of the four experimental groups has liver tissue samples from a total of 6 mice.
Changes in protein abundance with genotype were initially evaluated with sexes pooled (Figure 2B-C). Differences between normal male and normal female mice in circulating growth hormone levels and pulse patterns cause sex-specific differences in liver gene expression [30], and genetic ablation of growth hormone signaling significantly alters the sex differences in growth hormone-susceptible genes [30,31]. To account for sex effects on proteins changed by the ghr mutation, we performed a 2-factor analysis (sex vs. genotype), to identify proteins with a significant interaction effect, i.e., an effect of genotype on the sex differences (Figure 2B-D). Of the 394 proteins that changed (with p < 0.05) in ghr KO mice, 69 had a significant (p < 0.05) interaction p value.
To further evaluate the effects of genotype on sex differences in protein abundance, we compared the log2 fold change in protein abundance with sex (males/females) between the ghr KO and control genotypes (Figure 2D-E). Using the 2-factor analysis (sex vs. genotype), there were 340 proteins with a significant interaction p value. Thus, consistent with previous evidence on sex-specific GH effects, the effect of sex on the abundance of these proteins is altered by the ghr deletion; reciprocally, the effect of the ghr genotype on these proteins is sex-specific. This subset of 340 proteins included several xenobiotic metabolizing enzymes (Figure 2D), whose sex-specific differences in mRNA levels are altered by the ghr mutation [31]. All the changes observed in the protein levels of the xenobiotic metabolizing enzymes have the same directionality as the previously published changes in mRNA expression [31], suggesting that at least some of the effects of genotype on sex-specific differences in protein abundance are driven by sex-specific effects of the growth hormone signaling pathway on mRNA expression. The data shown in (Figure 2D) are presented as a heat map in (Figure 2E).
Finally, we compared the effects of the ghr KO genotype on protein abundance between males and females (Figure 2F). Consistent with the previous analyses, few of the proteins changed by ghr deletion in females showed similar changes in males, and vice-versa. Taken together, these data suggest that the ghr mutation significantly affects sex-specific differences in protein expression. Thus, ghr genotype effects on protein expression should be interpreted with caution, when pooling both sexes, or when evaluating one sex only.
We conducted a gene ontology (GO) analysis on proteins changed by ghr deletion, from sexes pooled, or considered separately. GO analysis indicated that the proteins increased in abundance in ghr KO mice were highly enriched for pathways involved in mitochondrial respiration and oxidoreductase activity (Fig. S2). Proteins decreased in abundance in ghr KO mice were highly enriched in pathways associated with translation/peptide synthesis (Fig. S2). Most of the top five GO terms derived from pooling the sexes were also significantly modified by the ghr mutation when the sexes were considered separately, i.e., the pathways most significantly changed by ghr deletion are changed in both sexes.
Candidate CMA target proteins belong to specific molecular pathways
To identify proteins putatively regulated by selective lysosomal proteolysis in ghr KO mice, we plotted the log2 fold-change in protein abundance in liver against the log2 fold-change in lysosomal protein enrichment in response to leupeptin (Figure 3A). The log2 fold-change in the liver was calculated with sexes pooled, as shown in (Figure 2B), with the understanding that this approach requires significant follow-up analysis and validation. The change in lysosomal protein enrichment in response to leupeptin was calculated by the 2-factor analysis of the effect of the ghr KO genotype on the leupeptin response, as shown in (Figure 1E-G).
Figure 3.
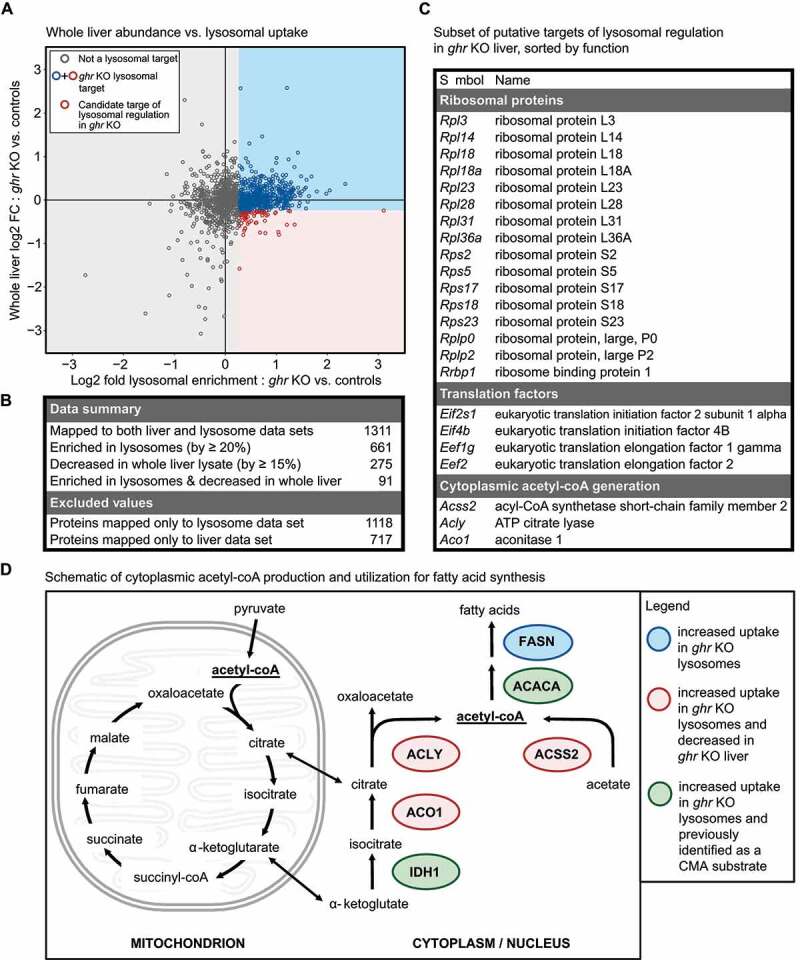
Identification of a subset of the ghr KO proteome putatively regulated by lysosomal degradation. (A) Scatter plot of whole liver log2 fold change against log2 fold enrichment in ghr KO lysosomes. Red circles represent proteins with > 20% enrichment in ghr KO lysosomes in response to leupeptin, and a decrease of >15% in whole liver lysates. Blue circles indicate proteins with > 20% enrichment in ghr KO lysosomes in response to leupeptin, but without a decrease of >15% in whole liver lysates. All remaining proteins are shown as gray circles (B) Summary table of proteins changed in liver and liver lysosomes. The numbers of proteins excluded from the analysis are also listed. (C) Table summarizing which of the candidate targets of lysosomal regulation in ghr KO mice are ribosomal constituents, translation regulation factors, or enzymes for cytoplasmic acetyl-coA production. (D) Schematic of cytoplasmic acetyl-coA production and utilization for fatty acid synthesis, with lysosomal targets highlighted.
As an exploratory attempt to categorize proteins as targets of selective lysosomal degradation in ghr KO mice, we assigned a log2 fold change threshold of 20%, similar to what was used in the previous studies [16,19]. All proteins with >20% increased enrichment into ghr KO lysosomes in response to leupeptin were termed “ghr KO lysosomal targets”. Proteins that met both criteria of being decreased in total abundance in ghr KO liver by 15% and 20% enrichment in ghr KO lysosomes over control, in response to leupeptin were termed “candidate targets of lysosomal regulation”. The use of thresholding based on log2 fold change requires substantial experimental validation, especially with the large number of sex/genotype interactions in protein abundance. The remainder of this paper focuses on experimental validation of results derived from Figure 3A.
We found 91 “candidate targets of lysosomal regulation” in ghr KO liver (Figure 3B). Many of these 91 proteins belonged to specific functional groups, especially ribosomal constituents, translation initiation and elongation factors, and enzymes involved in nucleocytosolic acetyl-coA production (Figure 3C). CMA is known to regulate many metabolic enzymes, translation initiation factors, and ribosomal proteins [16,19,20,32]. Thus, our results are consistent with the current understanding of CMA in regulating protein abundance. However, these data suggest a novel role for selective lysosomal proteolysis in regulating nucleocytosolic acetyl-coA production, translation elongation, and a large subset of ribosomal proteins. To visualize a possible role for lysosomal degradation in controlling translation in ghr KO, we re-labeled (Figure 3A) to highlight proteins belonging to the “ribosome” gene ontology (from AmiGO2) and the “translation initiation” gene ontology (Fig. S3A). There was a noticeable nonrandom distribution of the ribosome and translation initiation proteins indicating increased accumulation in ghr KO lysosomes (in response to leupeptin) and decreased total abundance in ghr KO liver tissue. This distribution was statistically nonrandom (p = 0.0001; Fig. S3B).
Previously, enzymes involved in glycolysis have been described as CMA substrates [16]. While we found that most enzymes mapping to the “Glycolytic Processes” GO term were more enriched in ghr KO lysosomes in response to leupeptin, compared to controls, these enzymes did not show any clear pattern of changing in total abundance in ghr KO liver (Fig. S3C-D). Determining the role of selective lysosomal proteolysis (or more specifically, CMA) in regulating the pathways identified by the proteomics will require extensive experimental validation. As an initial step, we decided to focus on the role of lysosomal proteolysis in regulating nucleocytosolic acetyl-coA-producing enzymes.
Acetyl-coA is best known for its role in mitochondrial energy production. Acetyl-coA is also an essential metabolite in the cytosol and nucleus, and it is exchanged between these two compartments. However, nucleocytosolic acetyl-coA is not directly exchanged with mitochondrial acetyl-coA [33]. In the cytosol, acetyl-coA is consumed for lipid biosynthesis (Figure 3D) and used in the acetylation of many proteins, and in the nucleus, it is used for histone acetylation. Thus, nucleocytosolic acetyl-coA abundance influences the activities of many proteins, patterns of gene expression (via histone modifications), and rates of triglyceride and cellular membrane synthesis [33]. Nucleocytosolic acetyl-coA is produced primarily by two enzymes found in both the cytosol and nucleus, ACLY and ACSS2 [33,34]. ACLY consumes ATP to convert citrate into acetyl-coA, yielding oxaloacetate as a byproduct. ACSS2 generates acetyl-coA from acetate [33]. A schematic of these pathways is shown in Figure 3D.
Our proteomics data suggest the hypothesis that lysosomal proteolysis reduces the abundance of ACLY, ACSS2, and ACO1 (which acts immediately upstream of ACLY) in the liver of ghr KO mice (Figure 3D). Western blots for these proteins are shown in (Fig. S4A-B). By western blot, ACLY and ACSS2, but not ACO1, showed significant effects of genotype on protein abundance (by 2-factor ANOVA). However, ACLY had a significant sex/genotype interaction (the magnitude of the decrease in ACLY in females was much larger than the magnitude of decrease in ACLY in males). IDH1, which acts upstream of ACO1, has been previously identified as a CMA substrate [16]. ACACA, which catalyzes the first step in lipid synthesis using nucleocytosolic acetyl-coA as a substrate, is also a known CMA substrate [16,35]. FASN, which acts downstream of ACACA in lipid synthesis, was enriched in ghr KO lysosomes, but did not decrease in tissue-wide abundance (Figure 3D) [35].
Many known CMA substrates, including ACADL, ENO1, and GAPDH, did not change in abundance in ghr KO liver (Fig. S4A-B), despite the increased enrichment of these proteins into ghr KO lysosomes in response to leupeptin (Figure 1E). In fact, most of the proteins that had increased leupeptin-induced enrichment in ghr KO liver lysosomes did not show changes in tissue-wide abundance, consistent with the emerging idea that CMA directly controls only a small subset of the proteome [19].
Mice with enhanced CMA, but reduced macroautophagy, have reduced liver abundance of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes
pou1f1 mutant/Snell dwarf mice, like ghr KO mice, live dramatically longer than their normal littermates [36]. Snell dwarf mice have reduced hepatic macroautophagy, but enhanced hepatic CMA [24]. We evaluated the protein levels of CMA target proteins in liver tissue from six male and six female Snell dwarf mice and equal numbers of littermate controls, all fed ad libitum. The Snell dwarf mice had significant reductions in IDH1, ACO1, ACLY, ACACA, FASN, and ACSS2 (Figure 4A-B). While this finding does not prove that CMA is downregulating nucleocytosolic acetyl-coA enzymes in Snell liver, it is consistent with the overall hypothesis that enhanced CMA is sufficient to downregulate these proteins. The downregulation of these proteins in Snell mice cannot be attributed to macroautophagy, which is decreased in these mice, relative to their sibling controls. Snell dwarf mice did not show a significant decrease in ACADL, ENO1, or GAPDH, (similar to the ghr KO mice) suggesting that enhanced CMA is not sufficient to reduce the abundance of these proteins in liver tissue (Figure 4A-B).
Figure 4.
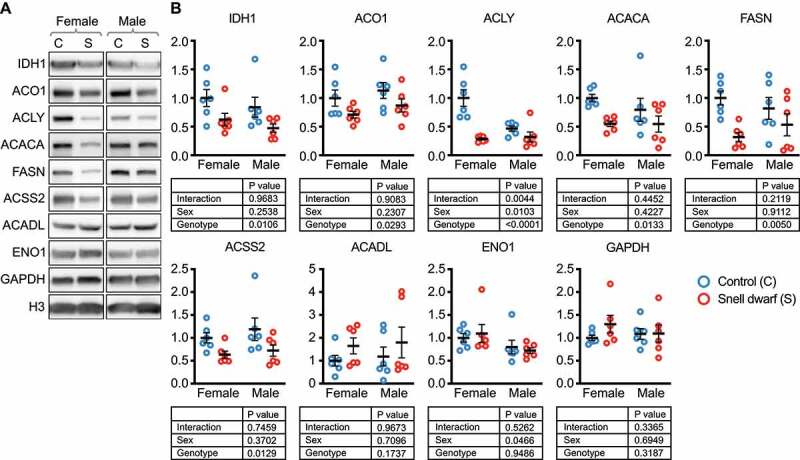
Nucleocytosolic acetyl-coA and fatty acid production enzymes are reduced in Snell dwarf liver. (A) Western blots for the indicated proteins in liver lysates from ad libitum fed Snell dwarf and control mice, age 5–6 months. (B) Quantification of western blots shown in (A). Results of a 2-way ANOVA are indicated below each graph. Black lines indicate mean and SEM. n = 6 for each group tested. Display images for westerns blots for males and females were run on the same gel, with intervening lanes cropped out. All proteins are adjusted for loading, using H3 as a control.
Lysosomal proteolysis, but not proteasomal proteolysis, is necessary to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in NIH3T3 cells
To determine if lysosomal proteolysis is necessary to regulate the abundance of nucleocytosolic acetyl-coA producing enzymes and enzymes involved in fatty acid synthesis, we inhibited lysosomal proteolysis in NIH3T3 cells with 100 μM leupeptin and 20 mM ammonium chloride for 48 h, and then measured the abundance of these proteins. The protein levels of LAMP2A and LC3-II increased in response to the leupeptin and ammonium chloride treatment, confirming successful inhibition of lysosomal proteolysis (Figure 5A). The protein levels of IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN significantly increased when lysosomal proteolysis was inhibited (Figure 5A-B). Interestingly, the previously identified CMA substrates, which did not decrease in whole-tissue abundance in ghr KO mice, ACADL, ENO1, and GAPDH, also did not change in abundance when lysosomal proteolysis was inhibited (Figure 5A-B).
Figure 5.
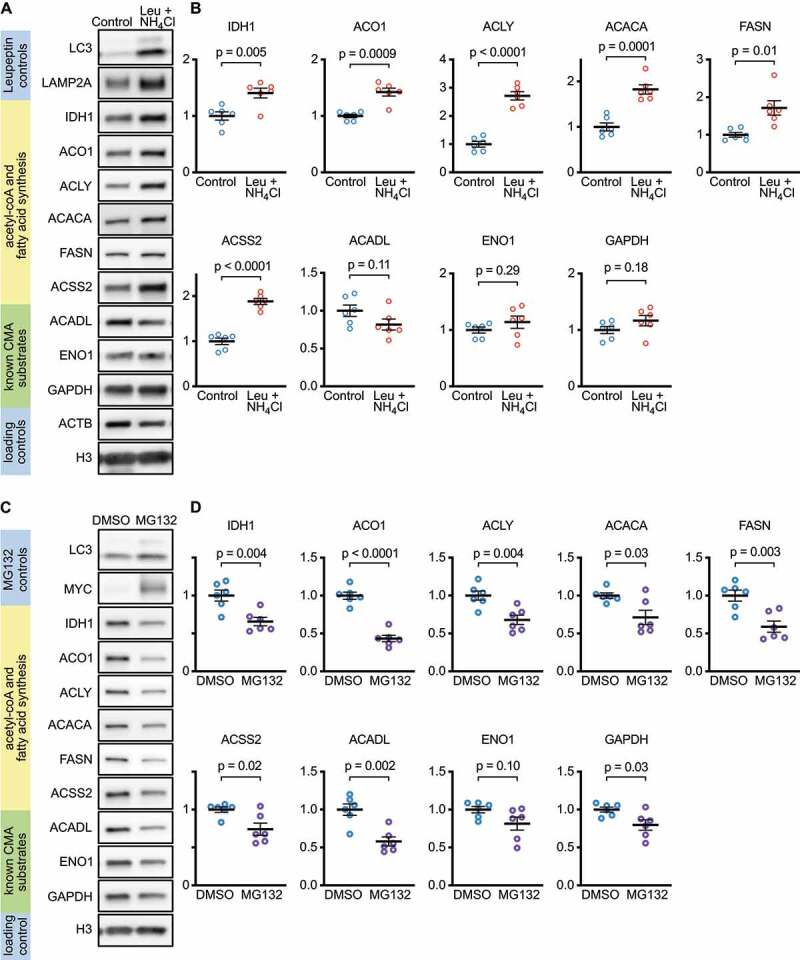
Lysosomal proteolysis, but not proteasomal proteolysis, is necessary to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid production enzymes. (A) Western blots for indicated proteins in lysates from NIH3T3 cells treated for 48 h with leupeptin and ammonium chloride to block lysosomal, but not proteasomal proteolysis. (B) Quantifications of western blots shown in (A). (C) Western blots for indicated proteins in lysates from NIH3T3 cells treated for 24 h MG132 to block proteasomal, but not lysosomal proteolysis (48 h treatments with MG132 were cytotoxic). (D) Quantifications of western blots shown in (C). Each experiment was repeated six times, with individual values shown on the graphs. Black bars indicate means and SEM. p values shown on graphs are derived by unpaired t-tests with Welch’s correction for unequal SDs. All proteins are adjusted for loading, using H3 as a control.
To test if proteasomal degradation is necessary to regulate the abundance of nucleocytosolic acetyl-coA producing enzymes and fatty acid synthesis enzymes in NIH3T3 cells, we inhibited proteasomal degradation with 10 μM MG132 for 24 h (48-h treatments were extremely cytotoxic). As expected, MYC, a proteasome target, dramatically increased in abundance, showing that the treatment was effective (Figure 5C). Interestingly, IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN all decreased significantly (Figure 5C-D). ACADL and GAPDH also decreased significantly, but there was no change in ENO1 (Figure 5C-D). The decreases in these proteins might be caused by an increase in lysosomal proteolysis as a compensatory response to the inhibition of the proteasome. These data suggest that, in NIH3T3 cells, the proteasome is not necessary to regulate the abundance of IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN.
CMA is both necessary and sufficient to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in NIH3T3 cells
To determine if CMA is necessary to control the abundance of nucleocytosolic acetyl-coA enzymes, we inhibited CMA in NIH3T3 cells by knocking down Lamp2a by siRNA. Ninety-six hours after siRNA treatment, IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN all showed significantly increased protein levels, suggesting that CMA is necessary to control the abundance of these enzymes (Figure 6A-B). ACADL, ENO1, and GAPDH did not change significantly (Figure 6A-B). Thus, even though these three enzymes are de facto CMA substrates [16,37], CMA is not necessary to control their abundance in NIH3T3 cells. The mRNA abundances of Idh1, Aco1, Acly, Acss2, and Acaca were not altered by siLamp2a treatment (Figure 6C). Fasn mRNA was decreased, despite the increase in FASN protein levels. This suggests that the increase in the abundance of these proteins when CMA is inhibited is not because of a transcriptional change.
Figure 6.
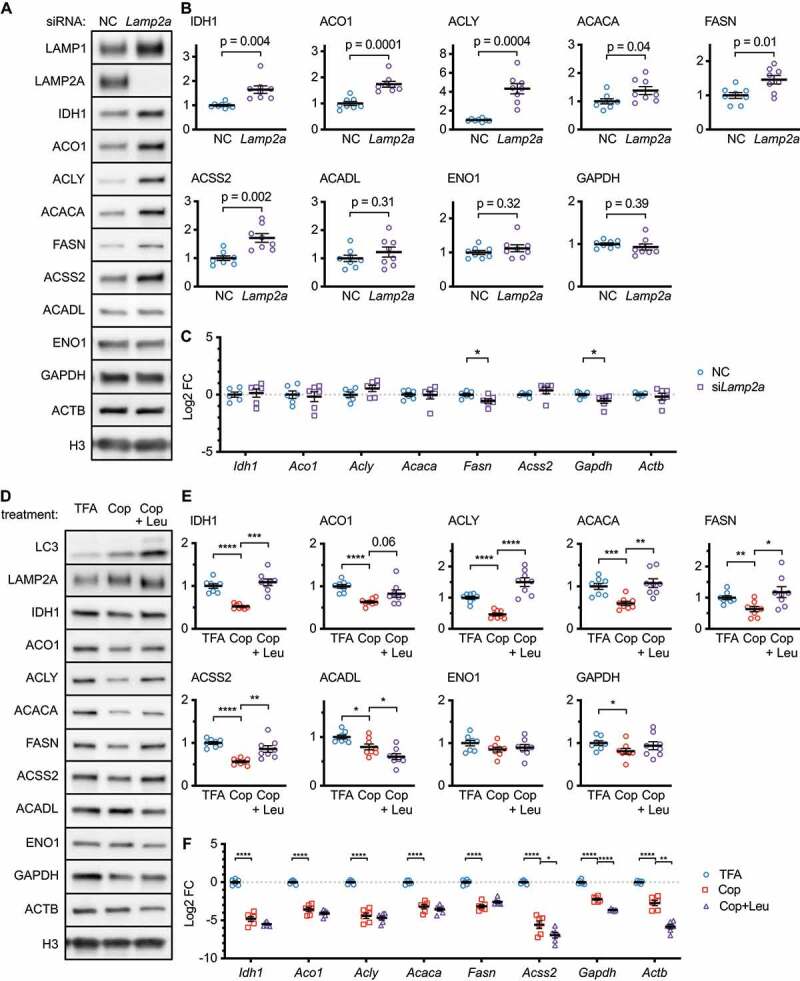
CMA is both necessary and sufficient to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid production enzymes. (A) Western blots for indicated proteins in lysates from NIH3T3 cells, collected 96 h after siRNA transfection. (B) Quantifications of western blots shown in (A). All proteins are adjusted for loading, using H3 as a control. (C) Relative mRNA expression of the indicated genes in NIH3T3 cells collected 96 h after transfection with the indicated siRNAs. (D) Western blots for indicated proteins in lysates from NIH3T3 cells treated for 48 h with 0.0005% TFA (solvent control), 100 nM copanlisib, or 100 nM copanlisib + leupeptin/ammonium chloride. (E) Quantifications of western blots shown in (D). (C) Relative mRNA expression of the indicated genes in NIH3T3 cells treated with the same conditions as (D-E). All proteins are adjusted for loading using the geometric mean of ACTB and H3 as a loading control. Individual values are shown for all experiments. Black bars indicate means and SEM. p values shown on graphs are derived by unpaired t-tests with Welch’s correction for unequal SDs. In (E), p-values are replaced with asterisks to save space: *p < 0.05, **p < 0.01, ***p < 0.001.
CMA and endosomal microautophagy each depend on HSPA8 for substrate selection and uptake [5,38]. To test the requirement of the different forms of autophagy to regulate these enzymes, we treated NIH3T3 cells with siRNA against Lamp2a, Atg5, or Tsg101, to block CMA, macroautophagy, or endosomal microautophagy, respectively. No proteins increased when macroautophagy was inhibited (Fig. S5A-B), suggesting that macroautophagy is not required to regulate any of the enzymes tested. IDH1, ACADL, ENO1, and GAPDH showed significant reductions when macroautophagy was inhibited, possibly resulting from compensatory upregulation in other forms of proteolysis (Fig. S5A-B). ACLY, ACSS2, and ACACA showed significant increases when endosomal microautophagy was inhibited, suggesting that both forms of HSPA8-mediated autophagy are required to regulate these proteins (Fig. S5A-B). ACADL, ENO1, and GAPDH did not change when endosomal microautophagy was inhibited, suggesting that neither form of HSPA8-mediated autophagy is required to regulate the abundance of these proteins (Fig. S5A-B).
We next tested whether CMA is sufficient to regulate the protein abundance of IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN in NIH3T3 cells. We have previously shown that inhibitors of the PIK3CA/p110 catalytic component of the class I phosphoinositide 3-kinase (PI3K) activate CMA in multiple cell types and in mouse liver lysosomes, without increasing macroautophagy [26]. Moreover, mice treated with these inhibitors have increased lysosomal enrichment of endogenous CMA substrates in response to leupeptin, but do not have increased enrichment of PPID, a substrate of endosomal microautophagy [26]. We hypothesized that treating NIH3T3 cells with inhibitors of class I PI3Ks would reduce the abundance of CMA target proteins. We treated NIH3T3 cells for 48 h with two doses of copanlisib or two doses of pictilisib, along with their respective solvent controls. As expected, both drugs showed dose-dependent effects on IDH1, ACO1, ACLY, ACACA, FASN, and ACSS2 (Fig. S6A-D).
We considered the possibility that the PI3K inhibitors could affect the levels of CMA target proteins through a mechanism other than lysosomal proteolysis, such as a transcriptional change. We performed a rescue experiment, where we treated NIH3T3 cells with 100 nM copanlisib (previously published dose [26]) with or without leupeptin/ammonium chloride co-treatment, for 48 h. We reasoned that if leupeptin/ammonium chloride blocked the effects of copanlisib on the changes in candidate CMA target proteins, and then the effects of copanlisib on the abundance of these proteins must be mediated by lysosomal proteolysis. Indeed, copanlisib treatment caused a significant reduction in IDH1, ACO1, ACLY, ACSS2, ACACA, FASN, and GAPDH, which was rescued by leupeptin/ammonium chloride co-treatment (Figure 6D-E). This suggests that, in NIH3T3 cells, CMA is both necessary and sufficient to regulate the protein levels of IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN. CMA is sufficient, but not necessary, to regulate the level of GAPDH. CMA is neither necessary nor sufficient to regulate the abundance of ACADL and ENO1. The mRNA abundance of every gene tested was decreased by copanlisib treatment. However, leupeptin did not rescue the mRNA levels of any genes (Figure 6F), suggesting that lysosomal proteolysis is required to reduce the levels of the tested proteins, regardless of the mRNA changes.
Our previous data suggest that copanlisib does not activate endosomal microautophagy [26]. To show that the effects of copanlisib on ACLY and ACSS2 are specific to CMA and not endosomal microautophagy, we treated NIH3T3 cells with siRNA targeting either Lamp2a or Tsg101. Seventy-two hours after transfection, the cells were treated with copanlisib for 48 h. We found that knocking down Lamp2a significantly rescued the effects of copanlisib on the abundance of ACLY and ACSS2 (Figure 7A-B). However, knocking down Tsg101 did not rescue the effects of copanlisib on either protein (Figure 7C-D). These data are consistent with the conclusion that the effects of copanlisib on ACLY and ACSS2 are mediated by CMA, rather than by endosomal microautophagy.
Figure 7.
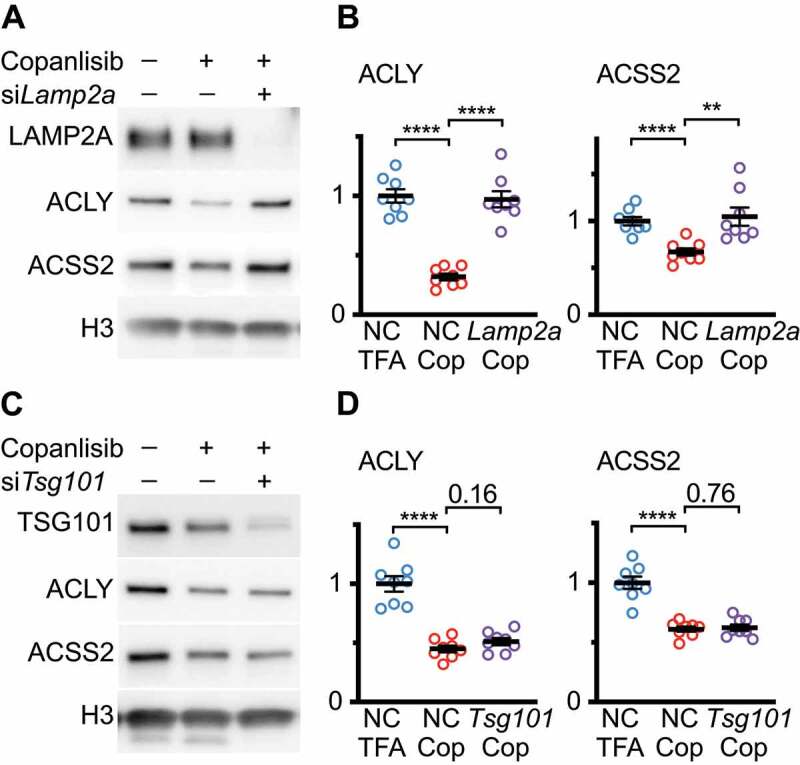
Blocking CMA, but not endosomal microautophagy, rescues the effects of copanlisib on ACLY and ACSS2 abundance. (A) Western blots for indicated proteins in lysates from NIH3T3 cells, that were treated with copanlisib for 48 h, starting 72 h after transfection with negative control (NC) or Lamp2a siRNAs. (B) Quantification of westerns shown in (A). (C) Western blots for indicated proteins in lysates from NIH3T3 cells, that were treated with copanlisib for 48 h, starting 72 h after transfection with negative control (NC) or Tsg101 siRNAs. (D) Quantification of westerns shown in (C). Black bars indicate means and SEM. p values shown on graphs are derived by unpaired t-tests. Significant p-values are replaced with asterisks to save space: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Mice treated with a CMA-activating drug have enhanced lysosomal enrichment of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in response to leupeptin treatment
We have previously reported that mice treated with CMA-activating drug buparlisib have increased enrichment of CMA substrates in liver lysosomes in response to leupeptin treatment, when compared to solvent treated mice [26]. We evaluated liver lysosomes from these mice for enrichment of enzymes involved in nucleocytosolic acetyl-coA and fatty acid synthesis (Figure 8A). We found that, as a general trend, buparlisib increased lysosomal enrichment of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in response to leupeptin, although this fell short of statistical significance for ACSS2 (Figure 8B). These data suggest that enhancing CMA increases the lysosomal uptake of IDH1, ACO1, ACLY, ACACA, and FASN.
Figure 8.
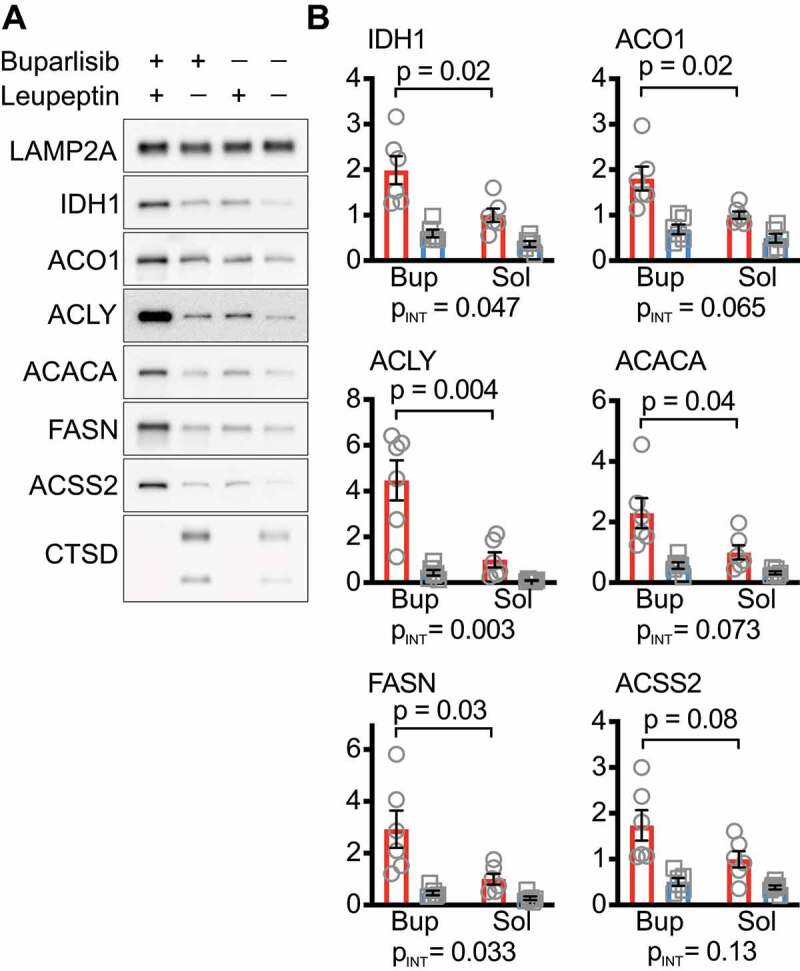
Mice treated with a CMA activating drug have enhanced lysosomal enrichment of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in response to leupeptin treatment. (A) Western blots for indicated proteins in liver lysosomes isolated from mice treated with buparlisib or solvent control that were also treated with leupeptin or solvent control 2 h before being sacrificed. LAMP2A is a loading control. CTSD is a control for successful administration of leupeptin. (B) Quantification of Western blots shown in (A). Black bars indicate means and SEM. PINT, shown below each graph, is the interaction term p-value from a 2-way ANOVA. p values shown on graphs in are derived from unpaired t-tests.
CMA is sufficient to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid synthesis enzymes in AML12 hepatocytes
To confirm that the effects of CMA activation on IDH1, ACO1, ACLY, ACSS2, ACACA, and FASN protein levels are not cell-type specific, we evaluated the effects of CMA activation on these proteins in AML12 hepatocytes. We began by seeking doses of copanlisib that are effective for activating CMA in AML12 cells. We first determined that a dose of 100 nM copanlisib significantly enhances CMA, using a KFERQ-Dendra2 reporter (Figure 9A-B). The KFERQ-Dendra2 reporter accumulates as lysosomal puncta, when CMA is active, and is often used as an initial test to identify CMA activating interventions [26,39,40]. This 100 nM dose of copanlisib did not affect the macroautophagic flux in AML12 cells (Figure 9C-D). We then treated AML12 cells for 48 h with 100 nM copanlisib, with or without leupeptin/ammonium chloride co-treatment. We found that copanlisib treatment caused a significant reduction in IDH1, ACO1, ACLY, ACACA, FASN, ACSS2, ACADL, and GAPDH. However, leupeptin treatment only rescued the levels of IDH1, ACLY, ACACA, FASN, and ACSS2 (ACO1 had a trend toward a rescue, but this was shy of significance, p = 0.06; Figure 9E-F). The protein abundances of ACADL and GAPDH were not significantly rescued by leupeptin. Taken together, these data suggest that CMA is sufficient to downregulate the abundance of IDH1, ACLY, ACACA, FASN, and ACSS2 in AML12 hepatocytes.
Figure 9.
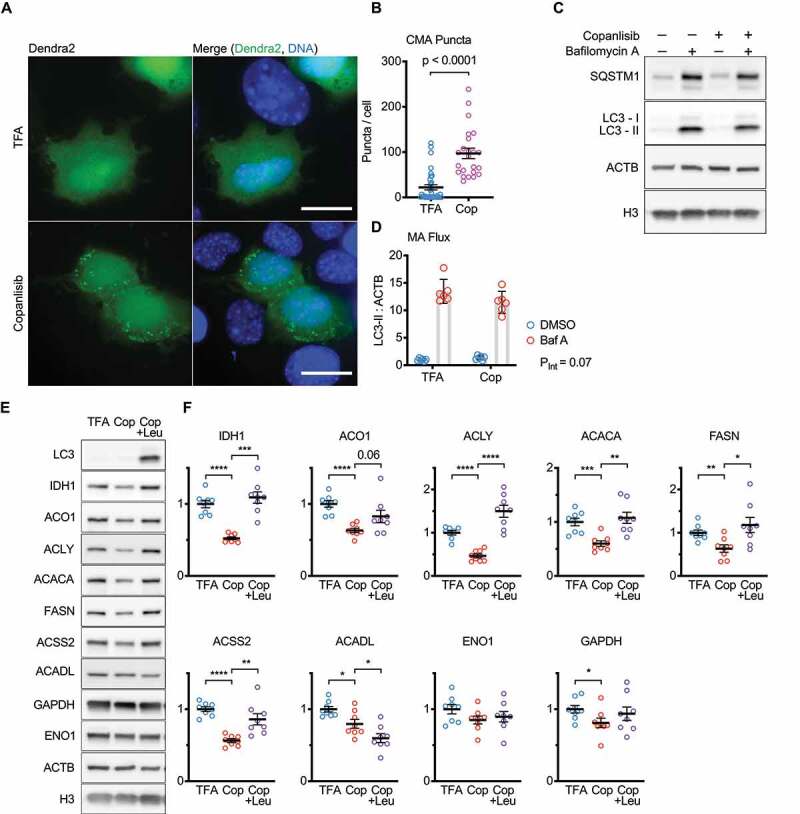
CMA is sufficient to regulate the abundance of nucleocytosolic acetyl-coA and fatty acid production enzymes in AML12 hepatocytes. (A) Z-projections of images of Dendra2 CMA reporter in AML12 cells treated with 100 nM copanlisib, or TFA solvent control for 10 h. (B) Dendra2 puncta per cell, from 4 frames from 3 separate experiments. (C) Western blots showing macroautophagic flux in AML12 cells treated with 100 nM copanlisib, or TFA solvent control for 10 h. (D) quantification of LC3-II flux, pertinent to (C); six replicates are shown on the graph. (E) Western blots for indicated proteins in lysates from AML12 cells treated for 48 h with 0.0005% TFA (solvent control), 100 nM copanlisib, or 100 nM copanlisib + leupeptin/ammonium chloride. (F) Quantifications of western blots shown in (E). All proteins are adjusted for loading, using ACTB as a loading control. For (E-F) each experiment was repeated eight times, with individual values shown on the graphs. Black bars indicate means and SEM. p value shown in (B) is from a Kolmogorov–Smirnov test. PINT, shown in (D) is an interaction term p-value from a 2-way ANOVA. p values shown on graphs in (F) are derived by unpaired t-tests with Welch’s correction for unequal SDs. In (F), p-values are replaced with asterisks to save space: *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: 20 μm.
CMA regulates lipid droplet accumulation through ACLY and ACSS2
The acetyl-coA produced by ACLY and ACSS2 can be used for a variety of intracellular functions, including histone acetylation and fatty acid synthesis [33,34,41]. We examined H3 acetylation in NIH3T3 cells treated with siRNA targeting Lamp2a, but we did not detect any consistent changes in H3 acetylation (data not shown). Previous studies have found that inhibition of CMA causes accumulation of lipid droplets in NIH3T3 cells [25]. We hypothesized that the increase in ACLY and ACSS2 caused by the inhibition of CMA could contribute to increased de novo lipogenesis, leading to an increase in lipid droplets. To test this hypothesis, we treated NIH3T3 cells with siRNA against Lamp2a, to induce lipid droplet accumulation (Figure 10A). We also co-treated cells with siRNA against Acly or Acss2, in addition to the Lamp2a siRNA treatment. Ninety-six hours after transfection, the cells were stained with BOPIDY to highlight lipid droplets, and fixed for microscopy (Figure 10A-B). We found, as expected, that a reduction in Lamp2a led to an increase in intracellular lipid droplets, and then found that a reduction in either ACLY or ACSS2 was sufficient to partially rescue the lipid droplet phenotype, suggesting that both of these enzymes contribute to lipid droplet accumulation (Figure 10C). These data suggest a mechanism where CMA reduces de novo lipid synthesis by negatively regulating the protein levels of ACLY and ACSS2.
Figure 10.
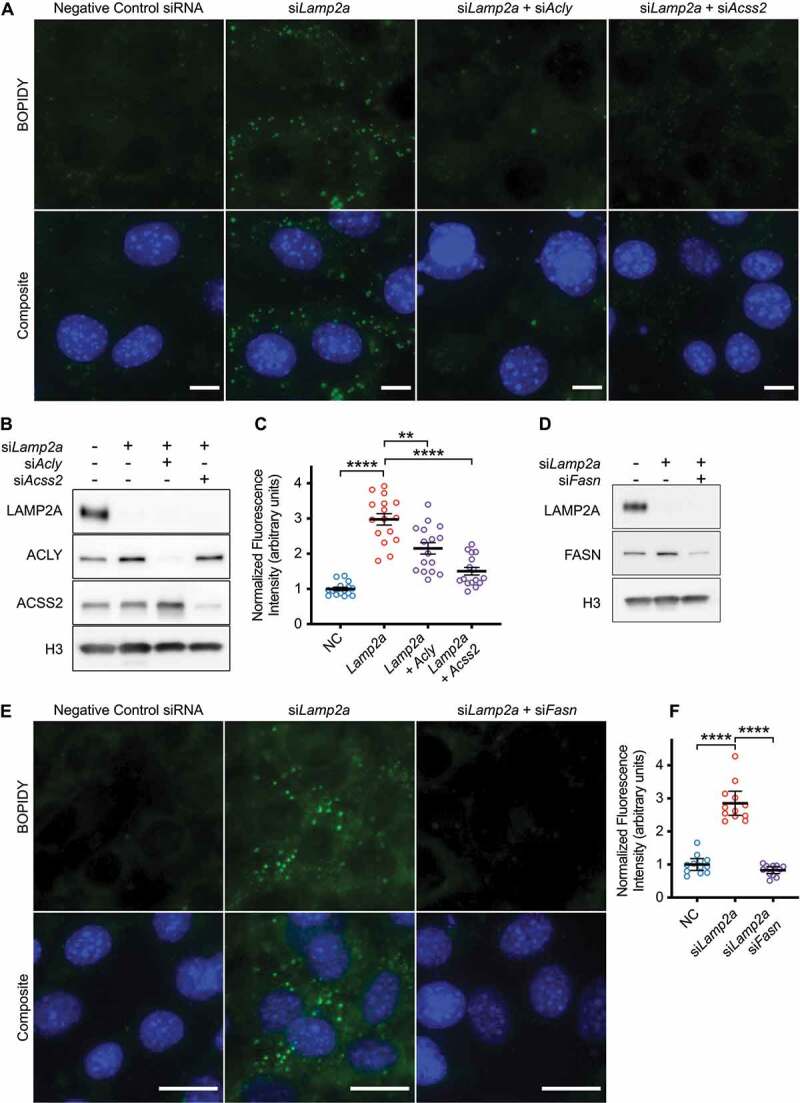
CMA regulates lipid droplet accumulation through ACLY and ACSS2. (A) NIH3T3 cells were stained with BOPIDY (lipid droplets) and Hoechst (DNA), 96 h after transfection with the indicated siRNAs. Scale bars: 10 μm. (B) Western blot validation for knockdown efficiency for siRNAs against Lamp2a, Acly, and Acss2. (C) Quantification of BOPIDY staining intensity across whole fields of confluent cells, imaged with identical exposure times, with no post-acquisition image adjustment. Four fields for each of four experimental repeats are pooled, for a total of 16 data points for each group. (D) Western blot validation for knockdown efficiency for siRNAs against Lamp2a and Fasn. (E) NIH3T3 cells were stained with BOPIDY and Hoechst 96 h after transfection with the indicated siRNAs. Scale bars: 20 μm. (F) Quantification of BOPIDY staining, using the same methods as (C). p-values are replaced with asterisks to save space: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. For (D) results of a 2-way ANOVA are shown in the table under the graph.
As a control, we showed that treating Lamp2a-depleted cells with siRNA targeting Fasn was sufficient to rescue the lipid droplet accumulation phenotype (Figure 10D-F). This confirms that lipid droplet accumulation in response to the inhibition of CMA is dependent upon de novo lipid synthesis.
Enzymes involved in nucleocytosolic acetyl-coA synthesis are reduced in white adipose tissue of ghr KO mice
Adipocytes in inguinal white adipose tissue (I-WAT) in ghr KO mice have a significant reduction in cell volume, and therefore, lipid droplet volume [42]. We hypothesized that this reduction in cell volume might be associated with a reduction in the enzymes responsible for de novo lipid synthesis. We measured the protein levels of IDH1, ACO1, ACLY, ACSS2, ACACA, FASN, ACADL, ENO1 and GAPDH in ghr KO I-WAT, finding a significant reduction in ACLY, ACC1, and FASN, in ghr KO, relative to controls (Fig. S7). ACSS2 was significantly decreased only in females. These data suggest the hypothesis that elevated CMA in ghr KO mice reduces de novo lipid synthesis, and these results could be the foundation for future work evaluating the role of CMA in adipose tissue.
Discussion
The role of protein degradation in maintaining health in young animals is often attributed to maintaining a “clean” proteome by removing proteins that are damaged, dysfunctional, or present in excess, thus maintaining the mouse proteome in normal working order [43,44]. According to this model, lysosomal proteolysis decreases with age, contributing to age-associated declines in proteostasis and the development of age-related disease [23,45]. Thus, restoration of youthful levels of CMA or macroautophagy in aging tissues has been tested as a strategy for delaying aging and associated morbidities [17,45–47].
Our own results suggest an alternative paradigm: when enhanced in adult mice, as it is in ghr KO and Snell dwarf mice, CMA promotes longevity, not only by “cleaning” the proteome or degrading proteins whose accumulation might lead to disease but instead by shaping molecular pathways that fundamentally influence the rate of aging. In this model, elevated CMA reduces the abundance of normal functioning proteins, through selective degradation, to influence specific molecular pathways. Snell dwarf mice, which have enhanced CMA, show a reduction of two CMA-sensitive proteins [18], CIP2A and MYC, in liver, kidney, and skeletal muscle, independent of mRNA changes [24]. CMA directly targets CIP2A, a positive regulator of MYC abundance, and thus CMA indirectly regulates MYC levels through CIP2A [18]. Mice hemizygous for the myc gene are long-lived [48], suggesting one possible connection between the elevation of CMA in Snell dwarf mice and their exceptional longevity [24,48]. We now report that ghr KO liver lysosomes have increased degradation of ACLY and ACSS2. These proteins decrease in ghr KO liver and I-WAT, and their abundance is highly sensitive to CMA function in NIH3T3 and AML12 cells. Loss of function in the homologs of either ACLY or ACSS2 is sufficient to extend lifespan in Drosophila [34,49,50], despite the partially overlapping roles for these proteins in nucleocytosolic acetyl-coA synthesis. There is currently intense interest in whether chemical inhibition of ACLY can promote healthy aging in mice [51], and additional characterization of the role of ACLY in mammalian longevity will help to define a role for CMA in regulating mammalian aging.
Our proteomics data also suggest that ghr KO mice show enhanced lysosomal degradation of EEF2, with a reduction in EEF2 protein levels in the liver. EEF2 is the primary regulator of translation elongation rate, and an important modulator of translation fidelity [52,53]. Decreasing EEF-2/EEF2 activity increases translation fidelity in C. elegans and cultured human cells [54]. While translation fidelity does not decrease as a function of age in mammals [55], enhanced translation fidelity has co-evolved with mammalian longevity, in the sense that cultured dermal fibroblasts from long-lived mammals have fewer translation errors than those from short-lived mammals [56]. Dampening EEF-2 activity to promote translation fidelity is necessary for lifespan extension in C. elegans in response to nutrient restriction [54]. A recent study identifying a role for CMA in regulating translation initiation factors did not find an effect of CMA on EEF2 levels in human breast cancer cells [19]. Whether the disparity between our results with mouse liver and the data on human breast cancer cells is attributable to species-specific or tissue-specific differences, or the effects of neoplastic transformation, will require additional analysis.
CMA regulates the abundance of many neurodegeneration-associated proteins, including SNCA/α-synuclein, MAPT/Tau, HTT (huntingtin), APP (amyloid beta precursor protein), LRRK2, and TARDBP/TDP-43 [23,57–61]. Rats with reduced CMA in the brain quickly exhibit death of dopaminergic neurons in the substantia nigra and display Parkinson's disease-like motor symptoms [22]. Mutant forms of SNCA (identified in human Parkinson's disease patients) resist degradation by CMA and bind to the LAMP2A receptor, blocking other peptides from being degraded by CMA, causing proteotoxic stress [62]. Overexpression of LAMP2A (the rate-limiting component of CMA) in the rat brain protects against SNCA mediated neurodegeneration [17]. CMA-activating chemical derivatives of atypical retinoids delay protein aggregation and behavioral changes in mouse models of Alzheimer’s disease [63]. The clear role of CMA in controlling the abundance of neurodegeneration-related proteins has driven efforts to discover CMA activating drugs, by our group and by others. Here, we have shown that CMA-activating drugs (inhibitors of class I PI3K) can reduce the abundance of proteins involved in nucleocytosolic acetyl-coA production, a pathway implicated in regulating the pace of aging [34,49,50]. Future studies using class I PI3K inhibitors or other CMA activating drugs to modulate pathways implicated in the basic biology of aging in vivo, and in the pathogenesis of age-dependent diseases, will be vital to address the fundamental question of whether or not enhancing CMA is a viable strategy for boosting human longevity.
Studies investigating CMA targets often emphasize the importance of KFERQ motifs in substrate recognition. While the majority of proteins in the mouse proteome contain a KFERQ motif [14], recent work suggests that CMA is sufficient to modulate the abundance of only a small subset of the proteome, clustering into specific pathways [19]. Our data support the model where CMA regulates a small and specific set of proteins. Our data are consistent with the idea that CMA may participate in the turnover of a substantial portion of the proteome but is not sufficient to create significant changes in the steady state abundance of most proteins (Figure 3A). This is likely to reflect multiple mechanisms of control in protein abundance at the levels of transcription, translation, and degradation. HSPA8 binds efficiently to proteins without binding partners [64]. Thus, CMA might play an important role in maintaining appropriate stoichiometry of protein complexes, without meaningfully changing the total abundance of the functional complex. However, additional analysis is needed to test this possibility.
HSPA8 is thought to be the primary substrate selector for both CMA and endosomal microautophagy [5,38]. Interestingly, we found that in cultured NIH3T3 cells, both CMA and endosomal microautophagy are required to regulate the abundance of ACLY, ACSS2, and ACACA. Based on our limited data from evaluating endosomal microautophagy substrate PPID, the pou1f1 and ghr KO mutations and the CMA-activating class I PI3K inhibitors do not seem to be sufficient to activate endosomal microautophagy [24,26]. However, a more thorough analysis of changes to endosomal microautophagy in response to CMA activating mutations and drugs could prove to be an interesting line of investigation for future studies. Additional work on whether endosomal microautophagy activating interventions are sufficient to regulate the CMA target proteins identified in this study could also prove valuable.
In conclusion, this study has identified a novel role for CMA in regulating nucleocytosolic acetyl-coA-producing enzymes ACSS2 and ACLY. Our findings suggest that CMA regulates lipid accumulation by controlling the abundance of ACLY and ACSS2. Even more significantly, the proteomics data generated in this study provide a wealth of possible directions for new research into how selective lysosomal proteolysis regulates the abundance of ribosomal proteins and translation elongation factors in long-lived mice. Differences in the lysosomal membrane composition between ghr KO and control mice also offers new research directions into possible regulators of CMA.
Materials and methods
Antibodies
Commercially available antibodies are listed below. Some antibodies were validated by the vendors by knock out (KO), siRNA knockdown, or overexpression of a tagged protein, and this is indicated where applicable:
ACACA/ACC1 (Cell Signaling Technology [CST]), 4190S; rabbit host; Lot 4; siRNA validated for specificity from ACACB/ACC2), ACADL (AbCam, 196,655; rabbit host; Lot GR224119-15), ACLY (AbCam, 40,793; rabbit host; Lot GR3244069-3; KO validated), ACO1 (AbCam, 183,721; rabbit host; Lot GR153729-5; KO validated), ACSS2 (CST, 3658S; rabbit host; Lot 2), ACTB/β-Actin (CST, 8457 L; rabbit host; Lot 7), ATG5 (AbCam, 108,327; rabbit host; Lot GR3202986-8; KO validated), CHMP3/VPS24 (AbCam, 175,930; rabbit host; Lot GR136727-4), CTSD/cathepsin D (AbCam, 75,852; rabbit host; Lot GR260148-28), ENO1 (CST, 3810S; rabbit host; Lot 2; KO validated), FASN (AbCam, 22,759; rabbit host; Lot GR3192402-1; KO validated), GAPDH (CST, 2118S; rabbit host; Lot 14), H3 (AbCam, 176,842; rabbit host; Lot GR3277361-2), HSPA8/Hsc70 (AbCam, 154,415; rabbit host; Lot GR307969-3), IDH1 (AbCam, 172,964; rabbit host; Lot GR130705-18; KO validated), LAMP1 (AbCam, 24,170; rabbit host; Lot GR3255586-1), LAMP2A (AbCam, 125,068; rabbit host; Lot GR23784-34), MAP1LC3B/LC3 (CST, 2775S; rabbit host; Lot 12), OXPHOS cocktail (AbCam, 110,413; mouse host; Lot L6699), SQSTM1/p62 (CST, 5114S; rabbit host; Lot 6; validated by overexpression of a tagged protein), TSG101 (AbCam, 125,011; rabbit host; Lot GR299332-27), VPS4A (AbCam, 180,581; rabbit host; Lot GR3258605-1).
For western blots, most antibodies detected a single band at the expected molecular weight. For the ACO1 antibody, two bands were detected, and the band near the 100-kDa predicted molecular mass was shown.
ATG5 is predominantly present as a covalently-joined heterodimer with ATG12 (55-kDa band). For most western images, the ATG12-ATG5 heterodimer band is shown, as indicated in the figures.
Antibodies for LAMP2A (AbCam, 125,068), TSG101 (AbCam, 125,011), and ACSS2 (CST, 3658S) were validated by siRNA knockdown in this manuscript. All other antibodies validated by siRNA knockdown in this study were previously validated by knockout.
The OXPHOS cocktail (AbCam, 110,413) and LAMP1 antibody are validated in this manuscript by the staining of purified organelle fractions.
Cell culture
In all cell culture experiments, cells were passaged no more than 20 times, after thawing the original stock from ATCC. Cells were checked for mycoplasma contamination upon thaw, returning a negative result each time. All cells were maintained in a humidified 37°C incubator with 10% CO2 and ambient O2. Recipes for cell culture growth media were obtained from the ATCC website. NIH3T3 cells were obtained from ATCC (CRL-1658) and maintained in DMEM (Gibco, 11,965,092) supplemented with 10% FBS (Corning: 35–016-CV) and penicillin–streptomycin (Gibco, 15,070,063). AML12 cells were obtained from ATCC (CRL-2254) and maintained in DMEM:F12 (Gibco, 11,320,033), supplemented with insulin-selenium-transferrin (Gibco, 51,500–056), 10% FBS (Corning, 35–016-CV), 40 ng/mL dexamethasone (Sigma: D4902), and penicillin–streptomycin (Gibco, 15,070,063).
The KFERQ-Dendra2 plasmid was a gift of Ana Maria Cuervo (Albert Einstein College of Medicine). The reporter consists of the N-terminus of RNASE/RNAase A fused to the photo-switchable fluorescent protein Dendra2, in a third-generation pCCL lentiviral backbone, similar to what has been reported in other studies [39,65,66]. Lentivirus for transgene delivery was created by the University of Michigan BRCF Vector Core. Cells were transduced with virus, using standard techniques to generate stable lines.
CMA reporter
AML12 cells stably expressing the Dendra2 reporter were seeded in 24-well plates with a cover-glass (Fisher, 1,254,580) dropped into each well. Cells were treated with copanlisib (SelleckChem, S2802) for 10 h, washed with PBS (diluted from 10x stock; Gibco, 70,011–044), and fixed in a solution of 1% paraformaldehyde (Fisher, AC416780250), 1% sucrose (Fisher, 50–165-7045), in PBS, pH 7.2, for 7 min at room temperature. Cells were rinsed twice with PBS, stained with Hoechst (Sigma, 94,403), and mounted onto microscope slides (Fisher, 12–550-343), using fluorescent protein mounting medium (GBI Labs, E18-18). Slides were dried overnight at 4°C and were imaged the next day.
Dendra2 puncta were counted blind, with images analyzed in random order, using our previously published methods [26]. First, images were subjected to the ImageJ Max Entropy thresholding algorithm to identify cells with sufficient fluorescent protein expression to be counted. Then, the number of puncta per cell were manually counted in unadjusted images and recorded (because of uneven background, automated counting is consistently less accurate than manual counting). Cells were excluded from analysis if they were on the edge of the image (such that a portion of the cytosol was outside the image, preventing an accurate count), or if their cytoplasm overlapped substantially with another cell, preventing accurate quantification. The final results were unblinded and tabulated by a different person than the one performing the analysis. For each experiment, data were pooled from at least three independent replicates.
Drugs for cell culture experiments
Drugs for cell culture experiments were obtained as follows: Bafilomycin A1 (Sigma, SML1661), MG132 (Sigma, M7449), leupeptin (Sigma, L2884), and ammonium chloride (Sigma, A4514).
Bafilomycin A1 was used at a concentration of 160 nM for all experiments. All other drug doses and incubation times are indicated in the figure legends or directly in the figures. Bafilomycin A1 and MG132 were dissolved in DMSO (Sigma, D2650). Copanlisib, which is not DMSO soluble, was initially solubilized in a 10% trifluoroacetic acid (Sigma, T6508)-90% water solution to a concentration of 2 mM and then diluted with water to appropriate working stock concentrations. Leupeptin (Sigma, L2884) was solubilized in sterile PBS.
For all drug treatments, the cells were maintained in a complete growth medium, with serum, according to the recipes specified above.
Drugs for mouse experiments
Leupeptin was dissolved in sterile PBS to a concentration of 25 mg/mL and administered by intraperitoneal injection at a dose of 100 mg/kg, in a final volume of approximately 100 μL, as previously described [24,26].
Buparlisib (MedChem Express, HY-70063) was administered at a dose of 50 mg/kg. Buparlisib was dissolved in NMP-PEG300 (10% N-methylpyrrolidone [Sigma, 270,458], 90% polyethylene glycol 300 [Sigma, 90,878]) to concentrations of 15 mg/mL so that the final volume delivered to each mouse was approximately 100 μL. To dissolve the drug, the solution was passed through a 23-gage needle repeatedly, until clear. Buparlisib solution was aliquoted, stored at −80°C, and thawed immediately before use. Drugs were administered by oral gavage at approximately the same time every day for one week prior to leupeptin injection and lysosome isolation.
Lysosome isolation
Mice were dissected at approximately the same time for each experiment (between 9 and 10 AM, with the dark period ending at 6 AM). All mice were allowed free access to food during the entire course of experimentation. Upon dissection, mice were qualitatively assessed for the presence of food in the stomach (and all mice used in the study had food in the stomach).
Lysosome isolation was performed as we have described previously [24,26]. Freshly dissected liver tissue was immediately washed in ice-cold PBS, until no visible blood remained. All subsequent steps were performed at 4°C. Tissue was diced with scissors and gently dounce homogenized (VWR, 62,400–595) in commercially available fractionation buffers (Thermo, 89,839). Nuclei, extracellular matrix, and unbroken cells were removed by 10-min centrifugation at 500 × g. The post-nuclear sample was mixed with iodixanol/OptiPrep (Sigma, D1556) to a concentration of 15% OptiPrep and loaded onto a discontinuous 17%–30% OptiPrep density gradient in ultracentrifuge tubes (Beckman Coulter, 344,057) for a 55Ti-SW rotor (Beckman Coulter, 342,194). A 10% Optiprep layer was laid over the sample, and the samples were subjected to ultracentrifugation at 145,000 × g for 2 h in a Beckman Coulter L-70 ultracentrifuge. During ultracentrifugation, the sample is resolved into visible bands in the density gradient. The visible band at the 10%–15% gradient interface is highly enriched for lysosomal markers and was used for all experiments. After the lysosome-containing band was removed from the density gradient, the sample was diluted in PBS to decrease the OptiPrep density, allowing the lysosomes to be pelleted, washed, and resuspended in an isotonic MOPS-sucrose buffer (10 mM MOPS [Sigma, M1254], 300 mM sucrose, pH 7.2). Protein concentrations were measured by BCA assay (ThermoFisher, 23,225), and all samples were adjusted to the same protein concentration.
Microscope image acquisition and analysis
Microscopy was performed in the UM BRCF Microscopy Core. Images were acquired with a Zeiss Axioplan2 microscope equipped with Zeiss ApoTome for optical sectioning. Image capture was performed with a Zeiss AxioCam MRm camera. The objective lens used was a Zeiss Plan-NEOFLUAR 40x, with a numerical aperture of 0.75 (this lens requires no immersion medium). The microscope was operated with Zeiss Axiovision software. All microscopy was performed on fixed samples at room temperature.
For each experimental replicate, equal numbers of images for each experimental group were acquired on the same day, using the exact same exposure settings and light intensity. Image stacks were acquired with a 0.5 μm z-plane slice-distance. Fluorescence quantification was performed in ImageJ, using raw, unadjusted images. Post-acquisition image adjustment was only performed for display images (i.e., only after analysis was complete), using ImageJ software.
Mouse stocks and husbandry
All animal experiments were approved by the University of Michigan Institutional Animal Care and Use Committee. Mice were housed in Specific Pathogen Free facilities, with sentinel animals checked quarterly for infection (all tests were negative). Mice had free access to food (5L0D; Lab Diet, 0067138) and water during the course of all experiments. The ghr KO mice were produced by from a breeding stock kindly provided by Dr. John Kopchick (Ohio University) and were maintained by crosses between heterozygotes, on a background that contains equal contributions from C57BL/6 J and C57BL/6 N. Snell dwarf homozygotes are produced by a cross between homozygous males (DW/J-dw/dw) that had been treated with hormones to promote sexual maturation, and heterozygous females on a (DW/J x C3H/HeJ) F1 background. In each case, littermates were used as control animals.
mRNA isolation and quantification
RNA was isolated from NIH3T3 cells using a cultured cell RNA isolation kit (Life Magnetics, CPC-0050). cDNA was synthesized with an iScript cDNA synthesis kit (BioRad,1,708,891). qPCR was performed with FAST SYBR Green Master Mix (Applied Biosystems, 4,385,612), in an Applied Biosystems Step One Plus Real-Time PCR Machine. Data acquisition was performed using the manufacturer’s software.
Quantification intracellular lipid droplets
For lipid droplet staining in NIH3T3 cells, cells were grown on coverslips and transfected with siRNA. Four days after transfection, 5 μM BODIPY dye (ThermoFisher, D3922) was added to the cell culture media, and cells were incubated under normal conditions for 30 min. Cells were quickly rinsed in PBS and then fixed in 4% PFA for 10 min. Coverslips were rinsed twice with PBS, stained with Hoechst (Sigma, 94,430), and then mounted to microscope slides with mounting medium (GBI Labs, E18-18). Slides were dried overnight and were imaged the next day.
Quantitative proteomics sample preparation
Lysosome samples were frozen/thawed twice before carrying out in-solution digestion. Briefly, 0.2% PPS Silent Surfactant (Protein Discovery, 21,011) in 50 mM ammonium bicarbonate was added to lysosome samples (at 1:1 ratio volume to volume). Proteins were heated at 95°C for 5 min, reduced with dithiothreitol (Sigma, D0632) and alkylated with iodoacetamide (Sigma, I1149). Sequencing grade modified porcine trypsin (Promega, V5111) was used to digest lysosome samples (1 ug trypsin to 50 ug lysosome protein ratio) for 16 h at 37°C. Peptides were desalted with Oasis MCX cartridges (Waters Corporation, 186,000,782) using the manufacturer’s instructions and dried with a vacuum concentrator. Peptides were reconstituted in 0.1% formic acid in water prior to mass spectrometry acquisition.
Liver tissue powder samples were processed and digested using S-Trap Micro Spin Column (Protifi, C02-micro-80) following the manufacturer’s protocol. SDS (5%) lysis and solubilization buffer were added to liver powder samples and homogenized on ice using a sonicator probe. Protein concentrations were determined using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, PI23227). Proteins were digested at 1 ug trypsin to 25 ug protein ratio on S-Trap at 47°C for 1 h. Peptides extracted from S-Trap were dried with a vacuum concentrator and reconstituted in 0.1% formic acid in water prior to mass spectrometry acquisition.
Quantitative proteomics data-independent acquisition mass spectrometry
Data were acquired using data-independent acquisition (DIA) on a Thermo EASY-nLC 1000 and a Thermo Q-Exactive HF orbitrap mass spectrometer. Peptides were separated using PicoFrit Self-Packed Columns (360 μm OD x 75 μm ID; New Objective, Woburn, MA) packed to 30 cm with 3 μm ReproSil-Pur C18 beads (ESI Source Solutions, r13.aq.0001). The trap column was packed to 2 cm with same C18 beads using 360 μm OD x 150 μm ID fused silica capillary fritted with Kasil on one end. Solvent A was 0.1% formic acid in water, and solvent B was 0.1% formic acid in 80% acetonitrile. For each injection, 1 µg of sample was loaded and eluted using a 90-min gradient from 5% to 45% B at 250 nL/min. DIA methods followed the chromatogram library workflow as described in detail previously [67]. All lysosome samples were pooled in equal ratio to create a lysosome library sample, and all liver tissue samples were pooled in equal ratio to create a liver tissue library sample. The library samples were used to generate chromatogram libraries (one for each sample type) using the strategy described by [68]. Thermo Q Exactive HF MS was used to acquire six gas phase fractionated runs spanning a total mass range of 400 to 1000 m/z, each with staggered 4 m/z narrow precursor isolation windows [69]. EncyclopeDIA software suite was used to generate a chromatogram library [67] and retention time models for all peptides detected at 1% FDR determined by Percolator [70].
For quantitative DIA run, equal amounts of protein were used for each sample. A 24 × 24 m/z staggered window from 400.43190 to 1000.7048 m/z was used as described previously [69]. EncyclopeDIA software suite was used to search the chromatogram library with calibrated retention time and quantify peptides and proteins in each sample.
RNA interference
siRNAs were acquired as follows: Medium GC content negative control (Invitrogen, 465,372), Atg5 (Invitrogen, Atg5MSS247019, 44,009,549), Tsg101 (Invitrogen, Tsg101MSS238580, 54,064,795), Acly (Invitrogen, AclyMSS200420, 54,479,077), Acss2 (Invitrogen, Acss2MSS226952, 54,713,652), Lamp2a (cocktail of two custom-designed Stealth siRNA duplexes from Invitrogen; sequences: CAGCUCUGGGAGGAGUACUUAUUCU and CAAGCGCCAUCAUACUGGAUAUGAG, complexed to the reverse complement antisense sequences). Transfections were performed using Lipofectamine RNAi MAX (Invitrogen, 56,532), with transfections prepared in OptiMEM (Gibco, 31,985–062), according to the manufacturer’s instructions.
Statistical analysis
Proteomics analysis was performed in R Studio, using the following packages, available from CRAN: Biobase, limma, readxl, ggplot2, naniar, tidyverse, imputeLCMD, stats, and PCAtools. The data were cleaned before expression analysis, by removing proteins that were identified by mapping to a single peptide. The data were normalized by the quantile method. Missing values, which were highly left censored (i.e., most commonly found in low abundance proteins), were imputed using the stochastic minimal value approach (in the imputeLCMD package). Only proteins where less than 50% of values are missing are considered by this method. Statistical comparisons were performed in limma, using empirical Bayes statistics (documentation available at https://www.bioconductor.org/packages/release/bioc/html/limma.html). To compute moderated t-statistics, moderated F-statistic, and log-odds of differential expression from a linear model fit, we employed limma’s eBayes method, an implementation of empirical Bayes moderation of the standard errors toward a global value. The sigma^2 prior (s2.prior) and degrees of freedom associated with s2.prior (df.prior) are assigned automatically by eBayes by an internal call to an additional limma function fitFDist, which provides a moment estimation of the parameters of a scaled F-distribution given one of the degrees of freedom.
Comparison strategies are shown in Figures 1A and 2A. Display items were created with ggplot2, with minor adjustments to fonts and legends in Adobe Illustrator. GO analysis was conducted in R, using the goana function contained in BioConductor.
KFERQ motifs were identified using KFERQ finder V0.8 (http://ec2-18-188-198-152.us-east-2.compute.amazonaws.com:3838/kferq/).
Statistical analyses and graph generation for cell culture data and mouse western blot data were performed with GraphPad Prism 8. Results of t-tests and 2-way ANOVAs are reported directly in the figures or in the figure legends. Unless otherwise stated, all t-tests are unpaired, with Welch’s correction for unequal SD.
Supplementary Material
Acknowledgments
Mouse husbandry was conducted by Roxann Alonzo, Asuman “Ilkim” Erturk, Natalie Perry, Lori Roberts, and Jacob Sheets. Xiaofang Shi shared mouse tissues. CMA reporter puncta were counted by Katherine Zhang.
Funding Statement
The funding information “NIH”, “University of Washington Nathan Shock Center”, “Paul F.Glenn Foundation for Medical Research”, “SJE and ACM” and “EIH” provided has been checked against the Open Funder Registry and we failed to find amatch. Please confirm if the Funding section is accurate and also confirm the funder name “NIH”, “University of Washington Nathan Shock Center”, “Paul F.Glenn Foundation for Medical Research”, “SJE and ACM” and “EIH”.
Data availability
All raw proteomics data are available at:
https://panoramaweb.org/1cbegN.url
Spreadsheets of analyzed data are available at:
Code availability
Code for proteomics analysis in R is available at:
Disclosure statement
The authors have no competing interests to disclose.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Murrow L, Debnath J.. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol Mech Dis. 2013;8:105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ryter SW, Cloonan SM, Choi AMK.. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2014;7:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aniento F, Roche E, Cuervo AM, et al. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J Biol Chem. 1993;268:10463–10470. [PubMed] [Google Scholar]
- [7].Cuervo AM, Knecht E, Terlecky SR, et al. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269:C1200–8. [DOI] [PubMed] [Google Scholar]
- [8].Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–2499. [DOI] [PubMed] [Google Scholar]
- [9].Chiang H-L, Terlecky SR, Plant CP, et al. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. [DOI] [PubMed] [Google Scholar]
- [10].Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. [DOI] [PubMed] [Google Scholar]
- [11].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. [DOI] [PubMed] [Google Scholar]
- [12].Bandyopadhyay U, Kaushik S, Varticovski L, et al. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell. 2008;28:5747–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. [DOI] [PubMed] [Google Scholar]
- [14].Kirchner P, Bourdenx M, Madrigal-Matute J, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019;17:e3000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dyson HJ, Wright PE, Scheraga HA. The role of hydrophobic interactions in initiation and propagation of protein folding. Pnas. 2006;103:13057–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014;20:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xilouri M, Brekk OR, Landeck N, et al. Boosting chaperone-mediated autophagy in vivo mitigates sαynuclein-induced neurodegeneration. Brain. 2013;136:2130–2146. [DOI] [PubMed] [Google Scholar]
- [18].Gomes LR, Menck CFM, Cuervo AM. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy. 2017;13:928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hao Y, Kacal M, Ouchida AT, et al. Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy. 2019;15:1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xia H-G, Najafov A, Geng J, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015;210:705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]..Brekk OR, Makridakis M, Mavroeidi P, et al. Molecular and Cellular Neuroscience. Mol Cell Neurosci. 2019;95:1–12. [DOI] [PubMed] [Google Scholar]
- [22].Xilouri M, Brekk OR, Polissidis A, et al. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy. 2016;12:2230–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Endicott SJ, Boynton DN, Beckmann LJ, et al. Long-lived mice with reduced growth hormone signaling have a constitutive upregulation of hepatic chaperone-mediated autophagy. Autophagy. 2020;00:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015;17:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Endicott SJ, Ziemba ZJ, Beckmann LJ, et al. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J Cell Biol. 2020;218:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Carroll B, Dunlop EA. The lysosome: a crucial hub for AMPK and mTORC1 signalling. Biochem J. 2017;474:1453–1466. [DOI] [PubMed] [Google Scholar]
- [28].Savini M, Zhao Q, Wang MC. Lysosomes: signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 2019;29:876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Deng L, Feng J, Broaddus RR. The novel estrogen-induced gene EIG121 regulates autophagy and promotes cell survival under stress. Cell Death Dis. 2010;1:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Waxman DJ, O Connor C. Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol. 2006;20:2613–2629. [DOI] [PubMed] [Google Scholar]
- [31].Li X, Bartke A, Berryman DE, et al. Direct and indirect effects of growth hormone receptor ablation on liver expression of xenobiotic metabolizing genes. AJP Endocrinol Metab. 2013;305:E942–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kacal M, Zhang B, Hao Y, et al. Quantitative proteomic analysis of temporal lysosomal proteome and the impact of the KFERQ- like motif and LAMP2A in lysosomal targeting. Autophagy. 2021;00:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pietrocola F, Galluzzi L, Pedro JMB-S, et al. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–821. [DOI] [PubMed] [Google Scholar]
- [34].Zhao S, Torres A, Henry RA, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016;17:1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song Z, Xiaoli A, Yang F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients. 2018;10:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Flurkey K, Papaconstantinou J, Miller RA, et al. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci USA. 2001;98:6736–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cuervo AM, Terlecky SR, Dice JF, et al. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem. 1994;269:26374–26380. [PubMed] [Google Scholar]
- [38].Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: jointed by a chaperone. J Biol Chem. 2018;293:5414–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Koga H, Martinez-Vicente M, Macian F, et al. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dong S, Aguirre-Hernandez C, Scrivo A, et al. Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat Commun. 2020;11:1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li X, Frazier JA, Spahiu E, et al. Muscle-dependent regulation of adipose tissue function in long-lived growth hormone-mutant mice. Aging (Albany NY). 2020;12:8766–8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3:a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Toyama BH, Hetzer MW. Protein homeostasis: live long, wont prosper. Nat Rev Mol Cell Biol. 2012;14:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015;21:1406–1415. [DOI] [PubMed] [Google Scholar]
- [46].Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–31513. [DOI] [PubMed] [Google Scholar]
- [48].Hofmann JW, Zhao X, De Cecco M, et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell. 2015;160:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Eisenberg T, Schroeder S, Andryushkova A, et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014;19:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Peleg S, Feller C, Forne I, et al. Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep. 2016;17:455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Catana C-S, Atanasov AG, Berindan-Neagoe I. Natural products with anti-aging potential: affected targets and molecular mechanisms. Biotechnol Adv. 2018;36:1649–1656. [DOI] [PubMed] [Google Scholar]
- [52].Hekman KE, Yu GY, Brown CD, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu S, Bachran C, Gupta P, et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNAtranslation and mouse development. Pnas. 2012;109:13817–13822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xie J, de Souza Alves V, von der Haar T, et al. Regulation of the elongation phase of protein synthesis enhances translation accuracy and modulates lifespan. Curr Biol. 2019;29:737–749.e5. [DOI] [PubMed] [Google Scholar]
- [55].Filion AM, Laughrea A. Translation fidelity in the aging mammal: studies with an accurate in vitro system on aged rats. Mech Ageing Dev. 1985;29:125–142. [DOI] [PubMed] [Google Scholar]
- [56].Ke Z, Mallik P, Johnson AB, et al. Translation fidelity coevolves with longevity. Aging Cell. 2017;16:988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang Y, Martinez-Vicente M, Kruger U, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang CC, Bose JK, Majumder P, et al. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. 2014;127:3024–3038. [DOI] [PubMed] [Google Scholar]
- [59].Qi L, Zhang X-D, Wu J-C, et al. The role of chaperone-mediated autophagy in Huntington degradation. Plos One. 2012;7:e46834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Orenstein SJ, Kuo S-H, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Park J-S, Kim D-H, Yoon S-Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016;49:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. [DOI] [PubMed] [Google Scholar]
- [63].Bourdenx M, Martin-Segura A, Scrivo A, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell. 2021;184:2696–2714.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ryu SW, Stewart R, Pectol DC, et al. Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. Plos Biol. 2020;18:e3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Arias E, Koga H, Diaz A, et al. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59:270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Anguiano J, Garner TP, Mahalingam M, et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013;9:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Searle BC, Pino LK, Egertson JD, et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun. 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pino LK, Just SC, MacCoss MJ, et al. Acquiring and analyzing data independent acquisition proteomics experiments without spectrum libraries. Mol Cell Proteomics. 2020;19:1088–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Amodei D, Egertson J, MacLean BX, et al. Improving precursor selectivity in data-independent acquisition using overlapping windows. A Am Soc Mass Spectrom. 2019;30:669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kall L, Canterbury JD, Weston J, et al. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw proteomics data are available at:
https://panoramaweb.org/1cbegN.url
Spreadsheets of analyzed data are available at: