

ABSTRACT
Lacking a self-contained metabolism network, viruses have evolved multiple mechanisms for rewiring the metabolic system of their host to hijack the host’s metabolic resources for replication. Newcastle disease virus (NDV) is a paramyxovirus, as an oncolytic virus currently being developed for cancer treatment. However, how NDV alters cellular metabolism is still far from fully understood. In this study, we show that NDV infection reprograms cell metabolism by increasing glucose utilization in the glycolytic pathway. Mechanistically, NDV induces mitochondrial damage, elevated mitochondrial reactive oxygen species (mROS) and ETC dysfunction. Infection of cells depletes nucleotide triphosphate levels, resulting in elevated AMP:ATP ratios, AMP-activated protein kinase (AMPK) phosphorylation, and MTOR crosstalk mediated autophagy. In a time-dependent manner, NDV shifts the balance of mitochondrial dynamics from fusion to fission. Subsequently, PINK1-PRKN-dependent mitophagy was activated, forming a ubiquitin chain with MFN2 (mitofusin 2), and molecular receptor SQSTM1/p62 recognized damaged mitochondria. We also found that NDV infection induces NAD+-dependent deacetylase SIRT3 loss via mitophagy to engender HIF1A stabilization, leading to the switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis. Overall, these studies support a model that NDV modulates host cell metabolism through PINK1-PRKN-dependent mitophagy for degrading SIRT3.
Abbreviations: AMPK: AMP-activated protein kinase; CCCP: carbonyl cyanide 3-chlorophenylhydrazone; ECAR: extracellular acidification rate; hpi: hours post infection LC-MS: liquid chromatography-mass spectrometry; mito-QC: mCherry-GFP-FIS1[mt101–152]; MFN2: mitofusin 2; MMP: mitochondrial membrane potential; mROS: mitochondrial reactive oxygen species; MOI: multiplicity of infection; 2-NBDG: 2-(N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl) amino)-2-deoxyglucose; NDV: newcastle disease virus; OCR: oxygen consumption rate; siRNA: small interfering RNA; SIRT3: sirtuin 3; TCA: tricarboxylic acid; TCID50: tissue culture infective doses.
KEYWORDS: Cellular metabolism, glycolysis, mitochondrial fission, mitophagy, newcastle disease virus, SIRT3
Introduction
Viruses are biological entities that are non-cellular life forms. It is obligate parasites that depend on host cells to provide the energy, molecular precursors, and specialized components needed to produce infectious virions [1]. Cellular metabolism caters to the energy and metabolite needs of cells [2]. During process infection of cells, most virus have evolved multiple mechanisms for rewiring the metabolic system of their host and hijack the host’s metabolic resources for replication [3]. For instance, an adenovirus infection induces increased glucose consumption and lactate production. Mechanistically, the adenovirus protein E4ORF1 binds to the transcription factor MYC to promote glycolytic target gene expression, including for HK2 [4]. The Zika virus induces differential metabolic reprogramming between human and mosquito cells, and a Zika virus infection differentially alters glucose metabolism and nucleotide triphosphates in HFF1 and C6/36 cells [5]. Similar observations have been reported for dengue virus (DENV) [6,7], EBV [8], SARS-CoV-2 [9,10], and KSHV [11]. In general, cellular metabolic reprogramming is a hallmark of viral infection and is largely beneficial for viral reproduction and not the host [12]. Thus, identifying factors that regulate cell metabolism will enhance our understanding of how viruses hijack metabolism for selective advantages, as well as provide innovative approaches for therapeutic targeting.
Most virus-infected cells commonly exhibit the Warburg effect (aerobic glycolysis) to satisfy the demands of increased proliferation of virions [4]. One of the key requirements for viral proliferation is metabolic reprogramming, which is intrinsically linked to an imbalance in cellular energetics [12]. Mitochondria are the important bioenergetics and biosynthetic hubs with metabolic network dynamically undergoing fusion and fission [13–15]. Several viruses have been implicated in targeting mitochondrial dynamics, selective mitophagy, and metabolic shift [16]. Mitophagy is a selective form of autophagy that eliminates damaged mitochondria and promotes the recycling and cellular balance of three key classes of metabolites: nucleotides, amino acids, and lipids [17–19]. AMPK is a master sensor for energy stress, with AMPK and MTOR playing principal roles in the regulation of metabolic programs [20,21]. Besides, AMPK as a direct regulator of mitochondrial dynamics via MFF-DNM1L mediates fission and mitophagy [22]. Previous studies found that DENV NS4B protein alters mitochondrial morpho-dynamics by inactivating DNM1L. Subsequently, DNM1L-mediated mitochondrial fission is followed by PINK1-PRKN-dependent mitophagy [23,24,25]. Currently, researchers have found that autophagy mediates the degradation of functional proteins. Such as STAT2 [26], SIRT1 (sirtuin 1) [27,28], and LMNB1 (lamin B1) [29]. Furthermore, hepatitis B virus X protein/HBx promotes cancer stemness in HCC cells by increasing mitophagy-mediated glycolysis metabolic reprogramming [30].
Sirtuins are a conserved family of NAD-dependent ADP-ribosyl transferases and/or protein deacetylases involved in metabolism and stress response [31,32]. Mammals express seven sirtuins (SIRT1 to SIRT7), three of which (SIRT3 to SIRT5) are localized to the mitochondrion [33]. As a class, SIRT3 is a major mitochondrial deacetylase that targets many enzymes involved in central metabolism, resulting in the activation of many oxidative pathways [34]. SIRT3 is involved in metabolic regulates that, upon activation, induce glycolysis and fatty acid biosynthesis [35]. Previous studies have revealed that SIRT3 regulates cellular metabolic reprogramming through the SIRT3-HIF1A axis [36]. SENP1-SIRT3 signaling modulates SIRT3 activation and mitochondrial metabolism during metabolic stress [37]. Currently, only a few studies have described the role that SIRT3 plays in virus infection. Furthermore, whether and how SIRT3 mediation manipulates the host’s cell metabolism is unknown in infectious cells.
NDV is a highly contagious avian pathogen belonging to the family Paramyxoviridae. It causes a disease that is responsible for significant economic losses and requires compulsory notification of the World Organization for Animal Health (OIE) [38]. NDV is also known to be an oncolytic virus [39–41], which can selectively replicate in tumor cells and trigger organelle-selective autophagy in mitochondria, peroxisome, and the Golgi apparatus [42–45]. Thus, NDV is an applicable model for further cell biology research. Due to the pivotal role of SIRT3 in cell metabolism [36], we hypothesized that SIRT3 can serve as an important checkpoint for the balance between glycolytic and mitochondrial oxidative metabolism in the regulation of energy production for viral replication. In this study, we provide evidence for metabolic reprogramming in NDV infectious cells, a process regulated by the energy metabolic switch, SIRT3.
Results
NDV elevates glucose uptake and glycolysis in infectious cells
Aerobic glycolysis is a well-known phenomenon in rapidly proliferative cells [13]. To determine whether NDV infection leads to changes in glucose metabolism, we infected A549 cells with NDV to monitor glucose uptake ability using a fluorescent glucose analog 2-(N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl) amino)-2-deoxyglucose (2-NBDG) labeling assay and lactate production at different time intervals following NDV infection. We found that NDV infection of A549 cells significantly increases glucose uptake compared to mock-infected cells at 6, 12, 18, and 24 hours post infection (hpi) (Figure 1A,B). To confirm glucose uptake for increased glycolysis, we measured lactate production. As expected, NDV-infected cells secreted more lactate into the medium than the mock cells in a time-dependent manner (Figure 1C). These findings suggest that NDV promotes glucose utilization and aerobic glycolysis in infectious cells.
Figure 1.
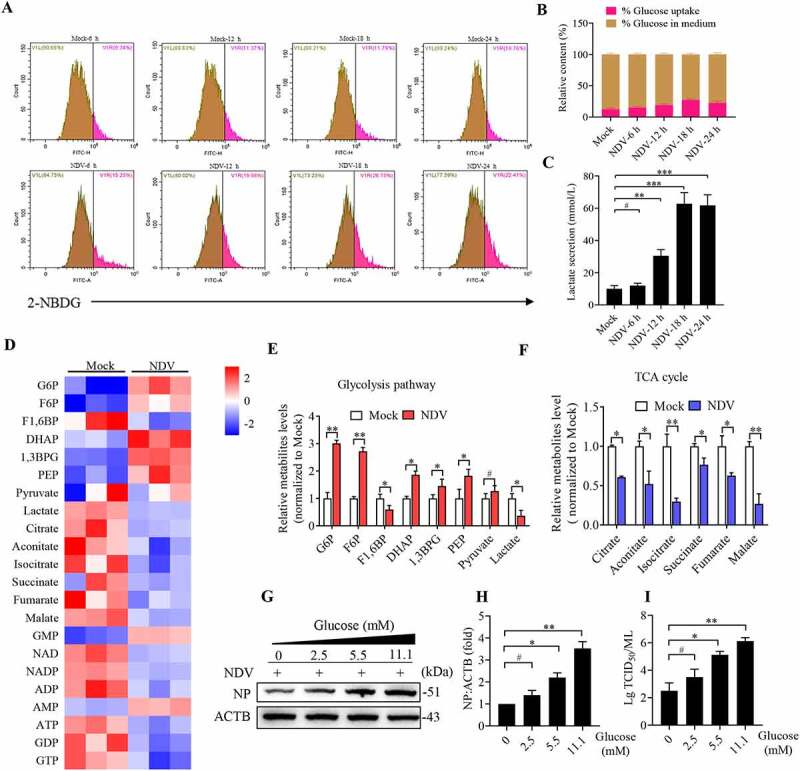
NDV elevates glucose uptake and glycolysis in infectious cells. (A) Glucose uptake was determined by flow cytometry with 2-NBDG at 6, 12, 18, and 24 hpi. (B) Histograms present a quantification of glucose uptake and rest via mean fluorescence intensity (MFI). (C) The lactate levels in the culture medium were determined by the lactate assay kit at 6, 12, 18, and 24 hpi. (D) Heatmap of energy related metabolites changed. (E) Levels of glycolytic intermediates. (F) Levels of TCA cycle intermediates. (G) A549 cells were infected with mock or NDV (MOI = 1) for 1 h under continuous agitation and rested for 18 h in medium containing different glucose concentrations (0, 2.5, 5.5, and 11.1 mM). Whole-cell extracts were prepared from mock-infected and NDV-infected cells at 18 hpi, NP was used as a maker for virus infection. (H) Quantification of NP:ACTB ratio. (I) Quantification of TCID50. Data are mean ± SEM, all data are from three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001, # p > 0.05.
Virus infection alters the host’s metabolic pattern [3]. We hypothesized that NDV can manipulate both glycolysis and OXPHOS to facilitate virus replication. We first examined the influence of NDV on metabolites using liquid chromatography tandem mass spectrometry (LC-MS/MS). The metabolic profile indicates a clear shift toward glycolytic metabolism when compared with mock-infected cells (Figure 1D). In NDV-infected cells, intermediates of glycolysis were elevated, whereas tricarboxylic acid (TCA) cycle metabolites were reduced (Figure 1E,F). Consistent with a pattern of increased glucose usage (Figure 1B). It should be noted that intracellular lactate decreased whereas extracellular lactate increased (Figure 1C,E), we further performed stable isotope tracing by using [U-13C6]-glucose or [1,2–13C2] -glucose in culture medium, these results show that A549 cells infected with NDV had significantly enhanced the 13C-enrichment of lactate [M + 3] and [M + 2] isotopologs derived from [U-13C6]-glucose (Figure S1A and B) and [1,2–13C2]-glucose (Figure S1C), respectively. These results also suggest that NDV infection leads to glycolysis increased, in conjunction with lactate concentration in medium, supporting our notion that NDV infection promotes lactate secretion into the medium in infectious cells. Next, we examined the glucose levels associated with NDV replication. The results show that glucose directly increases viral load in a dose-dependent manner (Figure 1G–I). Taken together, the data indicates that NDV infection enhances intermediate levels in glycolysis for biosynthesis to support viral replication.
NDV induces mitochondria membrane depolarization, fission, and ETC dysfunction
As mentioned earlier, a reduced TCA cycle was observed in NDV infectious cells (Figure 1F). Next, we asked whether NDV infection caused mitochondrial dysfunction. To this end, mitochondrial membrane potential (MMP) was used to evaluate the degree of mitochondrial dysfunction. The flow cytometry results show that there was a significant MMP reduction in the NDV-infected and carbonyl cyanide 3-chlorophenylhydrazone (CCCP) groups compared with the mock group, CCCP as a positive control for mitophagy (Figure 2A,B). To study the effects of NDV on mitochondrial dynamics, immunofluorescence was performed to determine mitochondrial morphological changes. Confocal microscopy images of MitoTracker staining reveal extensive fragmentation of mitochondria following NDV infection and CCCP treatment (Figure 2C,D). To further ascertain that NDV infection induces mitochondrial morphology to switch from fusion to fission, MFN1, MFN2, OPA1 (regulates mitochondrial inner membrane fusion), and COX4I1 were detected using western blot. As shown in Figure 2E–H, both MFN1 and MFN2 were reduced in a time-dependent manner, and promotes cleave long-OPA1 to generate short-OPA1, which mediated mitochondrial fission. Besides, DNM1L plays a vital role in mitochondrial fission [14,46,47], we found that NDV infection promoted DNM1L phosphorylation (Ser616) and inhibited DNM1L phosphorylation (Ser637) (Figure 2I–K). CCCP also reduced COX4I1, MFN1, and MFN2 (Figure 2L,M) and promoted mitochondria fission (Figure 2C). Mitochondrial respiratory chain complex I is the largest component of the OXPHOS system. The activity of mitochondrial respiratory chain complex I was also significantly decreased following NDV infection and CCCP treatment (Figure 2N).
Figure 2.
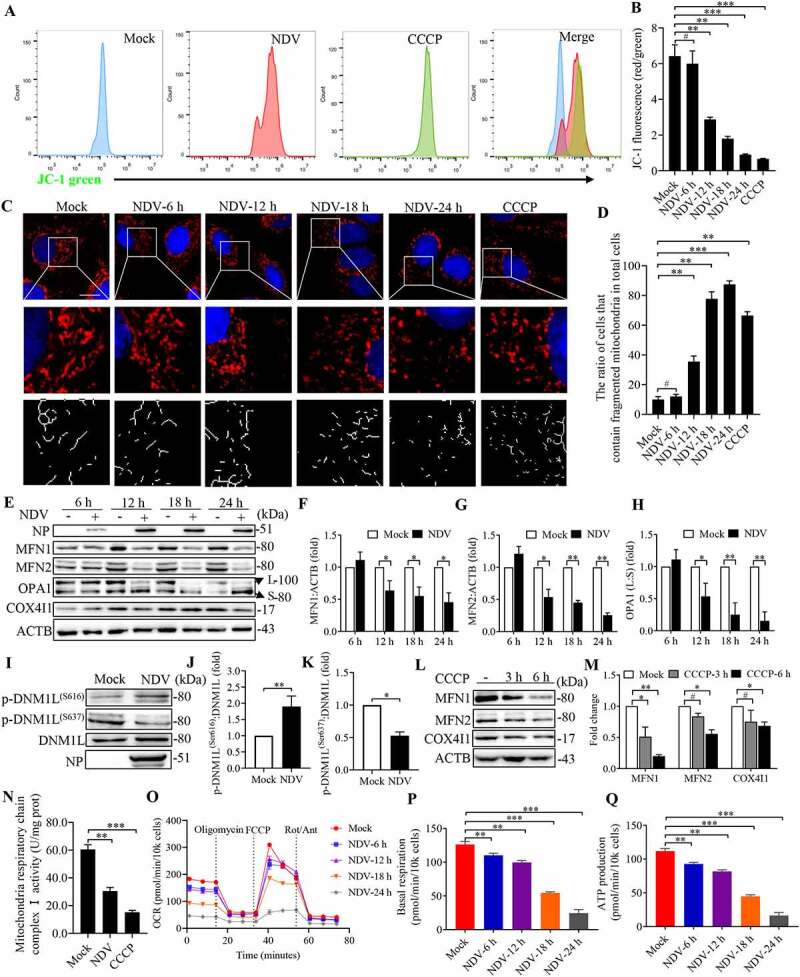
NDV infection induces mitochondria membrane depolarization, fission, and ETC dysfunction. (A and B) MMP were detected using flow cytometray by JC-1 stained, CCCP as a positive control. (C) Confocal microscopy images showing mitochondrial fragmentation at 6, 12, 18, and 24 hpi and CCCP group by MitoTracker stained. (D) Image-Pro Plus 6.0 software was used to calculate the ratio of cells that contain fragmented mitochondria in total cells. (E) MFN1, MFN2, OPA1, COX4I1, and ACTB protein levels were determined by western blot, NP was used as a maker for virus infection. (F-H) Quantification of MFN1, MFN2, and OPA1. (I) p-DNM1L (S616), p-DNM1L (S637), DNM1L and NP protein levels were determined by western blot at 18 hpi. (J and K) Quantification of p-DNM1L (Ser616):DNM1L ratio and p-DNM1L (Ser637):DNM1L ratio. (L) A549 cells were either mock or CCCP treated at 3 h and 6 h. MFN1, MFN2, COX4I1, and ACTB protein levels were determined by western blot. (M) Quantification of MFN1, MFN2, and COX4I1. (N) Mitochondria respiratory chain complex I activity was detected. A549 cells were prepared from mock-infected, NDV-infected cells at 18 hpi and treated with CCCP (10 μM). (O-Q) Real-time oxygen consumption rate (OCR) in mock-infected and NDV-infected cells was measured at 6, 12, 18, and 24 hpi. Statistical analysis of basal respiration and ATP production are shown on Figure P and Figure Q. White boxed regions in the panels are enlarged. Scale bar: 10 μm. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, *** p < 0.001, # p > 0.05.
The real-time oxygen consumption rate (OCR), a marker of mitochondrial stress, was directly measured using a Seahorse XF extracellular flux analyzer. As shown in Figure 2O, NDV-infected cells showed significantly decreased OCRs compared with mock-infected cells. Specifically, NDV infection reduced basal respiration and ATP production in a time-dependent manner (Figure 2P,Q). These results suggest that NDV infection not only is harmful for MMP but also induces mitochondrial morphology to switch from fusion to fission, thus resulting in energy metabolism dysfunction.
NDV reduces total ATP production with a higher proportion of glycolysis-derived ATP
The vast majority of ATP is produced by mitochondria. Upregulating glycolysis can compensate for insufficient ATP production in the OXPHOS [48]. To detect whether glycolysis-derived ATP is modulated by viral infection, we used three cell lines for real-time ATP synthesis research: A549 (tumor cells, glycolysis dependent), Hep G2 (tumor cells, glycolysis independent), and DF-1 (non-transformed cells, balance between OXPHOS and glycolysis). Real-time ATP synthesis rate assay was performed on a Seahorse extracellular flux analyzer, which revealed a lower total ATP production in NDV-infected cells than in the mock cells (A549, Hep G2, and DF-1) (Figure 3A,D,G). Instead, with an increase in the proportion of ATP derived from glycolysis, there was a decrease in the proportion of ATP derived from the OXPHOS in all three cell lines (Figure 3B,E,H). An energy map of the mitochondrial ATP production rate (mito-ATP) versus the glycolysis ATP production rate (Glyco-ATP) shows a metabolic switch from oxidative phosphorylation to glycolysis (Figure 3C,F,I). The overall energetic rate was higher in the Glyco-ATP production rate following NDV infection. Although these cells must rely energetically on OXPHOS primarily and tend to have a low metabolic flux rate, glycolysis appears to be more essential for NDV during infection than OXPHOS. These data indicates that NDV infection causes an increase in overall carbon metabolism, particularly glycolysis.
Figure 3.
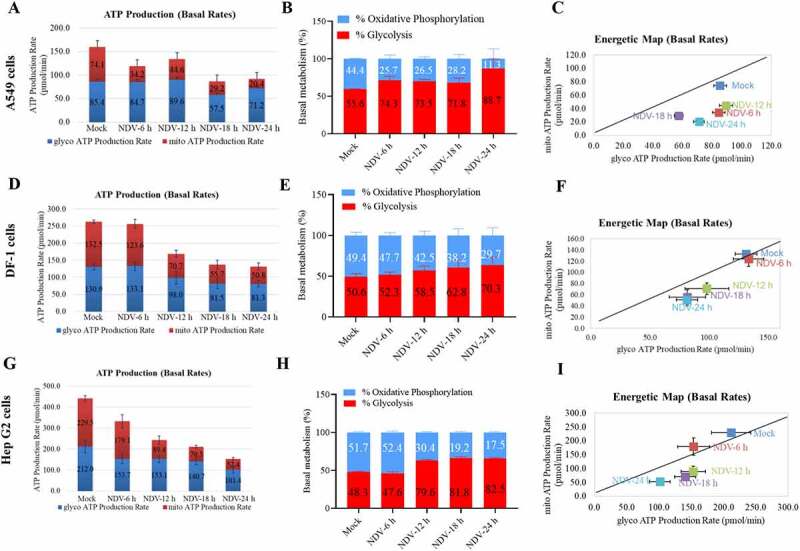
NDV reduces total ATP production, with a higher proportion of glycolysis-derived ATP. (A-C) Seahorse XF Real-time ATP rate analysis in A549 cells upon NDV infection at 6, 12, 18, and 24 hpi. Metabolic flux analysis on the Seahorse XF shows decrease in total ATP production in A549 cells compared to mock infection (A), with an increase proportion of ATP derived from glycolysis, decrease proportion of ATP derived from OXPHOS (B). Energetic map of the mitochondrial ATP production rate (mito-ATP) versus glycolysis ATP production rate (Glyco-ATP) (C). (D-F) Seahorse XF Real-time ATP rate analysis in DF-1 cells upon NDV infection at 6, 12, 18, and 24 hpi. Metabolic flux analysis on the Seahorse XF shows decrease in total ATP production in DF-1 cells at 6, 12, 18 and 24 hpi compared to mock infection (D), with an increase proportion of ATP derived from glycolysis, decrease proportion of ATP derived from OXPHOS (E). Energetic map of the mitochondrial ATP production rate (mito-ATP) versus glycolysis ATP production rate (Glyco-ATP) (F). (G-I) Seahorse XF Real-time ATP rate analysis in Hep G2 cells upon NDV infection at 6, 12, 18, and 24 hpi. Metabolic flux analysis on the Seahorse XF shows decrease in total ATP production in Hep G2 cells at 6, 12, 18, and 24 hpi compared to mock infection (G), with an increase proportion of ATP derived from glycolysis, decrease proportion of ATP derived from OXPHOS (H). Energetic map of the mitochondrial ATP production rate (mito-ATP) versus glycolysis ATP production rate (Glyco-ATP) (I). Data are mean ± SEM, all data are from three independent experiments.
NDV shifts cellular metabolism toward glycolytic signatures by stabilizing HIF1A, which facilitates NDV replication
Growing evidence shows that glycolysis occurs during viral infection [3,49,50]. Yet, to our knowledge, the specific mechanism involved has not been elucidated. HIF1A is a master transcription factor regulated in an oxygen-dependent manner and is a key regulator of glycolysis in cancer [51]. To further interrogate how NDV hijacks cellular metabolism, we examined whether NDV infection upregulates HIF1A, a key transcription factor under hypoxia. As expected, NDV infection engenders HIF1A stabilization by inhibiting HIF1A hydroxylation (Figure 4A,B, and S2H). Next, we investigated whether NDV induces mROS to regulate HIF1A stabilization. Compared with the mock group, the mROS levels in NDV-infected and rotenone-treated cells were significantly increased, which can be counteracted by ROS scavenger N-acetyl-L-cysteine (NAC) (Figure 4C,D).
Figure 4.
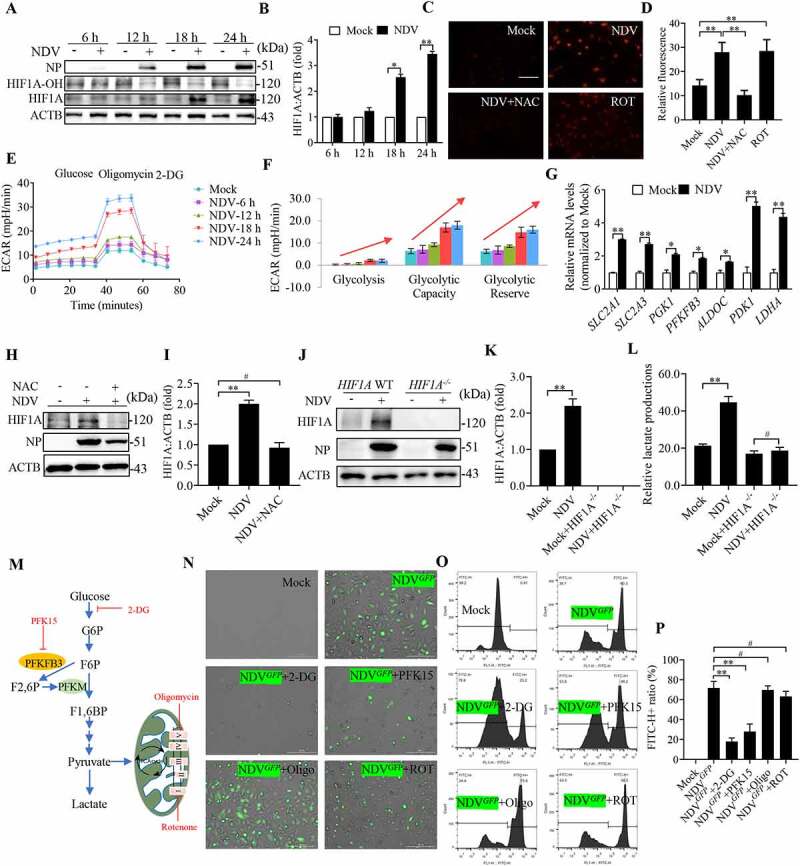
NDV infection shifts cellular metabolism toward glycolysis by stabilizing HIF1A, which facilitates NDV replication. (A) Cells were either mock treated or infected by NDV Herts/33 strain at an MOI of 1, whole-cell extracts were prepared from mock-infected and NDV-infected cells at 6, 12, 18, and 24 hpi. HIF1A-OH, HIF1A, and ACTB protein levels were determined by western blot, NP was used as a maker for virus infection. (B) Quantification of HIF1A. (C and D) A549 cells were mock treated or infected with NDV (MOI of 1) or cells were treated with ROS inhibitor (NAC) for final 3 h, and treated with ROT for 12 h as a positive control. mROS staining of A549 cells and quantification. (E and F) ECAR in mock-infected and NDV-infected cells was measured 6, 12, 18, and 24 hpi. (G) Cells were either mock treated or infected by NDV Herts/33 strain at an MOI of 1. mRNA levels were detected using qRT-PCR with HIF1A target genes. (H and I) Cells were mock treated or infected with NDV or Cells were treated with NAC for final 3 h, HIF1A and ACTB protein levels were determined by western blot and quantification of HIF1A. (J and K) HIF1A WT or HIF1A −/− of HeLa cells were infected at MOI = 1. HIF1A, NP, and ACTB protein levels were determined via western blot and quantification of HIF1A. (L) HIF1A WT or HIF1A −/− of HeLa cells were infected at MOI = 1. The lactate levels in the culture supernatants were determined by the lactate assay kit. (M) Schematic of glucose metabolism with metabolic modulators (PFKFB3 inhibitor PFK15, glycolysis inhibitor 2-DG, mitochondrial respiration complex I inhibitor rotenone and ATP synthase inhibitor oligomycin). (N) A549 cells were infected with mock or NDV-GFP (MOI 1) for 1 h under continuous agitation and rested for 18 h in media containing different inhibitors (PFKFB3 inhibitor PFK15, glycolysis inhibitor 2-DG, mitochondria respiratory chain complex I inhibitor rotenone and ATP synthase inhibitor oligomycin. GFP positive were infected cells based on microscopy. (O) NDV-GFP were detected using flow cytometry. (P) Quantification of NDV-GFP ration following either mock treated or infected at in media containing different inhibitors. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, # p > 0.05.
The extracellular acidification rate (ECAR), a marker of glycolysis, was directly measured using a Seahorse XF extracellular flux analyzer. As shown in Figure 4E, compared with the mock-infected cells, the NDV-infected cells showed significantly increased ECARs. Importantly, glycolysis, glycolytic capacity, and glycolytic reserve were all substantially increased in NDV-infected cells compared with mock cells (Figure 4F). Furthermore, the glycolytic genes and proteins levels were significantly higher than those in the mock cells (Figure 4G, and S2A to 2 G). Next, we treated cells with the NAC in order to probe the model that suppressing ROS could block the stabilization of HIF1A after NDV infection. we observed that NAC treatment reduced HIF1A in NDV infectious cells. (Figure 4H,I). To examine the contribution of increased HIF1A to altered energy metabolism in infectious cells. HIF1A WT or HIF1A −/− of HeLa cells were infected at multiplicity of infection (MOI) = 1. We found that HIF1A −/− cells no longer elevated lactate production, NDV viral load also be inhibited (Figure 4J–L, and S2I). These results suggest that NDV infection increases cellular HIF1A levels via a mechanism dependent on increased mROS levels.
Based on our findings that NDV infection increases the glycolytic rate in infected cells, we wanted to understand if the direct blockage of glycolysis can attenuate viral replication. We next used 2-DG to inhibit glucose flux, PFK15 to inhibit PFKFB3, rotenone to inhibit the mitochondrial respiratory chain complex I, and oligomycin to inhibit ATP synthase (complex V) (Figure 4M), respectively. Then, cells were infected with GFP- labeled NDV. As expected, the addition of 2-DG and PFK15 produced a marked reduction in GFP- labeled NDV fluorescence, but no such a change was observed with rotenone and oligomycin treatment (Figure 4N), respectively. Flow cytometry results also show that there was a marked reduction in the GFP signal when 2-DG and PFK15 were added, which is consistent with the pattern of GFP-labeled NDV fluorescence (Figure 4O,P). Moreover, 2-DG treatment inhibited NDV viral load in a dose-dependent manner (Figure S2J to 2 L). These data indicates that glycolysis is essential for NDV replication, whereas mitochondria respiratory chain is dispensable.
NDV reduces mitochondrial SIRT3, regulates HIF1A stability, and promotes the Warburg effect
Previous studies have found that SIRT3 is localized to the mitochondrion, and its loss increases glucose uptake and glycolysis by link between SIRT3 and HIF1A [36]. We hypothesized that SIRT3 is degraded following damage to mitochondria while SIRT3 is an energy metabolic checkpoint for the shift from OXPHOS to glycolysis in NDV infectious cells. To test this hypothesis, we first used immunofluorescence assays to confirm that SIRT3 was colocalized with mitochondria (Figure S3A). Previous studies have established SIRT3 as a regulator of ROS, and the loss of SIRT3 promotes HIF1A stabilization, which induces the Warburg effect. Thus, we sought to establish a relationship between SIRT3 and HIF1A targets during NDV infection. We then performed network analysis to confirm the close association of the identified SIRT3 and HIF1A in regulating the metabolic network (Figure 5A). Next, we investigated the relationship between SIRT3 and HIF1A targets in human breast cancer cells. Gene expression profiling of seven normal breast samples and 40 ductal breast carcinomas revealed that SIRT3 expression is significantly lower (p = 3.53e−8) in breast carcinomas [52] (Figure 5B). Several HIF1A target genes: SLC2A1/GLUT1, HK2, and LDHA, were significantly higher in the breast carcinomas than in the normal breast samples in the same data set [52] (Figure 5B). RNA-seq data showing HIF1A enrichment and glycolysis in NDV-infected cells recorded high expressions of HIF1A target genes (Figure 5C).
Figure 5.
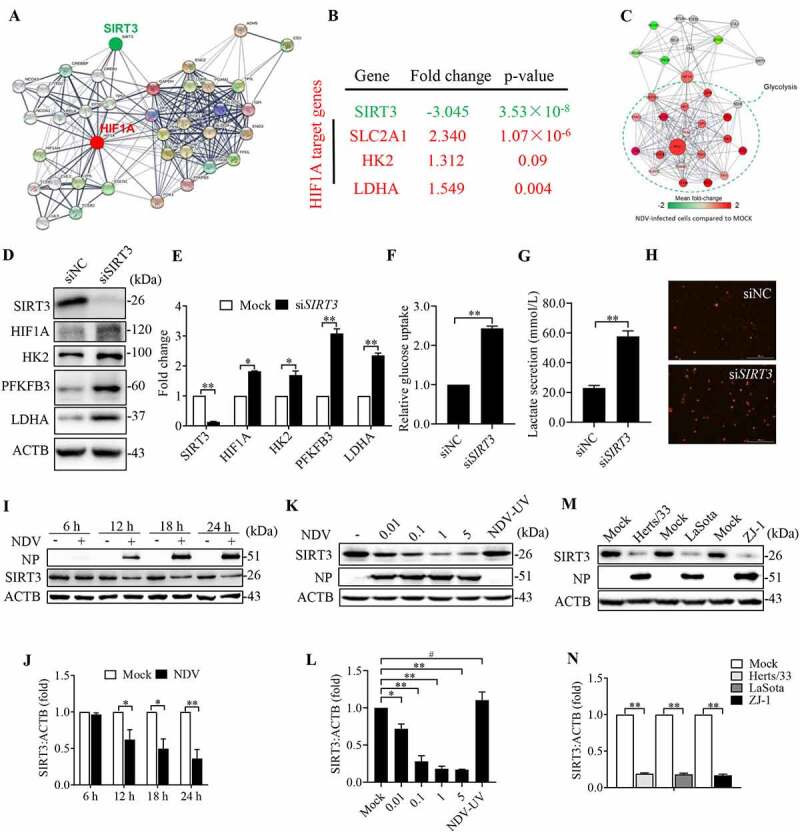
NDV reduces mitochondrial SIRT3, regulates HIF1A stability, and promotes the Warburg effect. (A) A network model describing interactions between the HIF1A and SIRT3-targeted metabolic genes. (B) SIRT3 expression levels and several HIF1A target genes were determined using the Oncomine cancer microarray database (http://www.oncomine.org) in normal breast samples versus breast cancer samples. (C) RNA-seq data showing enrichment of HIF1A, SIRT3, and glycolysis pathways in NDV-infected cells. (D and E) A549 cells were transfected with SIRT3 siRNA or siNC. SIRT3, HIF1A, HK2, PFKFB3, LDHA, and ACTB protein levels were determined via western blot and quantification. (F-H) A549 cells were transfected with SIRT3 siRNA or siNC. Glucose uptake (F), lactate levels in the culture supernatants (G) and mROS (H) were determined. (I and J) A549 Cells were either mock treated or infected by NDV Herts/33 strain at an MOI of 1, whole-cell extracts were prepared from mock-infected and NDV-infected cells at 6, 12, 18, and 24 hpi. SIRT3, NP, and ACTB protein levels were determined by western blot and quantification of SIRT3, NP was used as a maker for virus infection. (K and L) A549 cells were either mock treated or infected with NDV Herts/33 strain at an MOI of 0.01, 0.1, 1, and 5 or with UV-treated NDV Herts/33 strain. NP, SIRT3, and ACTB protein levels were determined via western blot and quantification of SIRT3. (M and N) A549 cells were either mock treated or infected with different NDV strains (Herts/33, Lasota, and ZJ-1). NP, SIRT3, and ACTB protein levels were determined via western blot and quantification of SIRT3. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, # p > 0.05.
We next investigated whether SIRT3 directly modulates HIF1A stability. To test this idea, we first examined the levels of HIF1A and its target in cells of SIRT3 knockdown. Strikingly, the induction of HIF1A and its target genes (HK2, PFKFB3 and LDHA) was markedly upregulated by SIRT3 knockdown (Figure 5D,E). Furthermore, SIRT3 knockdown promotes both glucose uptake (Figure 5F) and lactate production (Figure 5G), while mROS also significantly increased (Figure 5H). Together, our results demonstrated that SIRT3 directly modulates HIF1A function, increased mROS production contributes to enhanced HIF1A stability and its target genes expression.
We hypothesized that SIRT3 could serve as an important regulator of the balance between glycolytic and mitochondrial oxidative metabolism to regulate viral replication. Subsequently, the effect of NDV infection on SIRT3 in infectious cells was determined using cells that had been infected at various times. The MOI were 0.01, 0.1, 1, and 5, and the strains were Herts/33, LaSota, and ZJ-1. Western blot was performed to determine the SIRT3 protein level. The results show that NDV reduces SIRT3 in a time-dependent and dose-dependent manner in comparison to the mock-infected cells (Figure 5I–N). To rule out the possibility that the downregulation of SIRT3 protein was caused by NDV replication, we also determined the SIRT3 protein levels after UV-inactivated NDV infection and found that UV-inactivated NDV did not induces SIRT3 degradation, and the NP was also not detected (Figure 5K). Immunofluorescence assays in mock-infected and NDV-infected cells also confirmed that SIRT3 was reduced (Figure S3B). Using purified mitochondrial and cytosolic fractions of NDV infection cells. As shown in Figure S3C, SIRT3 is mostly expressed and decreased in the mitochondrial fraction. Furthermore, our results show that has no significant change in the abundance of SIRT3 mRNA after NDV infection. The result indicates that NDV infection has no affect in SIRT3 mRNA levels (Figure S3D). Collectively, these results suggest that NDV infection leads to the degradation of SIRT3 protein, regulates HIF1A stability, and promotes energy metabolic reprogramming.
AMPK-MTOR axis regulated autophagy contributes to the degradation of SIRT3 during NDV infection
Our previous research indicates that NDV induces autophagy for optimal replication [45,53,54]. To further identify cellular energy sensor signaling pathways and autophagic occurrences during NDV infection, we first utilized transmission electron microscopy (TEM) to observe it. Notably, NDV infection resulted in an increased number of mitochondria that were trapped by double or single membrane vesicles (Figure 6B). In contrast, autophagosome-like vesicles were rarely observed in the mock-infected cells (Figure 6A). Quantitative analysis showed the number of membrane vesicles containing mitochondria was markedly increased in NDV-infected cells (Figure 6C). These results show that NDV infection can induce mitophagy.
Figure 6.
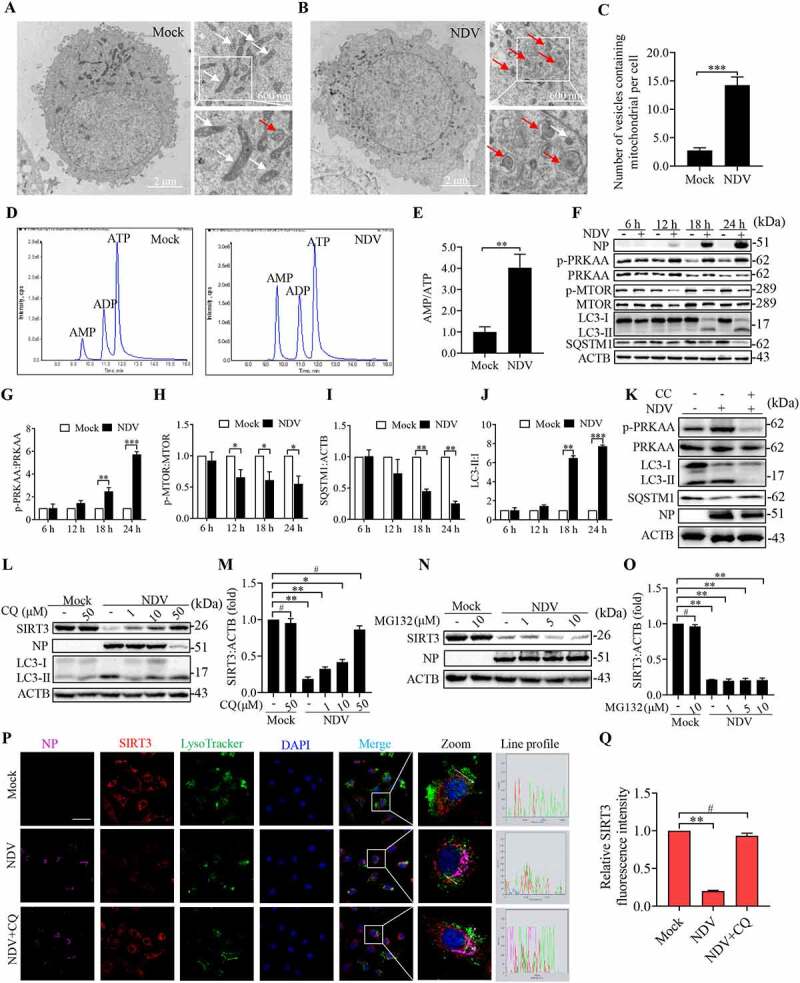
AMPK-MTOR axis regulated autophagy contributes to the degradation of SIRT3 during NDV infection. (A and B) A549 cells were either mock treated or infected with NDV Herts/33 strain at an MOI of 1. Electron microscopy images revealed the mitochondrial ultrastructure. In the zoomed images, typical elongated tubular mitochondria in mock cells and fragmented elliptic mitochondria engulfed with membrane-like vesicles in NDV- infected cells were observed. (C) Quantification of the mitophagosome-like vesicles per cell. Scale bar: 2 μm. (D) Adenylates AMP, ADP, and ATP were determined via HPLC following either mock treatment or NDV infection at 18 h. (E) AMP:ATP ratios in mock versus NDV-infected A549 cells. (F) p-PRKAA, PRKAA, p-MTOR, MTOR, LC3, SQSTM1, and ACTB protein levels were determined via western blot. (G-J) Quantification of p-PRKAA:PRKAA ratio, p-MTOR:MTOR ratio and quantification of SQSTM1, LC3-II:I ratio. (K) Western blot showing p-PRKAA, PRKAA, LC3 and SQSTM1 expression in NDV-infected cells (18 hpi) treated with AMPK inhibitors, CC. (L and M) A549 cells were either mock treated or infected with NDV Herts/33 strain at an MOI of 1. Western blot showing SIRT3 expression in NDV-infected cells (18 hpi) treated with autophagy inhibitors: CQ for the final 6 h. CQ was added at 1, 10, and 50 μM after NDV infection at an MOI of 1. Mock samples were treated in the presence or absence of CQ (50 μM) and quantification of SIRT3. (N and O) Treated with protease inhibitor MG132 for the final 6 h. MG132 was added at 1, 5, and 10 μM after NDV infection at an MOI of mock samples were treated in the presence or absence of MG132 (10 μM) and quantification of SIRT3. (P) Immunofluorescence of mock, NDV, and NDV+CQ (50 μM for the final 6 h) infected A549 cells stained for NP, SIRT3, and LysoTracker. (Q) Quantification of SIRT3 fluorescence intensity. White boxed regions in the panels are enlarged. Scale bar: 10 μm, Data are mean ± SEM, all data are from three independent experiments, ** p < 0.01, *** p < 0.001, # p > 0.05.
Recent studies have revealed that multiple newly discovered targets of AMPK are involved in various aspects of mitochondrial homeostasis, including mitophagy. We hypothesized that this imbalance in AMP, ADP, and ATP levels activates the AMPK in NDV-infected cells and its activities associated with energy metabolism and mitophagy. ATP, ADP, and AMP levels were measured using HPLC. The results show that NDV infection markedly increased the AMP:ATP ratio (4-fold) compared with a mock group (Figure 6D,E). Elevated AMP but not ADP levels result in an up to 10-fold allosteric activation of AMPK [22]. The western blot results show that NDV infection significantly enhanced AMPK phosphorylation. As mentioned previously, we found that MTOR phosphorylation was markedly downregulated (Figure 6F to 6H). In Figure 6F, it also can be seen that NDV induced the conversion of LC3-I to LC3-II (autophagy induction) and reduced SQSTM1 levels in a time-dependent manner (Figure 6I,J). Furthermore, compound C (CC, an AMPK inhibitor) effectively suppressed AMPK activation, the conversion of LC3-I to LC3-II, and the autophagic flux induced by NDV infection (Figure 6K, and S4A to 4C), indicating that NDV promotes autophagic flux via the AMPK-MTOR pathway. Together, these results suggest that the AMPK-MTOR axis regulates autophagy in NDV infectious cells.
It has long been assumed that the autophagy-lysosomal pathway (ALP) and the ubiquitin-proteasome system (UPS) are major pathways for protein degradation in cells [55,56]. To determine which degradation system dominantly controls the degradation of SIRT3 during viral infection, NDV-infected A549 cells were treated with the lysosome acidification inhibitor, chloroquine (CQ), or the autophagic maturation vacuole inhibitor, bafilomycin A1 (BAF-A1), and proteasome inhibitor, MG132. We found that the degradation of SIRT3 can be blocked by CQ (Figure 6L,M) and BAF-A1 (Figure S4E and F), but not MG132 (Figure 6N,O). Macroautophagy is a central lysosomal-mediated process activated following a viral infection [17]. Next, using siRNA to knock down ATG7 (upstream autophagy protein) in A549 cells and then examining the SIRT3 protein levels, we compared the siNC and siATG7-impaired SIRT3 degradation following an NDV infection (Figure S4G and H). We also used ATG7−/− HEK-293 cells, which is consistent with ATG7 knockdown (Figure S4I and J). Furthermore, immunofluorescence also showed that SIRT3 was colocalized with the lysosome, confirming the involvement of this pathway in SIRT3 degradation following NDV infection (Figure 6P,Q). These results confirm that SIRT3 expression in the infected cells was rescued only with CQ or BAF-A1 treatment, indicating that NDV-mediated SIRT3 reduction is via lysosomal proteolysis rather than the ubiquitin-proteasome pathway.
NDV induces SIRT3 degradation through PINK1-PRKN-dependent selective mitophagy
The damaged mitochondria underwent fission prior to mitophagy. Previous studies have demonstrated that mitochondrial depolarization leads to autophagy-mediated removal of mitochondria (mitophagy) [57]. We investigated whether NDV infection caused mitophagy, which is mediated by SIRT3 degradation. Mito-QC is a bona-fide endpoint mitophagy reporter in cultured cells and tissues [58,59]. The basic principle of the mito-QC model in vitro is presented in Figure 7A. HeLa cells were transfected with mito-QC reporter, followed by NDV infection and a CCCP treatment. The results show that NDV infection of mito-QC-transfected HeLa cells results in a shift from yellow to partially red fluorescence at 18 hpi. The CCCP as a positive control was consistent with the NDV-infected cells (Figure 7B,C). The data indicate that NDV infection induces selective mitophagy in NDV infectious cells.
Figure 7.
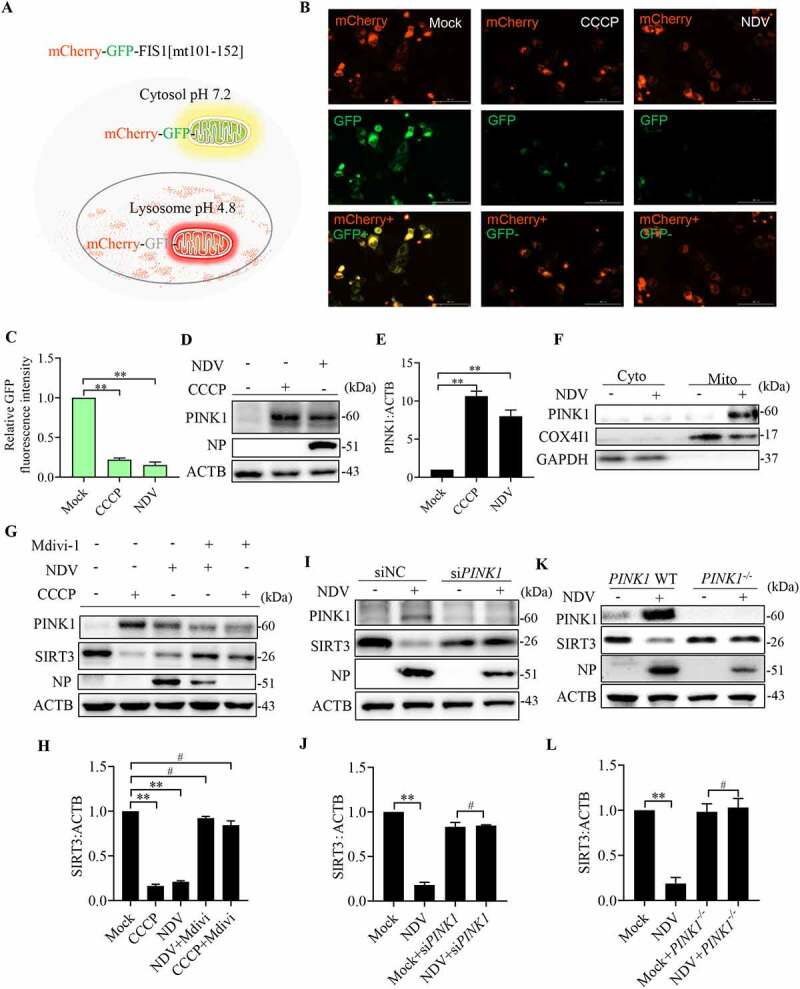
NDV induces SIRT3 degradation via PINK1-PRKN-dependent selective mitophagy. (A) Diagram of tandem-tagged mCherry-GFP-FIS1 based mitophagy assay. (B) HeLa cells expressing the mCherry-GFP-FIS based mitophagy assay reporter were treated with NDV for 18 h or CCCP for 6 h. Cells were fixed and subjected to immunofluorescence analysis to detect mitochondrial fluorescence (red and green). (C) Quantification of GFP fluorescence intensity. (D) A549 cells were treated with NDV for 18 h or CCCP for 6 h. PINK1 and ACTB protein levels were determined via western blot. (E) Quantification of PINK1:ACTB ratio. (F) Cytoplasm and mitochondria were separated and purified. After purification, COX4I1, PINK1, and GAPDH protein levels were determined via western blot. (G) A549 cells were mock treated or infected with NDV (MOI = 1) or treated with CCCP for the final 6 h and added in the presence or absence of the mitophagy inhibitor, mdivi-1. NP, PINK1, SIRT3, and ACTB protein levels were determined via western blot and quantification of SIRT3. (I and J) A549 cells were transfected with PINK1 siRNA or siNC. NP, PINK1, SIRT3, and ACTB protein levels were determined via western blot and quantification of SIRT3. (K and L) PINK1 WT or PINK1−/− of HeLa cells were infected at MOI = 1 or treated with mock infection. NP, PINK1, SIRT3, and ACTB protein levels were determined via western blot and quantification of SIRT3. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, # p > 0.05.
PINK1 is known to drive mitophagy following mitochondrial depolarization [57]. To assess the contribution of PINK1 to basal mitophagy after NDV infection. As expected, endogenous mitochondrial PINK1 levels responded robustly to increased following NDV infection and CCCP treatment (Figure 7D,E). Purified mitochondrial and cytosolic fractions (Figure 7F), PINK1 expression predominantly selectively accumulates on dysfunctional mitochondria, and it increased in the mitochondrial fraction. Together, these results demonstrate that NDV causes PINK1 translocation to mitochondria, initiating mitophagy activities. As we know, SIRT3 is localized to the mitochondrion. Next, we tested whether specific suppression of mitophagy via chemical treatment with Mdivi-1 would inhibit SIRT3 degradation. The results show that Mdivi-1 effectively alleviates SIRT3 degradation induced by mitophagy following NDV infection and CCCP treatment (Figure 7G,H). We used siRNA to knockdown PINK1 in A549 cells, and then to examine the SIRT3 protein levels. Compared with siNC, siPINK1 impaired SIRT3 degradation following NDV infection (Figure 7I,J). We also used PINK1−/− HeLa cells, which is consistent with PINK1 knockdown (Figure 7K,L). The results suggest that mitophagy-mediated SIRT3 reduction upon NDV infection is a PINK1-PRKN-dependent mitophagy manner.
SIRT3 degradation promotes glycolysis to support virus replication
Given the identification of SIRT3 as a potential mediator of glucose metabolism, we hypothesized that SIRT3 degradation results in a metabolic shift toward glycolysis essential for viral replication. Therefore, we first test whether the metabolic patterns shift is linked to SIRT3 loss, and as a compensatory to support virus replication, we measured glucose uptake, lactate production, and viral titers in SIRT3 knockdown cells upon NDV infection. As expected, in SIRT3 knockdown cells, glycolysis is increased (Figure 8A–C) and viral titers in SIRT3 knockdown cells is also increased (Figure 8D). These data demonstrate that NDV infection induces SIRT3 loss promotes glycolytic to support virus replication.
Figure 8.
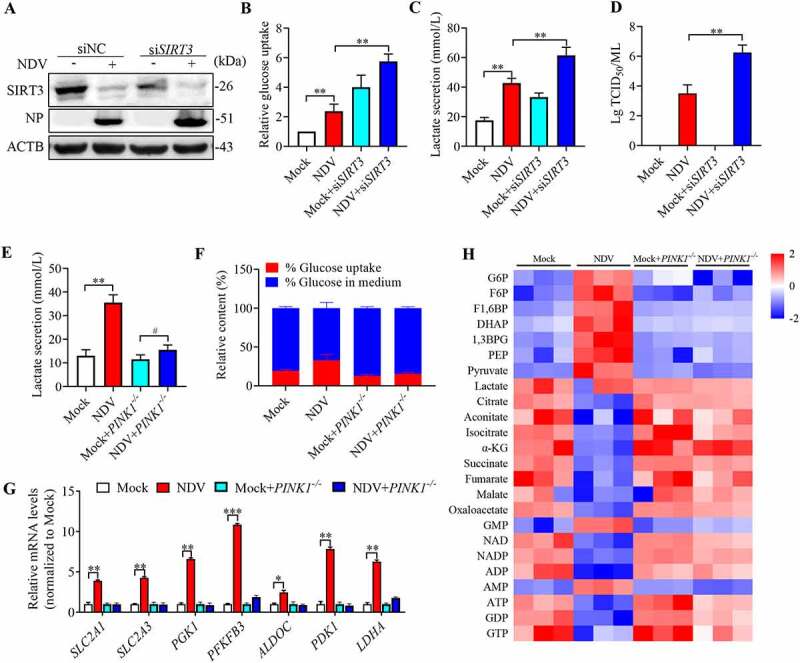
SIRT3 degradation promotes glycolysis to support virus replication. (A) siSIRT3 or siNC of A549 cells were infected at MOI = 1 or treated with mock infection. SIRT3, NP, and ACTB protein levels were determined via western blot. (B-D) siSIRT3 or siNC of A549 cells were infected at MOI = 1 or treated with mock infection. Glucose uptake (B), lactate levels in the culture supernatants (C) and viral titers (D) were determined. (E and F) PINK1 WT or PINK1−/− of HeLa cells were infected at MOI = 1 or treated with mock infection. The lactate levels in the culture medium (E) and glucose uptake (F) were determined. (G) PINK1 WT or PINK1−/− of HeLa cells were infected or treated with mock infection. mRNA levels were detected using qRT-PCR with HIF1A target genes. (H) PINK1 WT or PINK1−/− of HeLa cells were infected or treated with mock infection. Heatmap of altered energy-related metabolites. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, *** p < 0.001.
We next asked whether metabolite patterns reflect an increase in glycolysis linked to SIRT3 degradation by selective mitophagy, we measured glucose uptake, lactate production, and glycolysis gene expression in PINK1−/− cells. As expected, NDV infection induced lower levels of intracellular glucose and higher levels of glucose uptake in the PINK1−/− cells (Figure 8F). PINK1−/− reverses lactate production into the medium more than was observed for PINK1 WT cells with NDV infection (Figure 8E), and glycolysis genes expression was also reversed (Figure 8G). To provide direct evidence for a link between PINK1-regulated degradation of SIRT3 and cellular metabolism, we performed another set of global metabolic analysis on the PINK1−/− cells. Strikingly, our data confirms a global reversal in the PINK1−/− cells following NDV infection, with at least 20 metabolites specifically altered (Figure 8H). One of the most consistent metabolic alterations observed was an alteration of glucose metabolism. This supports the hypothesis that SIRT3 plays a significant role as a critical regulator of energy metabolism reprogramming. Together, these data clearly indicates that NDV infectious PINK1−/− cells can reverse the metabolic shift associated with the Warburg effect.
NDV induces SIRT3 degradation and reprogram energy metabolism in avian cells
To confirm that NDV induces SIRT3 degradation to reprogram energy metabolism in avian cells, we detected SIRT3 protein levels by western blot in DF-1 (Figure 9A,B) and CEF (Figure 9C,D) cells at 6, 12, 18, and 24 hpi. The results show that NDV reduces SIRT3 in a time-dependent manner in both DF-1 and CEF cells (Figure 9A–D), which is consistent with A549 cells (Figure 5I,J). Next, we measured lactate production, as expected, NDV-infected cells also extruded more lactate into the medium than the mock cells in a time-dependent manner in both DF-1 (Figure 9E) and CEF cells (Figure 9F). Furthermore, we also examined the influence of NDV on metabolites using liquid chromatography-mass spectrometry (LC-MS) in CEF cells. The metabolic profile indicates that intermediates of glycolysis were elevated, whereas tricarboxylic acid (TCA) cycle metabolites were reduced (Figure 9G,H). Consistent with a pattern of human cells (Figure 1D–F). Taken together, the data indicates that NDV infection upregulates glycolysis pathway to reprogram energy metabolism, and utilize host’s energy to support virus replication in both human and avian cells.
Figure 9.
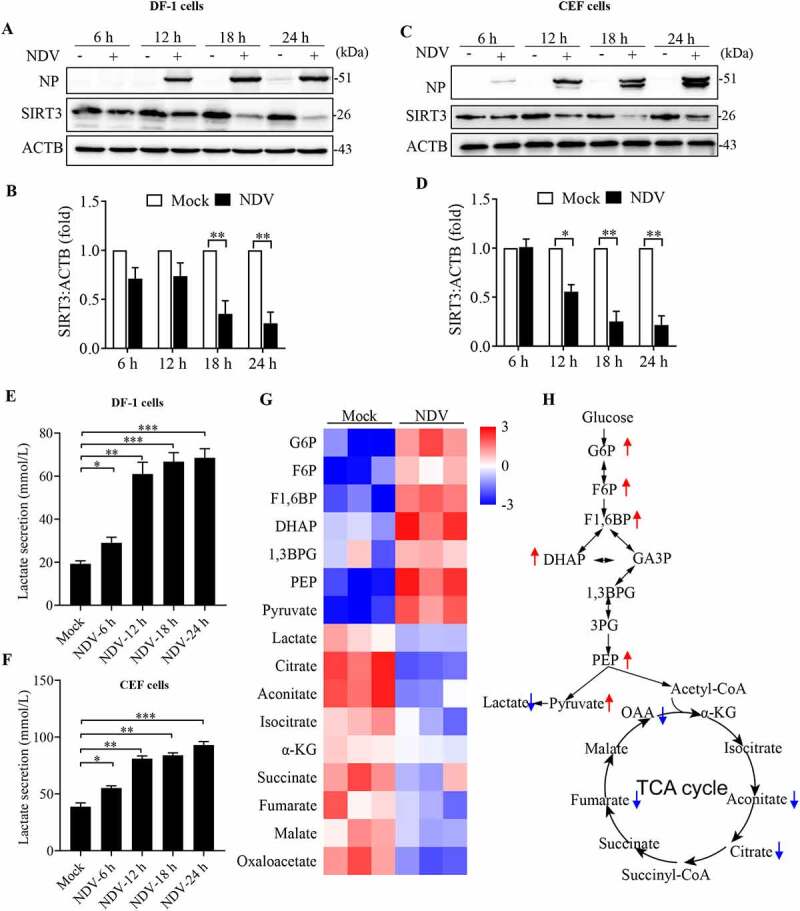
NDV induces SIRT3 degradation and reprogram energy metabolism in avian cells. (A, B, and E) DF-1 Cells were either mock treated or infected by NDV Herts/33 strain at an MOI of 1, whole-cell extracts were prepared from mock-infected and NDV-infected cells at 6, 12, 18, and 24 hpi. SIRT3 and ACTB protein levels were determined by western blot and quantification of SIRT3, NP was used as a maker for virus infection. The lactate levels of DF-1 in the culture medium were determined (E). (C, D, and F) CEF cells were either mock treated or infected by NDV Herts/33 strain at an MOI of 1, whole-cell extracts were prepared from mock-infected and NDV-infected cells at 6, 12, 18, and 24 hpi. SIRT3 and ACTB protein levels were determined by western blot and quantification of SIRT3, NP was used as a maker for virus infection. The lactate levels of CEF in the culture medium were determined (F). (G) Heatmap of energy related metabolites. (H) Schematic of glucose metabolism, red arrow show intermediates were up-regulated and blue arrow show intermediates were down-regulated. Data are mean ± SEM, all data are from three independent experiments, * p < 0.05, ** p < 0.01, *** p < 0.001.
Discussion
There is increasing evidence that viruses remodel cellular metabolism in a similar manner, it is known as the Warburg effect [1,3]. However, various viral engagements with the host’s metabolic regulation in different mechanisms, and not all infection causes mitochondrial dysfunction and autophagy. Our previous studies have reported that NDV induces endoplasmic reticulum stress, initiates the unfolded protein response and signaling cascades, and triggers apoptosis of the mitochondria-related caspase pathway [43]. This research is an in-depth study to characterize metabolic reprograming during NDV infection. We confirmed and extended these findings by demonstrating the role of SIRT3 as a checkpoint for energy metabolic reprograming [60]. We found that OXPHOS was decreased after mitochondrial stress, mROS accumulation, and reduced mitochondrial respiratory chain complex I activity, all of which accelerating mitochondrial damage. As energy compensation, NDV reprograms glucose metabolism in infectious cells by increasing glucose use in the glycolysis pathway, which facilitates NDV replication. Damaged mitochondria were degraded by selective autophagy. SIRT3 was degraded by autophagosome–lysosome fusion along with the damaged mitochondria. There is ample evidence suggesting that mitophagy is closely related to the PINK1-PRKN-dependent pathway [61]. Importantly, degradation of SIRT3 in damaged mitochondria via PINK1-PRKN-dependent mitophagy, leading to increased HIF1A stabilization and the expression of its target genes in NDV infectious cells, shifts mitochondrial bioenergetic metabolism toward aerobic glycolysis (Figure 10). Indeed, our data indicates that NDV infection can shift the bioenergetics of the infected host cell to a higher energetic state, benefiting viral replication.
Figure 10.
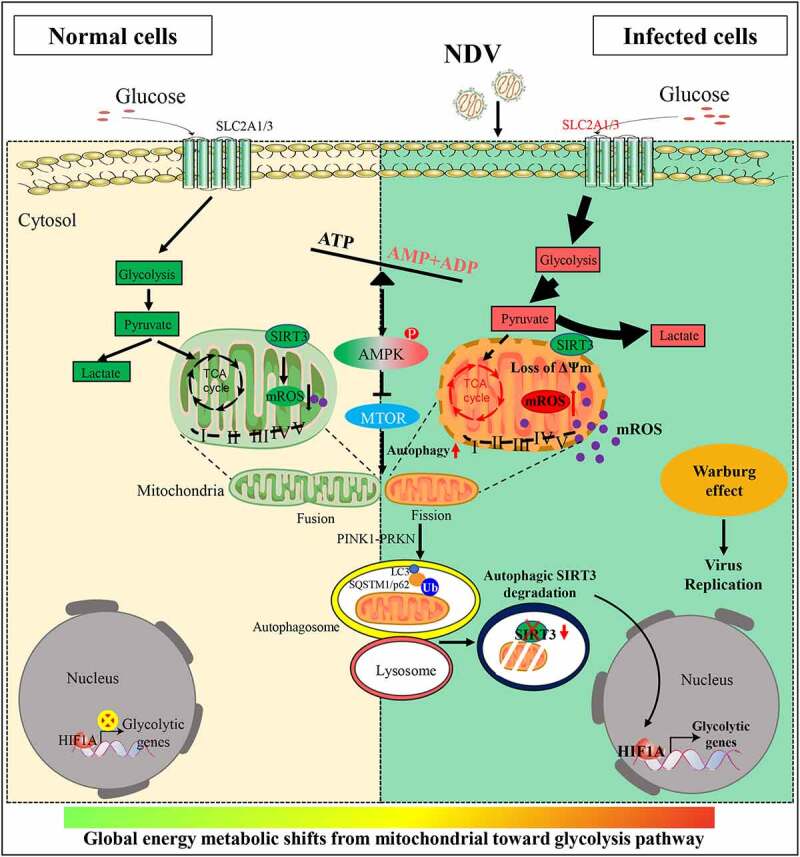
Diagram depicting NDV-driven energy metabolic reprogramming to facilitates virus replication. NDV promotes glucose utilization and aerobic glycolysis in infectious cells, Upregulating glycolysis (the Warburg effect) can compensate for the lack of ATP production by the OXPHOS. In brief, NDV infection is harmful not only for mitochondria but also promotes mitochondria from fusion to fission and energy stress (imbalance in ATP, ADP and AMP levels). Damaged mitochondria were degraded by selective autophagy, SIRT3 was degraded by PINK1-PRKN-dependent selective mitophagy. Consequently, degradation of SIRT3 in damaged mitochondria, increases cellular mROS levels, leading to increased HIF1A stabilization and its target genes expression in NDV infectious cells, shifts mitochondrial bioenergetic metabolism toward aerobic glycolysis.
Mitochondria are in a balance of constant fission and fusion, which is essential to cellular homeostasis [62,63]. As the energy power plants of cells, mitochondria generate ATP via the respiratory electron transport chain [64]. However, glycolysis proceeds rapidly, and under certain conditions, cells appear to utilize glycolysis over cellular respiration [65]. For instance, a virus infection requires a significant portion of the host’s energy, and most viruses shift mitochondrial bioenergetic metabolism toward aerobic glycolysis in vitro and in vivo [66]. DENV, a virus in the Flaviviridae family, has been shown to increase glucose consumption and glycolysis during an infection [6,7]. Studies show that Zika virus infection differentially reprograms glucose metabolism by increasing glucose use in the tricarboxylic acid cycle in human cells and in the pentose phosphate pathway in mosquito cells [5]. Thus, there should be further studies on host cell metabolism and mitochondrial mechanisms associated with autophagy and apoptosis, as well as immune responses associated with viral infections, to establish whether modulating metabolic networks to design therapeutic approaches to fight viral infections. Our group found that NDV induces ER stress and the release of a lot of calcium ions (Ca2+) from the cellular Ca2+ pool, which is transferred to mitochondria and quickly induces the mitochondrial stress response. Virus replication is an extremely energetically expensive process [66]. AMPK is a highly sensitive indicator of cellular energy status and has recently been reported to control various aspects of mitochondrial homeostasis, from mitochondrial dynamics and biogenesis to removal by mitophagy [67–69]. In this study, we show that autophagy is promoted by elevation of the AMP:ATP ratio during NDV infection, subsequent AMPK activation, and inhibition of MTOR. Autophagy is a conserved catabolic multistep process that non-selectively or selectively delivers large cytoplasmic proteins including damaged organelles into specific double-membrane autophagosome vesicles and shuttles them to the vacuole/lysosomes for degradation and recycling [70].
The activation of AMPK under energy stress not only promotes mitochondrial fission, but also regulates mitophagy [69,71]. Recent studies have shed light on the mechanism via which SIRT3 regulates cellular stress [36,37]. SIRT3 is colocalized to mitochondria and NDV utilizes selective mitophagy to degrade SIRT3, with the reduced SIRT3 activity acting as a signal to reprogram cellular metabolism. Loss of SIRT3 increases glycolytic metabolism by regulating the stability and activity of HIF1A [72], facilitating the extremely energetically expensive process of viral replication. In this study, we establish that NDV induces mitochondrial damage, and the subsequently elevated mROS contributes to increased HIF1A stabilization and activity, increasing glucose uptake and catabolism. Consequently, providing the metabolic precursors necessary to fuel a high rate of replication. We confirmed that NDV-induced metabolic reprogramming is mediated by the degradation of SIRT3, which occurs via PINK1-PRKN-dependent mitophagy [73]. Furthermore, SIRT3 loss is associated with increased expression of HIF1A target genes in glycolysis, providing a link between SIRT3 degradation and virus replication.
In summary, our study reveals the profound impact of SIRT3 function on glycolysis and virus replication. Essentially, NDV induces mitochondrial damage and SIRT3 is subsequently degraded via mitophagy, shifting mitochondrial bioenergetic metabolism toward glycolysis, which contributes to viral replication.
Materials and methods
Cells and viruses
HeLa (ATCC, CRL-12401), A549 (ATCC, CCL-185), Hep G2 (ATCC, HB-8065), HEK-293 (ATCC, CRL-1573), and DF-1 (ATCC, CRL-12203) cells were purchased from the American Type Culture Collection (ATCC). Chicken embryo fibroblast (CEF) primary cells were prepared from 9-day-old specific-pathogen-free (SPF) embryonated chicken eggs as described previously [45]. PINK1−/− HeLa and ATG7−/− HEK-293 cells were obtained from Qi-Ming Sun (Zhejiang University, Zhejiang, China). HIF1A−/− HeLa cells were obtained from Shi-Min Zhao and Wei Xu (Fudan University, Shanghai, China). These cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, C11995500BT) supplemented with 10% fetal bovine serum (FBS; Gibco, 10270106), in a humidified atmosphere containing 5% CO2 at 37°C. NDV strains Herts/33 and LaSota were obtained from the China Institute of Veterinary Drug Control (Beijing, China). NDV strain ZJ1 was provided by Xiufan Liu (Yangzhou University, Yangzhou, China). The UV-inactivated NDV Herts/33 strain was prepared as described in Sun et al. [74]. GFP-labeled NDV was constructed in our laboratory. Cells were infected with NDV at a MOI of 1 at 37°C. Following a 1-h absorption period, unattached viruses were removed and the cells were then washed three times with serum DMEM and cultured in maintenance medium at 37°C. Viral titers on DF-1 cells were determined as median tissue culture infective doses (TCID50) as described previously [75].
Chemical reagents
PFK15 (S7289), MG-132 (S2619), compound C (S7306), 2-deoxy-D-glucose (2-DG), (S4701), CCCP (S6494), acetylcysteine (N-acetylcysteine) (NAC) (S1623), oligomycin (S1478), and Mdivi-1 (S7162) were purchased from Selleck Chemicals. Rotenone (ROT) (R105077) and bafilomycin A1 (BAF-A1) (B101389) were obtained from Aladdin. chloroquine (CQ) was purchased from Sigma-Aldrich (C6628). MitoTracker™ Red CMXRos (M7512) and LysoTracker™ Red DND-99 (L7528) were purchased from Thermo Fisher Scientific. JC-1 (C2006) was purchased from Beyotime. 2-NBDG was purchased from (Cayman Chemical, B6035). Amplite™ Fluorometric 20S Proteasome Assay Kit (B13456) was purchased from AAT Bioquest. D-glucose (U-13C6, 99%) (CLM–1396–1) and D-glucose (1,2–13C2, 99%) (CLM–504–1) were obtained from Cambridge Isotope Laboratories, Inc.
Metabolite profiling
Cells were infected with or without NDV for the indicated time points. Metabolites were extracted in ice-cold methanol, and endogenous metabolite profiles were obtained using LC-MS methods as described. Data were acquired using a 4000 QTRAP mass spectrometer (Applied Biosystems/Sciex). Multi-Quant software (Applied Biosystems/Sciex) was used for analysis. Metabolite levels were normalized to protein content, which was determined by performing a BCA protein assay kit (Beyotime, P0012). on a duplicate set of cells treated identically to the experimental cells.
Glucose uptake assay
Cells were infected with or without NDV for the indicated time points. The DMEM culture medium was replaced by glucose-free medium with 10% FBS. After incubating the cells for another 30 min, 2-NBDG (Cayman Chemical, B6035) was added at a final concentration of 100 μM for 15 min and cells were collected and washed twice with PBS (Solarbio, P1010). The cells were ready to be analyzed by flow cytometry (Beckman) equipped with FlowJo software.
Measurement of lactate production
Cells were seeded in a 6-well plate, and infected with NDV. Mock-infected and NDV infected cells were cultured for 6, 12, 18, and 24 h. Lactate concentrations in the culture media was measured using the Lactate Assay Kit (Jiancheng, A019–2–1), respectively, according to the manufacturer’s instructions. For the detection of lactate, 450 nm was used, respectively, using an automated microplate reader (BioTek). Lactate measurements was normalized to cell numbers.
Seahorse XF Real-Time ATP rate assay
Seahorse XF Real-Time ATP rate test assay (103592–100) was used to test mitochondrial ATP production rate and glycolysis ATP production rate of DF-1, A549 and Hep G2 cells. Cells were seeded in XFe96 Cell Culture Microplates (Seahorse Bioscience, 102601–100) with concentration of 20,000 cells/80 μL per well, then allowed to grow overnight in a cell culture incubator. In the meantime, Seahorse XFe96 Sensor Cartridge were hydrated and placed in a non-CO2 37°C incubator overnight. Cells were washed twice with assay medium based on the type of XF assays were run, used XF Real-Time ATP rate Assay Medium (XF Base Medium [Seahorse Bioscience, 103575–100] with 5.5 mM glucose [Seahorse Bioscience, 103577–100], 1.0 mM pyruvate [Seahorse Bioscience, 103578–100], 2 mM L-glutamine [Seahorse Bioscience, 103579–100], adjusted pH to 7.4 at 37°C). Cells were covered by assay medium for a final volume of 180 μL /well, and were placed in a 37°C incubator without CO2 for 1 h prior to the assay. Meanwhile compounds were loaded into the XFe96 Sensor Cartridge. In Seahorse XF Real-Time ATP rate test assay, compounds consist of port A: oligomycin for a final concentration of 1.5 μM, port B: rotenone for a final concentration of 0.5 μM, and antimycin A (Seahorse Bioscience, 103592 − 100) for a final concentration of 0.5 μM. Subsequently, the assay was run on an Agilent Seahorse XFe96, and results were viewed and analyzed using Wave Desktop and Report Generator software.
Seahorse XF cell mitochondrial/glycolysis stress test assay
Seahorse Cell Mito/Glycolysis Stress Test Assay (103015–100, 103020–100) were used to test mitochondrial respiration capacity and glycolysis capacity of DF-1 cells. Brief, DF-1 cells were seeded in XFe96 Cell Culture Microplates, then allowed to grow overnight. In cell mitochondrial stress test assay, compounds consist of port A: oligomycin for a final concentration of 1 μM, port B: FCCP for a final concentration of 0.5 μM, port C: rotenone for a final concentration of 0.5 μM, and antimycin A for a final concentration of 0.5 μM. While in glycolysis stress test assay, compounds consist of port A: glucose for a final concentration of 10 mM; port B: oligomycin for a final concentration of 1 μM; port C: 2-DG (Seahorse Bioscience, 103020–100) for a final concentration of 50 mM. Subsequently, the assay was run on an Agilent Seahorse XFe96, and results were viewed and analyzed using Wave Desktop and Report Generator software.
Western blot analysis and antibodies
Immunoblot analysis was performed, as described below. Briefly. Cells were lysed in cell lysis buffer containing a protease inhibitor cocktail at 4°C for 30 min (Merck Millipore, P8340). The lysates were denatured and then subjected to SDS-PAGE and transferred to nitrocellulose membranes (Whatman, NC-a101-b105). The membranes were then blocked and reacted with primary antibodies overnight at 4°C and horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson, 148774) for 2 h at room temperature. The protein bands were visualized using the enhanced chemiluminescence (ECL) system (Tanon Science & Technology, 180–5001) and quantified using Image J software.
The following antibodies were used: ATG7 (8558S), SQSTM1/p62 (88588S), COX4I1 (4850 T), OPA1 (80471S), p-DNM1L/DRP1 (S616; 3455S), p-DNM1L/DRP1 (S637; 4867S), p-PRKAA/AMPKα (Thr172; 50081S), PRKAA/AMPKα (5831S), p-MTOR (Ser2448; 5536S), MTOR (2983S), SIRT3 (2627S), PINK1 (6946 T), hydroxy-HIF1A (3434 T), HIF1A (36169S), HK2 (2867S), PKM/PKM2 (4053S), SLC2A1/GLUT1 (12939S) and LDHA (3852S) from Cell Signaling Technology; SLC2A3/GLUT3 (20403–1–AP), PFKFB3 (13763–1–AP), ACTB (66009–1–Ig), and GAPDH (60004–1–Ig) from Proteintech; LC3B (NB100–2220), PINK1 (BC100–494), and HIF1A (NB100–105) from NOVUS; The dilution of all the primary antibody incubation in immunoblotting is 1:1000, except for the 1:5000 dilution of GAPDH and ACTB.
Immunofluorescence staining
The cells were seeded on cover slips in a 12-well plate. After being infected with NDV, NDV-infected and mock-infected cells were collected and fixed in 4% paraformaldehyde for 30 min, washed with PBS, permeabilized with 0.1% Triton-X100 (Merck, 9002–93–1), blocked in 5% BSA (Beijing ZEPING Bioscience & Technology, 02FC007780) in PBS, and incubated with primary antibodies (1:100) at 4°C overnight. Then, the cells were washed with PBS and incubated with Cy3-conjugated goat-anti-rabbit IgG (1:500; Thermo Scientific, A-21222) at 37°C for 1 h, followed by staining with DAPI. The slides were subsequently mounted using glycerin and were observed under a confocal microscope (Olympus, Tokyo, Japan). The images were analyzed using ImageJ.
Transmission electron microscopy (TEM)
To detect the effect of NDV on mitochondria, A549 Cells were seeded in a 10-cm dishes, and infected with NDV. Briefly, cells were collected at 18 h post-infection, and washed twice with PBS and fixed with 2.5% glutaraldehyde diluted in PBS at 4°C for 30 min. Cell pellets were dehydrated with an acetone series and embedded in epoxy resin (Sigma, SLCJ5080). Images of the ultrathin sections were observed using a CM-120 TEM (Philips) at 80 kV.
RNA isolation and RT-qPCR analysis
Total RNA was extracted with TRIzol reagent (Thermo Fisher Scientific, 15596026) and subjected to reverse transcription with PrimeScript® RT reagent Kit (Takara, RR014A). PCR reactions were performed with SYBR® Premix Ex Taq™ kit (Takara, RR420A) using ABI Prism VIIA7 Real-Time PCR System. All measurements were performed in duplicate and the arithmetic mean of the Ct-values was used for calculations: target gene mean Ct values were normalized to the respective housekeeping genes (ACTB), mean Ct-values (internal reference gene, Ct), and then to the experimental control. Obtained values were exponentiated 2−ΔΔCt to be expressed as n-fold changes in regulation compared with the experimental control 2−ΔΔCt by the method of relative quantification. The assay was performed in biological triplicates, and error bars represented SE. Primers used for PCR are list in Table S1.
Detection of mROS and MMP by flow cytometer
For mROS analysis, Cells were infected with or without NDV for the indicated time points. Then, cells were incubated in PBS containing 5 μM MitoSOX Red Mitochondrial Superoxide Indicator (ThermoFisher Scientific, M36008) at 37°C for 30 min. Emitted Fluorescence (Ex/Em = 510/580) was measured.
MMP was detected with JC-1 (Beyotime, C2006). After various treatments of each group, the cells were harvested with trypsin, then suspended in PBS, and immediately stained with JC-1 according to the manufacturer’s instructions. Then, the samples were analyzed with a flow cytometer (FAC Scan flow cytometer, Becton Dickinson). The results were analyzed using FlowJo software.
mCherry-GFP-FIS1[mt101–152] (mito-QC) assay
mito-QC as a bona-fide endpoint mitophagy reporter in cultured cells and tissues [58]. The mito-QC assay is based on a tandem tag construct of mCherry-GFP. This tag is targeted to mitochondria through the addition of a 51 amino acid mitochondrial targeting sequence derived from the outer mitochondrial membrane protein, FIS1[mt101–152]. The resultant mCherry-GFP-FIS1[mt101–152] (mito-QC) construct is observable as a red and green fluorescent signal, which when merged appears yellow. The engulfment of mitochondria by the phagophore and concomitant fusion of the mitophagosome with the lysosome result in the appearance of mCherry-only puncta, due to the acid-labile properties of GFP [59]. Cells were transfected with the indicated mito-QC. At 6 hpi, cells were washed three times with PBS and incubated for an additional 48 h before virus infection. The cells were observed under a confocal microscope (Olympus, Tokyo, Japan). The images were analyzed using ImageJ.
siRNA transfection
Small interfering RNA (siRNA) oligonucleotides for targeting endogenous PINK1, SIRT3, and ATG7 were purchased from Gene Pharma (Shanghai China). Cells were transfected with the indicated siRNA or scramble siRNA as described previously [74]. At 6 hpi, cells were washed three times with PBS and incubated for an additional 48 h before virus infection.
Statistical analysis
Significant differences among groups were determined with one-way analysis of variance (ANOVA) followed by a Tukey test using statistical software SPSS version 18.0. The data are presented as the mean ± SEM using Graphpad Prism (Graph Pad, San Diego, CA). The P values less than 0.05 were considered statistically significant. All results were confirmed from a minimum of three independent experiments.
Supplementary Material
Acknowledgments
We thank Dr. Pengwei Zhao (Zhejiang University, Zhejiang, China) for providing plasmid mito-QC. We thank Dr. Qi-Ming Sun (Zhejiang University, Zhejiang, China) for providing PINK1−/− HeLa and ATG7−/− HEK-293 cells. We thank Dr. Shi-Min Zhao and Dr. Wei Xu (Fudan University, Shanghai, China) for providing HIF1A−/− HeLa cells. We thank Dr. Xiufan Liu (Yangzhou University, Yangzhou, China) for providing NDV strain ZJ1. This work was funded by the National Natural Science Foundation of China (No. 32030108 to C.D., 91957120 to S.H.L. and 32122085 to Y.J.S.) and Natural Science Foundation of Shanghai (No. 18ZR1448700 to W.W.L.).
Funding Statement
This work was supported by the National Natural Science Foundation of China [32030108]; National Natural Science Foundation of China [91957120]; National Natural Science Foundation of China [32122085]; Natural Science Foundation of Shanghai [18ZR1448700].
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Goodwin CM, Xu S, Munger J.. Stealing the keys to the kitchen: viral manipulation of the host cell metabolic network. Trends Microbiol. 2015. Dec;23(12):789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fan S, Wu K, Zhao M, et al. LDHB inhibition induces mitophagy and facilitates the progression of CSFV infection. Autophagy. 2020 Sep 28;17(9):1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sanchez EL, Lagunoff M.. Viral activation of cellular metabolism. Virology. 2015. May;479–480:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Thai M, Graham NA, Braas D, et al. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014. Apr 1;19(4):694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thaker SK, Chapa T, Garcia G Jr., et al. Differential metabolic reprogramming by Zika Virus promotes cell death in human versus mosquito cells. Cell Metab. 2019. May 7;29(5):1206–1216 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Allonso D, Andrade IS, Conde JN, et al. Dengue Virus NS1 protein modulates cellular energy metabolism by increasing Glyceraldehyde-3-Phosphate dehydrogenase activity. J Virol. 2015. Dec;89(23):11871–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fontaine KA, Sanchez EL, Camarda R, et al. Dengue virus induces and requires glycolysis for optimal replication. J Virol. 2015. Feb;89(4):2358–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang J, Jia L, Tsang CM, et al. EBV infection and glucose metabolism in nasopharyngeal carcinoma. Adv Exp Med Biol. 2017;1018:75–90. [DOI] [PubMed] [Google Scholar]
- [9].Mullen PJ, Garcia G Jr., Purkayastha A, et al. SARS-CoV-2 infection rewires host cell metabolism and is potentially susceptible to mTORC1 inhibition. Nat Commun. 2021. Mar 25;12(1):1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/glycolysis-dependent axis. Cell Metab. 2020. Sep 1;32(3):498–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sanchez EL, Pulliam TH, Dimaio TA, et al. Glycolysis, glutaminolysis, and fatty acid synthesis are required for distinct stages of Kaposi’s Sarcoma-Associated herpesvirus lytic replication. J Virol. 2017. May 15;91(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thyrsted J, Holm CK. Virus-induced metabolic reprogramming and innate sensing hereof by the infected host. Curr Opin Biotechnol. 2020. Oct;25(68):44–50. [DOI] [PubMed] [Google Scholar]
- [13].Grasso D, Zampieri LX, Capeloa T, et al. Mitochondria in cancer. Cell Stress. 2020. May 11;4(6):114–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Polyakov VY, Soukhomlinova MY, Fais D. Fusion, fragmentation, and fission of mitochondria. Biochemistry (Mosc). 2003. Aug;68(8):838–849. [DOI] [PubMed] [Google Scholar]
- [15].de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal. 2008. Mar;10(3):621–633. [DOI] [PubMed] [Google Scholar]
- [16].Khan M, Syed GH, Kim SJ, et al. Mitochondrial dynamics and viral infections: a close nexus. Biochim Biophys Acta. 2015. Oct;1853(10Pt B):2822–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Anderson CM, Macleod KF. Autophagy and cancer cell metabolism. Int Rev Cell Mol Biol. 2019;347:145–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012. Feb 15;125(Pt 4):795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang L, Cho YL, Tang Y, et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018. Aug;28(8):787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018. Feb 6;27(2):299–313. [DOI] [PubMed] [Google Scholar]
- [21].Zhang CS, Hawley SA, Zong Y, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017. Aug 3;548(7665):112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018. Feb;19(2):121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chatel-Chaix L, Cortese M, Romero-Brey I, et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe. 2016. Sep 14;20(3):342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017. Apr;45:83–91. [DOI] [PubMed] [Google Scholar]
- [25].Matsuda N, Tanaka K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy. 2010. Oct;6(7):952–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Avia M, Rojas JM, Miorin L, et al. Virus-induced autophagic degradation of STAT2 as a mechanism for interferon signaling blockade. EMBO Rep. 2019. Nov 5;20(11):e48766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang L, Xu C, Johansen T, et al. SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy. 2021. Feb;17(2):593–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu C, Wang L, Fozouni P, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020. Oct;22(10):1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015. Nov 5;527(7576):105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen YY, Wang WH, Che L, et al. BNIP3L-Dependent mitophagy promotes HBx-Induced cancer stemness of hepatocellular carcinoma cells via glycolysis metabolism reprogramming. Cancers (Basel). 2020. Mar 11;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hirschey MD, Shimazu T, Huang JY, et al. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:267–277. [DOI] [PubMed] [Google Scholar]
- [32].Alhazzazi TY, Kamarajan P, Verdin E, et al. Sirtuin-3 (SIRT3) and the hallmarks of cancer. Genes Cancer. 2013. Mar;4(3–4):164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Alhazzazi TY, Kamarajan P, Verdin E, et al. SIRT3 and cancer: tumor promoter or suppressor? Biochim Biophys Acta. 2011. Aug;1816(1):80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015. Oct;15(10):608–624. [DOI] [PubMed] [Google Scholar]
- [35].Shi T, Wang F, Stieren E, et al. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005. Apr 8;280(14):13560–13567. [DOI] [PubMed] [Google Scholar]
- [36].Finley LW, Carracedo A, Lee J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011. Mar 8;19(3):416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang T, Cao Y, Zheng Q, et al. SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol Cell. 2019. Aug 22;75(4):823–834 e5. [DOI] [PubMed] [Google Scholar]
- [38].Ganar K, Das M, Sinha S, et al. Newcastle disease virus: current status and our understanding. Virus Res. 2014. May;12(184):71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tayeb S, Zakay-Rones Z, Panet A. Therapeutic potential of oncolytic newcastle disease virus: a critical review. Oncolytic Virother. 2015;4:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zamarin D, Palese P. Oncolytic newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol. 2012. Mar;7(3):347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Meng S, Zhou Z, Chen F, et al. Newcastle disease virus induces apoptosis in cisplatin-resistant human lung adenocarcinoma A549 cells in vitro and in vivo. Cancer Lett. 2012. Apr 1;317(1):56–64. [DOI] [PubMed] [Google Scholar]
- [42].Ren S, Ding C, Sun Y. Morphology remodeling and selective autophagy of intracellular organelles during viral infections. Int J Mol Sci. 2020. May 23;21(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li Y, Jiang W, Niu Q, et al. eIF2alpha-CHOP-BCl-2/JNK and IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of newcastle disease virus. Cell Death Dis. 2019. Nov 26;10(12):891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cheng JH, Sun YJ, Zhang FQ, et al. Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci Rep. 2016. Apr;21(6):24721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sun Y, Yu S, Ding N, et al. Autophagy benefits the replication of newcastle disease virus in chicken cells and tissues. J Virol. 2014. Jan;88(1):525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Santel A, Frank S. Shaping mitochondria: the complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life. 2008. Jul;60(7):448–455. [DOI] [PubMed] [Google Scholar]
- [47].Yu R, Liu T, Ning C, et al. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J Biol Chem. 2019. Nov 15;294(46):17262–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018. Jul;20(7):745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yu Y, Clippinger AJ, Pierciey FJ Jr., et al. Viruses and metabolism: alterations of glucose and glutamine metabolism mediated by human cytomegalovirus. Adv Virus Res. 2011;80:49–67. [DOI] [PubMed] [Google Scholar]
- [50].Levy P, Bartosch B. Metabolic reprogramming: a hallmark of viral oncogenesis. Oncogene. 2016. Aug 11;35(32):4155–4164. [DOI] [PubMed] [Google Scholar]
- [51].Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, et al. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009. Aug;9(9):1084–1101. [DOI] [PubMed] [Google Scholar]
- [52].Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006. Feb;9(2):121–132. [DOI] [PubMed] [Google Scholar]
- [53].Ren S, Rehman ZU, Shi M, et al. Syncytia generated by hemagglutinin-neuraminidase and fusion proteins of virulent newcastle disease virus induce complete autophagy by activating AMPK-mTORC1-ULK1 signaling>. Vet Microbiol. 2019. Mar;230:283–290. [DOI] [PubMed] [Google Scholar]
- [54].Meng C, Zhou Z, Jiang K, et al. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch Virol. 2012. Jun;157(6):1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017. Jun;20(86):193–224. [DOI] [PubMed] [Google Scholar]
- [56].Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019. Nov 15;366(6467):818–822. [DOI] [PubMed] [Google Scholar]
- [57].Yoboue ED, Valente EM. PINK1 and Parkin: the odd couple. Neurosci Res. 2020. May 15;159:25–33. [DOI] [PubMed] [Google Scholar]
- [58].Sugiura A, McLelland GL, Fon EA, et al. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014. Oct 1;33(19):2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].McWilliams TG, Ganley IG. Investigating mitophagy and mitochondrial morphology in vivo using mito-QC: a comprehensive guide. Methods Mol Biol. 2019;1880:621–642. [DOI] [PubMed] [Google Scholar]
- [60].Zu Y, Chen XF, Li Q, et al. PGC-1alpha activates SIRT3 to modulate cell proliferation and glycolytic metabolism in breast cancer. Neoplasma. 2021. Mar;68(2):352–361. [DOI] [PubMed] [Google Scholar]
- [61].Gladkova C, Maslen SL, Skehel JM, et al. Mechanism of parkin activation by PINK1. Nature. 2018. Jul;559(7714):410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yapa NMB, Lisnyak V, Reljic B, et al. Mitochondrial dynamics in health and disease. FEBS Lett. 2021. Apr;595(8):1184–1204. [DOI] [PubMed] [Google Scholar]
- [63].Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016. Feb 1;594(3):509–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ozawa S, Ueda S, Imamura H, et al. Glycolysis, but not Mitochondria, responsible for intracellular ATP distribution in cortical area of podocytes. Sci Rep. 2015. Dec;18(5):18575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009. May 22;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mahmoudabadi G, Milo R, Phillips R. Energetic cost of building a virus. Proc Natl Acad Sci U S A. 2017. May 30;114(22):E4324–E4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Seabright AP, Fine NHF, Barlow JP, et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020. May;34(5):6284–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhang CS, Lin SC. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016. Mar 8;23(3):399–401. [DOI] [PubMed] [Google Scholar]
- [69].Hu Y, Chen H, Zhang L, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021. May;17(5):1142–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sica V, Galluzzi L, Bravo-San Pedro JM, et al. Organelle-specific initiation of autophagy. Mol Cell. 2015. Aug 20;59(4):522–539. [DOI] [PubMed] [Google Scholar]
- [71].Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016. Jan 15;351(6270):275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wang X, Shen K, Wang J, et al. Hypoxic preconditioning combined with curcumin promotes cell survival and mitochondrial quality of bone marrow mesenchymal stem cells, and accelerates cutaneous wound healing via PGC-1alpha/SIRT3/HIF-1alpha signaling. Free Radic Biol Med. 2020. Nov 1;159:164–176. [DOI] [PubMed] [Google Scholar]
- [73].Yamada T, Dawson TM, Yanagawa T, et al. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy. 2019. Nov;15(11):2012–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sun Y, Zheng H, Yu S, et al. Newcastle disease virus v protein degrades mitochondrial antiviral signaling protein to inhibit host Type I interferon production via E3 ubiquitin ligase RNF5. J Virol. 2019. Sep 15;93(18):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Romer-Oberdorfer A, Werner O, Veits J, et al. Contribution of the length of the HN protein and the sequence of the F protein cleavage site to Newcastle disease virus pathogenicity. J Gen Virol. 2003. Nov;84(Pt 11):3121–3129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.