

Abstract
NMDA (N-methyl-d-aspartate) receptor antagonists are promising tools for the treatment of a wide variety of central nervous system impairments including major depressive disorder. We present here the activity optimization process of a biphenyl-based NMDA negative allosteric modulator (NAM) guided by free energy calculations, which led to a 100 times activity improvement (IC50 = 50 nM) compared to a hit compound identified in virtual screening. Preliminary calculation results suggest a low affinity for the human ether-a-go-go-related gene ion channel (hERG), a high affinity for which was earlier one of the main obstacles for the development of first-generation NMDA-receptor negative allosteric modulators. The docking study and the molecular dynamics calculations suggest a completely different binding mode (ifenprodil-like) compared to another biaryl-based NMDA NAM EVT-101.
The activity of biphenyl-based NMDA receptor allosteric modulator identified in virtual screening (IC50 = 5 μM) was improved 100 times (IC50 = 50 nM); SAR is consistent with ifenprodil analogs.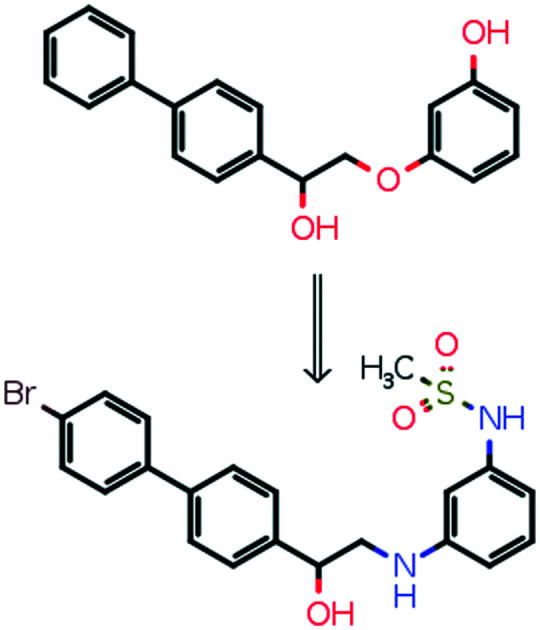
Introduction
NMDA receptors are heterotetrameric cationic channels activated by l-glutamate in the presence of glycine and play a key role in the control of the excitatory neurotransmission in the brain.1 These receptors, as well as other glutamate receptor types (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors), are involved in neural development, cell survival, and synaptic plasticity serving as a molecular basis for learning and memory formation. Localized postsynaptically, NMDA receptors are permeable for sodium and calcium ions upon activation causing membrane depolarization and, as a consequence, electric signal transduction.
The calcium permeation ability made these receptors an attractive target for the treatment of brain injury and neurodegenerative diseases. Overactivation of NMDA receptors by glutamate freed from damaged neurons leads to excessive calcium concentrations in a cell and launches apoptotic processes.2 This idea led to the development of a weak non-selective NMDA receptor channel blocker memantine as a drug for the treatment of stroke and Alzheimer's disease.3
NMDAR antagonists demonstrated rapid antidepressant effects in animal models, and FDA approved S-ketamine4 for treatment of major depressive disorder in 2019. One of the proposed mechanisms explains the antidepressant action by the direct blockage of NMDA receptors by ketamine which leads to inhibition of GABAergic inhibitory interneurons inducing the increase of the glutamate level in the prefrontal cortex.5 Unfortunately, ketamine being a dissociative anesthetic can cause side effects and has potential for abuse.
NMDA receptors similar to other glutamate receptors possess a modular architecture and are composed of an amino terminal domain (ATD), a ligand binding domain which binds to endogenous agonists, a transmembrane domain (TMD) which forms an ionic pore, and a regulatory C-terminal domain (CTD) which can be phosphorylated and can bind to extracellular effectors8 modulating the NMDA receptor activity (Fig. 1). Known NMDA-receptor subtypes consist of glycine binding subunits GluN1, GluN3A, and GluN3B as well as GluN2A-D subunits which are activated by glutamate. It should be noted that functional receptors should contain two GluN1 subunits and two subunits of other subtypes. The most abundant receptors in the adult forebrain are NMDARs containing the GluN2A and GluN2B subunits.9
Fig. 1. The architecture of the GluN1/GluN2B receptor complex. GluN1 and GluN2B subunits are shown in red and blue colors, respectively. The C-terminal domain is not shown due to its absence in the known structures. The figure was created using VMD 1.9.4 (ref. 6) based on Protein Data Bank (PDB) structure 4PE5.7.

GluN2B-containing receptors are of interest due to the unique binding site for allosteric modulators located on the interface between ATDs of GluN1 and GluN2B chains.10 It is not conserved in the other GluN2 subtypes and can provide selective inhibition of GluN2B-containing receptors. Ifenprodil (1) was the first ligand proved to bind to this site with high affinity. Firstly developed as an α-adrenergic receptor ligand10 it showed poor selectivity acting on 5-HT1A, 5-HT2, σ, NMDA receptors, and the hERG channel. The ifenprodil mechanism of action was studied in detail at a molecular level:11,12 ligand binding stimulates complex formation of an agonist bound LBD with an ATD preventing the conformational change in the TMD induced by the LBD. Ifenprodil demonstrated a neuroprotective,13 analgesic,14 and antidepressant15 effects in vivo. However, it served only as a lead compound for the development of more selective NMDA-receptor ligands.
Currently, a number of crystal structures of GluN1/GluN2B ATD complexes with various ligands have been published16,17 demonstrating at least two possible distinct binding modes for high-affinity NMDA receptor allosteric modulators. However, the ligands fill the hydrophobic binding pocket formed by Tyr109, Thr110 (GluN1) and Pro78, Ile111, and Phe114 (GluN2B) in all reported structures.16,17 Molecular dynamics simulations18 revealed that upon removal of a ligand from the binding site on the GluN1/GluN2B interface the GluN2B ATD undergoes an internal rotation between two lobes of the domain. These results were confirmed by an X-ray study12 demonstrating the “opening” and intersubunit rotation in GluN2B ATDs.
Plenty of allosteric modulators were developed for the ifenprodil-binding site (Fig. 2). The benzylpiperidine fragment of the ifenprodil is a source of additional off-target effects related to the action on aminergic receptors and, in addition, the basic nitrogen atom in this environment may significantly increase hERG channel inhibition.19 That is why compounds 2 and 320,21 with an improved NMDA receptor activity still have significant hERG blocking activity while compound 4 (ref. 22) does not show significant hERG inhibition. Efforts were also made to identify macrocycle-based ifenprodil analogs23 (e.g.5) which have high affinity for sigma receptors and a peptide-based ifenprodil analog.24
Fig. 2. Several chemotypes of the known GluN2B-selective allosteric modulators.

Compound 6 bearing a biaryl moiety was published as an NMDA receptor antagonist which competes with ifenprodil and has an unusual binding mode.15 At the same time, the docking pose of a biphenyl-containing compound 10a (identified during the virtual screening of the in-house virtual library containing about 30 000 compounds, Fig. 2) demonstrates high similarity to the ifenprodil binding mode. The virtual screening was performed with Autodock Vina with exhaustiveness equal to 10. The compound was selected for its ability to form a hydrogen bond with Glu236 and to fill the pocket formed by Tyr109, Thr110 (GluN1), Pro78, Ile111, and Phe114 (GluN2B) and its straightforward synthesis.25 Guided by molecular modeling results and published SARs for ifenprodil analogs, we managed to improve the inhibitory activity up to 50 nM (IC50). The potential antidepressant activity of compound 10h was evaluated in the Porsolt forced swimming test.
Results and discussion
Compounds 10a–j are accessible from commercially available α-bromoketones 7a and b which reacted with a variety of phenols 8a–c (Scheme 1) and anilines 8d–h (Scheme 2) to give the products of nucleophilic substitution 9a–i in good yields. Then the corresponding alcohols 10a–i were obtained from the ketones 9a–e, h and i by treatment with LiAlH4 (10a–c, Scheme 1) or NaBH4 (10d, e, h and i, Scheme 2). In the case of compound 10g the simultaneous reduction of the ketone and amide groups of 9g with NaBH4 proceeds in the presence of iodine which serves to initiate the reaction. Also, the Boc protecting group was removed under acidic conditions for compound 9e (Scheme 2). Compound 10j was synthesized by a two-step reductive amination process including the oxime formation and subsequent reduction with NaBH4 initiated by molecular iodine (Scheme 1).
Scheme 1. Synthetic path to compounds 10a–c and ja. aReagents and conditions: (a) K2CO3, MeOH, rt, 12 h; (b) LiAlH4, THF, reflux, 3 h; (c) (i) NH2OH·HCl, Py, rt, 48 h; (ii) NaBH4, I2, THF, reflux, 3 h.

Scheme 2. Synthetic path to compounds 10d–ia. aReagents and conditions: (a) K2CO3, MeOH, rt, 24 h; (b) TFA, DCM, rt, 24 h; (c) in the case of 9d, e, h and i: NaBH4, THF, H2O, rt, 3 h; in the case of 9g: NaBH4, I2, THF, reflux, 3 h.

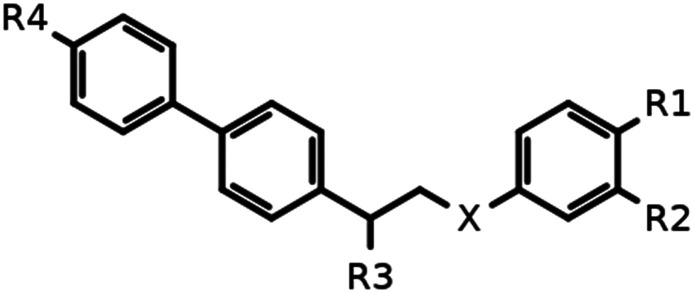
The goal of compound 10a SAR exploration was to prove that the inhibitory activity is caused by the binding to the ifenprodil binding site and realize which binding mode it adopts (ifenprodil-like or EVT-101-like, Fig. 3A). The results of the electrophysiological evaluation and radioligand studies for the set of synthesized ligands (10a–j) are summarized in Table 1. The radioligand binding results for the initially identified hit compound (10a) demonstrated a decreased ability to substitute ifenprodil compared to inhibition of NMDA/Gly-induced currents. But the subsequent SAR exploration led to correlated results between functional and radioligand assays. Further synthesized compounds which showed low activity in electrophysiological studies (10b–d and g) were also unable to substitute [H3]-ifenprodil, and Fig. 4A shows high correlation between the mentioned assays for compounds 10a, e, and h–j. Thus, based on these results we believe that NMDA receptor inhibition by compounds 10a–j is caused by the binding to the well-known ifenprodil binding site on the interface between GluN1 and GluN2B ATDs.
Fig. 3. The difference between the docking results of compounds 10a and c and a possible binding mode of compound 10i (S-isomer): (A) the superposition of docking poses for compounds 10a and c, the green color marks a hydrogen bond with Glu236 and magenta lines mark a distance in Ångström to the carbonyl oxygen of Ser132 (GluN1); (B) LIGPLOT37 representation of the 10i binding site, hydrogen bonds are shown by green dotted lines, residues having van der Waals contact with the ligand are represented by red arcs; (C) the comparison of binding modes of 10i, ifenprodil (pink, PDB: 5ewj) and EVT-101 (green, PDB: 5ewm); (D) residues forming the binding site, the blue color marks the residues from the GluN2B subunit and the red one indicates the residues from the GluN1 subunit, hydrogen bonds between protein and compound 10i are shown in green dashed lines.

Results of the radioligand and electrophysiological studies conducted for compounds 10a–j and ifenprodil.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | X | R1 | R2 | R3 | R4 | NMDA/Gly-induced current inhibition | [H3]-ifenprodil substitution IC50, μM | |
| IC50, μM | E max (%) | |||||||
| 10a | –O– | –H | –OH | –OH | –H | 5.0 ± 2.0 | 62 ± 5 | 30.0 ± 10.0 |
| 10b | –O– | –H | –H | –OH | –H | >30a | — | >100 |
| 10c | –O– | –OH | –H | –OH | –H | >30b | — | >100 |
| 10d | –NH– | –H | –H | –OH | –H | >30c | — | >100 |
| 10e | –NH– | –H | –OH | –OH | –H | 0.45 ± 0.12 | 72 ± 9 | 4.2 ± 0.9 |
| 10g | –NH– | –H | –NHC2H5 | –OH | –H | >30d | — | >100 |
| 10h | –NH– | –H | –NHSO2CH3 | –OH | –H | 0.1 ± 0.03 | 83 ± 6 | 5.4 ± 1.10 |
| 10i | –NH– | –H | –NHSO2CH3 | –OH | –Br | 0.05 ± 0.02 | 78 ± 7 | 1.0 ± 0.3 |
| 10j | –O– | –H | –OH | –NH2 | –H | 5.0 ± 2.0 | 65 ± 6 | 41.0 ± 6.4 |
| Ifenprodil | 0.5 ± 0.2e | 62 ± 2e | — | |||||
37 ± 10% inhibition at 30 μM.
36 ± 7% inhibition at 30 μM.
42 ± 9% inhibition at 30 μM.
10 ± 7% inhibition at 30 μM.
High affinity component.
Fig. 4. (A) The relationship between the results of the radioligand binding assay and the results of the electrophysiological studies; (B) the relationship between the computed MM–GBSA binding free energy and the results of the electrophysiological studies. Points are labeled with the compound numbers (Table 1).

It should be noted that the understanding of the correct binding mode for compound 10a was not a trivial task since compounds 10a–d can be docked like both ifenprodil and EVT-101 (see Fig. S1†). We designed analogs taking into account the previously published activity landscape of ifenprodil-like compounds.20–23 First, the elimination of the meta-hydroxy group from compound 10a leads to a dramatic activity decrease (about 10 times) for compound 10b comparable to the activity change when the phenolic hydroxyl group is removed from ifenprodil.10 Interestingly, the position change of the phenolic hydroxyl group in compound 10a from meta to para (10c) leads to an activity decrease while the opposite behavior is observed for the other ifenprodil-like compounds.26 This fact can be explained by the docking results where to maintain the correct hydrogen bond geometry the biphenyl moiety significantly changes its position (Fig. 3A) and the oxygen atom bound to the aryl fragment gets closer to the Ser132 carbonyl oxygen forming a close contact. The distance between these two oxygen atoms (2.97 Å) is slightly lower than a favorable van der Waals distance (3.04 Å). The increase in activity was caused by a substitution of oxygen to nitrogen (Table 1, atom labeled X) which is able to donate hydrogen bonds and is explained by the molecular docking results: the hydrogen atom bound to this nitrogen atom can form a hydrogen bond with the backbone carbonyl oxygen from Ser132 (GluN2B) as shown in Fig. 3B and D. It should be noted that the synergistic effect is observed when the previously described structural changes (compounds 10a and 10b; 10b and 10d) are applied simultaneously leading to compound 10e, which is as active as ifenprodil itself. Interestingly, compound 10g, which is closely related to active compound 10e, showed the lowest activity in this series. Probably, the ethyl group may weaken the hydrogen bond donating ability of the meta-amino group. Several studies of ifenprodil analogs26,27 suggest that the phenolic hydroxyl can be beneficially substituted to the alkylsulfonamide group and, in this way, the activity of compound 10h increases five times as compared to that of compound 10e. Taking into account the X-ray structure of compound EVT-101 in complex with GluN1/GluN2B ATDs the docking results of compound 10h (see docking results of 10i (Fig. 3B and D) which are nearly identical to those of 10h with a bromine atom changed to a hydrogen) demonstrated the overlap of distal phenyl rings, and this observation made us think that the para substitution in the unsubstituted phenyl ring of a biphenyl moiety can be beneficial for the activity. The introduction of a bromine atom in the para position which demonstrates a good overlap with the difluoromethyl group of EVT-101 (Fig. 3C) led to compound 10i, which is two times more active. It should be noted that the substitution of the hydroxyl group (R3, Table 1) to the amino group does not have any observable effect on the activity and, due to the possible increase of hERG inhibition, we did not synthesize any other derivatives with this type of substitution.
Following the docking results, we performed molecular dynamics simulations of all complexes and carried out MM–PBSA binding free energy calculations which have driven the chemical space exploration of the considered biphenyls. It should be noted that the studied compounds were tested as racemic mixtures and that is why each enantiomer has been docked with subsequent binding free energy estimation. The isomer with the highest score was selected for comparison in each pair of enantiomers. The computed binding free energy values for the R- and S-isomers for the studied compounds do not significantly differ and cannot show which enantiomer is preferable for high activity. The computed binding free energy values demonstrated a strong correlation with the logarithmically scaled results of the electrophysiological studies (the best Pearson correlation coefficient value was observed for the MM–GBSA approach (r = −0.82), Fig. 4B). However, the MM–PBSA approach often shows better behavior for larger datasets.28 These results allow us to explain the activity variation in the set of studied compounds by their ability to form hydrogen bonds with Glu236 (GluN2B) and Ser132 (GluN1), and to fill the hydrophobic pocket formed by amino acid residues including Tyr109 (GluN1) and Phe114 (GluN2B).
All studied compounds inhibited the stationary component of NMDA/Gly-induced currents in a concentration-dependent manner. It should be noted that we did not observe the full inhibition of the induced currents for any compound. Ifenprodil shows similar behavior under the same conditions29 with a bimodal inhibition curve. The ifenprodil bimodality is caused by binding to the high affinity site in ATDs and the low affinity site. This low-affinity inhibition demonstrates voltage dependency and can also be observed for the other NMDA receptor subtypes with the affinity from 30 to 75 μM.30Fig. 5A shows the change in agonist-induced stationary current upon the application of different concentrations of the inhibitor. The inhibition curve for compound 10h (Fig. 5B) does not show the bimodality up to 30 μM concentrations which is clearly visible for ifenprodil (Fig. 5B, red curve). Unfortunately, we could not check the larger concentrations because concentrations above 30 μM caused a strong destabilizing effect on the patch and did not allow us to record meaningful results but as this compound does not have positively charged groups it cannot bind to the low-affinity voltage-dependent site.
Fig. 5. (A) The results of the whole-cell current measurements for applications of three different concentrations (0.03, 0.3 and 3 μM) of compound 10h. (B) The concentration dependence curves of NMDA/Gly-induced current inhibition shown for compound 10h (circles) and ifenprodil (1) (triangles).

To explore the other possible binding modes for biaryl-based NMDA antagonists we performed an additional computational study for the most active compound 10i (similar to Dohrke et al.).31 Twenty top-ranked conformations obtained for each enantiomer of compound 10i in the thorough docking procedure (for detailed description see the ESI†) were selected and 25 ns molecular dynamics simulations were performed for each complex. The structure of the best complex in terms of the scoring function is provided in Fig. 3B and D. The biaryl moiety of the ligand is accommodated in a hydrophobic binding pocket formed by Tyr109 (GluN1) and Ile82, Ile111, Phe114, and Gln110 (GluN2B). The hydroxyl and amino groups of the ligand form hydrogen bonds with the backbone of Ser132 (GluN1) while the sulfonamide group forms a hydrogen bond with the sidechain carboxyl group of Glu236 (GluN2B). The performed principal component analysis (PCA) allowed three clusters of conformations to be separated (Fig. S1†): ifenprodil-like cluster, EVT-101-like cluster, and the other one containing all different conformations which demonstrated low docking scores. As a result of molecular dynamics simulations we computed the MM–GBSA binding free energy, the average number of hydrogen bonds between the ligand and protein, and the RMSD of the ligand for the last 5 ns of the trajectory compared to the first frame (Table S1†). We selected four conformations which demonstrated the computed MM–GBSA binding free energy below −50 kcal mol−1 (Fig. S2†) which were reasonably separated from all other conformations in terms of the MM–GBSA energy. The positions of biphenyl and mesylaminophenyl groups are almost identical for the first two best conformations which correspond to the R- and S-isomers of compound 10i (Fig. S2 (A and B)†). The other two conformations (Fig. S2 (C and D)†) which have slightly lower computed affinity compared to the two conformations described above share several structural features of the best conformations. One of them has absolutely the same position of the biphenyl fragment while the mesylaminophenyl group is turned around and the methanesulfonamide group is unable to form a hydrogen bond with Glu236. And the latter conformation we are going to describe is presented in Fig. S2 (D).† The position of the mesylaminophenyl group is exactly the same as that we observed for the best two conformations but the biphenyl moiety is not bound to the hydrophobic pocket near Phe114 (GluN2B) and Tyr109 (GluN1) which is filled by all known crystal structures of GluN1/GluN2B ATDs with ligands. This feature enhances the hydrogen bond formation ability between the ligand hydroxyl group and the backbone oxygen atom of Pro177 (Table S1†). Interestingly, no EVT-like conformation was observed among the top ranked simulation results and all described complex structures were stable.
Additionally, we computed several relevant pharmacokinetic properties including blood–brain barrier penetration (logBB)32 because the target is located in the brain, human intestinal absorption (HIA),33 and hERG34 pIC50. The calculation results for all the compounds (Table 2) demonstrated high intestinal absorption, and the logBB values for the most active compounds (10h and i) suggest only a five–six times decrease of the brain concentration compared to the plasma one. The predicted hERG activities fall in the micromolar range of concentrations. Thus, the prediction results demonstrate the plausible values of certain pharmacokinetic parameters relevant to CNS drugs. The results of predictive model application to memantine are added as a reference and demonstrate good prediction power.
The results of MM–PBSA binding energy calculations for compounds 10a–j and the estimation of several pharmacokinetic parameters by previously published QSAR models.
| Comp. | MM–GBSA ± Std. dev., kcal mol−1 | MM–PBSA ± Std. dev. (ε = 2), kcal mol−1 | MM–PBSA ± Std. dev. (ε = 4), kcal mol−1 | MM–PBSA ± Std. dev. (ε = 8), kcal mol−1 | logBBa | HIA,b % | hERG (pIC50)c | |
|---|---|---|---|---|---|---|---|---|
| 10a | R | −35.9 ± 2.8 | −6.8 ± 2.3 | −13.7 ± 2.8 | −17.6 ± 2.7 | −0.57 | 80 | 6.2 |
| S | −28.4 ± 1.8 | −7.1 ± 2.4 | −14.2 ± 2.0 | −17.9 ± 2.0 | ||||
| 10b | R | −31.7 ± 2.5 | −7.9 ± 3.7 | −14.3 ± 3.0 | −17.6 ± 2.8 | 0.05 | 90 | 6.3 |
| S | −35.1 ± 3.3 | −13.3 ± 2.7 | −18.9 ± 2.9 | −21.8 ± 3.1 | ||||
| 10c | R | −36.3 ± 3.0 | −4.8 ± 3.9 | −13.3 ± 3.5 | −17.6 ±3.5 | −0.25 | 76 | 6.4 |
| S | −37.9 ± 2.6 | −6.9 ± 3.2 | −13.7 ± 2.9 | −17.1 ± 2.9 | ||||
| 10d | R | −34.3 ± 2.5 | −8.8 ± 2.7 | −14.4 ± 2.4 | −17.0 ± 2.2 | 0.08 | 91 | 6.2 |
| S | −31.1 ± 2.4 | −9.1 ± 2.9 | −15.9 ± 2.7 | −19.4 ± 2.6 | ||||
| 10e | R | −35.6 ± 2.8 | −7.9 ± 2.8 | −13.8 ± 2.6 | −17.2 ± 2.5 | −0.61 | 81 | 6.0 |
| S | −39.5 ± 2.7 | −8.6 ± 2.7 | −16.2 ± 2.3 | −19.2 ± 2.4 | ||||
| 10g | R | −45.9 ± 3.0 | −10.2 ± 3.3 | −19.3 ± 2.5 | −23.4 ± 2.3 | 0.11 | 92 | 6.5 |
| S | −37.8 ± 3.3 | −7.0 ± 4.0 | −16.1 ± 3.4 | −20.6 ± 3.1 | ||||
| 10h | R | −53.8 ± 3.1 | −17.9 ± 3.2 | −24.0 ± 2.9 | −27.8 ± 2.9 | −0.77 | 99 | 5.8 |
| S | −51.2 ± 2.8 | −15.8 ± 3.2 | −23.3 ± 2.9 | −27.6 ± 2.8 | ||||
| 10i | R | −60.7 ± 3.4 | −18.9 ± 3.2 | −25.0 ± 2.9 | −28.0 ± 2.8 | −0.77 | 99 | 5.9 |
| S | −59.1 ± 3.0 | −20.3 ± 3.3 | −26.8 ± 2.8 | −30.3 ± 2.7 | ||||
| 10j | R | −32.1 ± 3.2 | −13.3 ± 3.9 | −20.3 ± 2.7 | −24.2 ± 2.3 | −0.27 | 86 | 6.5 |
| S | −31.5 ± 2.6 | −13.5 ± 2.9 | −18.4 ± 2.6 | −20.4 ± 2.7 | ||||
| Memantine | 0.55 (∼0)35 | 100 (∼100)35 | 4.5 (<5.0)35 | |||||
Recent literature data have suggested the possibility of a rapid antidepressant effect for GluN2B-selective NMDA antagonists.36 Therefore we performed a Porsolt forced swim test with fluoxetine as a positive control and two doses of compound 10h (1 mg kg−1 and 5 mg kg−1, Fig. S3†). Fluoxetine (160 mg kg−1) strongly decreased the immobility time in the positive control group, while a statistically significant difference was not observed for groups with 10h injected. Note that higher concentrations were not studied due to solubility issues. However, it still could be possible that the elevated dose and/or prolonged administration of the drug could result in a more prominent effect since the clinical effects of existing antidepressant drugs are also manifested after several weeks of treatment.
Conclusion
In conclusion, the structure–activity relationships of biphenyl-based ifenprodil analogs are reported for the first time. We provided the description of the structure–activity relationships explained by molecular modeling results which suggest a similarity to the ifenprodil binding mode in contrast to other literature-derived NMDA antagonists binding to the ifenprodil site and containing a biaryl moiety (EVT-101).
Abbreviations
- NMDAR
N-Methyl-d-aspartate receptors
- hERG
Human ether-a-go-go-related gene
- AMPAR
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors
- FDA
Food and Drug Administration
- MDD
Major depressive disorder
- ATD
Amino terminal domain
- LBD
Ligand binding domain
- TMD
Transmembrane domain
- CTD
C-terminal domain
- PDB
Protein Data Bank
- 5-HT1A
5-Hydroxytryptamine receptor 1A
- 5HT2
5-Hydroxytryptamine receptor 2
- SAR
Structure–activity relationships
- MM–GBSA
Molecular mechanics–generalized Born surface area
- MM–PBSA
Molecular mechanics–Poisson–Boltzmann surface area
- PCA
Principal component analysis
- RMSD
Root mean square deviation
- logBB
Logarithmic value of compound concentration in the brain divided by compound concentration in plasma with base 10
- HIA
Human intestinal absorption
- CNS
Central nervous system
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- QSAR
Quantitative structure–activity relationships
- THF
Tetrahydrofuran
- TFA
Trifluoroacetic acid
- Py
Pyridine
- Boc
tert-Butyloxycarbonyl protecting group
Author contributions
D. S. K., V. A. P., E. B. A., P. A. P., and M. V. F. wrote the manuscript and designed the compounds for synthesis; D. S. K. performed the molecular modeling; N. S. T. and D. A. V. performed the chemical synthesis; O. I. B., A. S. Z., and M. Y. D. carried out the electrophysiological studies; V. V. G. and A. V. G. conducted the radioligand studies; V. A. A. and N. V. G. performed the in vivo studies; Y. K. G. carried out the NMR spectra deciphering; S. V. O. and Y. I. K. performed the purity evaluation.
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was supported by the Russian Science Foundation (grant no. 18-75-00077). The authors thank Prof. Alexander V. Kurkin for the chemical library to perform virtual screening. Development of a general technique of NMDA-receptor negative modulator radioligand studies was supported by the Institute of Physiologically Active Compounds RAS State Targets, topic no. 0090_2019_0005.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00001f
References
- Traynelis S. F. Wollmuth L. P. McBain C. J. Menniti F. S. Vance K. M. Ogden K. K. Hansen K. B. Yuan H. Myers S. J. Dingledine R. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. J. Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Reisberg B. Doody R. Stöffler A. Schmitt F. Ferris S. Möbius H. J. N. Engl. J. Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Berman R. M. Cappiello A. Anand A. Oren D. A. Heninger G. R. Charney D. S. Krystal J. H. Biol. Psychiatry. 2000;47:351–354. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Moghaddam B. Adams B. Verma A. Daly D. J. Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W. Dalke A. Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Karakas E. Furukawa H. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman Z. A. Gakhar L. Sorensen B. R. Hell J. W. Shea M. A. Structure. 2007;15:1603–1617. doi: 10.1016/j.str.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K. B. Ogden K. K. Yuan H. Traynelis S. F. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. Jiang X. Zu Y. Yang Y. Liu Y. Sun X. Xu Z. Ding H. Zhao Q. Eur. J. Med. Chem. 2020;200:112447. doi: 10.1016/j.ejmech.2020.112447. [DOI] [PubMed] [Google Scholar]
- Zhu S. Stein R. A. Yoshioka C. Lee C.-H. Goehring A. Mchaourab H. S. Gouaux E. Cell. 2016;165:704–714. doi: 10.1016/j.cell.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima N. Karakas E. Grant T. Simorowski N. Diaz-Avalos R. Grigorieff N. Furukawa H. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley B. Canney M. Clune B. Caldwell M. Leonard B. E. Junien J.-L. Neurochem. Int. 1996;28:509–521. doi: 10.1016/0197-0186(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Bernardi M. Bertolini A. Szczawinska K. Genedani S. Eur. J. Pharmacol. 1996;298:51–55. doi: 10.1016/0014-2999(95)00778-4. [DOI] [PubMed] [Google Scholar]
- Poleszak E. Wośko S. Serefko A. Szopa A. Wlaź A. Szewczyk B. Nowak G. Wlaź P. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2013;46:29–35. doi: 10.1016/j.pnpbp.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Stroebel D. Buhl D. L. Knafels J. D. Chanda P. K. Green M. Sciabola S. Mony L. Paoletti P. Pandit J. Mol. Pharmacol. 2016;89:541–555. doi: 10.1124/mol.115.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan M. C. Zhu Z. Yuan H. Myers S. J. Menaldino D. S. Tahirovic Y. A. Liotta D. C. Traynelis S. F. Furukawa H. Nat. Commun. 2019;10:321. doi: 10.1038/s41467-019-08291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlov D. S. Radchenko E. V. Zefirov A. N. Palyulin V. A. Pentkovski V. M. Zefirov N. S. Biochem. Biophys. Res. Commun. 2012;424:687–690. doi: 10.1016/j.bbrc.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Kratz J. M. Schuster D. Edtbauer M. Saxena P. Mair C. E. Kirchebner J. Matuszczak B. Baburin I. Hering S. Rollinger J. M. J. Chem. Inf. Model. 2014;54:2887–2901. doi: 10.1021/ci5001955. [DOI] [PubMed] [Google Scholar]
- Tamiz A. P. Cai S. X. Zhou Z.-L. Yuen P.-W. Schelkun R. M. Whittemore E. R. Weber E. Woodward R. M. Keana J. F. W. J. Med. Chem. 1999;42:3412–3420. doi: 10.1021/jm990199u. [DOI] [PubMed] [Google Scholar]
- Borza I. Kolok S. Gere A. Nagy J. Fodor L. Galgóczy K. Fetter J. Bertha F. Ágai B. Horváth C. Farkas S. Domány G. Bioorg. Med. Chem. Lett. 2006;16:4638–4640. doi: 10.1016/j.bmcl.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Garner R. Gopalakrishnan S. McCauley J. A. Bednar R. A. Gaul S. L. Mosser S. D. Kiss L. Lynch J. J. Patel S. Fandozzi C. Lagrutta A. Briscoe R. Liverton N. J. Paterson B. M. Vornov J. J. Mazhari R. Pharmacol. Res. Perspect. 2015;3:e00198. doi: 10.1002/prp2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister S. Schepmann D. Wünsch B. Bioorg. Med. Chem. 2020;28:115245. doi: 10.1016/j.bmc.2019.115245. [DOI] [PubMed] [Google Scholar]
- Karlov D. S. Barygin O. I. Dron M. Y. Palyulin V. A. Grigoriev V. V. Fedorov M. V. SAR QSAR Environ. Res. 2019;30:683–695. doi: 10.1080/1062936X.2019.1653965. [DOI] [Google Scholar]
- Temnyakova N. S. Vasilenko D. A. Barygin O. I. Dron M. Y. Averina E. B. Grishin Y. K. Grigoriev V. V. Palyulin V. A. Fedorov M. V. Karlov D. S. Mendeleev Commun. 2020;30:342–343. doi: 10.1016/j.mencom.2020.05.027. [DOI] [Google Scholar]
- Tahirovic Y. A. Geballe M. Gruszecka-Kowalik E. Myers S. J. Lyuboslavsky P. Le P. French A. Irier H. Choi W. Easterling K. Yuan H. Wilson L. J. Kotloski R. McNamara J. O. Dingledine R. Liotta D. C. Traynelis S. F. Snyder J. P. J. Med. Chem. 2008;51:5506–5521. doi: 10.1021/jm8002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. L. Gregory T. F. Bigge C. F. Boxer P. A. Serpa K. Meltzer L. T. Wise L. D. Cai S. X. Hawkinson J. E. Konkoy C. S. Whittemore E. R. Woodward R. M. Zhou Z. L. J. Med. Chem. 1999;42:2469–2477. doi: 10.1021/jm990148x. [DOI] [PubMed] [Google Scholar]
- Karlov D. S. Lavrov M. I. Palyulin V. A. Zefirov N. S. J. Biomol. Struct. Dyn. 2018;36:2508–2516. doi: 10.1080/07391102.2017.1360208. [DOI] [PubMed] [Google Scholar]
- Kew J. N. C. Richards J. G. Mutel V. Kemp J. A. J. Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S. D. Daggett L. P. Deal C. Lu C.-C. Johnson E. C. Veliçelebi G. J. Neurochem. 1998;70:1269–1279. doi: 10.1046/j.1471-4159.1998.70031269.x. [DOI] [PubMed] [Google Scholar]
- Dohrke J.-N. Watson J. F. Birchal K. Greger I. H. J. Biol. Chem. 2020;295:14565–14577. doi: 10.1074/jbc.RA120.014135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyabina A. S. Radchenko E. V. Palyulin V. A. Zefirov N. S. Dokl. Biochem. Biophys. 2016;470:371–374. doi: 10.1134/S1607672916050173. [DOI] [PubMed] [Google Scholar]
- Radchenko E. V. Dyabina A. S. Palyulin V. A. Zefirov N. S. Russ. Chem. Bull. 2016;65:576–580. doi: 10.1007/s11172-016-1340-0. [DOI] [Google Scholar]
- Radchenko E. V. Rulev Y. A. Safanyaev A. Y. Palyulin V. A. Zefirov N. S. Dokl. Biochem. Biophys. 2017;473:128–131. doi: 10.1134/S1607672917020107. [DOI] [PubMed] [Google Scholar]
- https://www.lundbeck.com/upload/ca/en/files/pdf/pm/Ebixa.pdf (accessed December 27, 2021)
- Garner R. Gopalakrishnan S. McCauley J. A. Bednar R. A. Gaul S. L. Mosser S. D. Kiss L. Lynch J. J. Patel S. Fandozzi C. Lagrutta A. Briscoe R. Liverton N. J. Paterson B. M. Vornov J. J. Mazhari R. Pharmacol. Res. Perspect. 2015;3:e00198. doi: 10.1002/prp2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A. Swindells M. B. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.